Week-06-hw-genetic-circuits-part-i
DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
- Phusion DNA Polymerase: Pyrococcus-like enzyme that contains a fused processivity-enhancing domain. It provides more than 50 gold higher fidelity than Taq polymerase.
- dNTPs: contains dATP, dCTP, dGTP, and dTTP that are required for extension reaction of the PCR.
- Buffers: MgCl2 as a cofactor for polymerase, KCl and TAPS-HCl ([tris(hydroxymethyl)methylamino]propanesulfonic acid) to maintain ionic strength and pH respectively, and beta-meracaptoethanol to maintain enzyme stability.
- Some other components that are provided seperately: DMSO (Dimethyl sulfoxide) to improve denaturation and primer binding, and nuclease free water as a solvent and matrix to avoid denaturation of the DNA.
What are some factors that determine primer annealing temperature during PCR?
Primer melting temperature: annealing temperature must be set around 3 to 5 degree celcius below the lowest melting temperature. So. anything that affects melting temperature also affects annealing temperature. Melting temperature is in turn affected by GC conent and primer length. Higher the GC content, and longer the length of the primer, the higher will be the melting temperature. For short primers (<20 bps) Wallace rule can be used to find the approximate primer melting temperature:
$$T_m (°C) = 2(A + T) + 4(G + C)$$Salt and ion concentration: monovalent cations like Na+ and K+ reduce the repulsion between two DNA strands by nutralizing the negative charge of the phosphate backbone. Mg2+ concentration, which is a cofactor for the polymerase, also increases the stability of the double helix, increasing the melting temperature.
Presence of Denaturants like DMSO and Formamide: They disrupt hydrogen bonds, and reduce the melting temperature.
Degenarate primers (primers that are not 100% match to the template) reduce the melting temperature, and complexity of template DNAs (Eg.; humans as opposed to bacteria) also require a higher annealing temperature to avoid ‘mispriming’.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR vs. Restriction Enzyme Digest
| Feature | Polymerase Chain Reaction (PCR) | Restriction Enzyme Digest (e.g., DpnI) |
|---|---|---|
| Mechanism | Uses primers and DNA polymerase to amplify specific target regions from a template. | Uses site-specific endonucleases to cleave DNA at specific recognition sequences. |
| Input DNA | Requires a template plasmid (e.g., mUAV) and synthetic oligonucleotides (primers). | Requires DNA containing specific recognition sites (e.g., methylated GATC for DpnI). |
| Protocol Steps | Thermal cycling: Includes initial denaturation, followed by cycles of denaturation, annealing, and extension. | Isothermal incubation: Typically a single-step incubation at a specific temperature (e.g., 37°C for 30–60 minutes). |
| Modifications | Can introduce intentional mismatches for mutagenesis (e.g., amilCP color mutations). | Precise cutting only; cannot “create” new sequences or mutations during the digest. |
| Selectivity | Amplifies only the region of interest flanked by the forward and reverse primers. | Selectively digests DNA based on sequence and methylation status (e.g., removing parental templates). |
When to Prefer PCR vs. Restriction Digest
| Situation | Prefer PCR | Prefer Restriction Enzyme Digests (e.g., DpnI) |
|---|---|---|
| Creating Mutants | You need to change the color-generating chromophore of a protein by introducing mismatches. | |
| Preparing for Assembly | You are generating linear fragments for Gibson or HiFi assembly that require specific 5’ overhangs. | |
| Targeted Amplification | You need to isolate specific regions like the origin of replication, promoters, or antibiotic resistance genes. | |
| Eliminating Background | You need to remove the original template DNA (mUAV) to ensure only the newly created PCR mutants are used. | |
| Targeting Methylation | You need to distinguish between DNA propagated in E. coli (methylated) and DNA synthesized via PCR (unmethylated). |
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Verification of Primer Design: The success of the assembly depends on the specific architecture of the primers:
Overlaps: Each primer must include a 20–22 bp overhang complementary to the adjoining fragment.
Binding Region: The core binding region should be 18–22 bp.
Melting Temperature (Tₘ): The Tₘ should be between 52–58°C.
Pair Compatibility: Primer pairs should have Tₘ values within 5°C of each other.
GC Content: Aim for 40–60% GC content with a GC clamp (1–2 G/C bases) at the 3′ end.
Secondary Structure: Use software to ensure Gibbs free energy is above –10 kcal to avoid strong hairpins or dimers.
- Post-PCR Processing Before assembly, fragments must be cleaned and templates removed:
DpnI Digestion: Treat PCR reactions with DpnI to eliminate the original mUAV plasmid (digests methylated DNA, preserves unmethylated PCR products).
DNA Purification: Use a purification kit (e.g., Zymo) to remove salts and enzymes.
Quantification: Measure DNA concentration with Nanodrop or Qubit; should be >30 ng/µL.
- Quality Control (Diagnostic Gel)
Run samples on an agarose gel at 100 mV for 15 minutes.
Verify that bands match the predicted size calculated on Benchling.
- Reaction Parameters
Molar Ratio: Use a 2:1 (insert:vector) molar ratio for optimal efficiency.
Orientation: Confirm fragments have correct 5′ → 3′ orientation with matching overlaps.
- How does the plasmid DNA enter the E. coli cells during transformation?
It enters through pores in the cell wall. The pores can be created using CaCl2 treatment, followed by heat shock (mixture kept on ice bath is suddenly incubated at 42 degree celcius for 30-90 seconds). Electroporation is another method, where a high-voltage electric pulse applied for a very short duration brefily disrupts the phospholipid bilayer, and simultaneously pushes the DNA molecules through the pores.
- How does the plasmid DNA enter the E. coli cells during transformation?
6.1 Describe another assembly method in detail (such as Golden Gate Assembly). Explain the method in 5–7 sentences plus diagrams (either handmade or online).
Modular Cloning Method: It is a method based on Golden Gate Assembly. It utilized Type IIS restiction enzymes that cut outside their restrcition site and create non-palindromic overhangs. The final product doesn’t contatin restriciton site, preventing the enyme from double-cutting.
Steps:
Step 0: Removal of the internal recognition sites so that the enzyme being used will not cleave it internally, addition of standard 4-bp overhangs and inserting the thus-modified sequence into storage vector. This has to be done seperately for all the units of transcription, i.e., promoter, 5’ UTRs, rbs, cds, terminator.
Step 1: The components of step 0 are added into the reaction vessel, along with the destination vector, restriciton enzyme, T4 ligase, buffer, and ATP. The temperature is cylced to and fro from a higher temperature (~37 degree celcius) for cutting, and a lower temperature (~16 degree celcius) for sticking. The restriction enzyme leaves behind the specific 4-bp overhangs. The DNA ligase binds the 4-bp overhangs in the order of Promoter -> 5’ UTR -> RBS -> CDS -> Terminator in the insertion site of the destination vector, which already contains selection and screening genes.
Step 2: In case a complex metabolic pathway involving multiple genes is to be synthesised, the final desitnation vector of the step 1 is used as storage vector for the step 2, and step 1 is repeated using other genes.
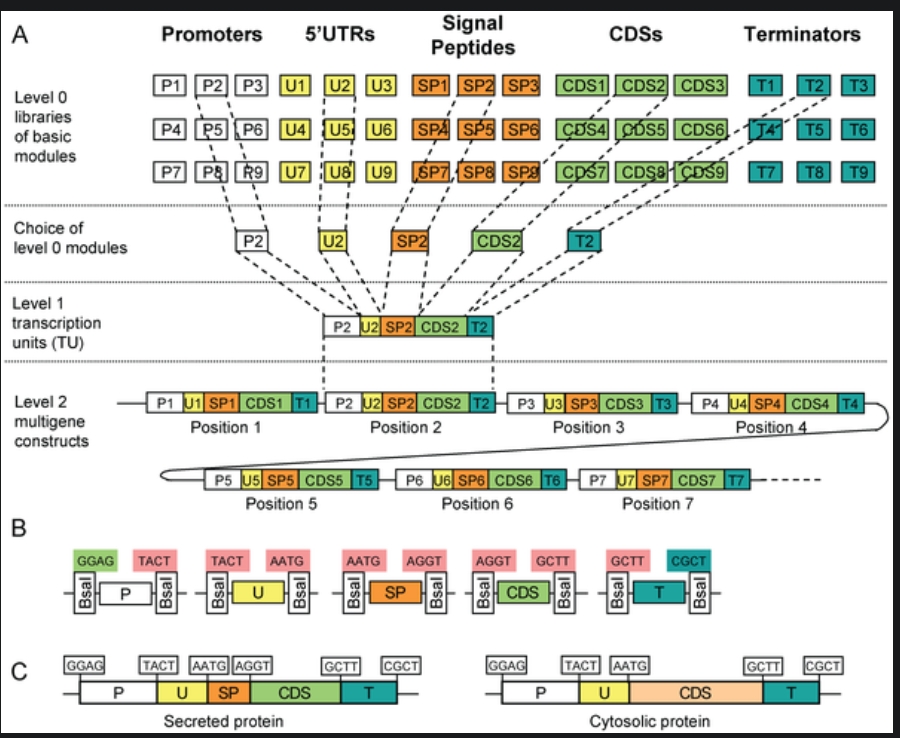 (Credit: https://www.addgene.org/cloning/moclo/)
(Credit: https://www.addgene.org/cloning/moclo/)
6.2 Model this assembly method with Benchling or a similar tool!
I got the following error.
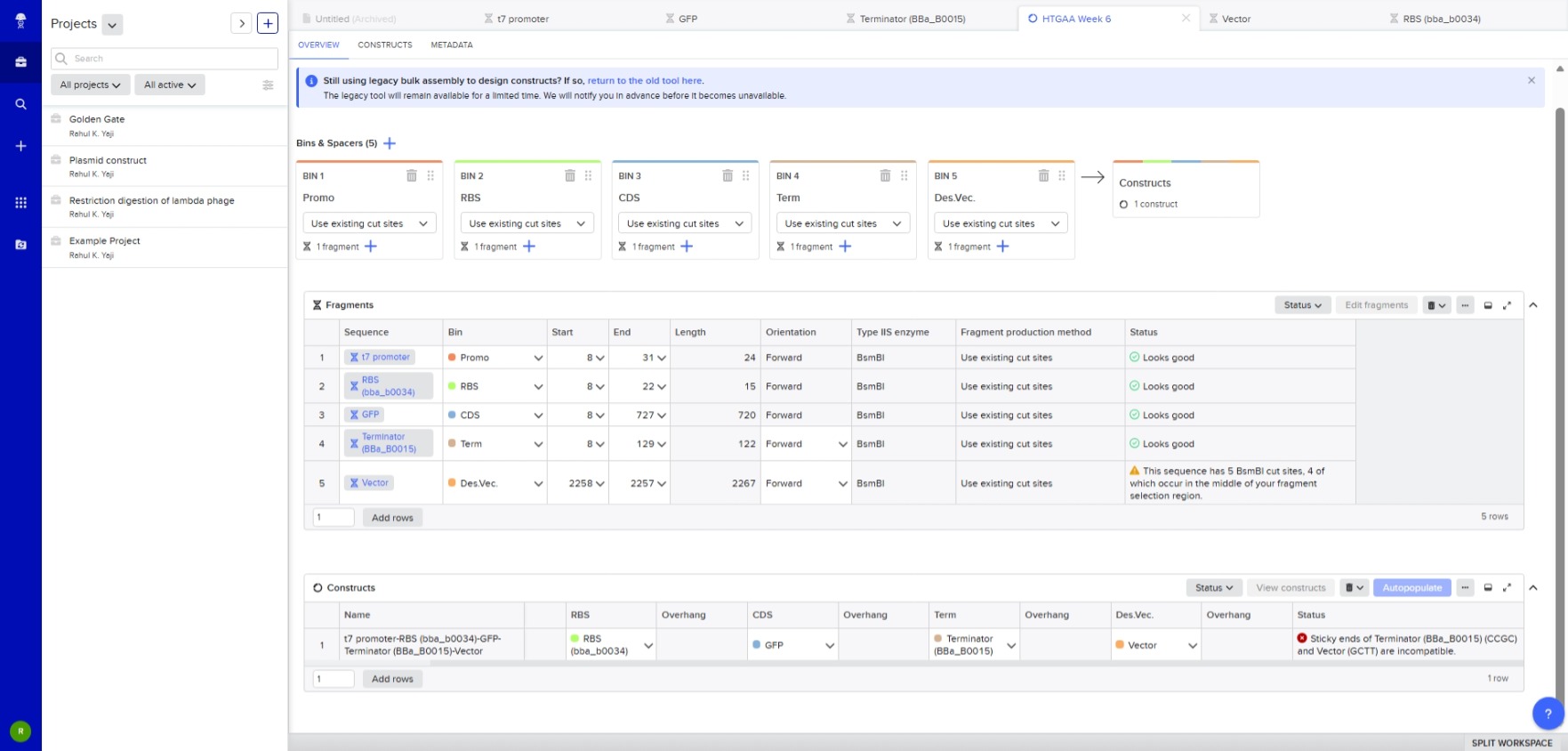
Asimov Kernel
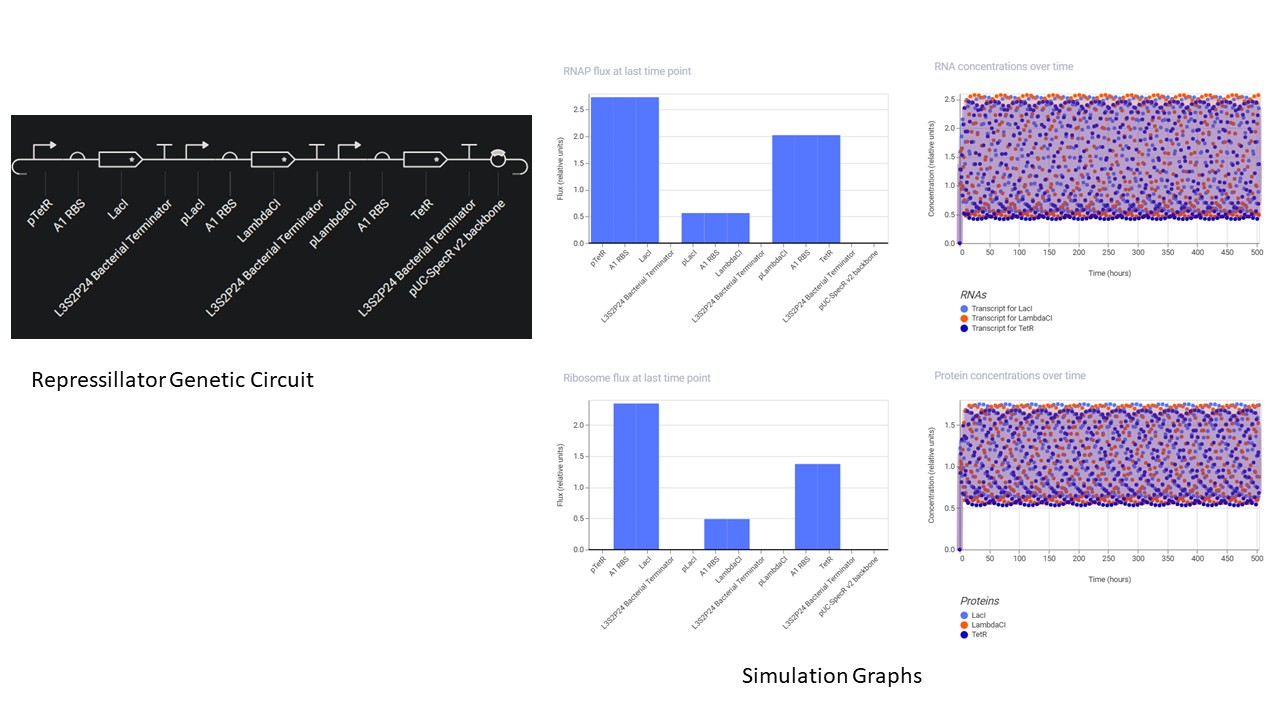
Construct and simulate the repressillitaor.
- A Repository was created using the “New” button.
- A Notebook was created using the same button to document the homework.
- In the notebook, a blank construct was created, the repressillator found in the Demo was recreated part by part.
- “Search bar” was used to search for the parts, and they were dragged and dropped at the desired location
- Using the simulation option, the repressillator was simulated using the following parameters:
- Chassis: E. coli
- Duration: 504 hours
- Timestep: 60 minutes
- Transfection: Transient Transfection
- The following output was recorded:
Build three of your own devices using the parts in the Characterized Bacterial Parts Repo and explain how you think the devices should function in an Electronic Notebook Entry.
First Part: Overexpression of lactic acid- The nucleotide sequence of the Ldh gene was copied from NCBI, and the start sequence ATG and Stop sequence TAG were manually inserted in the “Create part” option
- T7 promoter and terminator were used by creating new parts with the respective sequences taken from Vector Builder
- The effect of increasing the number of copies of gene was simulated using the parameters:
- Chassis: E. coli
- Duration: 24 hours
- Timestep: 10 minutes
- Transfection: Transient Transfection.
- It was found that the more the gene copies, the higher the protein levels.
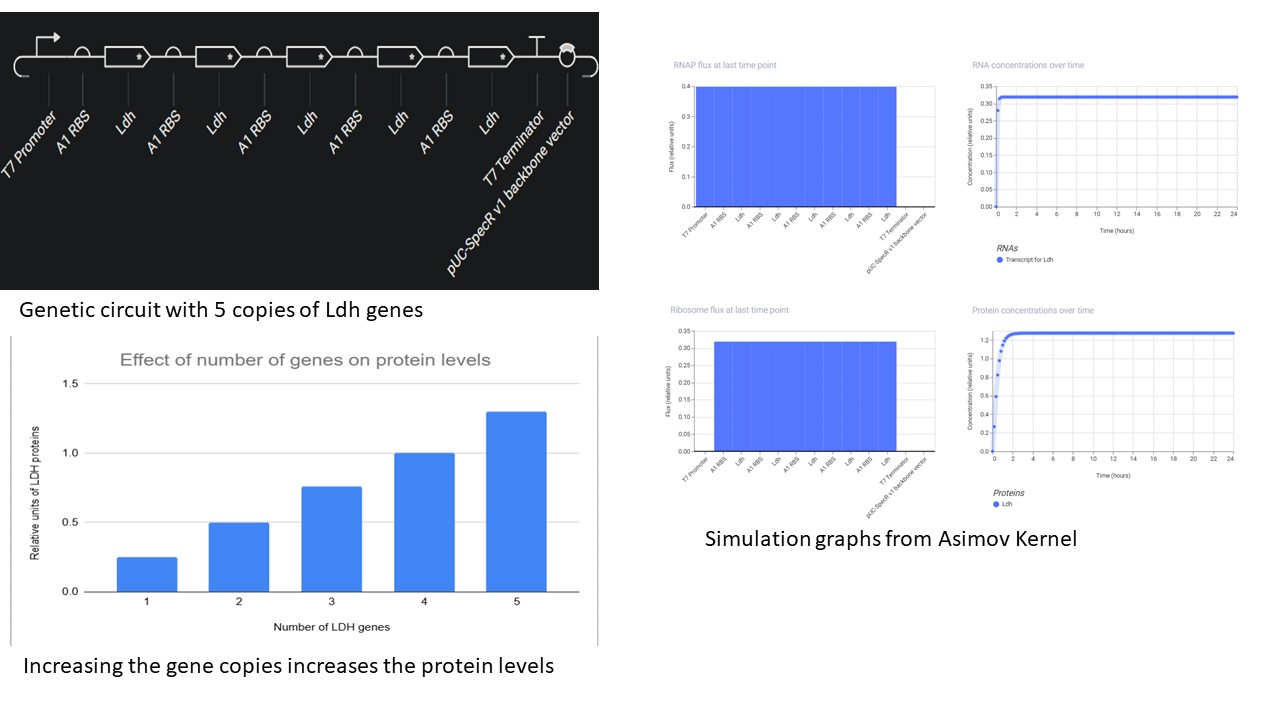
- Another interesting observation was that, the CDS must be followed by an RBS for each copy, even if they are flanked by the same promoter and terminator. Without RBS, it will not be translated and thus the protein levels stay down.
Second Part: Inducerless NOT Gate
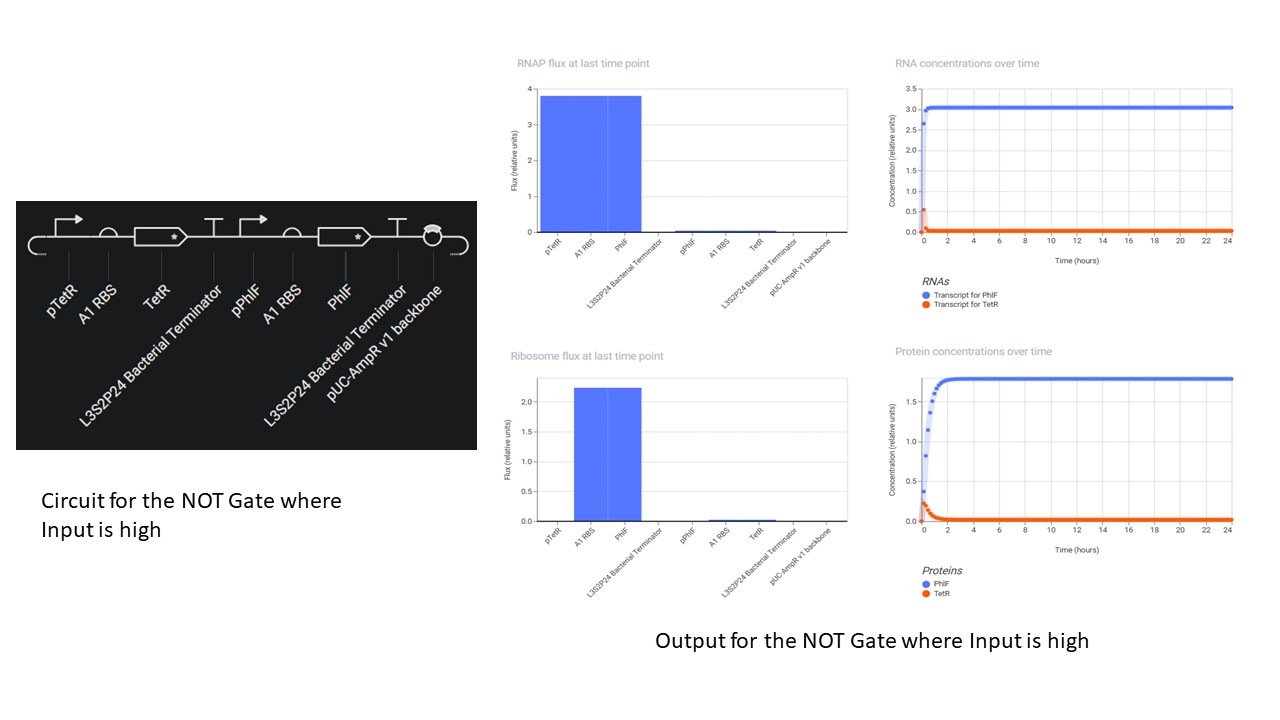
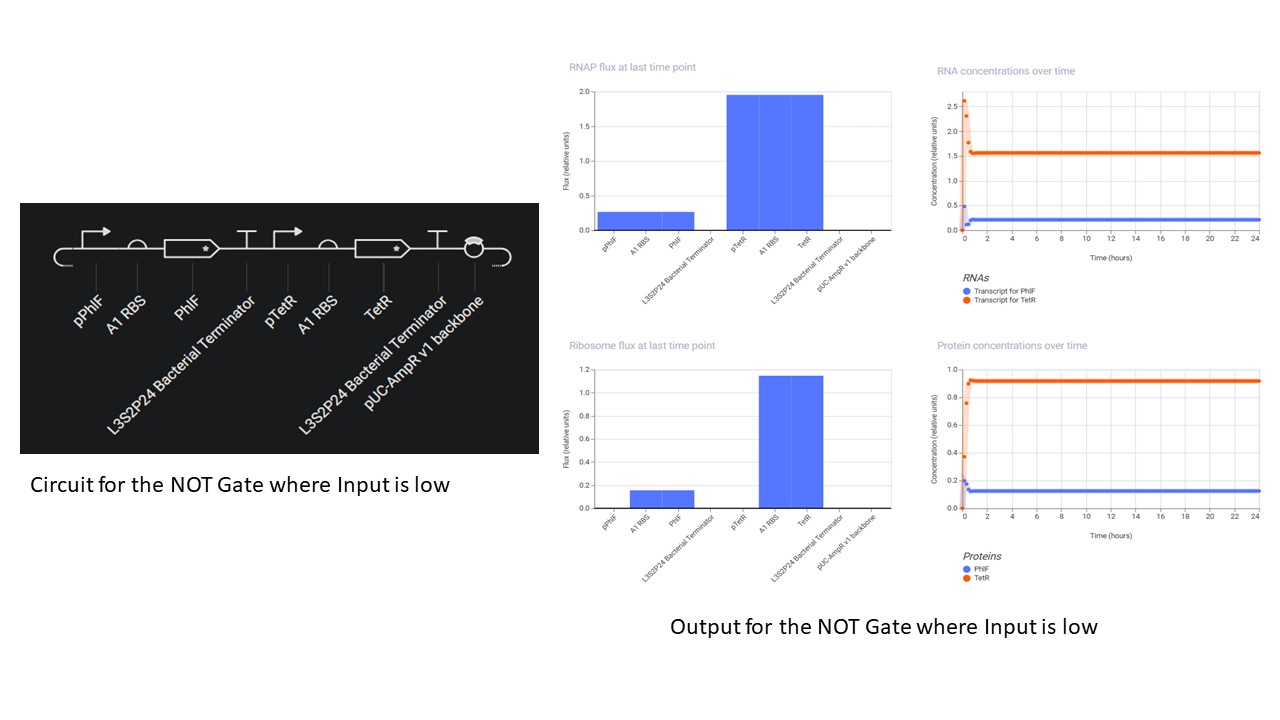
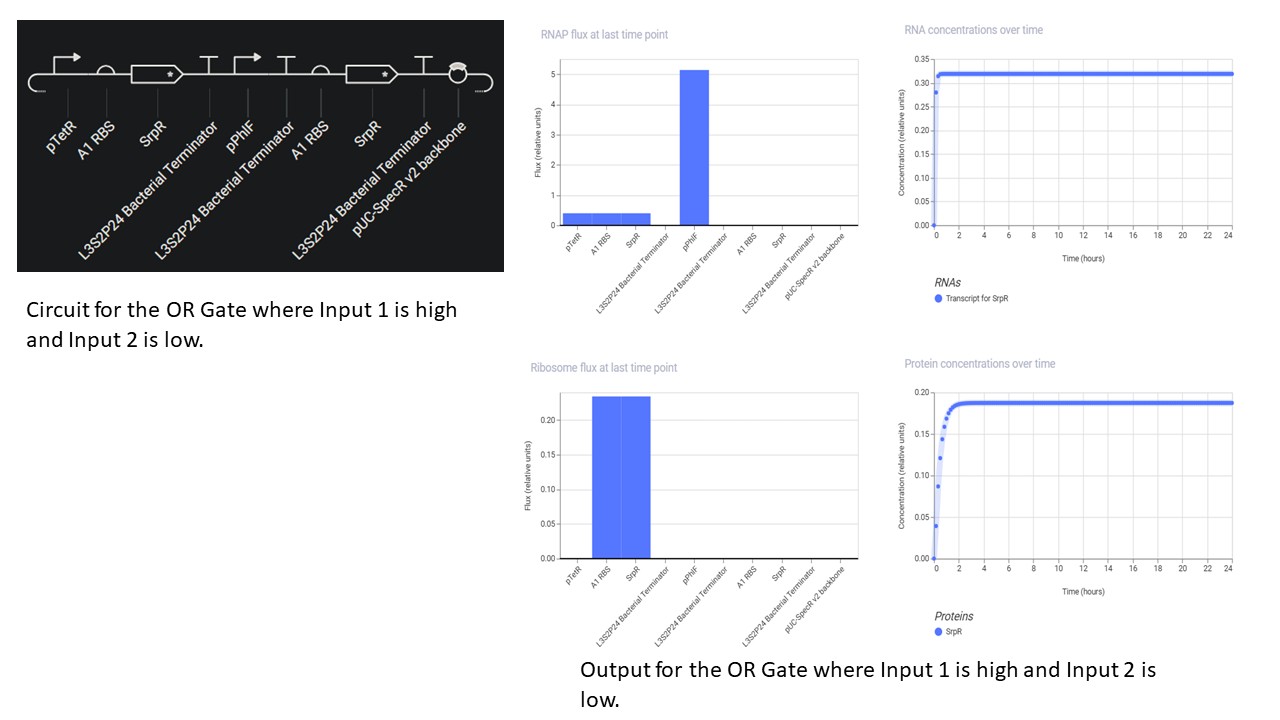
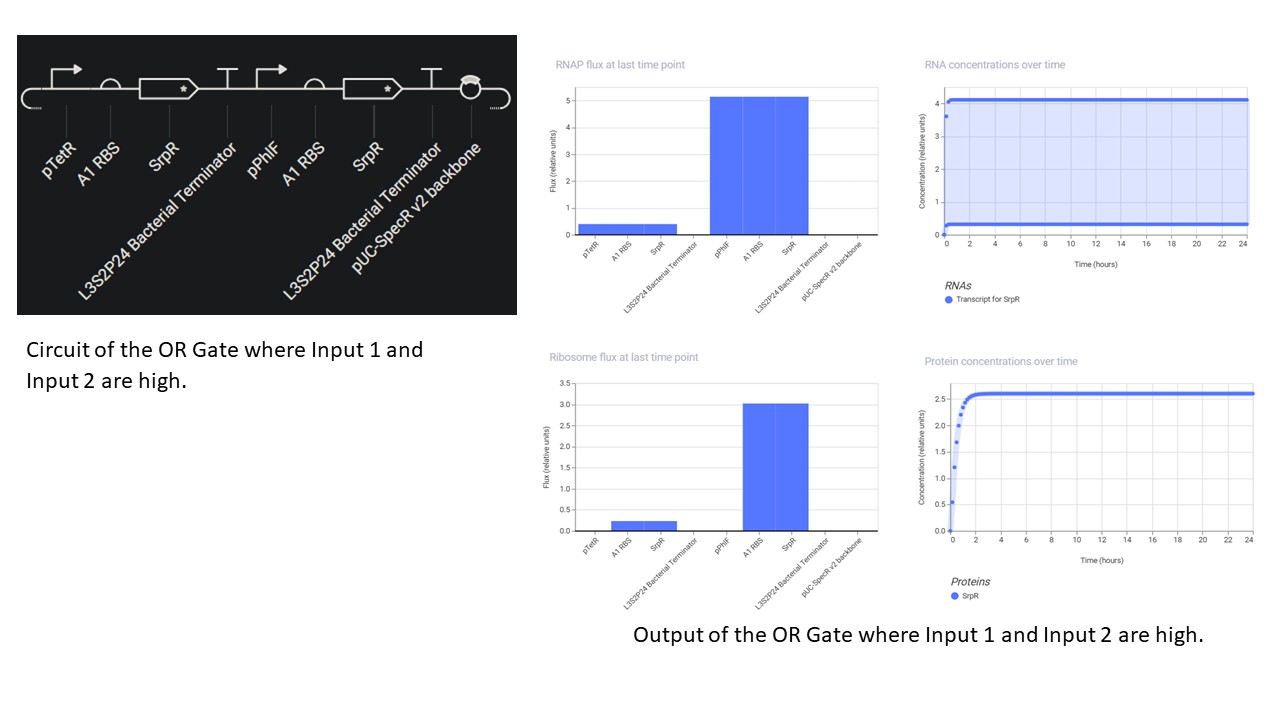
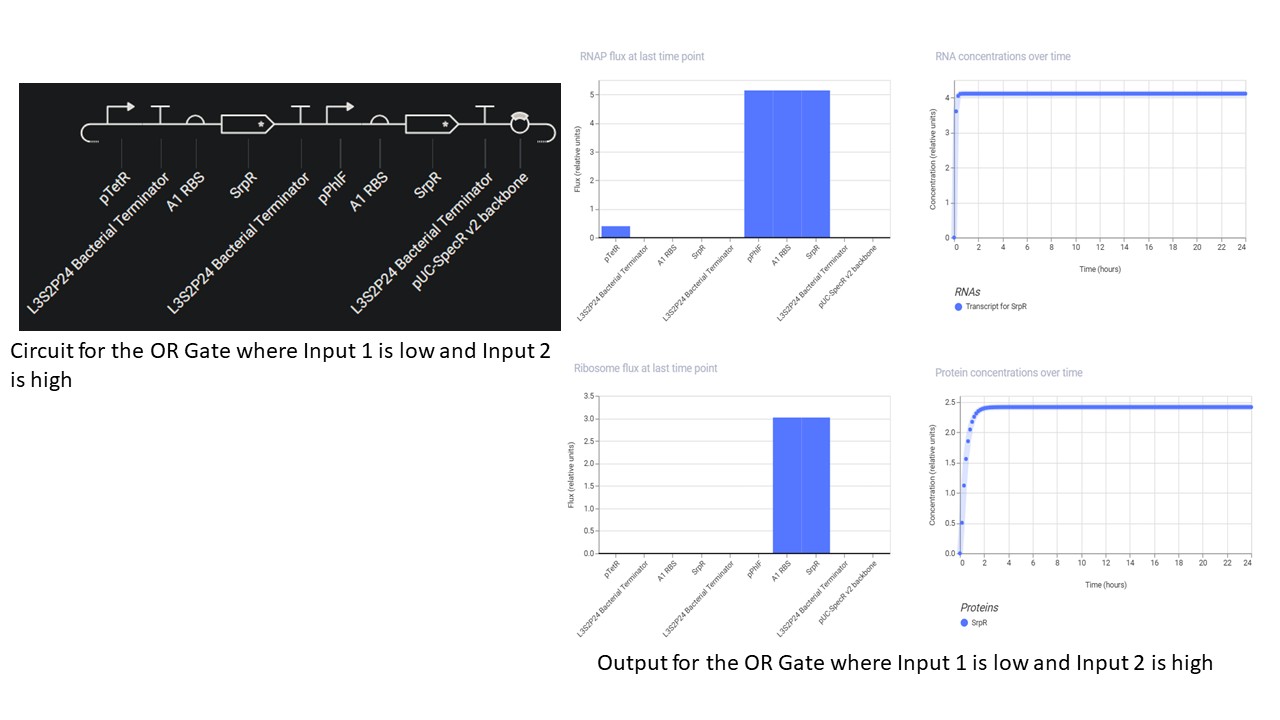
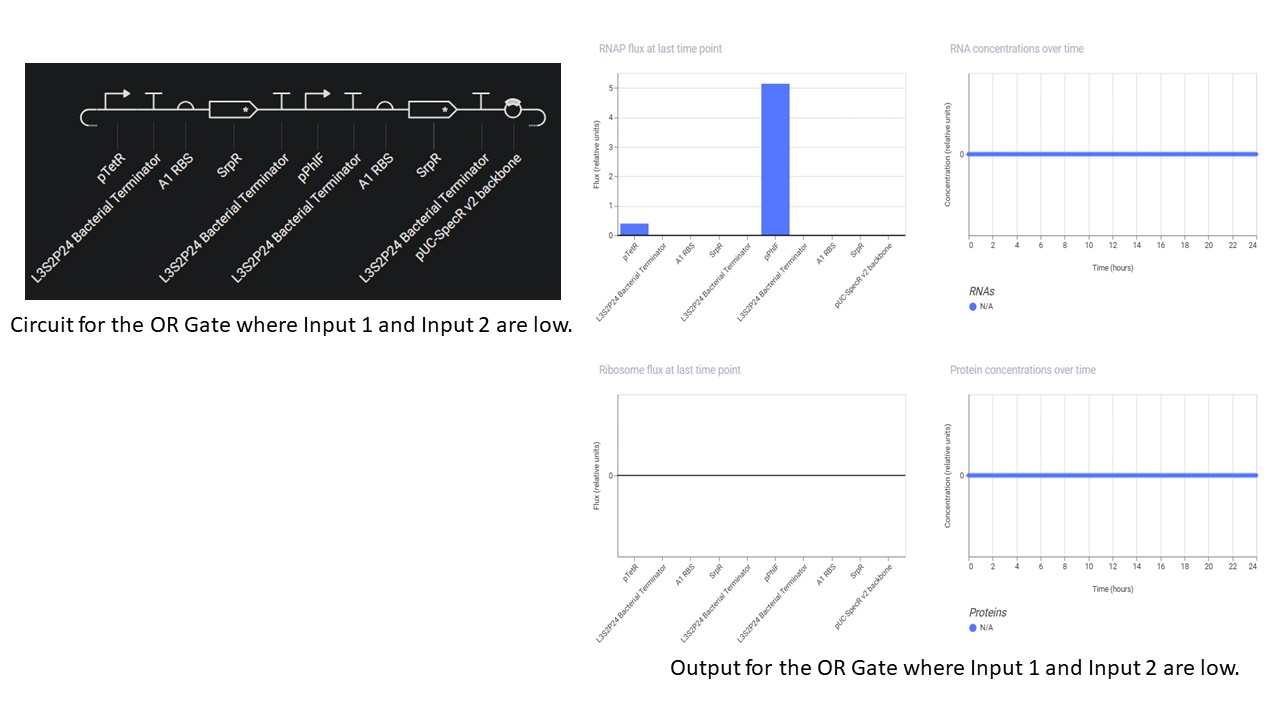
Third Part: Inducerless OR Gate