Greetings! I am Rahul Yaji, from Karnataka, India. My interest in extraterrestrial habitation drew me towards biotechnnology and I completed my graduation from NMAM Institute of Technology, with a major in biotechnology engineering. My curriculum was a mix of biosciences as well as engineering topics on bioreactor design, modelling reactions and simulations. My areas of interest include synthetic biology, metabolic engineering, and biomanufacturing.
A biological engineering application or tool I want to develop and why:
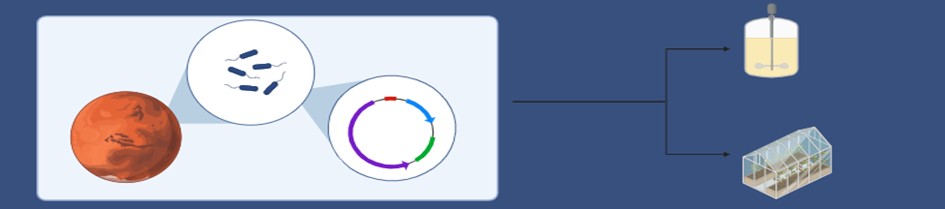
I want to develop an engineered consortium of microorganisms for pilot-scale biomanufacturing on Mars. The microbes will be engineered for self-sufficent surival subject to the multitude of constraints of the red planet. This insitu resource utilization (ISRU) will be a key step towards the goal of the eventual colonization of Mars, by reducing the import from Earth. The current methods of ISRU, although in their rudimentary stages, rely on high energy chemical conversion process. My application aims at providing an alternative to this, and pave way for sustainable biomanufacturing away from the Earth.
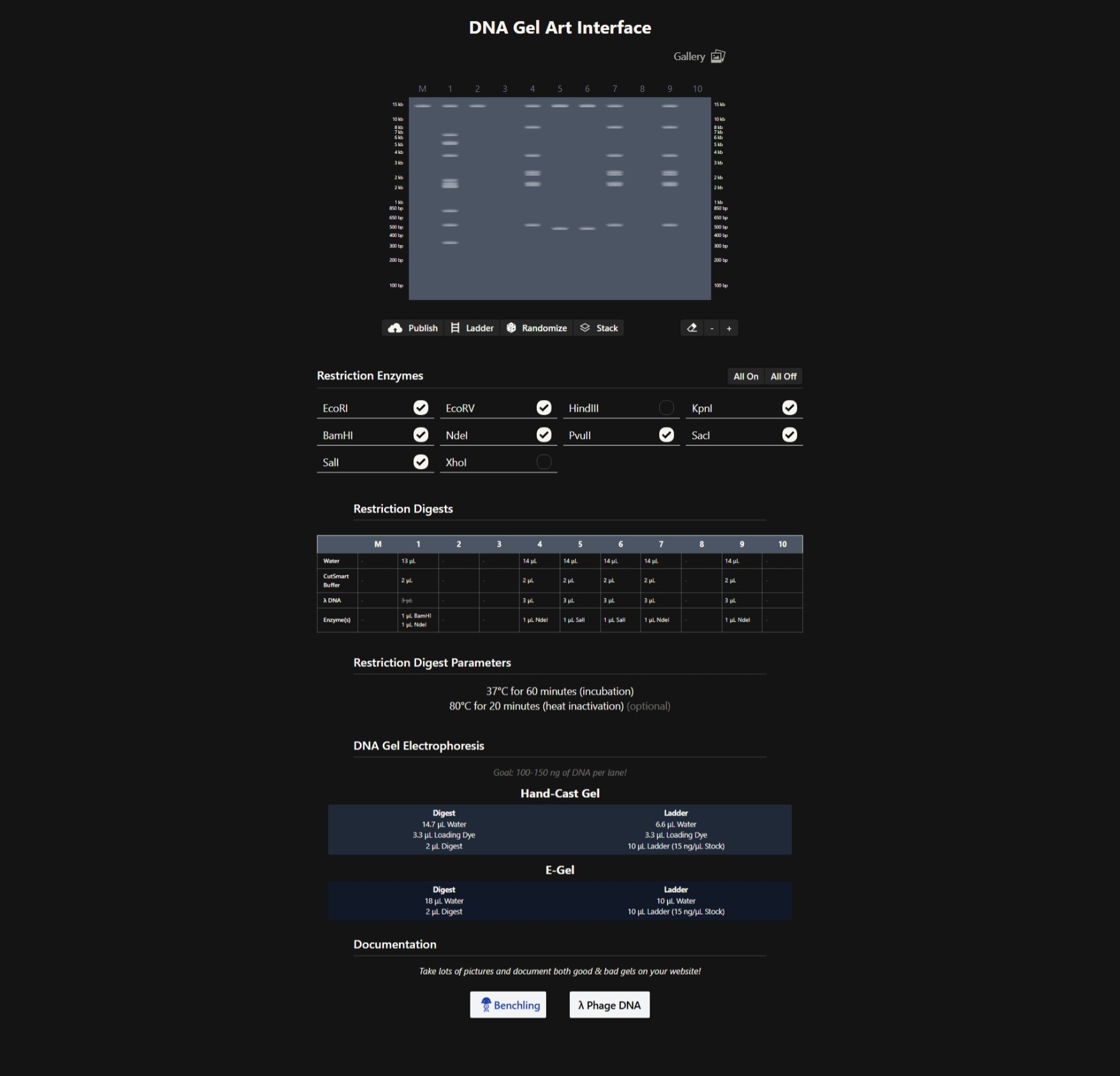
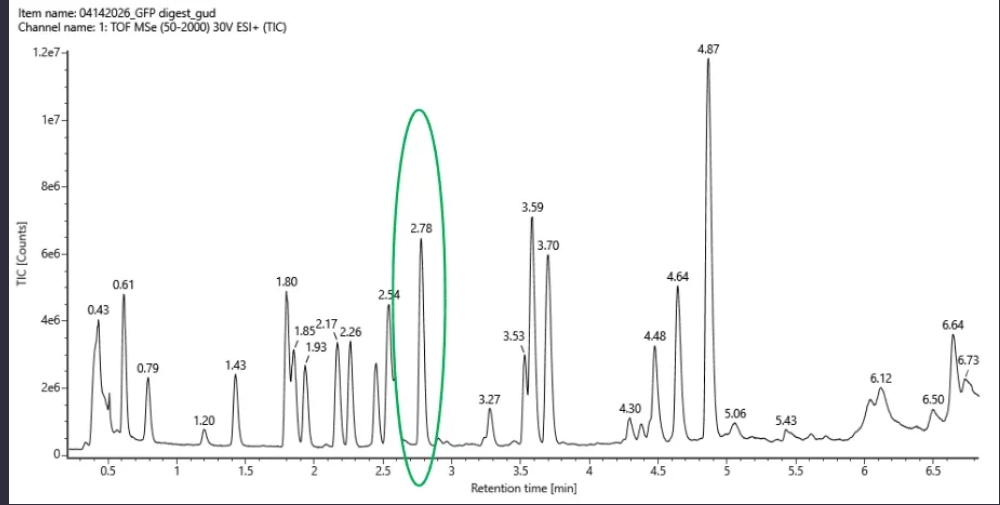
Part 1: Benchling & In-silico Gel Art 1.1 Restriction Digestion Simulation in Benchling: 1.2 DNA Gel Art Using Automation Art: Part 2: Laboratory Work on Gel Electrophoresis Skipped due to lack of access to lab.
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications. The paper: Slowpoke:An Automated Golden Gate Cloning Workflow for Opentrons OT‑2 and Flex
Part A. Conceptual Questions Answer any NINE of the following:
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) Assume that the mass of other components like fat, collagen etc. are negligible compared to proteins in meat.
Average wight of 1 molecule of amino acid = 100 Dalton = 1.7 * $10^{-24}$ g
Weight of piece of meat = 5 * $10^{2}$ g
Therefore, number of amino acid molecules = (5 * $10^{2}$) / 1.7 * $10^{-24}$ = 2.94 * $10^{26}$
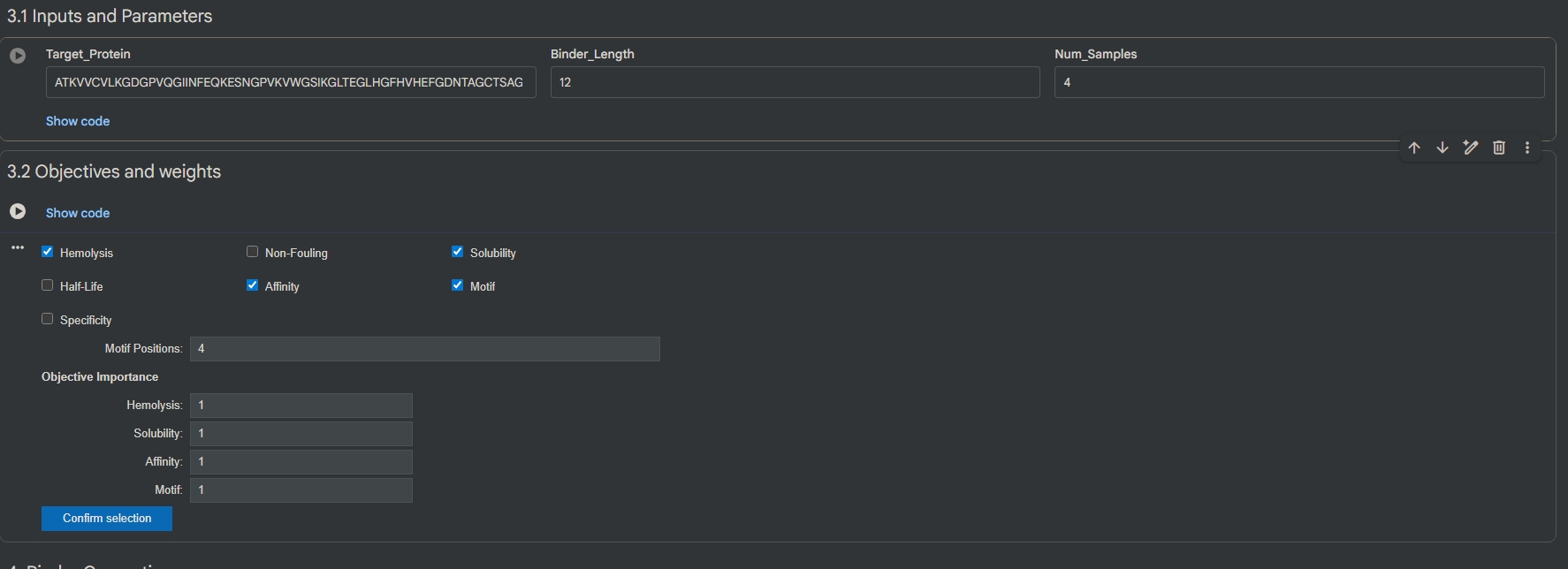
Part 1: Generate Binders with PepMLM Retrieve sequence and introduce mutation: (Pasted the sequence from UniPort, deleted M at 1st position, changed A to V at 4th position.) ATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Structure of the native sequence- predicted vs actual:
Generate 4 peptides using PepMLM Colab: index Binder Pseudo Perplexity 1 WRSPAVAVAHWE 7.76721411356481 2 WRVGWVGVELKE 24.2058244561383 3 WRSPAAXIEHKX 11.243453670563373 4 WRVYAAXIEWGK 20.449723821548965 Known binder: FLYRWLPSRRGG Perplexity score: 22.5252 A note about perplexity score: A key evaluation metric for language models that measures how well a probability model predicts a sample. Lower the score, higher the confidence of the model that the output satisfies the criteria.
DNA Assembly What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion DNA Polymerase: Pyrococcus-like enzyme that contains a fused processivity-enhancing domain. It provides more than 50 gold higher fidelity than Taq polymerase. dNTPs: contains dATP, dCTP, dGTP, and dTTP that are required for extension reaction of the PCR. Buffers: MgCl2 as a cofactor for polymerase, KCl and TAPS-HCl ([tris(hydroxymethyl)methylamino]propanesulfonic acid) to maintain ionic strength and pH respectively, and beta-meracaptoethanol to maintain enzyme stability. Some other components that are provided seperately: DMSO (Dimethyl sulfoxide) to improve denaturation and primer binding, and nuclease free water as a solvent and matrix to avoid denaturation of the DNA. What are some factors that determine primer annealing temperature during PCR?
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
They can interpret a range of inputs as opposed to the 0, 1 inputs of traditional genetic circuits. This allows them to aggregate multiple signals and apply the activation fucntion to filter biological noise. Traditional circuits often require a cascade of genetic logic gates, which lead to metabolic burden and competition for substrates. By utilizing weighted interactions, IANNs can accomplish the same task using fewer biolocial components. Nonlinear descision making is a struggle for tradional genetic circuits. They struggle to take into accout the relative ratios and thresholds of a multitude of proteins simultaneously, limiting themselves to simple linear logics. However, using ReLU and sigmoid -like activation behaviours, IANNs can perform complex tasks. Eg: A cell may be engineered to apoptosize only when a commplex profile of cancer markers are met, as oppossed to the presence of some of those markers that may not be cancerous. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
A useful applicaiton of IANN would be rapid plant cell response when it is infected by a pathogen.
Part A: General & Lecturer-Specific Questions General Homework Questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables.
Rapid Iteration and Throughput
Direct Use of Linear DNA Templates
Traditional methods require time-consuming cloning of DNA into circular plasmids before they can be inserted into a host cell.
In CFPS, you can use raw PCR products directly as the instruction manual, allowing you to move from a genetic design to a functional protein in just a few hours.
Homework: Final Project 1. Identify at least one aspect of your project that you will measure.
Answer:
The expression level of the L lactate dehydrogenase Gene The concentration of lactic acid Cell growth 2. What technologies will you use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry)?
Answer: RNA sequencing, RT qPCR, and OD600 etc.
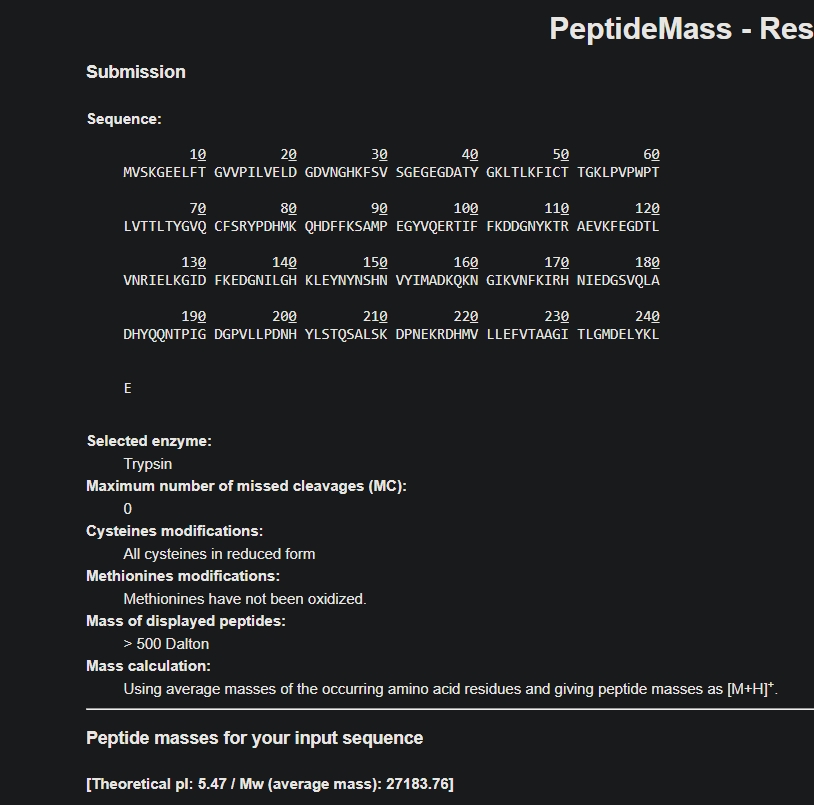
Homework: Waters Part 1 — Molecular Weight 1. Based on the predicted amino acid sequence of eGFP, what is the calculated molecular weight?
Answer: 27183.76 kda (after removing the H tag)
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork 1. Contribute at least one pixel to the global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST. 2. On your HTGAA webpage, note: 1. What you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”). Contriubted 4 different fluorscent proteins in the bottom of the art.
Subsections of Homework
Week 1 HW: Principles and Practices
1. A biological engineering application or tool I want to develop and why: I want to develop an engineered consortium of microorganisms for pilot-scale biomanufacturing on Mars. The microbes will be engineered for self-sufficent surival subject to the multitude of constraints of the red planet.
This insitu resource utilization (ISRU) will be a key step towards the goal of the eventual colonization of Mars, by reducing the import from Earth. The current methods of ISRU, although in their rudimentary stages, rely on high energy chemical conversion process. My application aims at providing an alternative to this, and pave way for sustainable biomanufacturing away from the Earth.
2. Governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm):
Goal 1- Prevention of forward contamination: Great care must be ensured in making sure that only the right microorganisms will colonize the desired niche. Since this may very well fit into the definition of ‘forward contamination,’ a thorough conformation of the non-existence of native Martian microbes shall guide the policy decision. International collaboration is going to be of prominence, because this goal will be of no consequence if even one of the space-capable nation refuses to abide by this. Goal 2- Address dual use concerns: It is inevitable that any microbe that has been engineered to tolerate Martian conditions would have multiple survival mechanisms that grant it an upper hand over its Earthen coutnerparts.Therefore, any type of microorganism that may even remotely prove to be pathogenic to humans must be avoided at all costs. Goal 3- Level playing fields: Monopolies and oligopolies should be prevented to the largest possible extent, especially in the early days of the settlement plan. If such imbalanced playing fields get established, it will stiffle innovations for generations to come by restricting know-hows and resources.
3. Next, describe at least three different potential governance “actions” by considering the four aspects below: 3.1 Technical Goverenance: The policy must ensure that whoever wants to set up biomanufacturing on Mars has suitably demonstrated the presence of kill switches (auxotrophic, toxin-anti-toxin etc.) to prevent accidental release into the environment. Completely orthogonal biological systems may be used in place of kill switches, but given today’s biotechnology, the former is more likely than the latter. Purpose: To prevent forward contamination. Design: Genetic circuits can be embedded with toxic-anti-toxic systems like CcdB-CcdA, MazF-MazE, and hok-soc etc. Strains auxotrophic for Glucosamine-6-phosphate Synthase ((\Delta glmS)) can be used as auxotrophic chassis organisms. Assumptions: The assumptions here would be that the strain will not bypass these kill-switches by any means, and also these kill-switches will not interefere with the organisms’ ability to synthesize the product of interest. Risks of Failure & Success: Failure to meet these parameters may lead to forward contamination, preventing the study of ‘pristine’ Martian grounds. However, the success in this context would not be permanent and require repeated peroidic demonstrations. There is also the possibility of false trigerring of kill-switch, leading to a wasted batch of products.
3.2 Regulatory Governance: A system to inventory and track all the organisms, genetic components, and manufacturing methods becomes important. This will provide a starting point to study the evolution of the microorganisms that might arise in the future. A high degree of match to the inventoried parts can help rule out any fasle-positivity regarding native Martian microbe claims. Purpose: To track any suspicious new microbes in the vicinity and beyond. Design: A robust inventory software, and the adherence of the players to documentation. Assumptions: All the players will abide by the regulations, and will not send any undocumented organisms to gain a competitive edge. Risks of Failure & Success: Failure would mean lots of undocumented and potentially unsafe microorganisms on Mars. It would also prevent any means of studying weather Mars had evolved any microbes independent of the Earth. On the other hand, a policy that is too transparent will hinder intellectual property safeguard.
3.3 Economic Incentive Governance: For this, I envision a system of “Biosecurity Bonds.” Any entity that wants to carry out biotechnology research on Mars would need to furnish a bond of a certain amount (probably in millions of dollars). If, after a period of time, no contamination can be established, the amount is refuned. If any contamination is found, the bonded amount can be utilized to ameliorate the spread. Purpose: To incentivize players to adhere to high standards of biosecurity. Design: A techno-legal framework in the form of an international treaty or agreement, among all the spacce-faring nations and also similar incentives at national level. Assumptions: None of the players will take this bond as an opportunity to “pay to pollute” and think that forfeiting the bond amount is cheaper than adhereing to the standards of biosecurity. Risks of Failure & Success: Failure can lead to an incentiveless, haphazard business models, that would aim towards establishing monopolies for profit. If this aspect is successfully governed, then there is still the risk of wealthy corporations outcompeting the not-so-wealthy ones.
4. Score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against the rubric of policy goals.
Does the option:
Option 1
Option 2
Option 3
Planetary Protection (forward)
1
3
3
• By preventing incidents
1
2
1
• By helping respond
1
3
1
Biosafety
1
3
1
• By preventing incident
1
3
1
• By helping respond
1
3
1
Redundancy and backup plans
1
2
1
• By preventing incidents
1
3
2
• By helping respond
3
1
1
Other considerations
• Minimizing costs and burdens to stakeholders
2
3
3
• Economic feasibility
3
2
3
• Not impede research
2
3
3
• Promote constructive applications
1
1
1
5. Drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties. Based on these parameters, I would priortize option 1, i.e, Technical Governance, and option 2, Economic Incentive Governance. Both of these would go hand in hand to cover the technical and the financial safeguards agianst the forward contamination, establishment of monopolies, and an imbalanced playing fields. However, the main trade off in not prioritizing option 2, i.e., regulatory governing would be the existence of loopholes to evade accountability. The uncertainty of non-adhering players will always remain as a looming threat in establishing a stable policy towards extraterrestrial resource utilization.
Homework questions from Dr. LeProust:
What’s the most commonly used method for oligo synthesis currently? Solid phase phosphoaramidite method is the most widely used method to synthesize oligonucleotides. Nucleoside phosphoramidites are used as the precursor molecules. It proceeds through 4 steps:
Detritylation: dimethoxytrityl group is removed from the 5’ end of the last nucleotide attached to the support using triacetic acid, activating the -OH group.
Coupling: Phosphoaramidite monomers are added along with an activator (usualy tetrazole), that protanates the phosphoaramidite. Now, the 5’ hydroxyl end of the growing chain can form a phosphite triester linkage at the 3’ phosphorous.
Oxidation: The unstable phosphite triester linkage is oxidized using iodine solution form a stable phosphate triester bond.
Capping: Once the required number of nucleotides have been synthesised using the above 3 steps, the unreacted 5’ ends are capped using an acetylation mix of acetic anhydride and N-methylimidazole. This is done to prevent wrong reactions in further cycles.
Why is it difficult to make oligos longer than 200nt via direct synthesis? If oligos are synthesized using phosphoaramidite method, the yield follows the equation Y = $C^{n}$; where Y is the yield %, C is the coupling efficiency, and n is the number of couplings. A diakósiamer (200mer) will have 199 couplings. This implies, even with a success rate of 99%, the yield would be $0.99^{199}$, which is around 13.5%. The rest of the sequences would be truncated at random lenghts less than 200 bps.
Why can’t you make a 2000bp gene via direct oligo synthesis? Using the same equation as above, we get the yield of only 1.88 * $10^{-7}$ percent, which is as good probability as nil in order to synthesize a 2 kb gene.
Homework Questions from Professor Jacobson:
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy? DNA polymerarases are accurate with upto $10^{−6}$ mutations/bp. Since human genome is around 3.2 * $10^{9}$ bp long, it would imply 3200 mutations per generation. Biology deals with this descrepancy by having a multitude of proofreading mechanisms like 3’-5’exonuclease activity in the polymerase that cleaves incorrect nucleotides, mismatch repair post replication where a protein complexes can recognize the template strand and the newly-synthesized strand due to the presence of nicks in the latter, and cleave the ‘wrong’ base pairs. Then, DNA ligase joins the correct nulceotides.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest? Considering an average protein to be 375 amino acids long, and each amino acid requiring 3 codons, there can be $3^{375}$ DNA sequences for an average protein. But in reality, the number of translatable codon is limited by the properties of mRNA and the availability of tRNA. Certain DNA sequenes transcript into an mRNA that will have haripin loop, tendency to form dsRNA and other difficult-to-translate structures. And also, the translational machinery possesses a limited number of tRNA, which is the limiting factor for the number of amino acids that can be translated, and thus protein that can be synthesized.
Homework Questions from George Church:
What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”? The nutritionally essential amino acids in all animals are:
Cystine, Leucine, Lysine, Methionine, Histidine, Phenylalanin, Tyrosine, Threonine, Tryptophan, and Valine. Since lysine is already an essential amino acid, meaning, it cannot be synthesized by reptiles on their own, lysine contingency does not make any sense. It can be easily obtained by feeding on the plant matter, and the orgnaisms that feed on the plant matter, readily. The scientists of the Jurassic Park were better off in making the dinosaurs auxtorphic to certain enzymes that are very much necessary for metabolic reactions.
AJ006456.2 Acidithiobacillus ferrooxidans cyc1, cyc2, coxA, coxB, coxC, coxD and rus genes and open reading frame
TTGGCATGTCGATTTTTGGACCTCTAGTGATCACGGCCTATAATTAAACGGCATGGTTAACATGATAAAATAACGTTAGCACATAATTCTTTTCTTATGTTCGTTATTTACTTTATTGCATTTTACTGGATCGATATTCTGGCAACTATGCGCAAAATATTGATTATAAAAGCATTATAGTTATGACCATCGAGGCGATCGCGAGATGCATGGATGAGGTAGCCATGCATTTTAATGAGCGCATAAAAAGATGTTGCAAAGCATCGCGGTTTGTATTAAATAGAACGTGTGGGTATTGTTAACAACGCAACAACATTGGTTAAAGGTCGAGGCTAATTGGCATCGCGTTGTTGTGGTTTGGTGTTACCAGCCTGGCAGGAAGACCGGGCGCATGAGCGTATTTTGTTTATCTAATATGCCTGAAAGCGCATACCGCTATGGAGGGGGTTATGGTGTCATCGTCCGTTGGTTTTAAAAAGAAAAGGTTGATCGTAGCATTAGCAGCAGTTGGTGGAATGGCGTTATCTTCCAGTGCCTGGGCACTGCCATCCTTTGCGCGCCAGACCGGTTGGTCGTGCGCCGCCTGTCACACATCCTACCCGCAGTTGACGCCCATGGGCAGAATGTTCAAATTGCTCGGGTTCACGACCACAAACCTGCAACGGCAGCAGAAGCTCCAAGCCAAGTTCGGGAACAGCGTCGGTCTGCTCATATCCCGCGTGTCACAATTTTCTATCTTCCTGCAGGCCTCGGCGACCAATGTTGGTGGCGGGCAGGCGGTGTTTGGTTCTGGTAACTCTAATGCGAATGCTTCTCCCAACAATAATGTTCAGTTTCCACAACAGGTGAGCTTGTTCTATGCCGGTGAAATCACTCCGCATATCGGCTCGTTTCTGCATATCACCTACTCGGGCGGCGGCAGTGGTACCGGCGGCGGAGGATTTAGTTTTGACGACTCCAGCATTGTCTGGGCCCATCCATGGAAGTTGGGCACCAACAATCTTTTGGTTACGGGCGTAGACGTCAACAATACCCCGACTGCTATGGACTTGTGGAATACCACACCTGATTGGCAGGCACCATTTTTCTCCTCGGATTATTCGTCTTGGGGCCACGTACCTCAGCCATTCATTGAAAGTTCAGCGGGCGCGGGTTACCCATTAGCGGGTGTTGGTGTCTATGGGGCGGATATTTTTGGGCCAAACCGGGCAAACTGGCTGTACGCAGACGCCGATGTCTATACCAACGGTCAAGGAACCCAAGTCAACCCGGTTGGCGGTTTTACTGCAGCTGGCCCCCAGGGCAGGCTTTCAGGGGGCGCTCCTTATGTTCGTCTTGCCTATCAGCACGATTGGGGTGACTGGAACTGGGAGGTCGGCACCTTTGGCATGTGGTCCAGCGTGTACGATAACACCCTAAATAATCCTCTCAATAATATCAGCAAAGCAGGCGGCCCCATTGATACCTTCGATGATTATGATTTAGATACTCAGCTCCAATGGCTTGATACCAACGACAACAATAACGTGACGATCCGTGCCGCATGGGTAAACGAGCAGCAGCAATTTGGAGCGGGGAATATCATATCTTCGAACTCCTCCGGTAACTTAAATTTCTTCAATGTTAACGCCACCTACTGGTATCATGACCACTACGGCATTCAGGGCGGATACCGGAATGTGTGGGGGTCCGCTAACCCCGGTCTCTACACTACCACATACACTAATAGTGGTTCTCCAGATACCAGCAATGAATGGATAGAGGCTTCCTATCTGCCGTGGTGGAATACCCGCTTCTCCTTGCGATATGTCGTATACAACAAGTTCAATGGCGTTGGTTCGGCGTCGTCCAACAACCTTGGATATGGGGCGTCTGCGTATAACACCCTTGAACTGCTGGCCTGGATATCATACTAGGAGCCGATGCCATGACGACATACTTAAGCCAAGACCGGTTGCGCAATAAAGAGAACGACACGATGACCTATCAACATAGCAAGATGTATCAGTCGAGAACCTTCCTTCTGTTCAGCGCACTCTTGCTGGTGGCCGGGCAGGCGAGTGCTGCAGTCGGCAGCGCCGACGCGCCGGCACCATACCGCGTCTCCAGTGATTGCATGGTATGCCACGGGATGACGGGCCGTGACACGCTCTATCCGATCGTCCCCCGCCTGGCCGGACAGCATAAGAGTTATATGGAAGCGCAGTTGAAAGCGTATAAGGATCACTCGCGTGCGGATCAGAATGGCGAGATCTACATGTGGCCCGTGGCGCAAGCGCTGGACAGTGCGAAAATCACGGCGCTGGCAGATTACTTCAACGCCCAGAAGCCGCCGATGCAAAGCAGCGGCATCAAGCATGCCGGTGCGAAAGAAGGAAAGGCCATATTCAACCAAGGGGTTACCAACGAACAAATCCCTGCCTGTATGGAATGCCACGGATCGGATGGCCAAGGGGCGGGCCCGTTCCCCCGGCTGGCGGGCCAGCGTTACGGCTACATCATTCAGCAGTTGACCTACTTCCACAACGGCACACGGGTAAATACCCTGATGAACCAGATTGCGAAGAATATCACCGTGGCGCAGATGAAGGATGTGGCGGCTTATCTTTCATCGCTGTAAGCGTTGTAATTGGTCAATAGAAGTTTTCCTGGCAGGCTGAAGTTTATAAAAATGGGTCTGCCAGGCATTTGCACCGTCAGGTTTATGTGCTTCTCAAAGGAGGTAGAGGTATGGCAGCAAAAAAAGGTATGACTACGGTGCTTGTATCCGCCGTGATATGCGCGGGGGTAATTATAGGTGCCCTGGAGTGGGAAAAAGCGGTAGCCCTGCCCAATCCTTCCGGGCAGGTCATTAATGGGGTACATCATTATACGATCGATGAGTTCAACTATTATTATAAACCGGATCGCATGACCTGGCATGTCGGGGAAAAAGTGGAGTTGACGATTGATAACCGATCGCAATCAGCGCCCCCGATTGCGCATCAGTTCTCCATCGGCAGAACGCTGGTATCCCGGGACAATGGCTTTCCAAAATCACAGGCTATCGCCGTGGGATGGAAAGATAACTTCTTTGATGGTGTGCCGATTACCAGCGGGGGACAGACAGGGCCAGTACCGGCGTTTTCCGTCAGCCTCAACGGTGGACAAAAGTACACCTTCAGTTTTGTGGTGCCCAATAAGCCCGGAAAATGGGAATATGGGTGTTTTCTGCAGACGGGTCAACACTTCATGAATGGGATGCATGGTATTCTTGACATACTACCTGCTCAGGGAAGCTAATTTAGGGAGGGCATATGAACGCAGCAAAAGAAAACTTATGGAAAGCTTTCCGCGGCTTGGTGGTGGTCTGGATTATTGGCCTGGCGATTTTCGAAACGCTGATGGCCTGGGGTATCGGTAACTGGCCAATTTTGGGGAGTATTCAGGCGCATATTACCGCAGATGCCACCACATACCTGTTGTGGCAGGCCGTATTCATCTATGTGCTGGTCGGCGGTGCGATTGTATATAGCGCATTTCGTTTCCGCGCATCATCCATGTCAGACACCGCGGCGCCGGCTTATCAAAAACGGACCTGGGCGCCTTTCGTGGTGACCTGGCTGGTTTTGGCCATAGGCATCAACCTGGCAAATACCATTTATCCGGGTATGGTGGGTCTGGAACAACTTTGGGGTATCCAGTTAGATACGAAGAACCCATTGGTGATCGATGTTACCGCGCAACAGTGGAAGTGGACGTTCTCTTATCCTAAGCAGGGCGTAACGGATGTGTCACAACTGGTGGTTCCCGAGGGCCGCACCATATACTTCGTTCTGCGGACAAAGGATGTCATGCACGATTTTTGGGTGCCTGCCTGGGGTGAGAAAAAAGATGTGATCCCCAATGAAGTGCGGCACTTGTTTATTACACCCACCATGTTGGGGACAACCGCTACAAACCCCATGCTGCGTGTACAGTGTTCCTTGATTTGTGGCAACGGACATCCGTTGATGCGCGCTCCGGTGAAAGTGGTAACGCCAGCGGACTTCAAGGCTTGGGTGGCAAACAATAGCTTCTAGTAAAGCCAACGGAAGGCTTGCCAGCACCCAACGTTAAATGTACTAAGGAGTAAGTAATGGCAACTAACGAAATTCAGGAAAATGCGTTGAACAATACGGGAGTGGACAAGACCCCATTTGCGGCTAGCATGCTGTTTCCGCTGTTCCGTGCGACGCTTTGGGGACTAACCGGCTATTTTGCTGCGGCATGGATCACTGCTTTATTGCTCCACACGGTAATCGTAAACCCTTTACCCGCGACAGTGGGTTATGTGGCCGGCTTGGTCTGCTGGCTGATGGGCAGCGGTGTATGGGAGGGATGGATACGACGCGCATTTGGAGGAAAAGAAGCTCCAACTTACACGGGTATCGAACGTTATTTTCGCTTTGGTCCCGATTCAAAATCCGCAGCCGTACGCTACGTAATCTTAAATATACTAACGTTCTGCTTTGCCGGCATGGCCGCCATGGCGATCCGCATTGAACTGTTGACGCCAGACTCCACCAGTTGGTGGCTGTCAGAAATCCAGTACAACCAAACGTTCGGTATTCATGGATTGATGATGTTGTTGGGTGTGGTGGCCTCTGCCATCGTCGGCGGTGTTGGCTACTATCTTATCCCGTTGATGCTTGGCACGAGAAATGTAGTATTCCCAAAACTTCTTGGCCTAAGTTGGTGGCTTTTGCCACCGGCGACCTTCGCTGTTTTTATGAGTCCTACGACCGGTGGGTTTCAGACGGGATGGTGGGGATATCCGCCGTTGGCGCAAAACAGTGGTAGCGGTATTGTGTGGTATGTCCTCGGTGCCGCCACCATTCTTGTTGCGTCGCTACTTGGAGCCATCAATATCGCCGGAACCATGGTGTACATGCGCGCCAAGGGCATGAGCCTGGGTCGCGTTCCGATTTTTGTGTGGGGTTTATTTGCGGCAGCCACCACTCTCGTCGTAGAGTCGCCAGCAACCTATACCGGCGCGCTCATGGACTTATCCGACATGATCGCCGGATCGCATTTTTATACCGGTCCCACCGGCCACCCGTTAGCGTATCTCGATCAGTTCTGGTTTTTGTTCCATCCAGAGGTCTACGTTTTCATTCTGCCCGCTTTTGCCATATGGCTGGAGATTCTTCCTGCCGCGGCCAAGCGGCCGTTGTTTGCTAGGGGTTGGGCCATCGCCGGACTGGTTGGCGTTTCCATGTCGGGTGCAATGTCGGGTGTCCATCACTACTTCACTGCGGTGAGTGACGCGCGTATGCCCATATTCATGACCATAACGGAAACTGTATCCATTCCGACAGGGTTCATTTATTTGTCCGCCATCGGAACGATATGGGGTGGTCGTTTAAGAATTAATGCTGCGGTATTGCTCGTACTGATGGCGATGATGAACTTCCTGATCGGTGGGCTGACGGGCATATTCAATGCCGACGTTCCCGCCGACCTTCAGCTGCACAACACCTACTGGGTTATTGCGCATTTCCATATACGATGCTTTGGTGGAGTGATCTTTACCTGGATTGCCGCGCTATACTGGTGGTTTCCCAAGGTTACTGGACGGAAGATCAATGAATTTTGGGGAAAGTTTCACGCATGGTGGTCCTTCGTATTCTTCAATTGTACGTTCTTTCCCATGTTTATAGCTGGACTAGATGGAATGAACAGGAGAATTGCGATATATCTTCCTTACCTGCATGACATCAACCTGTTTATGTCTATTTCATCCTTTTTCTTGGGCGCAGGGTTTCTCATTCCGCTGGCCAATCTTTTATACAGTTGGCGCTATGGGCCAAAGGCCGAAGCTAACCCTTGGGGCAGCAACGGCCTGGAATGGCAAATAAAATCGCCAACACCGTATGTGCCATATCCAGCAGGAACGGAGCCAGAGGTTGTGGGCCCGAACGATAACTACGCGGCGGAAGCAAAAGACCCCTTTATTTGGGTGTCTACGCCCAGCAAGTAAATTAGAAGGAGTTGAACCATGACAGACAACAGTTATGCCAAGCTAATGGATCCGGCCTCGGAGCGTGCAAAAAGGGGTGCGTTCTTTTTCCTGATGCTTTTTGCAGCCATCATTTTTGCGATGTGGGACCTCGCGCGTTTTCTGTGGGGGCACTCGGTGCCCGCTACATTGAGCATGGGCGTGGGTGTTGCGCTGACTGTTCTGATGCTCGTCAGCCTGGTGCCGGTGATGACGGCCCGCAAAAAACTGGATCAGGGCGATGATGCCGGTATCGTGAGCAGTCTGGCAACCCTGATGGTGGTCTCGTTGGTGATGGCGGGTGGAATCGTCTACAACTGGACTACCTTAACCATCGGTAGTGGTTATGGCGGGATTTATGACATCACCAGCTTGTGGTTTCTGGTACATTTCGTGGCGGCCATCCTGGCGCTGCTGGCGAGTATCATGAAAATCACTCGCACTCCAGAGCGCGCGAAACGCGAGCGATGGGTGTCGTATAACGTGTTAACCTTCTGGGGCGGTGTGATTGTTCTATGGGTTGCATTTTTTATTGTTTTCTATATTGCGTAATGCAGTTTAGAAGATTCTCTAATGGAGTGAGGGTTAGATAATGGATATGTCACATTTATCGTTCGTTATCCCGTCTGGAGCTGATGATCCGACGTTTTTCTGGCTGACGGGGTACATTGGGTTTCCTGTGGTGTTTCTGAGTGCATACTTTTGGTGGGTATTAAAGGAGGCAAGCAAGGAAGATCGGCTGCGTATTCTAAAAAAGGGAGAAGACGGCGCATCTGGAAACGCATGATGTTCCACGGATGGTCGTGCGAGTACCGGGCGGCCATCCGGAGTTGTTTTGCGTTTTACTGTTGCGACGTCGTTATCCATGCTTCAAAGGAGGTAAATCATGAACAAGGAAGGCTGTTTAATTTCTCACGATGATCGCGATGATGGCGCATGGGATGGAAACATCGTGTTGATCATAGGATTATTGTGGGCTATTATTGCTCTGGGTGGCTATTATGTTACCCTTAGAGTGCTGTTTTGAGACAATTCCCCGGCTGGATAGGGCGATGAATACCATGTAGTAGCATATTAAAATGCCAGAGGGCCCGGTGATGGTTTTGTAGGGCGGCTGGTTCTACTCAGGTTAAACGTTAAGGAGAAGGGATAACTTATGTATACACAGAACACGATGAAAAAGAACTGGTATGTGACTGTTGGTGCGGCTGCGGCTCTGGCGGCAACGGTCGGCATGGGTACCGCGATGGCCGGCACGCTGGATTCCACATGGAAAGAGGCGACGCTTCCCCAAGTTAAGGCCATGCTGGAGAAAGATACCGGGAAAGTCAGTGGCGATACAGTTACCTACAGCGGCAAGACTGTACATGTGGTCGCGGCGGCCGTGCTCCCGGGATTTCCGTTCCCGAGCTTTGAAGTTCATGACAAAAAGAACCCGACCTTGGAGATTCCCGCAGGGGCAACCGTAGACGTGACCTTCATTAACACCAACAAGGGATTTGGTCATAGTTTTGACATCACTAAAAAAGGACCGCCTTATGCGGTTATGCCGGTGATTGACCCCATTGTCGCAGGAACTGGATTTAGCCCGGTCCCAAAAGACGGCAAGTTCGGATATACGGATTTCACCTGGCATCCGACGGCGGGTACTTACTACTACGTATGTCAGATACCGGGGCATGCCGCCACCGGTATGTTTGGTAAAATCATTGTCAAGTAAGTCCTGGATGGTTGTTGTCTGGGCAGCTGTGCTTTGCTAGTGTAGGTCCTGGTGGCCAGGGCAAATGGTTATCTTGCCCTGGCCATTGGTATTTATTATAAAATACGAATTTCATGTATTGCGTTATGCTTTGTATGATGTTATGAGTATGTTTGCATGCAACATATGATGATTGATCTAGTTTATTAAGCTATGGACCACGAAAACACGCTGCCTCGGTACATATATTAATTCATTCAGATAAAGTCCCAAACTCAGATATCCTGACG
The sequence is over 8 kb long. So, I would suggest the use of cosmids for cloning. The cosmid can be inserted into E. coli, and be cloned. Inside the E. coli, the sequence replicates, transcripts, and finally translates into protein. The protein from this gene is found on the outer membrane of Acidithiobacillus ferrooxidans. But, since signal peptides and chaperone proteins for the desired protein is missing in the sequence, my educated guess is that it will be found intracellularly, and must be extracted and purified for further investigations.
Alternatively, the cell free method PURE (Protein synthesis Using Recombinant Elements) can also be used because of its faster turn around times. The DNA template strand is incubated in the presence of specific enzymes and cell extracts. The protein obtained must be purified through affinity chormatography.
a. What DNA would you want to sequence (e.g., read) and why? I would love to sequence the antifreeze protein gene from Leucosporidium sp..The protein has a lot of applications in food technology, and medicine, and I would love to produce it commercially.
I
b. What technology or technologies would you use to perform sequencing on your DNA and why? I would use SMRT (Single Molecule Real Time) sequencing technology from PacBio. It can generate long reads (10-25 kb) with Q40+ accuracy. It is also best used for de novo genome assembly.
c. Is your method first-, second-, or third-generation (or other)? It is a third generation sequencing method.
d. What is your input? How do you prepare your input (fragmentation, adapter ligation, PCR)? i) DNA has to be extracted and must be purified to make it free from proteins and RNA. Long and unbroken molecules are considered to be ideal. Freshly exracted DNA is preferred over stored one. ii) The DNA is enzymatically sheared into 10-25 kb long fragments iii) SMRTbell library format is preferred for the preparation of library, where hairpin adapters are ligated to both 5’ and 3’ ends to create a circular template of DNA fragment.
iv) Sequencing primers and the appropriate polymerases are added to the buffer containing the DNA.
v) It is then loaded on to SMRT cell, that contain zero-mode waveguides. Each ZMW captures a single DNA molecule for sequencing.
e. List the essential steps. Answered above.
f. How does your chosen sequencing technology decode bases (base calling)? Each nucleotide contains a unique fluorescent labels, which get excited with a laser whenever a new base is added. The instrument then records the color and timing of each flash, which corresponds to the base that has been added.
g. What is the output? HiFi reads, usually 10-25 kb long are obtained as output.
5.2 DNA Write
a. What DNA would you want to synthesize (e.g., write) and why? I want to synthesize the Temporin 1 CE A gene found in frogs. It is a small peptide antimicrobial, and can be used to combat antibiotic-resistant bacteria.
b. What technology or technologies would you use to perform DNA synthesis and why? Phosphoaramidite method, followed by Gibson assembly can be used to synthesize it.
c. Essential steps of chosen synthesis methods See Homeowrk 1 for the steps of phosphoaramidite synthesis. Steps of Gibson Assembly:
Mix the pure, synthesized fragments into the reacton mix containing 5’ exonuclease, DNA polymerase, and DNA ligase. It is essential to ensure that the synthesized fragments have 15-30 bp overalaps to prevent random ligations.
Incubate the samples at 50 degree celcius. This ensures that only cannonical base pairing (A=T and G≡C) occurs and non canonical bonds are prevented due to their instability at this temperature.
exonuclease cleaves the -OH group, polymerase adds the nucleotides, and ligase binds the sequneces together.
d. Limitations (speed, accuracy, scalability) Phosphoaramidite method is limited by its inability to synthesise fragments longer than 200 bp, poor yields for longer fragments, and relatively higher cost per synthesized base pairs. Gibson assembly is limited by its dependency on overlaping fragments that need to be precise. The assembled sequences must be sequenced again to make sure that it is accurate and misjoins and mutations have not occured.
5.3 DNA Edit
a. What DNA would you want to edit and why? I would edit RSL4 gene in plants, since its overexpression increases root hair length. Longer root hairs allow the plant to uptake more nutrients.
b. What technology or technologies would you use to perform DNA edits and why? I would use CRISPR/Cas9 because it allows precise, targeted edits and can be adapted for either gene activation or promoter replacement to drive RSL4 overexpression.
c. How does your technology edit DNA? CRISPR/Cas9 uses a guide RNA to direct the Cas9 nuclease to a specific DNA sequence, where it introduces a double‑strand break. Repair pathways or engineered activators then modify or enhance gene expression.
d. Essential steps
Design guide RNAs targeting the RSL4 promoter or coding region
Clone them into a CRISPR vector
Deliver the construct into plant cells
Select transformed cells and regenerate whole plants
e. Preparation needed (design steps) Identify target sites in the RSL4 promoter, ensure PAM sequences are present, and design guide RNAs with minimal off‑target potential. Choose a strong promoter or CRISPR activation system to boost expression.
f. Inputs (DNA template, enzymes, plasmids, primers, guides, cells) Inputs include the RSL4 gene sequence, Cas9 enzyme, guide RNAs, plasmid vectors with promoters, plant cells for transformation, and primers for verification PCR.
g. Limitations (efficiency, precision) CRISPR editing efficiency can vary across plant species, and off‑target effects may occur. Regeneration of edited plants is time‑consuming, and overexpression may cause unintended growth trade‑offs.
Keywords I would use to describe it: Opentrons OT-2, automation, standradization, synthetic biology.
Summary of the paper: The authors developed an open-source software called ‘Slowpoke’ to automate the Golden Gate assembly process. Opentrons were used to carry out bacterial transformation, GG assembly, and plating. After a few manual steps in between, Opentrons were used once again to perform cPCR. It demonstrated the feasiblity of automating GG assembly.
Opentrons were used to handle liquid transfers, reaction mixtures, and parameters. By integrating pipetting, transformataion, plating, and cPCR screening into a single pipleline. The validation was carried out manually using flow cytometry with transformed yeast cells. Using Slowpoke interface along with Opentrons, the authors achieved high assembly efficiencies, over 90% with Yeast Toolkit (YTK) and 60% with Subtilis Toolkit (STK), consistent with values reported for manual Golden Gate assemblies using these toolkits.
To conclude, this paper designed a tool (Slowpoke) that generates Opentrons-ready protocols in the form of CSV files, mitigating the expertise needed in coding to a great extent. However, it must be noted that human input was still necessary to collect the DNA fragments for running cPCR.
v
2. Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
I would like to automate the prototyping of a novel Bio In-situ Resource Utilization (Bio-ISRU) on Mars that comprises of two trophic levels. The producer level utilizes photoautotrophic organisms to convert Martian CO2 (and of course, sunlight) to produce the nutrients required for the primary consumer level. The latter would consist of a ‘biominer’- a bacteria that can precipitate, from the Martian regolith, metals- cheifly iron in the form of Fe3O4- for easier metallurgical applications.
The automation tools would be utilized in the following ways:
3D printing of a photobioreactor in gyroid shape to maximize the surface area for photosynthesis. This has to be validated in Martain gravity. Probably, can be carried out on a space station using a centrifuging apparatus to mimic the higher gravitational pull on Mars compared to Low Earth Orbit (LEO)
Bioreactor management (i.e, addition of nutrient media etc.)
Sensing if the maximum biomass has been achieved, and if yes, lysing the cells so that they may be utilized by the biominers
Efficient mixing of the lysed biomass with the Martian Regolith at appropriate ratio to maximize the precipitation of Fe3O4 by Acidithiobacillus spp.
Sensing the maximum quantity of Fe3O4 precipitaed, and removing it by operating a magnetic arm to separate the magnetite.
Sterilization of equippment, as well as decontamination, using gamma radiation.
Pseudocode (The following was the output of Gemini 3 for the prompt: “Write a pseudocode for the following”, and the above block was pasted.):
import ginkgo_nebula_api as nebula
# Configuration Constants
MARS_GRAVITY_RPM = 24.5 # Calculated RPM for centrifuge to mimic 0.38g
BIOMASS_THRESHOLD = 0.85 # OD600 value for harvest
MINING_RATIO = 0.4 # Ratio of Lysate to Regolith
IRON_SATURATION_POINT = 0.92 # Signal from Magnetic Flux sensor
class BioISRU_System:
def __init__(self):
self.stage = "PRODUCER_GROWTH"
self.centrifuge_active = True
def run_lifecycle(self):
# 1. GRAVITY VALIDATION
# Maintain Mars-simulated gravity via centrifugation
if self.centrifuge_active:
mars.centrifuge.set_speed(MARS_GRAVITY_RPM)
# 2. PRODUCER STAGE (Cyanobacteria)
if self.stage == "PRODUCER_GROWTH":
mars.pbr.inject_media(interval="6h")
current_od = mars.pbr.read_sensor("OD600")
# Check if gyroid surface area is maximized with biomass
if current_od >= BIOMASS_THRESHOLD:
print("Maximum biomass achieved. Initiating Lysing...")
self.stage = "LYSING"
# 3. LYSING & HANDOVER
if self.stage == "LYSING":
mars.pbr.activate_ultrasonic_lyser(duration="10m")
# Transfer to the Miner Tank
lysate_volume = mars.pbr.get_volume()
mars.transfer_pump.move(to="MINER_TANK", volume=lysate_volume)
self.stage = "BIO_MINING"
# 4. CONSUMER STAGE (Biominers + Regolith)
if self.stage == "BIO_MINING":
# Automated mixing of lysed biomass and Martian Regolith
regolith_amount = mars.transfer_pump.last_volume * MINING_RATIO
mars.regolith_feeder.add(regolith_amount)
# Efficient mixing to maximize Acidithiobacillus contact
mars.miner_tank.start_agitation(pattern="vortex", speed="medium")
# 5. SENSING & MAGNETITE HARVEST
iron_yield = mars.miner_tank.read_sensor("MAGNETIC_FLUX")
if iron_yield >= IRON_SATURATION_POINT:
print("Magnetite precipitation peaked. Deploying Magnetic Arm.")
mars.magnetic_arm.extend()
mars.magnetic_arm.energize()
mars.magnetic_arm.extract_to(bin="IRON_STORAGE")
self.stage = "DECONTAMINATION"
# 6. STERILIZATION & RESET
if self.stage == "DECONTAMINATION":
print("Initiating Gamma Radiation Sterilization cycle...")
mars.gamma_source.expose(duration="30m", target="ALL_CHAMBERS")
# Uplink yield data to Ginkgo Nebula for strain optimization
nebula.upload_log(yield_data=iron_yield, efficiency=1.2)
# Reset for next cycle
self.stage = "PRODUCER_GROWTH"
print("System Reset. Starting new ISRU cycle.")
# Initialize and Loop
isru_unit = BioISRU_System()
while True:
isru_unit.run_lifecycle()```
### Final Project Ideas:
[Google Slides](https://docs.google.com/presentation/d/1FAFN4YYisOcso3CI5F3W3Z7hj6_n9D1vAhVUywQXKPU/edit?slide=id.g3ca9627a0a6_624_27#slide=id.g3ca9627a0a6_624_27)
Week 04 HW: Protein Design Part 1
Part A. Conceptual Questions
Answer any NINE of the following:
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Assume that the mass of other components like fat, collagen etc. are negligible compared to proteins in meat. Average wight of 1 molecule of amino acid = 100 Dalton = 1.7 * $10^{-24}$ g Weight of piece of meat = 5 * $10^{2}$ g Therefore, number of amino acid molecules = (5 * $10^{2}$) / 1.7 * $10^{-24}$ = 2.94 * $10^{26}$
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
All the biomolecules found in foods get broken down into their constituent molecules via different enzymes and get resynthesized into the required biomolecules in various metabolic cycles as per the requirement of the body. This is vastly different from the synthesis of proteins through genetic translation. However, if we can encode bovine genes into the human genome, there is a possibility that some of the proteins synthesised via translation resemble that of the cow rather than human proteins. However, this is not enough for a human to become a cow because amino acids alone do not maketh species!
3. Why are there only 20 natural amino acids? Can you make other non-natural amino acids?
To answer the second part of the question, I would say yes. It is indeed possible to make non-natural amino acids. The first part however, is interesting. This paper lists out several reasons so as to why only 20 proteogenic amino acids exists. Some of the interesting takeaways I found are:
Some amino acids are highly “expensive to produce”. So, if two amino acids are (almost) similar in properties, life would favour the one with less productin cost. Case in point: Leucine requires 1 ATP, but Isoleucine requires 11.
Some of the possible side chains, especially the aromatic ones, would make it completely insoluble in water- which is detrimental for reactions. Therefore, any amino acid would have to be at least as soluble, if not more than the least soluble amino aicd (at pH 7), Tyrosine.
Some other side chains like esters and anhydrides are easily hydrolysed; ketones and aldehydes are suseptible to oxidation, reduction, and nucleophilic attacks; and carbon-carbon double and triple bonds are more reacctive than their single bond counterparts. Therefore, amino acids with those side chains are best avoided.
Secondary structure that does not form bonds with other amino acids; molten globules (non-polar parts hidden inside) will have flexible side chains that msut be frozen into fixed positions, costing energy; and aggregated clumps of amino acids, especially beta sheets, are useless and oftentimes even toxic (amyloids). Therefore, these can’t be overly favoured.
Incorporating other elements beside C, H, O, N also comes with a high energy costs. S containing amino acids (methionine and cysteine) are energetically expensive than other amino acids of the similar size. Therefore, evolution didn’t favour the ones with other elements.
4. Design some new amino acids.
5. Where did amino acids come from before enzymes that make them, and before life started?
The RNA world hypothesis can explain this to an extent. If we consider the first catalysing molecules to be RNAs, then it could very well have been possible for them to catalyse the synthesis of amino acids. Clay particles arranging themeselves in such a geometry that they would catalyse the synthesis is also a proposed hypothesis. Since these are biotic synthesis, only one of the racemic form would have been favoured. If abiotic synthesis of amino acds is considered, then the Miller-Urey experiment proved that high energy discharge in the form of lightining can lead to the formation of amino acids. Some other ways for abiotic snthesis would be; undersea volcanic eruptions; and meteorite impact etc. Although in these cases, equal proportion of L and D forms are likely to be formed.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
I would expect it to be left-handed helix. Since D-amino acids are the mirror images of L-amino acids, the steric and geometric parameters would flip, leading to the left-handed helix.
7. Can you discover additional helices in proteins?
Apart from $\alpha$ helices, $3_{10}$ helices, and $\pi$ helices, polyproline helices, collagen helices are known to exist. So, yes, it is possible to discover additonal helices in proteins.
8. Why are most molecular helices right-handed?
Due to the potential for steiric hindarance, biologically, either the biomolecules can all be either right-handed or left-handed helices. It is not possible for some molecules to be right-handed and some others to be left-handed. The right-handedness of proteins is due to the existence of L-amino acids, and the right-handedness of nucleic acids is due to D-sugars. Computation simulations show that L-amino acids consistently fold into stable structures than their D coutnerparts in ambient conditions. The same can be inferred for the stability of nucleic acids, although left-handed stuctures are known to exist under specific conditions.
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
Extensive hydrogen bonding
Hydrophobic side chains intergigitated with hydrophilic ones
Flat planarity as opposed to curved nature of helices
Strucutral complementarity of the edges, that favour inter-strucutral bond formation.
10. Why do many amyloid diseases form β-sheets?
Skipped.
11. Can you use amyloid β-sheets as materials?
Skipped.
12. Design a β-sheet motif that forms a well-ordered structure.
Skipped.
Part B. Protein Analysis and Visualization
Pick any protein with a 3D structure and answer:
1. Briefly describe the protein you selected and why you selected it.
Proline-Betaine Transporter is the protein I have selected, and it is because it acts as an osmoprotectant for bacteria under high salt concentrations.
2. Identify the amino acid sequence of your protein. How long is it? What is the most frequent amino acid?
3. How many protein sequence homologs are there for your protein? (Hint: Use Uniprot’s BLAST tool)
Lorem Ipsum
4. Does your protein belong to any protein family?
It is a transport protein.
5. Identify the structure page of your protein in RCSB. When was the structure solved? Is it a good quality structure?
Yes, the structure was solved. It is a good quality structure too.
6. Are there any other molecules in the solved structure apart from protein?
None
7. Does your protein belong to any structure classification family?
I don’t think it does.
8. Open the structure in PyMol (or similar).
Visualize as “cartoon”, “ribbon”, “ball and stick”.
Color by secondary structure. Does it have more helices or sheets?
Color by residue type. Distribution of hydrophobic vs hydrophilic residues?
Visualize surface. Does it have binding pockets? It has only helices.
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
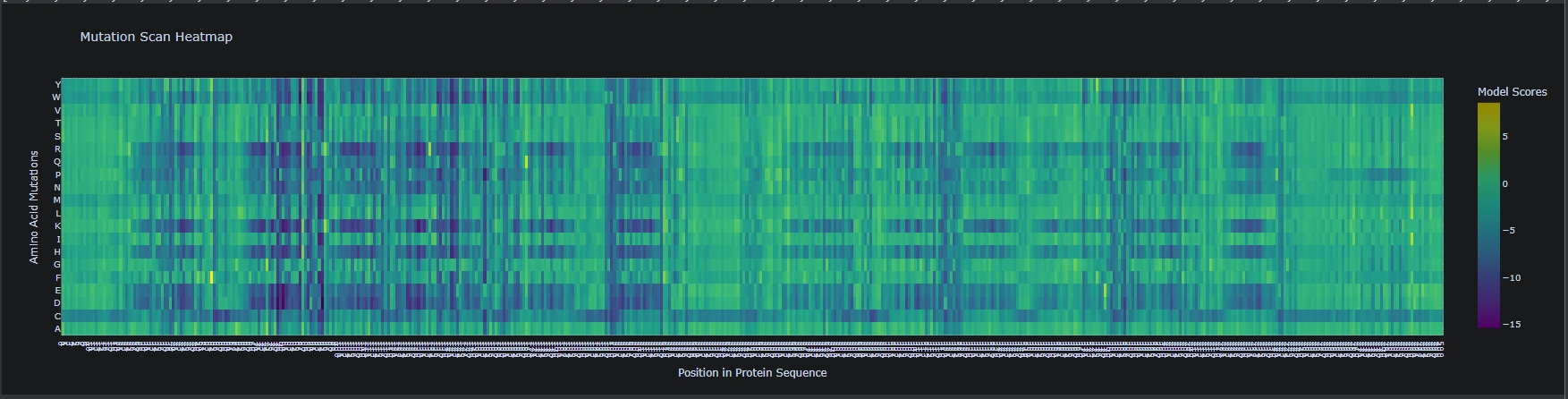
Generate deep mutational scan with ESM2. Can you explain any particular pattern?
(Bonus) Compare predictions to experimental scans. Skipped
Latent space analysis: Place your protein in the map and explain its position. Skipped
C2. Protein Folding
Fold your protein with ESMFold. Do predicted coordinates match original? The predictied coordinates do not match the original.
Try mutations and large sequence changes. Is structure resilient? The structure is not resilient as the conformation changes. The mutation carried out was: 5-10 H were replaced with A, and 242-247 were replaced with H.
C3. Protein Generation
Inverse-folding with ProteinMPNN. Compare predicted vs original sequence. The predicted sequence is 50.33% similar to the orignial sequence according to this python program
Input sequence into ESMFold and compare predicted structure.
Part D. Group Brainstorm on Bacteriophage Engineering
1. Choose one or two main goals (e.g., stabilize lysis protein, disrupt interaction with E. coli DnaJ).
My group consisted of 4 people: @lorem ipsum, @lorem ipsum, @lorem ipsum, and @lorem ipsum.
After brainstorming, we decided to focus on stabilizing the lysis protein.
We decided to focus on the main area of increasing the stability of the MS2 phage lysis protein L, with a possible secondary goal of reducing the dependency on host DnaJ, while still maintaining the lysis action.
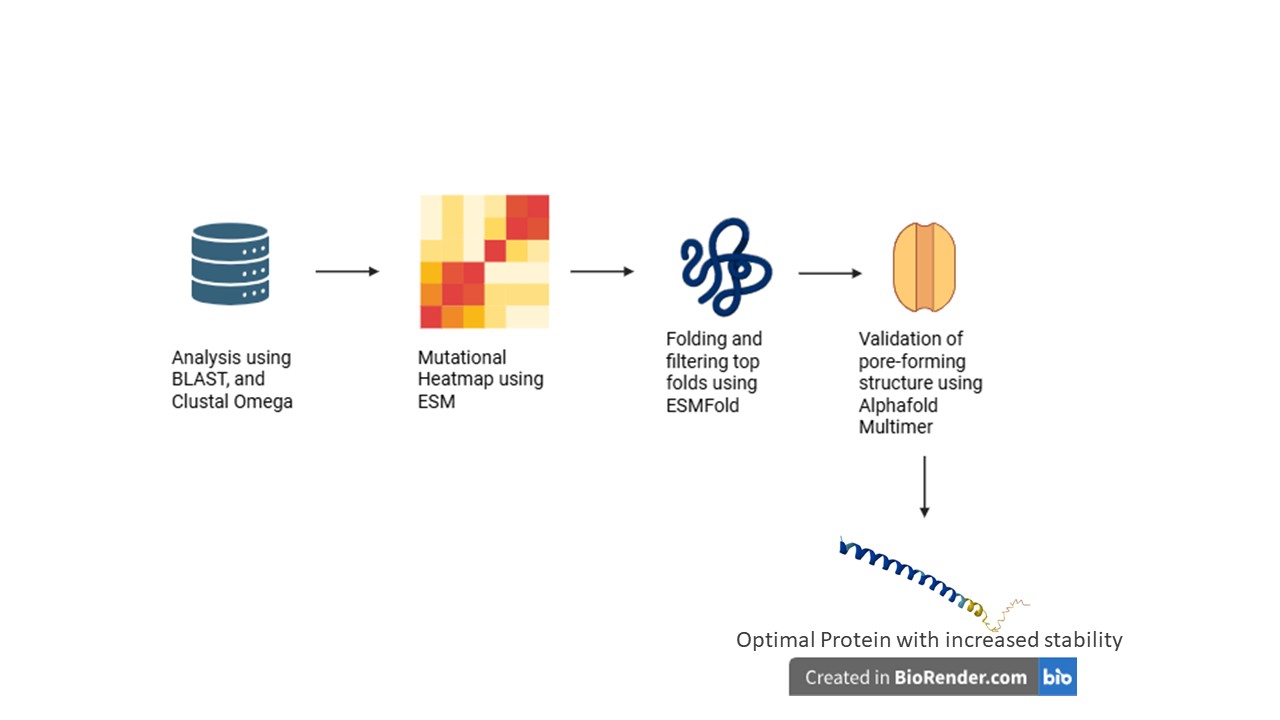
The tools AlphaFold, Clustal Omega, BLAST, ESM, and ESMFold were discussed.
BLAST can pull out homologous lysis proteins from the databases.
Clustal Omega can create MSAs to identify essential L48-S49 residues, and the pore-forming regions that must not be mutated.
ESM can create mutation heatmaps, which can guide the use of ESMFold to obtain highest score foldings in mutatable regions.
AlphaFold Multimer predicts whether the subunits of our protein can successfully create a pore in the host membrane, and also to check whether N-terminus can break the interaction with DnaJ.
We also identified a few pitfalls, with majors ones dealing with limited training datasets, that may not be properly aligned towards creating a transmembrane lysis protein.
Some other pitfalls include the lack of proper annotations for amurins; the possibility of an over-stable protein to form non-functional aggregates; and the vulnerability of modified protein to host proteases.
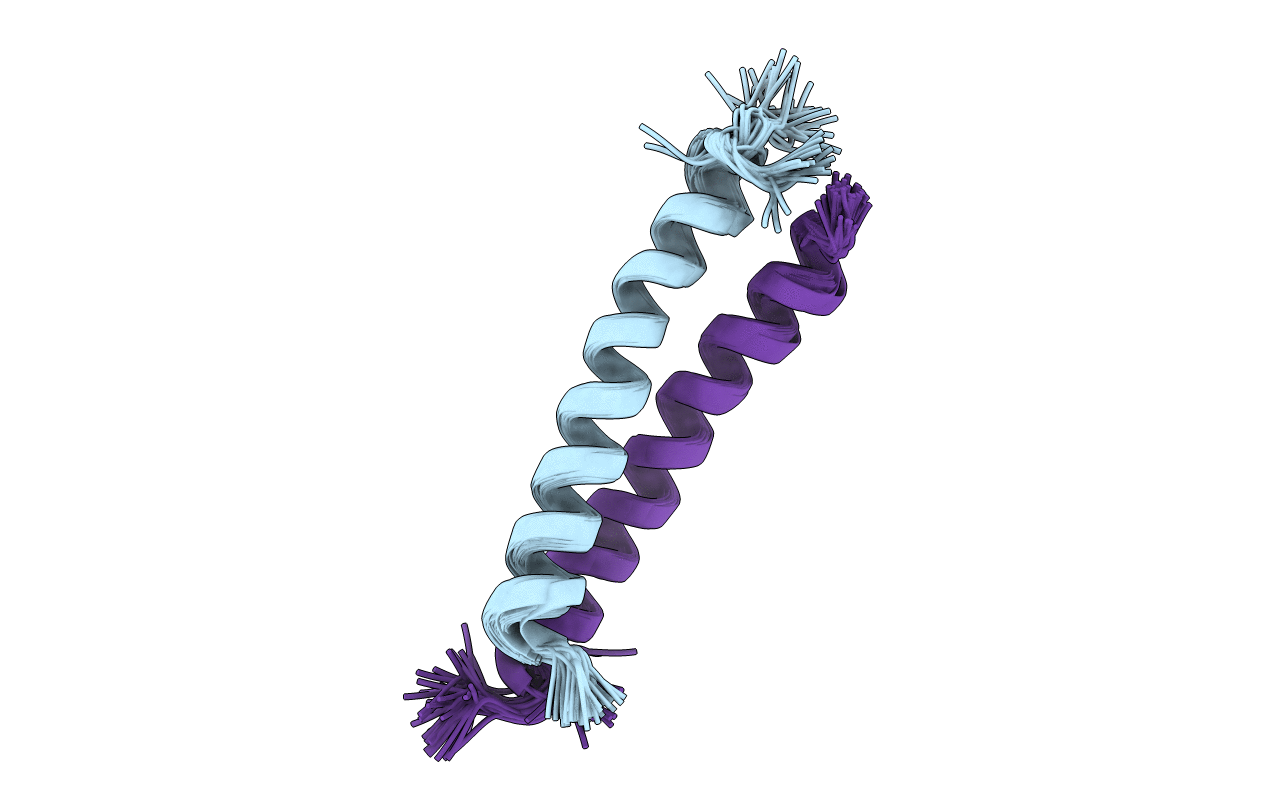
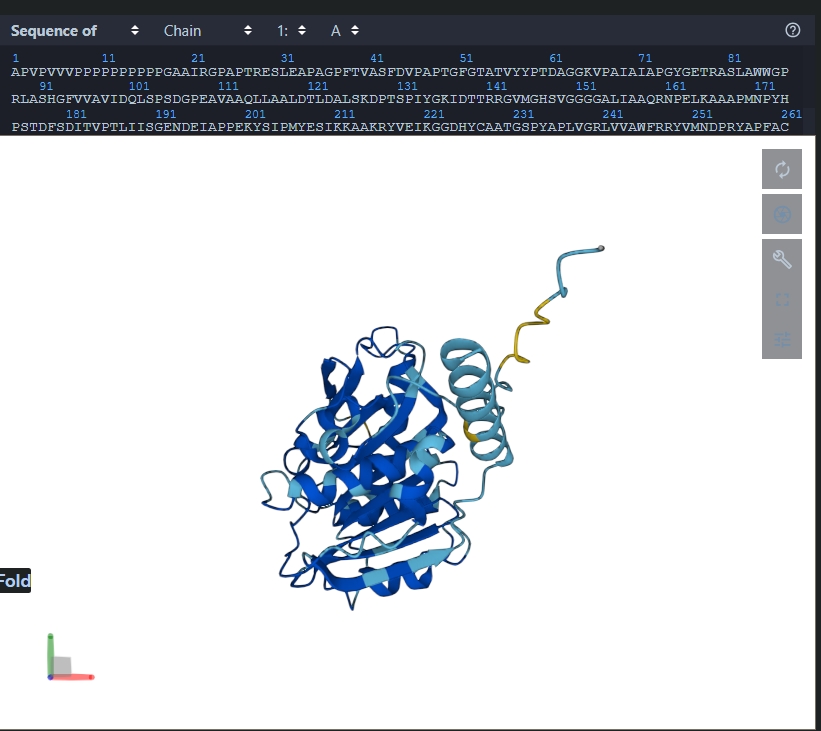
Structure of the native sequence- predicted vs actual:
Generate 4 peptides using PepMLM Colab:
index
Binder
Pseudo Perplexity
1
WRSPAVAVAHWE
7.76721411356481
2
WRVGWVGVELKE
24.2058244561383
3
WRSPAAXIEHKX
11.243453670563373
4
WRVYAAXIEWGK
20.449723821548965
Known binder: FLYRWLPSRRGG
Perplexity score: 22.5252
A note about perplexity score: A key evaluation metric for language models that measures how well a probability model predicts a sample. Lower the score, higher the confidence of the model that the output satisfies the criteria.
Part 2: Evaluate Binders with AlphaFold3
Peptide
Binding location
ipTM score
WRSPAVAVAHWE
None
0.28
WRVGWVGVELKE
None
0.35
WRSPAAXIEHKX
None
0.33
WRVYAAXIEWGK
None
0.34
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Peptide Comparison Results:
Input Sequence
Solubility
Hemolysis (Prob.)
Binding Affinity
Length (aa)
Mol. Weight (Da)
Net Charge (pH 7)
Isoelectric Point (pH)
Hydrophobicity (GRAVY)
WRSPAVAVAHWE
1.0
0.044 (Non-hemolytic)
5.361 (Weak)
12
1408.6
-0.14
6.76
-0.13
WRVGWVGVELKE
1.0
0.117 (Non-hemolytic)
7.089 (Medium)
12
1457.7
-0.23
6.28
-0.13
WRSPAAXIEHKX
1.0
0.011 (Non-hemolytic)
4.645 (Weak)
12
1158.5
0.85
8.76
-0.86
WRVYAAXIEWGK
1.0
0.043 (Non-hemolytic)
6.724 (Weak)
12
1360.7
0.76
8.59
-0.26
(Known) FLYRWLPSRRGG
1.0
0.047 (Non-hemolytic)
5.962 (Weak)
12
1507.7
2.76
11.71
-0.71
The best peptide I would chose for wet lab validation would be WRVGWVGVELKE due to its relatively high binding affinity.
Part 4: Generate Optimized Peptides with moPPIt
Parameters:
Binder
Hemolysis
Solubility
Affinity
Motif
SVKTKCCTTYQS
0.96447
0.916667
6.5756
0.890471
DDTKKCSCIQTH
0.974932
0.916667
6.31426
0.914592
ENGETFQCTKKV
0.970342
0.833333
6.04386
0.934673
KKSKKAFVCCVC
0.963174
0.666667
8.17171
0.613892
For the very long execution time, and the computational resources this program took, the only significant advantage it has (in this particular context) over PepMLM is the motif score, since there was no option to check for the motif specificity in the Peptiverse. All the other properties of the PepMLM generated sequences (predicted using Petptiverse) and those of the moPPIt peptides are comparable.
Part B: BRD4 Drug Discovery Platform Tutorial (Optional)
Skipped
Part C: Group Project: L-Protein Mutants
I chose the third option- Generating random mutations in the Lysis protein while avoiding the loss of function or non sense codons.The Python script was generated solely by the Google Gemini 2.5 Flash, that is in-built in Google Colab. The prompt was:
Develop a Python program in Google Colab that processes an amino acid sequence and generates mutated versions of it based on experimental data. The program should perform the following steps:
The data contains information about amino acid changes and their associated ‘Lysis’ activity.
Filter the mutation data to include only ‘active’ mutations (where ‘Lysis’ is not 0). Extract the ‘Original_Residue’, ‘Position’, and ‘Mutated_Residue’ from the relevant columns (e.g., ‘Amino Acid Change’ and ‘Amino Acid Position’ or a ‘Mutation’ column like ‘X###Y’).
Create a helper function to format amino acid sequences by inserting a space after every 5 amino acids for better readability.
Implement a function generate_random_mutation_combinations(sequence, mutation_df, num_mutations) that takes an original amino acid sequence, the filtered active mutations DataFrame, and the desired number of mutations as input.
This function should:
Identify all valid mutation sites where the original residue in the sequence matches an original residue in the mutation_df.
Ensure that the num_mutations are applied to unique positions in the sequence. If there are fewer available unique mutation positions than num_mutations, it should apply all available unique mutations.
Randomly select mutations from the available options for the chosen unique positions.
Return the new mutated sequence and print the applied mutations.
Generate Multiple Mutated Sequences: Prompt the user for the number of mutated sequences they wish to generate. For each requested sequence:
Call the generate_random_mutation_combinations function.
Display the generated sequence with a clear heading (e.g., ‘Sequence 1:’, ‘Sequence 2:’, etc.).
Print both the original and the mutated sequences, using the formatting function defined in step 5.
In a separate code block, display each generated mutated sequence individually using display() so that each sequence is easily copyable by the user.
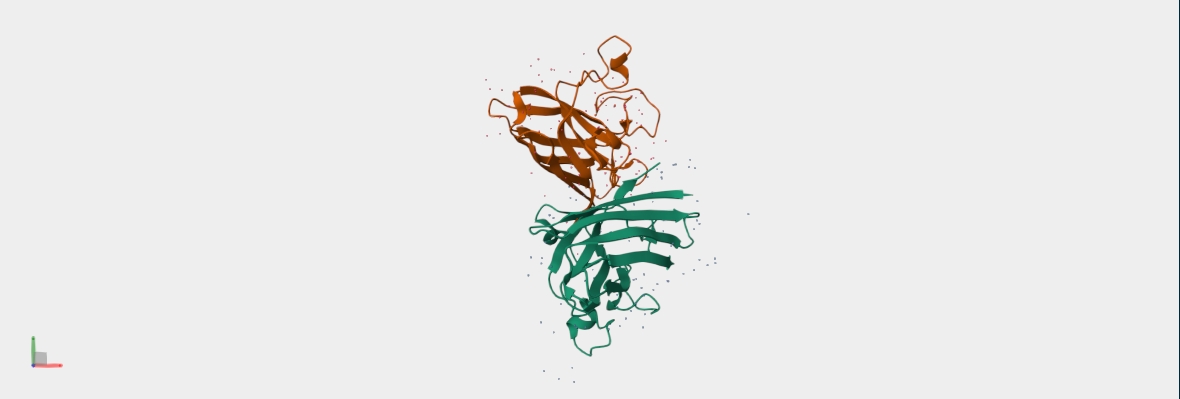
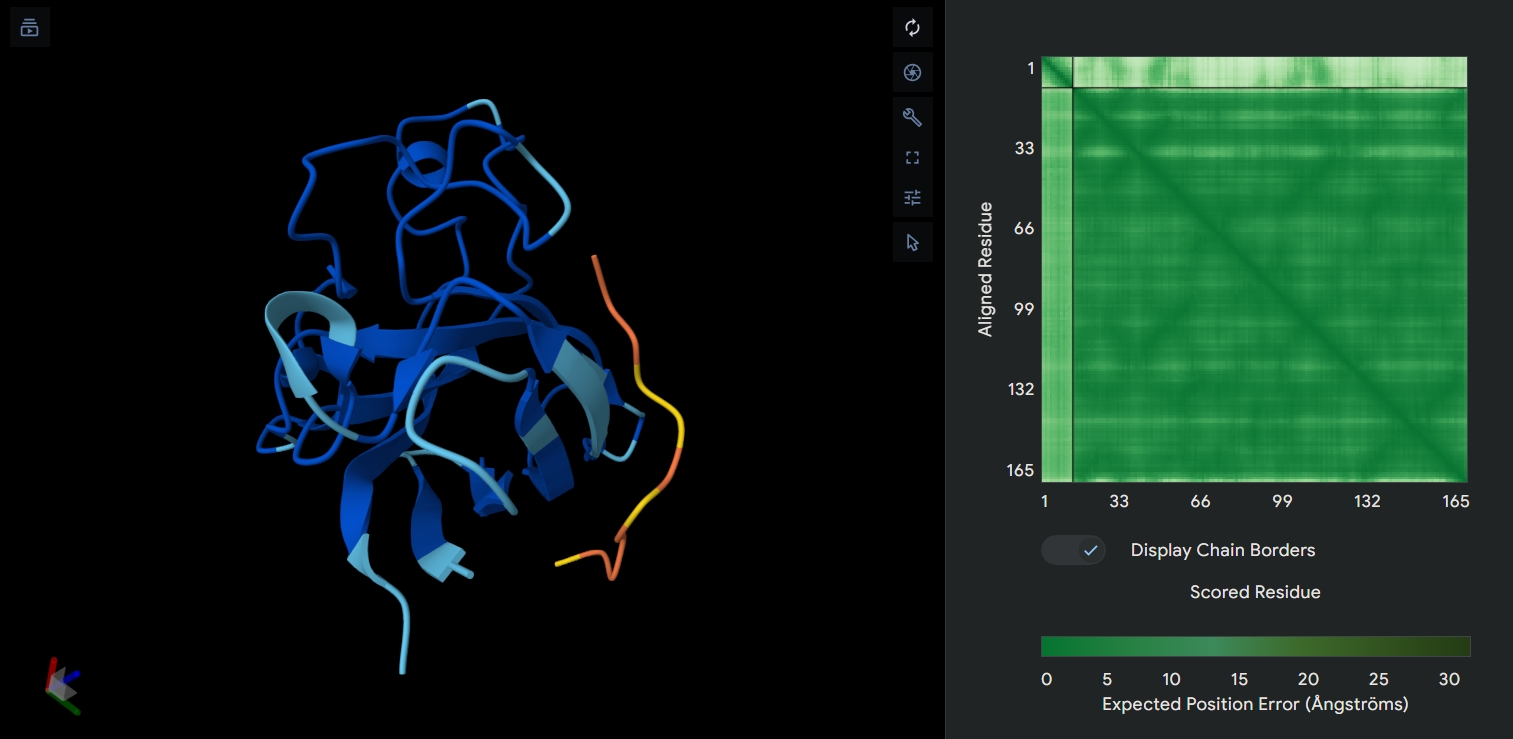
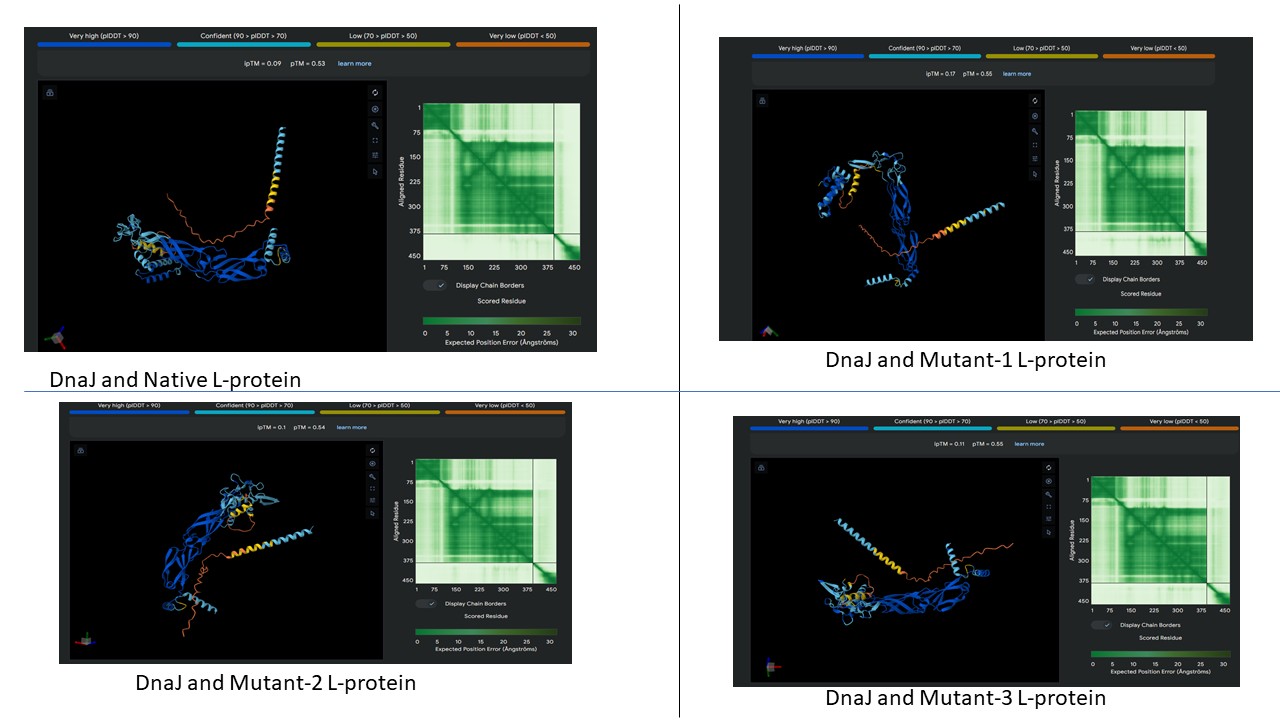
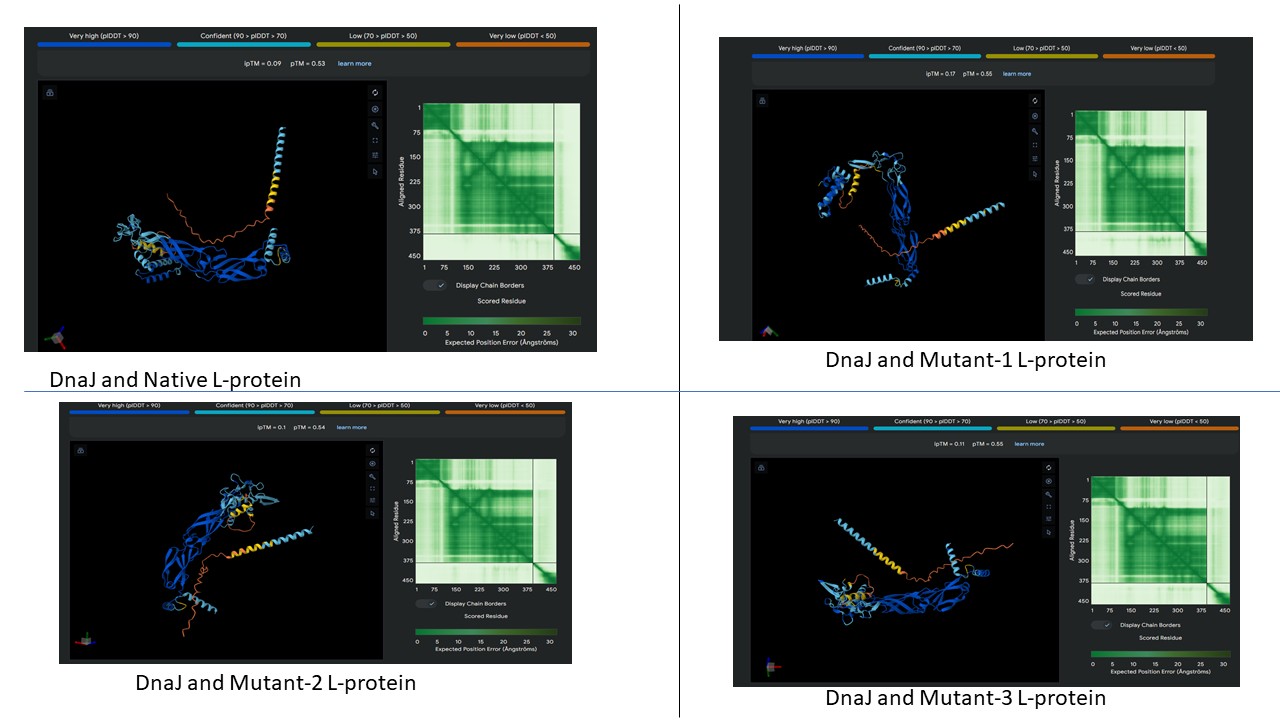
AF2 Multimer was used to co-fold the mutant Lysis protein (METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAFFLSKFTNQLLLSLLEAVIRTVTTLQQLLT) and DnaJ:
Cofolding was not performed for the other two sequences as my laptop started getting stuck while running the program.
The plDDT score indicates that the model is not confident about the folding of the input random mutated L protein.
Overall, it suggests that the random mutation approach is very time consuming to obtain leads.
Later, cofolding was performed using Alphafold server, and the results obtained are shown below:
Week-06-hw-genetic-circuits-part-i
DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion DNA Polymerase: Pyrococcus-like enzyme that contains a fused processivity-enhancing domain. It provides more than 50 gold higher fidelity than Taq polymerase.
dNTPs: contains dATP, dCTP, dGTP, and dTTP that are required for extension reaction of the PCR.
Buffers: MgCl2 as a cofactor for polymerase, KCl and TAPS-HCl ([tris(hydroxymethyl)methylamino]propanesulfonic acid) to maintain ionic strength and pH respectively, and beta-meracaptoethanol to maintain enzyme stability.
Some other components that are provided seperately: DMSO (Dimethyl sulfoxide) to improve denaturation and primer binding, and nuclease free water as a solvent and matrix to avoid denaturation of the DNA.
What are some factors that determine primer annealing temperature during PCR?
Primer melting temperature: annealing temperature must be set around 3 to 5 degree celcius below the lowest melting temperature. So. anything that affects melting temperature also affects annealing temperature. Melting temperature is in turn affected by GC conent and primer length. Higher the GC content, and longer the length of the primer, the higher will be the melting temperature. For short primers (<20 bps) Wallace rule can be used to find the approximate primer melting temperature: $$T_m (°C) = 2(A + T) + 4(G + C)$$
Salt and ion concentration: monovalent cations like Na+ and K+ reduce the repulsion between two DNA strands by nutralizing the negative charge of the phosphate backbone. Mg2+ concentration, which is a cofactor for the polymerase, also increases the stability of the double helix, increasing the melting temperature.
Presence of Denaturants like DMSO and Formamide: They disrupt hydrogen bonds, and reduce the melting temperature.
Degenarate primers (primers that are not 100% match to the template) reduce the melting temperature, and complexity of template DNAs (Eg.; humans as opposed to bacteria) also require a higher annealing temperature to avoid ‘mispriming’.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR vs. Restriction Enzyme Digest
Feature
Polymerase Chain Reaction (PCR)
Restriction Enzyme Digest (e.g., DpnI)
Mechanism
Uses primers and DNA polymerase to amplify specific target regions from a template.
Uses site-specific endonucleases to cleave DNA at specific recognition sequences.
Input DNA
Requires a template plasmid (e.g., mUAV) and synthetic oligonucleotides (primers).
Requires DNA containing specific recognition sites (e.g., methylated GATC for DpnI).
Protocol Steps
Thermal cycling: Includes initial denaturation, followed by cycles of denaturation, annealing, and extension.
Isothermal incubation: Typically a single-step incubation at a specific temperature (e.g., 37°C for 30–60 minutes).
Modifications
Can introduce intentional mismatches for mutagenesis (e.g., amilCP color mutations).
Precise cutting only; cannot “create” new sequences or mutations during the digest.
Selectivity
Amplifies only the region of interest flanked by the forward and reverse primers.
Selectively digests DNA based on sequence and methylation status (e.g., removing parental templates).
When to Prefer PCR vs. Restriction Digest
Situation
Prefer PCR
Prefer Restriction Enzyme Digests (e.g., DpnI)
Creating Mutants
You need to change the color-generating chromophore of a protein by introducing mismatches.
Preparing for Assembly
You are generating linear fragments for Gibson or HiFi assembly that require specific 5’ overhangs.
Targeted Amplification
You need to isolate specific regions like the origin of replication, promoters, or antibiotic resistance genes.
Eliminating Background
You need to remove the original template DNA (mUAV) to ensure only the newly created PCR mutants are used.
Targeting Methylation
You need to distinguish between DNA propagated in E. coli (methylated) and DNA synthesized via PCR (unmethylated).
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Verification of Primer Design:
The success of the assembly depends on the specific architecture of the primers:
Overlaps: Each primer must include a 20–22 bp overhang complementary to the adjoining fragment.
Binding Region: The core binding region should be 18–22 bp.
Melting Temperature (Tₘ): The Tₘ should be between 52–58°C.
Pair Compatibility: Primer pairs should have Tₘ values within 5°C of each other.
GC Content: Aim for 40–60% GC content with a GC clamp (1–2 G/C bases) at the 3′ end.
Secondary Structure: Use software to ensure Gibbs free energy is above –10 kcal to avoid strong hairpins or dimers.
Post-PCR Processing
Before assembly, fragments must be cleaned and templates removed:
DpnI Digestion: Treat PCR reactions with DpnI to eliminate the original mUAV plasmid (digests methylated DNA, preserves unmethylated PCR products).
DNA Purification: Use a purification kit (e.g., Zymo) to remove salts and enzymes.
Quantification: Measure DNA concentration with Nanodrop or Qubit; should be >30 ng/µL.
Quality Control (Diagnostic Gel)
Run samples on an agarose gel at 100 mV for 15 minutes.
Verify that bands match the predicted size calculated on Benchling.
Reaction Parameters
Molar Ratio: Use a 2:1 (insert:vector) molar ratio for optimal efficiency.
Orientation: Confirm fragments have correct 5′ → 3′ orientation with matching overlaps.
How does the plasmid DNA enter the E. coli cells during transformation? It enters through pores in the cell wall. The pores can be created using CaCl2 treatment, followed by heat shock (mixture kept on ice bath is suddenly incubated at 42 degree celcius for 30-90 seconds). Electroporation is another method, where a high-voltage electric pulse applied for a very short duration brefily disrupts the phospholipid bilayer, and simultaneously pushes the DNA molecules through the pores.
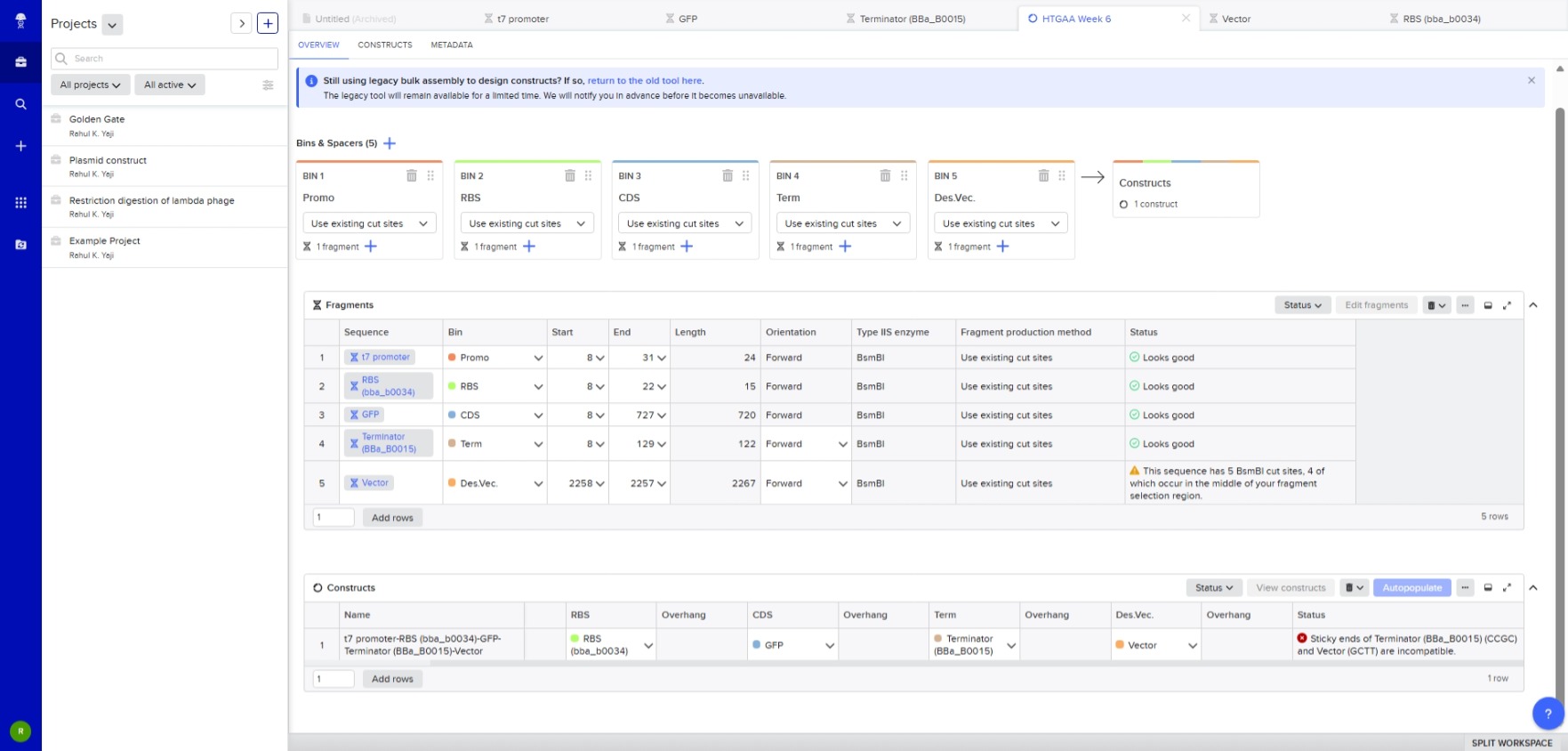
6.1 Describe another assembly method in detail (such as Golden Gate Assembly). Explain the method in 5–7 sentences plus diagrams (either handmade or online). Modular Cloning Method: It is a method based on Golden Gate Assembly. It utilized Type IIS restiction enzymes that cut outside their restrcition site and create non-palindromic overhangs. The final product doesn’t contatin restriciton site, preventing the enyme from double-cutting.
Steps: Step 0: Removal of the internal recognition sites so that the enzyme being used will not cleave it internally, addition of standard 4-bp overhangs and inserting the thus-modified sequence into storage vector. This has to be done seperately for all the units of transcription, i.e., promoter, 5’ UTRs, rbs, cds, terminator.
Step 1: The components of step 0 are added into the reaction vessel, along with the destination vector, restriciton enzyme, T4 ligase, buffer, and ATP. The temperature is cylced to and fro from a higher temperature (~37 degree celcius) for cutting, and a lower temperature (~16 degree celcius) for sticking. The restriction enzyme leaves behind the specific 4-bp overhangs. The DNA ligase binds the 4-bp overhangs in the order of Promoter -> 5’ UTR -> RBS -> CDS -> Terminator in the insertion site of the destination vector, which already contains selection and screening genes.
Step 2: In case a complex metabolic pathway involving multiple genes is to be synthesised, the final desitnation vector of the step 1 is used as storage vector for the step 2, and step 1 is repeated using other genes.
A Notebook was created using the same button to document the homework.
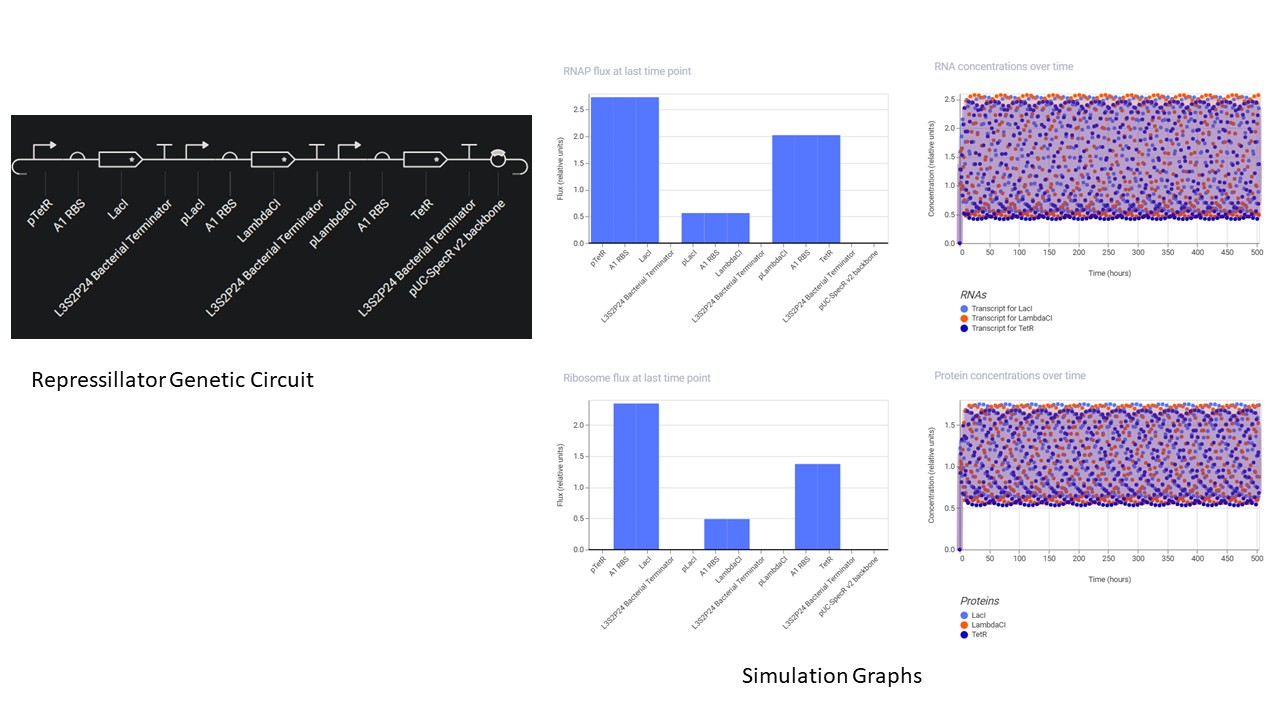
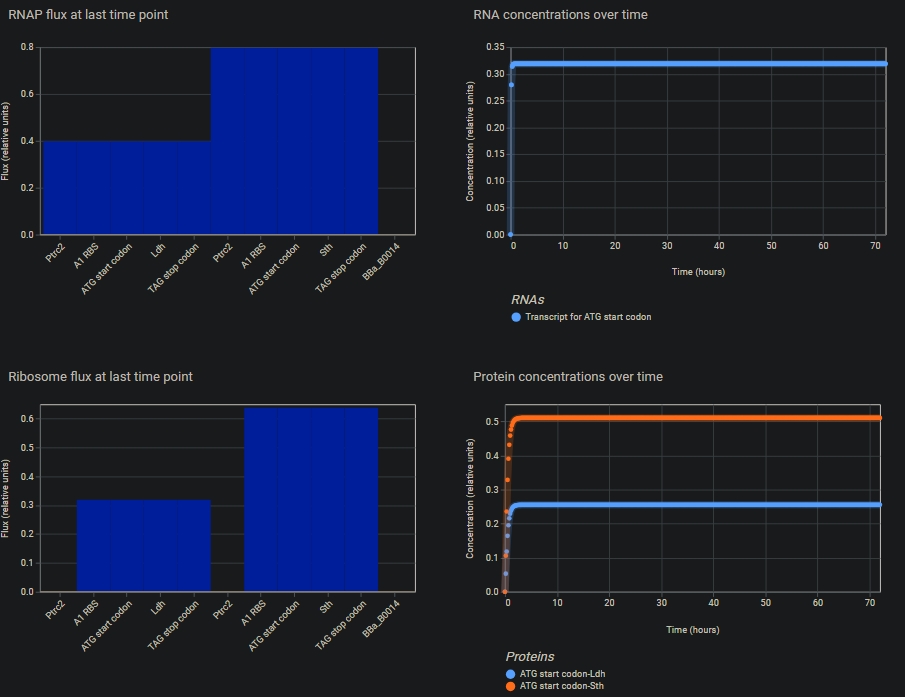
In the notebook, a blank construct was created, the repressillator found in the Demo was recreated part by part.
“Search bar” was used to search for the parts, and they were dragged and dropped at the desired location
Using the simulation option, the repressillator was simulated using the following parameters:
Chassis: E. coli
Duration: 504 hours
Timestep: 60 minutes
Transfection: Transient Transfection
The following output was recorded:
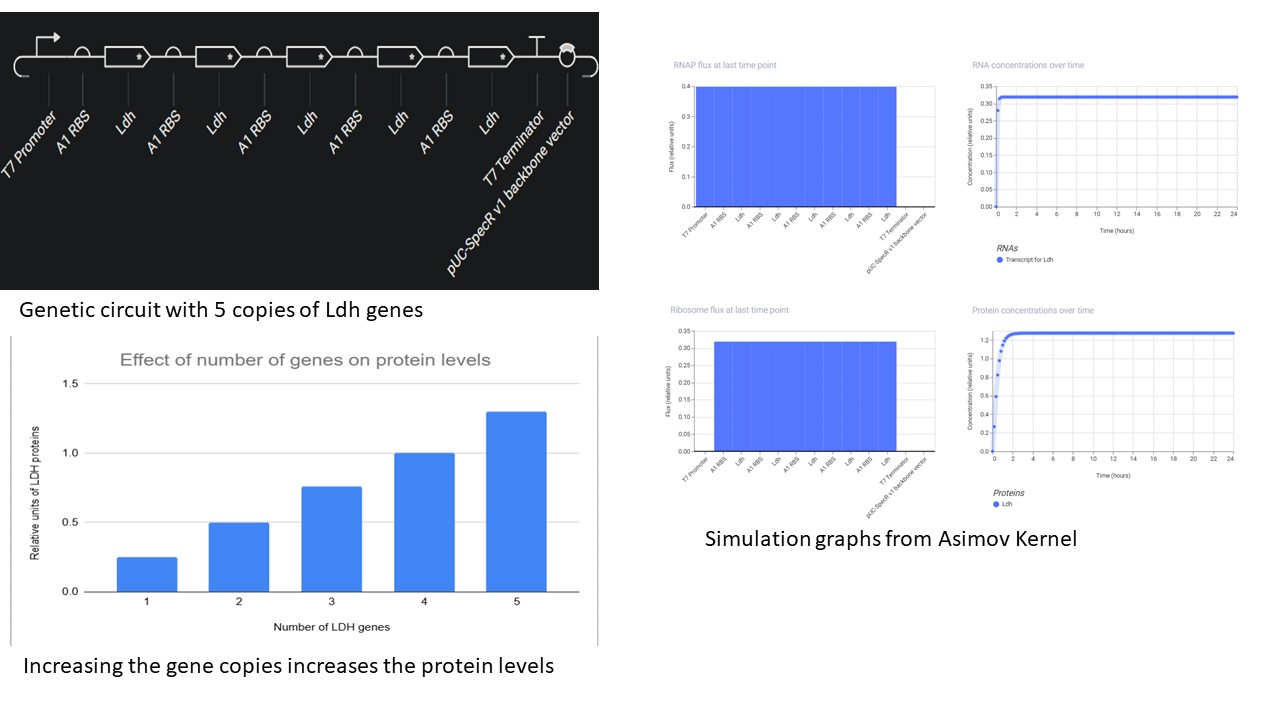
Build three of your own devices using the parts in the Characterized Bacterial Parts Repo and explain how you think the devices should function in an Electronic Notebook Entry. First Part: Overexpression of lactic acid
The nucleotide sequence of the Ldh gene was copied from NCBI, and the start sequence ATG and Stop sequence TAG were manually inserted in the “Create part” option
T7 promoter and terminator were used by creating new parts with the respective sequences taken from Vector Builder
The effect of increasing the number of copies of gene was simulated using the parameters:
Chassis: E. coli
Duration: 24 hours
Timestep: 10 minutes
Transfection: Transient Transfection.
It was found that the more the gene copies, the higher the protein levels.
Another interesting observation was that, the CDS must be followed by an RBS for each copy, even if they are flanked by the same promoter and terminator. Without RBS, it will not be translated and thus the protein levels stay down.
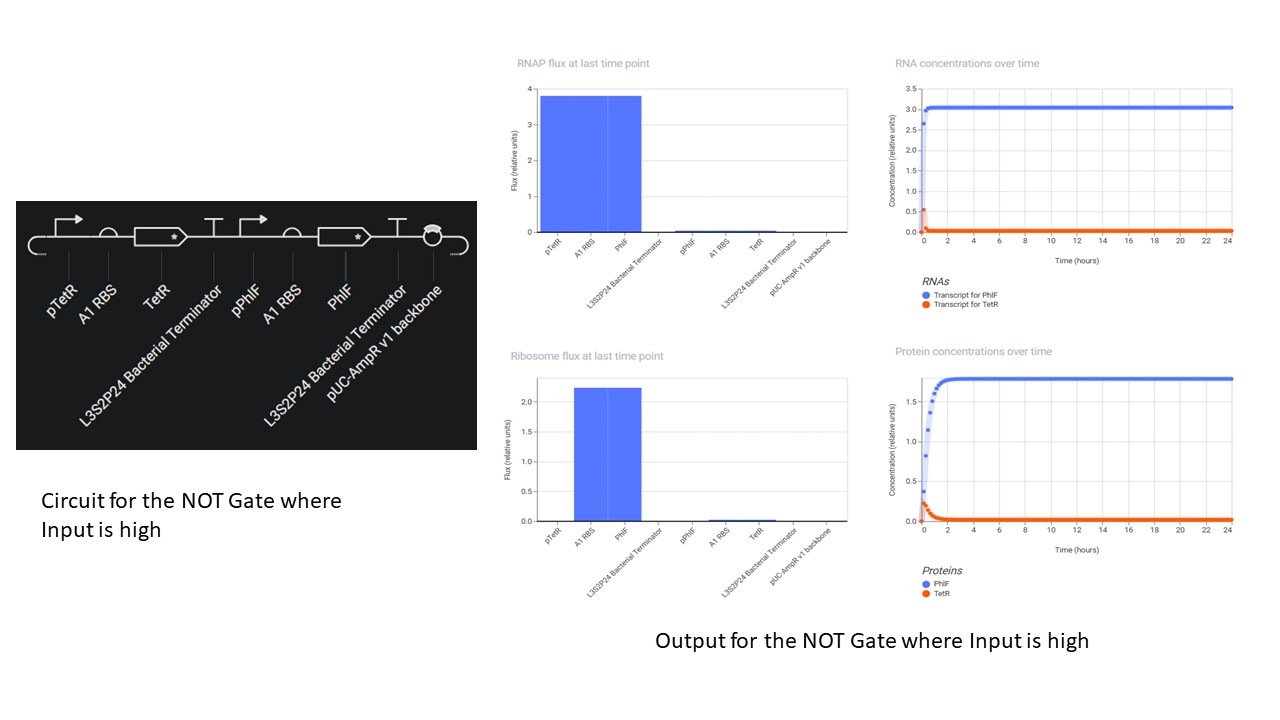
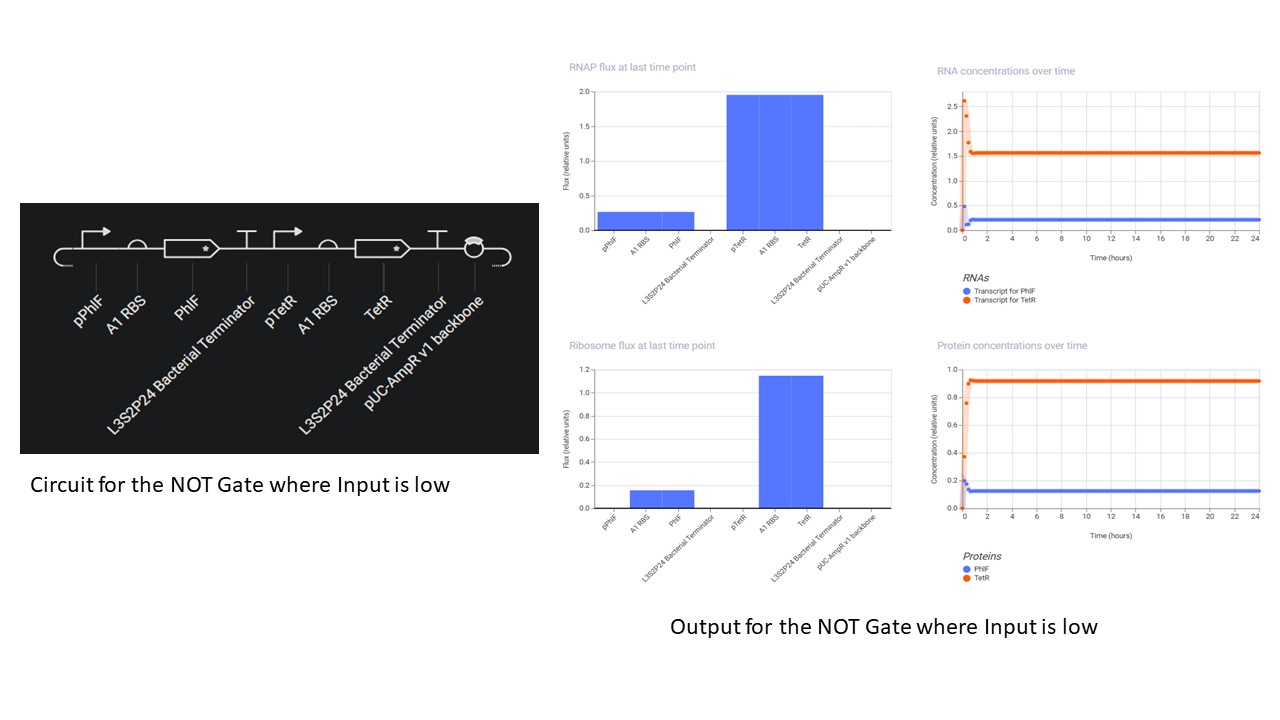
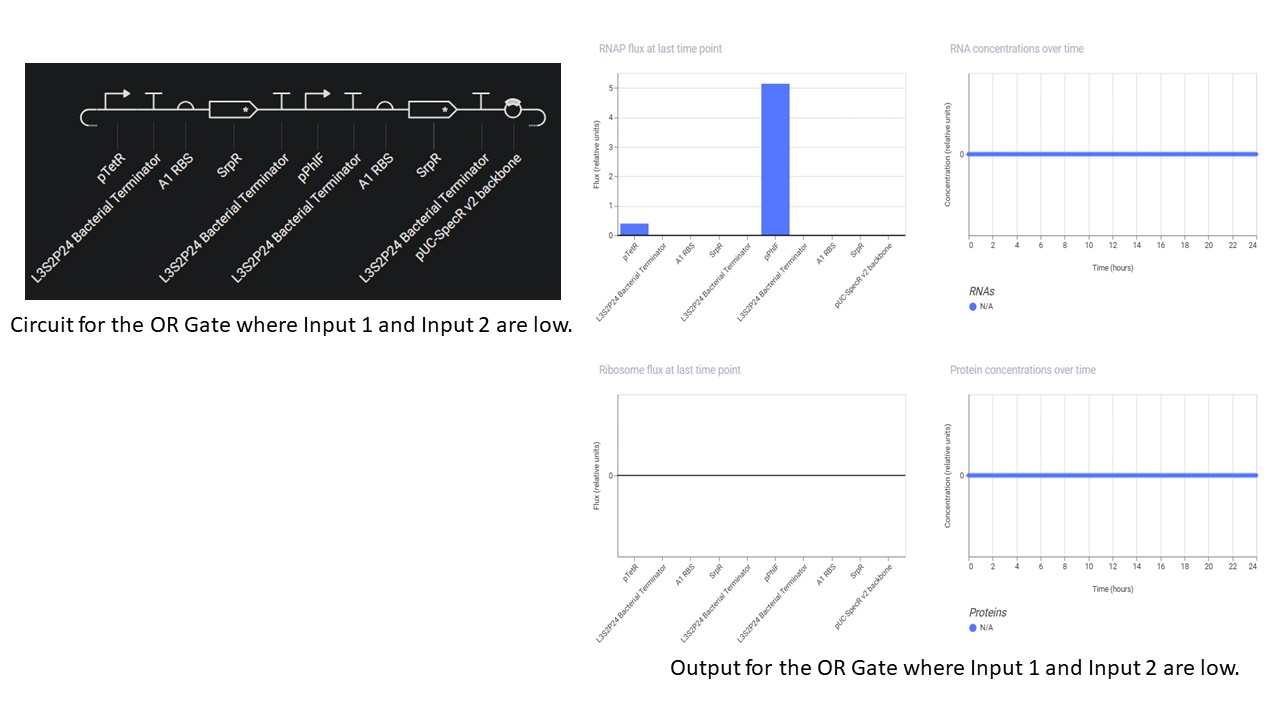
Second Part: Inducerless NOT Gate
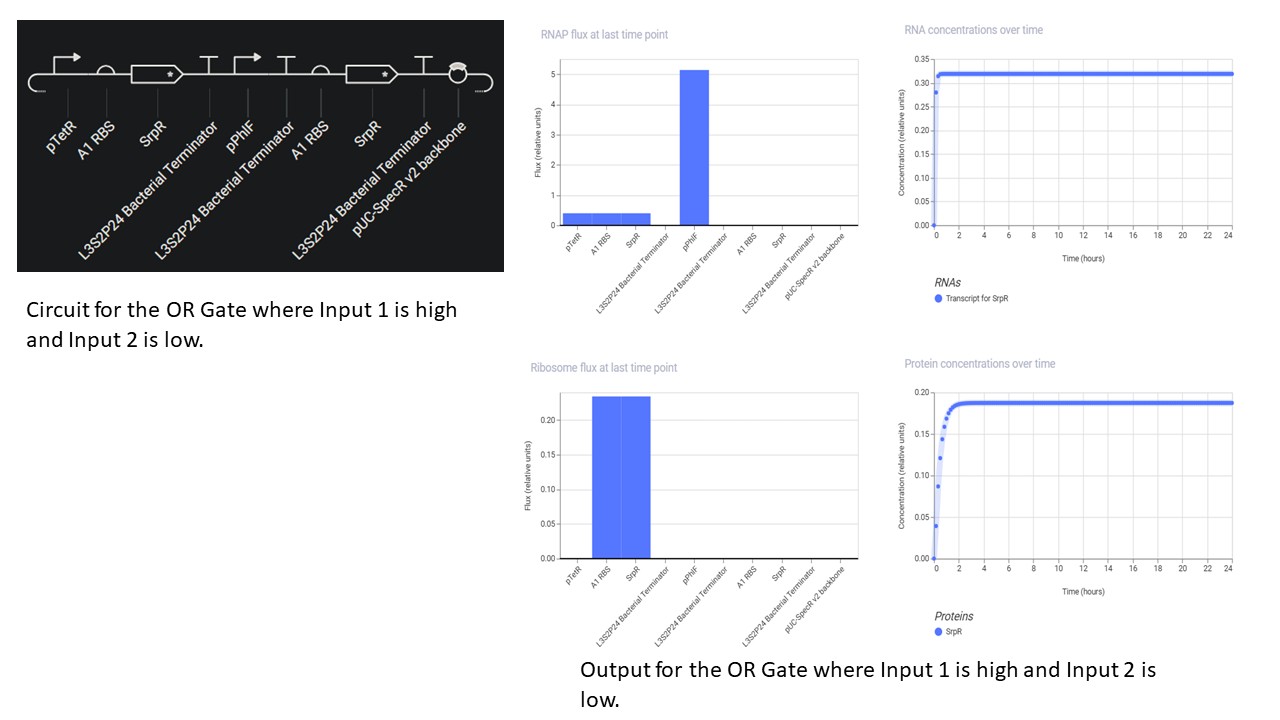
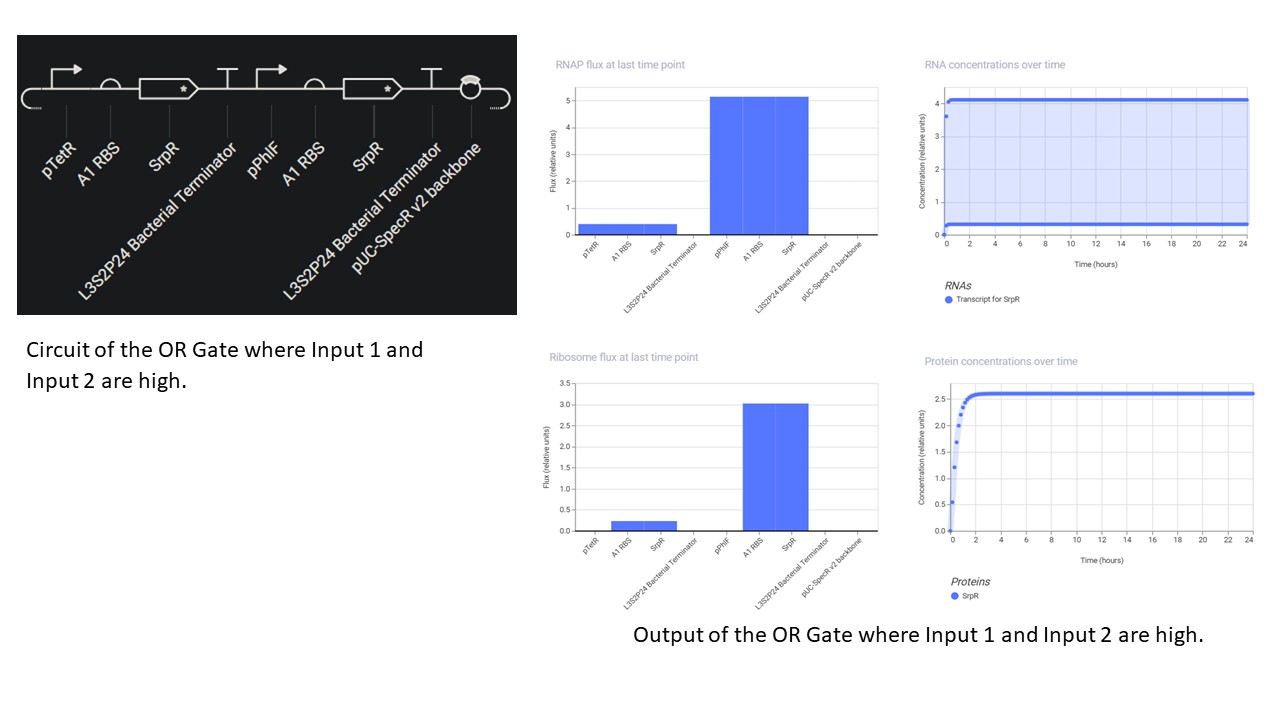
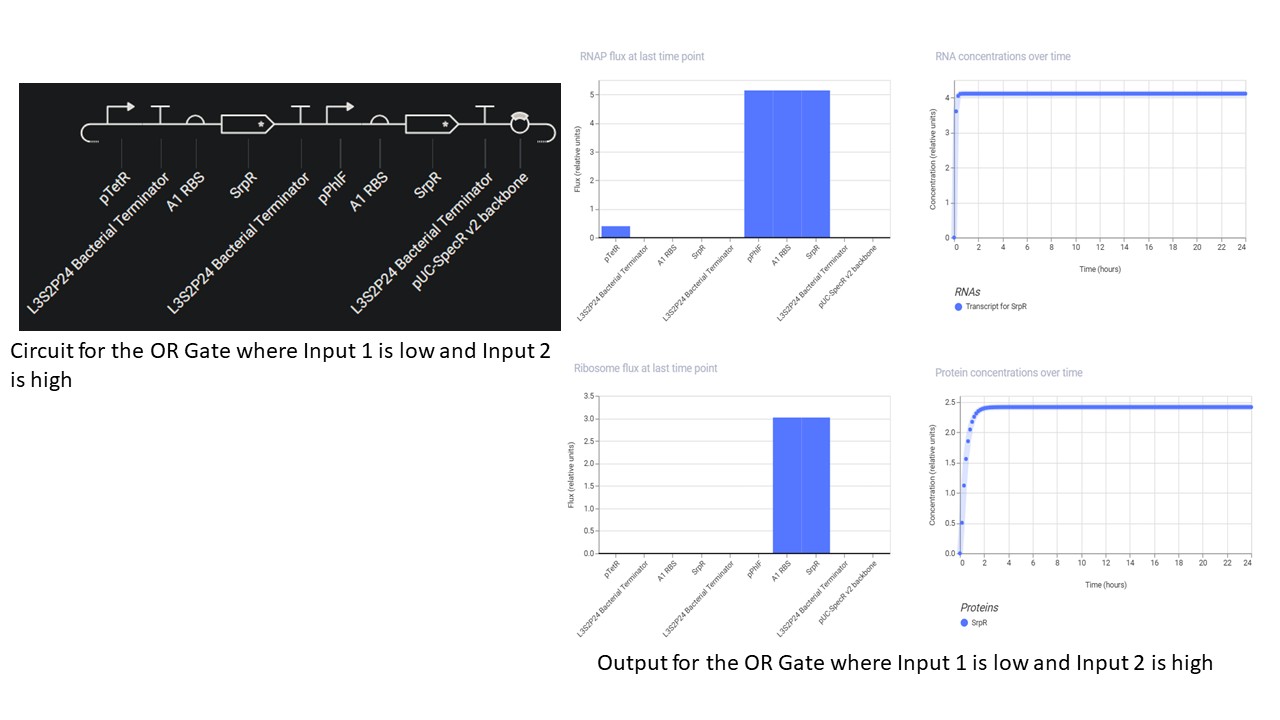
Third Part: Inducerless OR Gate
Week-07-HW-genetic-circuits-part-ii
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
They can interpret a range of inputs as opposed to the 0, 1 inputs of traditional genetic circuits. This allows them to aggregate multiple signals and apply the activation fucntion to filter biological noise.
Traditional circuits often require a cascade of genetic logic gates, which lead to metabolic burden and competition for substrates. By utilizing weighted interactions, IANNs can accomplish the same task using fewer biolocial components.
Nonlinear descision making is a struggle for tradional genetic circuits. They struggle to take into accout the relative ratios and thresholds of a multitude of proteins simultaneously, limiting themselves to simple linear logics. However, using ReLU and sigmoid -like activation behaviours, IANNs can perform complex tasks. Eg: A cell may be engineered to apoptosize only when a commplex profile of cancer markers are met, as oppossed to the presence of some of those markers that may not be cancerous.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal. A useful applicaiton of IANN would be rapid plant cell response when it is infected by a pathogen.
Input 1: Detection of Pathogen Associated Molecular Patterns. Chitin would be a good choice given that fungi are the most damaging plant pathogens.
Input 2: Detection of Plant Stress Volatiles like Methyl Salicylate. This adds one more layer of confirmation that the plant is under attack.
Input 3: Detection of Effector Proteins like Avr4 that are used by fungi to protect itself fromm plant defense mechanisms.
Different weights need to be assigned for different inputs. In this case, input 3 may be given more weightage compared to input 1.
The output may be in the form of a targeted release of antifungal peptide or apoptosis.
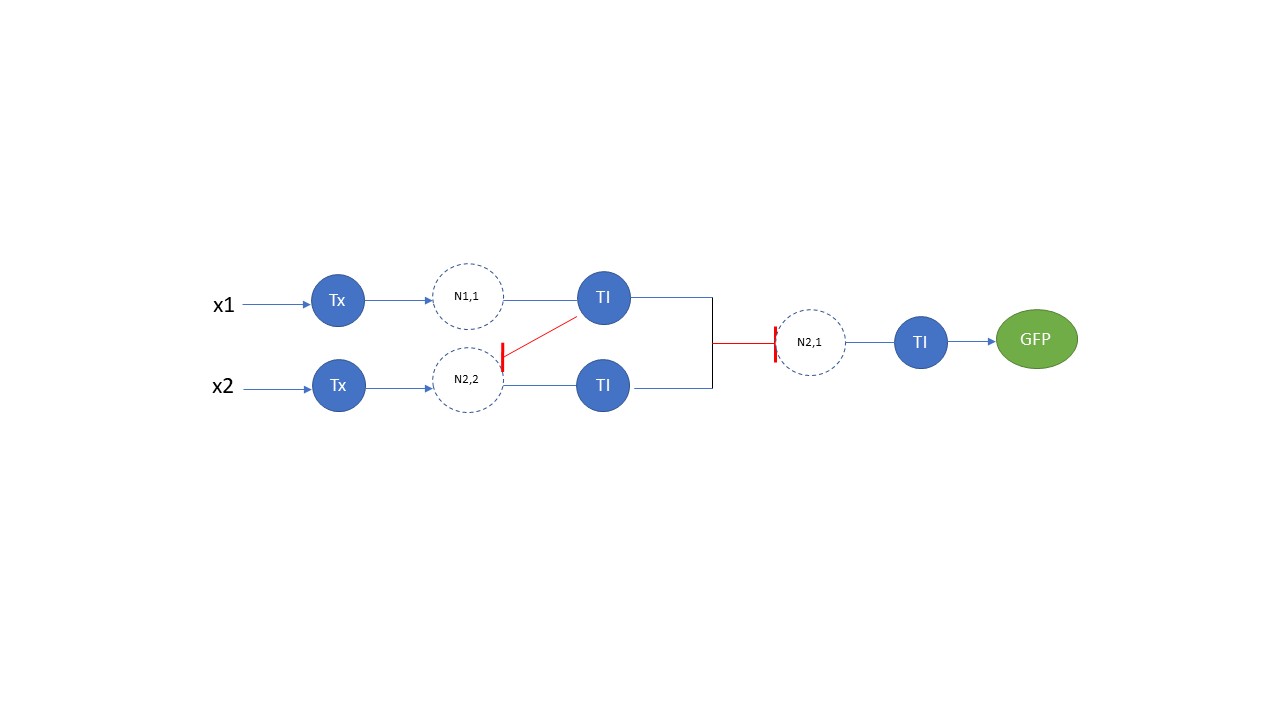
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Mycelium Leather: A sustainable alternative to traditional leather. Unlike the latter, it can be produced in as little as 5 days, and is biodegradable too. It can be treated with different chemicals to make it waterproof, weatherproof, and damage resistant. The chief disadvantage is that it is less robust than the animal-based ones and since it requires a controlled enviornment, the production costs will be on a higher side.
Mycellium-based composites: Organic wastes, especially agriculture wastes like wood chips, straws etc. are used as substrates to grow the fungi. The fungl mycellium holds the substrate together and the resulting material, after killing the fungi by baking, is called ‘MBC’. It finds its use in numerous fields such as packaging, insulation, construction materials etc. The primary disadvantage is that it is difficult to scale, and therefore, is not cost-effective. Some companies like Evocative, Mycoworks, and Mogu are working on MBCs.
Martian shelters: NASA is working on a system where the astronauts carry dormant fungi and a mould. When activated with water, the fungi grows around the mould, forming a fully functional human habitat. Prototypes have been built using Ganoderma lucidum, and have shown significant potential for water filtration, bioluminescent lighting, and self-repair. Additionally, pound for pound, mycellium-based builidng materials can outperform concrete in terms of strength.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria? Fungi can be better chassis organisms for genetic engineering than bacteria as they possess eukaryotic cell machinery, and are capable of post-translational modificaitons. The latter can be exploited for glycosylation of proteins, especially of antibodies; and phosphorylation and acetylation for protein modification. This, coupled with the fact that they are capable of advanced protein folding makes them the organism of choice to produce complex human proteins.
Additionally, fungi have superior secretion capacity that is complemented by compartmentalization. They can sequester toxic intermediates in organelles like vacuoles and peroxisomes and allow the cell to secrete high concentrations of desired chemicals that would have been lethal if it were to be found in cytoplasm. Their superior secretion capacity allows them to produce chemicals in “grams per litres” concentration. Moreover, the chemicals are usually secreted outside the cell, saving us the cost of cell-disruption and simplyfying the purification.
Even the growth requirement of fungi is more robust and adaptive compared to the bacteria. They can be grown on solid substrate with minimal additions, and can tolerate acidic enviornments better.
I would love to engineer fungi to produce biological selenomelanin- a type of melanin that incorporates selenium instead of sulphur. Fungi can be engineered to utilize selenocystine for the bioproduciton of selenomelanin. Also, fungal mycellium can be engineered to produce selenomelanin to proivde superior radiation protection to be used as martian shelters.
Assignment Part 3: First DNA Twist Order
Review the Individual Final Project documentation guidelines. Submit this Google Form with your draft Aim 1, final project summary, HTGAA industry council selections, and shared folder for DNA designs.
Lorem ipsum
Review Part 3: DNA Design Challenge of the week 2 homework. Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above.
Lorem ipsum
Document the backbone vector it will be synthesized in on your website.
Lorem ipsum
week-09-hw-cell-free-systems
Part A: General & Lecturer-Specific Questions
General Homework Questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables.
Rapid Iteration and Throughput
Direct Use of Linear DNA Templates Traditional methods require time-consuming cloning of DNA into circular plasmids before they can be inserted into a host cell. In CFPS, you can use raw PCR products directly as the instruction manual, allowing you to move from a genetic design to a functional protein in just a few hours.
High-Throughput Screening Compatibility Because the reaction occurs in a simple liquid phase without the need for incubator space or shaking flasks, it can be easily scaled down into 96-well or 384-well plates. This allows robotic systems to simultaneously test hundreds of different protein variants or reaction conditions under identical parameters.
Elimination of Cell Recovery and Lysis In living systems, you must wait for the culture to reach a specific density and then physically break the cells open to harvest the protein. CFPS skips these steps entirely because the protein is synthesized
Name at least two cases where cell-free expression is more beneficial than cell production.
Production of Cytotoxic Proteins In traditional in vivo production, the target protein often interferes with the host cell’s survival. For example, if you are trying to produce antimicrobial peptides (AMPs) or pore-forming toxins, the protein will kill the E. coli or yeast “factory” as soon as it is expressed, leading to zero yield. CFPS Benefit: Since there is no living cell to keep alive, the system is indifferent to the toxicity of the product. This allows for the high-titer production of potent toxins, lytic enzymes, and other proteins that are normally “undruggable” or unproduceable in living hosts.
Incorporation of Non-Canonical Amino Acids (ncAAs) If you want to create a “designer” protein with chemical properties not found in nature—such as adding a fluorescent tag, a “click-chemistry” handle, or a post-translational modification—you must use ncAAs. In a living cell, this requires complex metabolic engineering to ensure the cell doesn’t accidentally incorporate the synthetic amino acid into its own essential proteins, which would be lethal. CFPS Benefit: You can directly manipulate the translation machinery by adding pre-charged tRNAs and orthogonal synthetases without worrying about cross-reactivity with the host’s proteome. This provides a high degree of “chemical site-specificity,” allowing for the production of sophisticated protein-drug conjugates and advanced biomaterials.
Describe the main components of a cell-free expression system and explain the role of each component. Main Components of a Cell-Free Expression System
The Crude Extract (The Machinery) The extract is the heart of the system, typically derived from cells like E. coli, wheat germ, or rabbit reticulocytes that have been physically lysed. Role: Provides the essential molecular “hardware” required for translation, including ribosomes, tRNAs, aminoacyl-tRNA synthetases, and translation factors. It also contains endogenous enzymes needed for energy regeneration.
The DNA Template (The Instructions) This is the genetic blueprint for the protein you want to synthesize. Unlike in vivo methods, this can be a circular plasmid or a simple linear PCR product. Role: Contains promoter and terminator sequences that tell the machinery where to start and stop. Serves as the instruction manual for mRNA production (transcription) and subsequent protein synthesis (translation).
Energy Regeneration System Protein synthesis is energetically expensive. Since the system is no longer part of a living cell, it cannot “eat” or perform cellular respiration to stay powered. Role: Typically consists of high-energy phosphate compounds (like phosphoenolpyruvate or creatine phosphate) and corresponding kinases. Acts as a “battery pack” to continuously regenerate ATP and GTP, which are consumed during amino acid chain assembly.
Substrates and Cofactors (The Building Blocks) These are the raw materials added to the reaction buffer to facilitate biochemical reactions.
Amino Acids: The 20 standard building blocks or even non-canonical ones, used to assemble the protein chain.
Nucleotides (NTPs): Used for transcribing DNA into mRNA and as energy carriers.
Salts and Buffers: Magnesium ($Mg^{2+}$) and potassium ($K^{+}$) ions are strictly required for ribosome stability and enzymatic activity, while buffers maintain a stable pH.
Why is energy provision/regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy Demand of Translation Each amino acid added to a growing peptide chain consumes two ATP (for tRNA charging) and two GTP (for ribosome movement), making protein synthesis one of the most energy-intensive processes in biochemistry.
Risk of Rapid Depletion In a closed system without recycling, the initial ATP/GTP pool would be exhausted within minutes, stalling protein production. Accumulated phosphate byproducts can also bind magnesium ions, destabilizing the reaction.
Enzymatic Regeneration Pathways We can add high-energy donor molecules (e.g., phosphoenolpyruvate or creatine phosphate) with kinases like pyruvate kinase. These enzymes recycle ADP back into ATP, acting as a biological “battery chagrer.”
Dialysis-Based Continuous Supply Advanced setups use semi-permeable membranes to allow fresh nutrients and ATP to diffuse in while removing inhibitory byproducts. This maintains chemical equilibrium and enables sustained protein synthesis for days.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why. Comparison of Prokaryotic vs. Eukaryotic Cell-Free Systems
Example 1: Prokaryotic System (E. coli)
Protein to Produce: Green Fluorescent Protein (GFP)
Reasoning: GFP is small, robust, and does not require post-translational modifications to fluoresce.
Efficiency: E. coli extracts have the highest translation rates, enabling vast quantities of GFP production within hours.
Monitoring: Fluorescence can be tracked in real-time. GFP serves as an ideal reporter protein for testing new cell-free reaction conditions or energy regeneration strategies, since its folding is simple enough for bacterial machinery.
Example 2: Eukaryotic System (CHO Cells)
Protein to Produce: Tissue Plasminogen Activator (tPA)
Reasoning: tPA is a complex human enzyme used to dissolve blood clots and is difficult to produce in bacteria.
Disulfide Bonding: Contains 17 disulfide bonds. Bacterial cytoplasm is highly reducing and fails to form these correctly. Eukaryotic extracts with microsomal membranes provide the oxidative environment and chaperones (e.g., Protein Disulfide Isomerase) for proper folding.
Glycosylation: Requires specific sugar chains for stability and activity in the human body. Eukaryotic cell-free systems can be supplemented with microsomes (ER-derived vesicles) to perform these modifications, which are impossible in E. coli systems.
I have tried to sum up the advantages and disadvantages comparision of both expression systems here:
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
The key factors required to design a cell-free experiment for membrane proteins are:
1. Artificial Lipid Environments Membrane proteins are usually hydrophobic and require a lipid bilayer to fold correctly. In cell-free systems, researchers introduce artificial lipid structures such as liposomes, nanodiscs, or microsomes to mimic the natural membrane. These environments stabilize the protein during synthesis and facilitate proper insertion and folding. The MEMPLEX platform, for example, generates thousands of lipid-protein combinations to identify optimal conditions for each membrane protein.
2. Controlled Chemical Interactions Since CFPS allows precise control over the chemical environmentm, we can independently vary lipid composition, ionic strength, redox potential, and chaperone concentrations. This enables the fine-tuning of protein-protein and protein-lipid interactions, which are critical for membrane protein stability and functionality. MEMPLEX uses machine learning to predict and optimize these combinations, accelerating the design of functional synthetic environments.
The following problems may be encountered:
Challenge A: Protein Aggregation and Misfolding Hydrophobic transmembrane helices tend to clump together or stick to the tube walls without a lipid bilayer. Solution: Implement nanodiscs — small, uniform discoidal bilayers wrapped in membrane scaffold proteins (MSPs). Unlike liposomes, nanodiscs keep membrane proteins soluble and monomeric, making them ideal for structural studies such as Cryo-EM.
Challenge B: Low Yields due to Resource Depletion Membrane protein synthesis is slower and consumes more energy than soluble protein synthesis, leading to rapid depletion of ATP and accumulation of inhibitory byproducts. Solution: Use the Continuous-Exchange Cell-Free (CECF) method. A dialysis membrane provides a constant supply of ATP and nutrients while removing inorganic phosphate, sustaining the reaction for complex protein folding.
Challenge C: Maintaining Correct Orientation In vitro systems lack the natural “inside-outside” topology of living cells, so proteins may insert incorrectly into synthetic membranes. Solution: Adjust the physicochemical environment by tuning lipid ratios (e.g., phosphatidylethanolamine [PE] or phosphatidylglycerol [PG]) to encourage the Positive-Inside Rule. Supplement with purified chaperones (e.g., DnaK, GroEL) to keep proteins flexible until proper orientation is achieved.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Common Reasons for Low Protein Yield in CFPS and Fixes
Rapid Template Degradation Crude extracts tend to contain nucleases that degrade linear DNA templates before transcription. Fixes:
Add nuclease inhibitors (e.g., GamS to block RecBCD).
Switch to circular plasmid DNA, which is more resistant to degradation, but is slower to replicate.
Magnesium Ion ($Mg^{2+}$) Imbalance Magnesium stabilizes ribosomes and enzymes, but its optimal range is narrow (8–15 mM). Too little causes ribosome collapse; too much causes mRNA aggregation. ATP breakdown also sequesters magnesium mid-reaction. Fixes:
Perform magnesium titration (e.g., 2 mM increments in 96-well plates).
Use stronger buffers (HEPES or Tris) to maintain pH and magnesium solubility.
Inefficient Protein Folding or Aggregation This is usually the main culprit when complex proteins with disulfide bonds are involved. They may misfold or aggregate at high local concentrations. Fixes:
Lower reaction temperature (e.g., from 37°C to 25–20°C) to slow synthesis and allow proper folding.
Add molecular chaperones (e.g., DnaK, DnaJ, GroEL/ES) to prevent aggregation and assist folding.
Kate Adamala’s Question
Design an example of a useful synthetic minimal cell:
Pick a function and describe it (input/output).
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Could this function be realized by a genetically modified natural cell?
Describe the desired outcome of your synthetic cell operation.
Design all components (membrane composition, encapsulated molecules, Tx/Tl source organism).
How will your synthetic cell communicate with the environment?
Provide experimental details (lipids, genes, measurement method).
Designing a Microsynthetic Methanogen
1. Function, Input, and Output
Function: Acetoclastic Methanogenesis (converting acetate to methane).
Input: Acetate (from pre-processed biomass slurry). (I thought of going through just puliverized biomass. But the engineering becomes too complex or even unviable.)
Output: ($CH_4$) and Carbon Dioxide ($CO_2$).
2. Can this be realized by cell-free Tx/Tl alone?
No. Methanogenesis requires a proton motive force ($\Delta p$) across a lipid bilayer. In open Tx/Tl systems, ions dissipate and energy cycles fail.
3. Can this be realized by a genetically modified natural cell?
Yes, but inefficient. Natural methanogens are slow-growing, strictly anaerobic, and spend energy on survival. A synthetic cell directs all flux toward gas production.
4. Desired Outcome
A stable “biocatalytic bead” added to anaerobic digesters to accelerate acetate-to-methane conversion, bypassing microbial growth limitations.
Small Molecules: Coenzyme M (HS-CoM), Coenzyme B (HS-CoB) pre-loaded for methane production.
Communication (Permeability)
Substrate Entry: Acetate transporter AatP.
Product Exit: Methane ($CH_4$) diffuses naturally through the bilayer.
6. Experimental Details
Category
Component / Gene
Role
Lipids
POPC / Cholesterol (80:20)
Structural bilayer with gas retention
Gene 1
ackA (Methanosarcina)
Acetate Kinase → Acetyl-P
Gene 2
pta (Methanosarcina)
Phosphotransacetylase → Acetyl-CoA
Gene 3
mcrA, mcrB, mcrG (MCR Operon)
Methyl-coenzyme M reductase → $CH_4$
Gene 4
aatP (B. subtilis)
Acetate transporter
7. Measuring Function
Gas Chromatography (GC): Quantify methane volume and purity from headspace.
pH Fluorescent Probes: Encapsulate Pyranine (HPTS) to detect proton movement across the membrane.
Radioactive Labeling ($^{14}C$): Track conversion of $^{14}C$-acetate into labeled methane for definitive proof.
Peter Nguyen’s Question
Freeze-dried cell-free systems can be incorporated into materials. Choose one field (Architecture, Textiles/Fashion, or Robotics) and propose an application: 1. Write a one-sentence pitch: A “living” Martian masonry system composed of regolith bricks held together by mycellium, embedded with freeze-dried, cell-free biosensors that detect structural micro-fractures and signal repair needs via bioluminescence before catastrophic failure occurs.
2. Explain how the idea works in detail:
The core of this application is the integration of freeze-dried cell-free (FD-CF) machinery into the binder of Martian 3D-printed regolith. During the manufacturing of these mycellium-based bricks, a stabilized E. coli lysate containing the genetic instructoins for Luciferase is mixed with a porous substrate.
These components remain dormant in the hyper-arid, cold Martian environment. If a structural micro-crack forms, it allows a small amount of pressurized “habitat air” (containing water vapor and localized heat) to reach the FD-CF pocket. This moisture acts as the trigger, rehydrating the system. Upon activation, the machinery translates the bioluminescent protein, causing the crack to glow brightly against the dark Martian regolith, acting as an autonomous, self-powered alarm system for astronauts.
4. Identify the societal challenge or market need addressed.
The primary challenge in Martian architecture is human safety in extreme environments. Unlike Earth, a hairline crack on Mars can lead to explosive decompression or lethal radiation exposure. Current electronic sensors require extensive wiring, constant power, and are prone to radiation interference.
There is a need for passive, zero-energy monitoring systems that are lightweight to transport. By using cell-free systems, we eliminate the need to keep biological organisms alive during the 7-month space transit, providing a “just-expose-to-moisture” safety net for the first Martian colonies.
5. Discuss how you would overcome limitations of cell-free reactions (activation, stability, one-time use).
Activation Control
To prevent accidental activation from ambient habitat humidity, the FD-CF components are encapsulated in hygroscopic wax microspheres. These spheres only melt or dissolve when exposed to the specific temperature and moisture profile of a localized structural leak, ensuring the “bio-sensor” only fires when a true breach occurs.
Stability and Longevity
Space radiation is the biggest threat to biological molecules. We will try to address this by incorporating lyoprotectants (like trehalose) and polyphenolic antioxidants into the freeze-drying mix. Whether these can stabilize the protein machinery and DNA templates in a “glassy state,” allowing them to remain viable for years in the Martian crust without denaturing, must be validated experimentally.
One-Time Use to Repeatable Use
While a single cell-free reaction is typically “one-shot,” we can think of designing the material with modular “casings.” Each brick contains hundreds of isolated micro-pockets of extract. If one pocket is used to signal a crack and the crack is then patched, the surrounding unused pockets remain dormant. This “redundancy-by-design” ensures the material provides monitoring capabilities throughout the lifespan of the building, despite the one-time-use nature of each individual biochemical reaction. However, we can expect the cost to be very high, and must be adressed suitably.
Ally Huang’s Question
Develop a mock Genes in Space proposal using BioBits® (and optionally miniPCR® and P51 viewer):
Background information (≤100 words): Monitoring structural integrity in space-grown “myco-architecture” is vital for long-duration missions. While mycelium-regolith composites are promising, they face structural stress from internal pressurization and radiation. BioBits®—a freeze-dried cell-free (FD-CF) system—enables biological sensing without the logistical burden of keeping cells alive. This experiment, designed for the ISS, seeks to validate whether FD-CF machinery, embedded in a fungal-mineral matrix, can undergo autonomous rehydration and protein synthesis in microgravity. Proving this confirms the feasibility of using “living” bricks that glow to warn astronauts of pressure loss or radiation spikes.
Molecular/genetic target (≤30 words): A DNA plasmid encoding T7 RNA Polymerase and the mCherry fluorescent protein under a T7 promoter, optimized for detection via the P51 viewer and miniPCR® validation.
Relation to space biology challenge (≤100 words): The primary challenge is the stability of biological hardware in a high-radiation, microgravity environment. Traditional sensors rely on electronics that are heavy and sensitive to galactic cosmic rays. This project tests if BioBits® can survive the “launch-to-activation” timeline while embedded in a porous, fungal-regolith matrix. Validating this on the ISS addresses the need for low-mass, zero-power safety systems. It also explores how microgravity affects the diffusion of rehydrating fluids within the unique capillary structures of desiccated mycelium, a critical factor for sensor response time in orbit.
Hypothesis/research goal with reasoning (≤150 words): Hypothesis: Microgravity will not significantly inhibit the rehydration-induced activation of BioBits® within a mycelium-regolith matrix, and the fungal chitin will provide a protective micro-environment against ISS-level ionizing radiation.
Reasoning: In microgravity, fluid dynamics are dominated by surface tension rather than gravity-driven flow. We can hypothesize that the natural porosity of the mycelium will facilitate uniform rehydration of the FD-CF pellets via capillary action. Annd also, the molecular density of the regolith and the melanin content in the fungal cell walls should shield the DNA and ribosomes from radiation damage during their “dormant” phase. The goal will be to compare the fluorescence kinetics (speed and brightness) of the space-activated samples against Earth-based controls to determine if the lack of convection in microgravity slows down the metabolic-like reaction of the cell-free system.
Experimental plan (samples, controls, data collection, ≤100 words):
Samples: Three mycelium-regolith cubes containing embedded BioBits® pellets and the mCherry DNA circuit.
Controls: One “Dry” brick (unactivated) and one “Wet” brick (Earth-activated) as baselines.
Execution: Use a sealed MWA (Maintenance Work Area) to inject 100 μL of rehydration buffer into the bricks via syringe to simulate a localized atmospheric leak.
Data Collection: Cubes are placed into the P51 viewer; astronauts take time-lapse photos to track fluorescence development. Finally, miniPCR® will amplify the mCherry gene from the “Dry” brick to assess DNA degradation during the flight.
Part B: Individual Final Project
Decide and write down Aim 1 of your final project. Answer:
Add your chosen final project slide to the appropriate deck. (Attach or link slide here)
Submit the Final Project selection form if not already done. (Confirmation note here)
Begin planning documentation based on provided guidelines. (Notes here)
Prepare your first DNA order and place it in the correct Twist tab (deadline varies by group). (Details here)
week-10-hw-Imaging-and-Measurement
Homework: Final Project
1. Identify at least one aspect of your project that you will measure. Answer:
The expression level of the L lactate dehydrogenase Gene
The concentration of lactic acid
Cell growth
2. What technologies will you use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry)? Answer: RNA sequencing, RT qPCR, and OD600 etc.
Homework: Waters Part 1 — Molecular Weight
1. Based on the predicted amino acid sequence of eGFP, what is the calculated molecular weight? Answer: 27183.76 kda (after removing the H tag)
2. Select two charge states from the BioAccord data and determine z for each. Answer: I chose the peaks 903.7148 and 933.7349.
3. Calculate the MW of the protein using the relationship between m/z, MW, and z. Answer: z = (903.7148-1)/(933.7349-903.7148) = 30.07 Mw = 30.07 * (933.7319-1) = 28047.25 kda
4. Calculate the mass accuracy of the measurement. Answer: Mass Accuracy = (|28047.25-27183.76| / 28047.25) * 1000000 = 35464.8 PPM
Homework: Waters Part 3 — Peptide Map
1. How many Lysines (K) and Arginines (R) are in eGFP? Answer: 20 and 6
2. How many peptides will be generated from tryptic digestion of eGFP? Answer: 19
3. Based on LC-MS data, how many chromatographic peaks do you see between 0.5 and 6 minutes? Answer: 19
4. Does the number of peaks match the number of peptides predicted? Answer: Yes
5. Identify the m/z of the peptide shown in Figure 3b. What is the charge (z) of the most abundant charge state of the peptide? Calculate the mass of the singly charged form of the peptide ([M+H]+).
Answer:
Identify m/z
Observed peak: 525.76
Determine charge state (z)
Isotope spacing ≈ 0.5 Da
Charge state: z = 2
Calculate singly charged mass ([M+H]+)
$$
M = (m/z \times z) - (z - 1) \times H^+
$$
$$
M = (525.76 \times 2) - (1 \times 1.0073)
$$
$$
M \approx 1050.51 , \text{Da}
$$
Final Answer
m/z of peptide: 525.76
Charge state: z = 2
[M+H]+ mass: ~1050.5 Da
6. Identify the peptide based on MS/MS fragmentation spectrum. What is the mass accuracy of the measurement? What percentage of the sequence is confirmed by peptide mapping? Answer:
Identify the peptide
From the PeptideMass output, the peptide with experimental m/z ~1050.5 Da corresponds to the theoretical peptide:
Peptide mapping confirms 89.6% of the sequence is covered by identified peptides
Final Answer
Identified peptide: FEGDTLVNR
Mass accuracy (error): ~603 ppm
Sequence coverage: 89.6%
Homework: Waters Part 3 — Oligomers
**1. We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS:
Due to such a large error, I conclude that I did not make GFP.
Week 11-hw-Bioproduction
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
1. Contribute at least one pixel to the global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST.
2. On your HTGAA webpage, note:
1. What you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”).
Contriubted 4 different fluorscent proteins in the bottom of the art.
A. B.
2. What you liked about the project.
The massive collaboration among the committed listener community. I think we had dedicated students from all the continents except anatartica. 3. What could be improved for next year.
The students can be given the opportunity to print it in their nodes.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
1. Referencing the cell-free protein synthesis reaction composition, provide a 1–2 sentence description of each component’s role in the reaction:
E. coli LysateBL21 (DE3) Star Lysate (includes T7 RNA Polymerase): Provides the essential molecular machinery (ribosomes, tRNAs, and enzymes) and T7 RNA Polymerase required for coupled transcription and translation of the target DNA.
Salts/Buffer
Potassium Glutamate: Acts as the primary potassium source and a biocompatible anion that maintains osmotic balance and stabilizes protein-nucleic acid interactions. HEPES-KOH pH 7.5: Functions as a stable buffering agent to maintain a constant physiological pH, which is critical for enzymatic activity throughout the reaction. Magnesium Glutamate: Supplies $Mg^{2+}$ ions, which are essential cofactors for ribosome assembly, mRNA stability, and the catalytic function of polymerases. Potassium phosphate monobasic: Serves as a source of inorganic phosphate and contributes to the acidic component of the phosphate buffering system. Potassium phosphate dibasic: Acts as the basic component of the phosphate buffer to help stabilize the system and provide additional ionic strength.
Energy / Nucleotide System
Ribose: Serves as a carbon source and a structural backbone precursor for the de novo synthesis of nucleotides.
Glucose: Acts as a primary energy substrate that can be metabolized to regenerate ATP through glycolytic pathways within the lysate. AMP, CMP, GMP, UMP: These monophosphate nucleotides serve as the building blocks for RNA synthesis and are phosphorylated into active triphosphates (e.g., ATP, GTP) to drive the reaction. Guanine: Provides a specific purine base precursor to ensure an adequate supply of guanosine nucleotides for transcription and translation initiation.
Translation Mix (Amino Acids)
17 Amino Acid Mix: Provides a concentrated blend of standard amino acids required for the elongation of the polypeptide chain during translation.
Tyrosine: Supplemented separately due to its lower solubility to ensure it reaches the necessary concentration for efficient protein synthesis. Cysteine: Added individually to prevent its degradation or oxidation and to ensure precise control over disulfide bond formation if required.
Additives
Nicotinamide: Serves as a precursor to $NAD^{+}$ and $NADP^{+}$, which are vital redox cofactors for the metabolic pathways that regenerate energy within the system.
Backfill
Nuclease Free Water: Used to adjust the final volume of the reaction mixture while ensuring no exogenous enzymes degrade the DNA template or RNA products.
Homework Question
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2–3 sentences)
The main difference is in the energy strategy: the 1-hour mix utilizes PEP and NTPs for immediate, high-rate transcription and translation, while the 20-hour mix uses Ribose, Glucose, and NMPs to support a sustained protein production via secondary metabolic pathways. Structurally, the 20-hour mix utilizes Potassium phosphate buffering system and modifies salt concentrations, such as lowering HEPES and slightly increasing Magnesium Glutamate, to optimize for long-term stability rather than initial speed. In contrast, the 20-hour mix simplifies the additive profile by focusing on Nicotinamide for redox balance, while the 1-hour mix requires a broader range of boosters like Spermidine, DMSO, and Folinic Acid.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
1. For each of the 6 fluorescent proteins used (sfGFP, mRFP1, mKO2, mTurquoise2, mScarlet_I, Electra2), identify and explain at least one biophysical or functional property affecting expression/readout in cell-free systems (1–2 sentences each):
sfGFP: Exceptionally fast folding kinetics and high thermodynamic stability allow it to mature quickly in the simplified, non-chaperone-rich environment of cell-free lysates.
mRFP1: While it provides a distinct red readout, it matures slowly gives a lower quantum yield compared to second-generation variants can lead to a delayed or weaker signal in short-duration cell-free reactions.
mKO2: It features a high molar extinction coefficient and rapid maturation at $37$°C, making it an excellent high-brightness reporter for tracking protein synthesis rates.
mTurquoise2: This protein possesses a remarkably high quantum yield and long fluorescence lifetime, significantly improving the signal-to-noise ratio in systems with high background autofluorescence.
mScarlet_I: Extreme brightness and high acid stability make mScarlet-I specifically optimized for efficient folding, which minimizes the formation of non-fluorescent aggregates during high-yield cell-free production.
Electra2: This variant is engineered for ultra-rapid chromophore maturation, making it the ideal candidate for “real-time” reporting where the delay between translation and fluorescence detection must be kept to a minimum.
2. Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific property to maximize fluorescence over a 36-hour incubation. Clearly state:
The protein
The reagent(s)
The expected effect
To achieve a stable and productive 36-hour “Artwork” incubation, the master mix must not be a flash of lightining- a high-intensity burst of energy- but a sustained lantern of slow-release metabolic states. By significantly increasing the concentration of Ribose and Glucose and also elevating the Potassium Phosphate buffer strength, the system can sustain the secondary metabolism required to recycle NMPs into NTPs over several days. This high-capacity phosphate buffering is essential because the extended metabolic activity generates significant acidic byproducts that would otherwise crash the pH and halt translation within the first few hours. And also, adjusting the Magnesium Glutamate levels upward compensates for the gradual sequestration of magnesium ions by the accumulating inorganic phosphate, ensuring that the ribosomes remain structurally intact and functional for the duration of the long-term protein synthesis. This shift from the immediate energy of PEP-based systems to a precursor-fed ribose system allows for the sustainable production of fluorescent proteins, making it both cost-effective and ideal for biological art that develops over time.
Carbon Forge Red: Engineering a Photoautotrophic System for the Conversion of CO₂ into L-Lactic Acid as a Raw Material for Poly Lactic Acid on Mars.
HTGAA 2026: Individual Final Project Documentation SECTION 1: ABSTRACT Abstract: Sustainable Mars settlement requires In-Situ Resource Utilization (ISRU) to reduce dependence on Earth-based supply chains. This project addresses the critical need for manufacturing materials on Mars by engineering a biological system to convert atmospheric $CO_2$ into Polylactic Acid (PLA), a versatile bioplastic for 3D printing. The broad objective is to create a photoautotrophic platform using Chlorella vulgaris for carbon fixation and polymer precursor production. We hypothesize that by redirecting metabolic flux from pyruvate to lactate via the introduction of $L$-lactate dehydrogenase ($Lldh$) and pyruvate kinase ($pk$), while knocking down phosphoenolpyruvate carboxylase ($ppc$), significant yields of $L$-lactic acid can be achieved. Specific aims include genetically modifying the algae, validating lactate accumulation, and refining extraction protocols. Methods involve CRISPR-based metabolic engineering, cell lysis, and chromatography for purification, followed by chemical polymerization. This system bridges the gap in Martian ISRU by providing a renewable source for construction and tool fabrication. SECTION 2: PROJECT AIMS Define three aims for your final project (minimum one sentence per aim).
Subsections of Projects
Group Final Project
Group Name: Phage Forge
Group Members:
@2026a-nourelden-rihan, @2026a-ritika-saha, @2026a-rahul-yaji, and @2026a-keerthana-gunaretnam
We decided to focus on the main area of increasing the stability of the MS2 phage lysis protein L, with a possible secondary goal of reducing the dependency on host DnaJ, while still maintaining the lysis action.
The tools AlphaFold, Clustal Omega, BLAST, ESM, and ESMFold were discussed.
BLAST can pull out homologous lysis proteins from the databases.
Clustal Omega can create MSAs to identify essential L48-S49 residues, and the pore-forming regions that must not be mutated.
ESM can create mutation heatmaps, which can guide the use of ESMFold to obtain highest score foldings in mutatable regions.
AlphaFold Multimer predicts whether the subunits of our protein can successfully create a pore in the host membrane, and also to check whether N-terminus can break the interaction with DnaJ.
We also identified a few pitfalls, with majors ones dealing with limited training datasets, that may not be properly aligned towards creating a transmembrane lysis protein.
Some other pitfalls include the lack of proper annotations for amurins; the possibility of an over-stable protein to form non-functional aggregates; and the vulnerability of modified protein to host proteases.
The study shows that the MS2 phage lysis protein L requires the host chaperone DnaJ for efficient host cell lysis. A missense mutation (P330Q) in the highly conserved C-terminal domain of DnaJ blocks MS2 L-mediated lysis at 30 °C and delays lysis at higher temperatures, without affecting overall L protein synthesis. The defect is specific to L-mediated lysis and does not affect lysis by other phage lysis proteins.
Genetic suppressor screening identified Lodj alleles of the L gene that bypass the DnaJ requirement. These alleles encode truncated L proteins lacking the highly basic N-terminal domain, indicating that this domain confers dependence on DnaJ. Biochemical assays demonstrated that wild-type L forms a membrane-associated complex with DnaJ, whereas the P330Q DnaJ variant cannot interact with L.
The authors propose that DnaJ functions as a chaperone that facilitates proper folding or conformational activation of full-length L, preventing steric interference from the N-terminal domain and allowing L to interact with its unknown cellular target. Removal of the dispensable N-terminal domain eliminates the need for chaperone assistance and accelerates lysis.
The work identifies DnaJ as a host factor regulating MS2 lysis timing and suggests that chaperone-dependent modulation of lysis may be an evolutionary strategy to optimize phage replication cycles.
This study performed comprehensive mutational and genetic analyses of the MS2 phage lysis protein L to identify residues and domains required for function. Random mutagenesis of the 75-aa L protein showed that most loss-of-function mutations cluster in the C-terminal half of the protein, especially around a conserved Leu-Ser (LS) dipeptide motif. Many inactivating mutations were conservative amino-acid substitutions and did not affect protein accumulation or membrane association, suggesting that L function depends on specific protein–protein interactions rather than nonspecific membrane disruption.
Functional studies demonstrated that L-mediated lysis requires interaction with the host chaperone DnaJ. The highly basic N-terminal domain of L is dispensable for lytic activity but mediates DnaJ dependence. Truncation of this domain or certain suppressor mutations bypassed the chaperone requirement and restored rapid lysis.
Biochemical and genetic data support a model in which L is an integral membrane protein whose essential domains (including the LS motif and neighboring regions) form a helical structure that likely engages a host membrane target protein. The interaction may occur near sites of membrane curvature associated with peptidoglycan biosynthesis rather than by forming nonspecific membrane lesions.
The work, supported in part by the Center for Phage Technology and associated laboratories including research by Ry Young, suggests that MS2 L functions through a specific heterotypic protein–protein interaction mechanism and that chaperone-dependent regulation helps control lysis timing during infection.
The study refines the mechanistic model of MS2 lysis, proposing that conserved structural motifs rather than general membrane disruption drive lytic activity.
This study provides detailed in vitro and in vivo characterization of the MS2 lysis protein MS2-L, focusing on its membrane insertion mechanism, oligomerization behavior, and interaction with the host chaperone DnaJ.
Key findings show that MS2-L is a 75-amino-acid phage toxin whose essential lytic activity resides in the C-terminal ~35 amino acids, which form a hydrophobic transmembrane region. The N-terminal soluble domain is not required for bacterial killing but modulates folding, membrane insertion efficiency, and chaperone interaction.
Biochemical assays demonstrate that MS2-L interacts directly with DnaJ, primarily through the soluble N-terminal domain. However, this interaction does not significantly affect membrane insertion, solubilization, or oligomerization of the toxin, suggesting that DnaJ functions more as a folding or stabilization partner rather than being essential for lytic activity.
Native mass spectrometry revealed that MS2-L assembles into high-order oligomeric complexes (≥10 monomers) after insertion into lipid nanodiscs, and oligomerization is driven mainly by the transmembrane domain. In detergent environments, oligomer formation is reduced, indicating that membrane lipid context is important for stable assembly.
Fluorescence microscopy and cryo-electron microscopy showed that MS2-L expression in bacteria leads to peripheral membrane clustering, followed by sequential lesion formation beginning in the outer membrane, then disruption of the peptidoglycan layer, and finally inner membrane disintegration with cytoplasmic leakage.
The data support a model in which MS2-L functions as a pore-forming phage toxin that kills cells through higher-order oligomerization within the bacterial membrane, rather than by directly inhibiting peptidoglycan biosynthesis. Chaperone DnaJ binds MS2-L but is not required for membrane insertion or pore assembly, suggesting its role is mainly in modulating toxin folding or stability.
These findings strengthen the concept that MS2-L belongs to the amurin/single-gene lysis protein family and may be useful for bioengineering applications such as bacterial ghost cell production and antimicrobial design.
This review from Elsevier surveys the biological mechanisms, clinical development, and future directions of phage therapy as a strategy to combat antimicrobial resistance. It explains that therapeutic phages should ideally be strictly lytic, highly host-specific, and thoroughly characterized to ensure safety and efficacy.
The article describes how phages kill bacteria through mechanisms such as inhibition of essential cellular processes, expression of lysis proteins, or disruption of bacterial membranes. It also discusses advances in phage engineering, including synthetic genome construction and modification of phage host range and virulence.
Clinical applications of phage therapy are highlighted, particularly for treating drug-resistant infections where antibiotics are ineffective. However, challenges remain, including bacterial resistance to phages, regulatory hurdles, manufacturing standardization, and the need to understand phage–host interactions.
Future directions include the use of genetically modified or synthetic phages, computational prediction of therapeutic candidates, and integration of phage therapy with conventional antimicrobial strategies. Overall, phage therapy is presented as a promising but still developing alternative to antibiotics in the fight against antimicrobial resistance.
This preprint reports the first experimental demonstration of generative design of complete bacteriophage genomes using genome language models (Evo 1 and Evo 2). The authors fine-tuned models on about 15,000 Microviridae phage genomes to enable autoregressive generation of full viral genomes guided by template-based prompts and biologically motivated design constraints.
The workflow involved computational generation followed by multi-tier filtering for sequence quality, host tropism specificity, and evolutionary diversity. Constraints included genome length (4–6 kb), GC content, absence of long homopolymers, preservation of phage-like gene architecture, and spike protein similarity to the template phage to maintain host targeting.
Experimental validation showed that about 285 of 302 synthesized genome candidates could be assembled, and 16 produced viable infectious phages that inhibited growth of the target host strain. These generated phages displayed substantial sequence novelty, containing hundreds of mutations relative to natural Microviridae genomes, while preserving functional genome organization.
Structural and functional analyses indicated that some generated phages possessed altered protein interfaces but maintained compatible capsid–protein interactions. Cryo-electron microscopy and structure prediction suggested context-dependent co-evolution of structural proteins such as capsid and packaging proteins.
Fitness assays showed that several AI-generated phages matched or exceeded the replication and lytic performance of the template phage, and phage cocktail experiments demonstrated rapid suppression of resistant bacterial strains through recombination and mutation-driven adaptation.
The study was conducted with biosafety considerations, including restricting model training to bacteriophage genomes and using well-characterized laboratory strains. The work was supported by researchers affiliated with institutions such as the Stanford University and the Arc Institute.
Overall, the paper proposes a framework for generative genome engineering, showing that AI models can design biologically viable and evolutionarily novel bacteriophages, potentially enabling future synthetic biology and phage-based therapeutic development.
Overview of the Project Proposal: Engineering the MS2 Phage Lysis Protein L
Our primary goal is to increase the structural stability of the MS2 bacteriophage lysis protein (L) while maintaining its ability to lyse bacterial cells.
Our secondary goal is to reduce the dependency of L on the host chaperone DnaJ, which normally assists the protein in folding or activation. Reducing this dependency could allow the lysis protein to function more efficiently and independently in engineered systems.
The MS2 L protein is a 75-amino-acid single-gene lysis toxin whose C-terminal region forms a hydrophobic transmembrane domain responsible for membrane disruption and pore formation, while the basic N-terminal domain interacts with host factors such as DnaJ. Previous studies show that truncation of the N-terminal region can bypass the DnaJ requirement while preserving lysis activity.
Therefore, our design strategy focuses on:
Stabilizing the transmembrane and oligomerization regions
Maintaining essential functional motifs such as the L48–S49 motif
Exploring modifications to the N-terminal region to reduce DnaJ dependence
2. Computational Tools and Approaches
We will use a multi-step computational protein engineering pipeline combining sequence analysis, machine-learning mutagenesis predictions, and structural modeling.
2.1 BLAST – Homolog Discovery
First, we will use BLAST to identify homologous lysis proteins from related bacteriophages.
Purpose:
Identify evolutionarily conserved residues
Discover natural sequence variations that maintain function
Build a dataset for multiple sequence alignment
This will help determine which regions are functionally constrained vs mutable.
Select Candidate Mutations (stability or N-terminal modifications)
Structure Prediction (ESMFold)
Complex/Oligomer Prediction (AlphaFold Multimer)
Final Mutant Candidates (stable + functional lysis protein)
3. Proposed Engineering Pipeline
Computational workflow we will follow.
4. Expected Outcomes
Our pipeline aims to produce engineered variants of the MS2 L protein with:
Increased structural stability
Reduced aggregation risk
Maintained transmembrane insertion
Potentially reduced dependency on host DnaJ
These optimized proteins could be useful in applications such as:
Synthetic phage engineering
Bacterial ghost cell production
Antimicrobial protein development
5. Potential Pitfalls
5.1 Limited Training Data
Most protein language models and structural predictors are trained primarily on globular proteins, not small transmembrane phage toxins.
This may reduce prediction accuracy for MS2 L.
5.2 Risk of Over-Stabilization
Mutations designed to increase stability may cause:
Protein aggregation
Improper membrane insertion
Loss of functional oligomerization
Thus stability must be balanced with function.
5.3 Poor Annotation of Amurin Proteins
Single-gene lysis proteins (also called amurins) are poorly annotated in sequence databases.
This may limit the quality of homologous sequences used for alignment and training.
5.4 Host Protease Sensitivity
Mutations may unintentionally expose protease cleavage sites, making the engineered protein less stable inside bacterial cells.
6. Future Work
If promising mutants are identified computationally, the next steps would include:
Experimental expression in E. coli
Measuring lysis timing
Measuring protein stability
Testing DnaJ independence
This would validate whether computational predictions translate into improved biological function.
Week 5
Execution of workflows:
@2026a-rahul-yaji
Created a Python Script- L-Protein Mutagenisis to create random mutations at two distinct, non-conserved location L-protein, subject to the constraint that there should be no loss of lysis function due to the mutation.
The data of the L-Protein Mutants document was used to avoid loss-of-lysis (Hereafter known as LoL) mutatioins.
Key assumptions:
All the LoL mutations occur in the conserved areas
0 indicates LoL, and 1 indicates intact lysis function
If assumption 1 is true, MSA becomes redundant and therefore irrelavant
The generated mutant sequences were cofolded with host DnaJ chaperone protein to analyze interactions
So far, no mutant was found to have significant iPTM score, or interaction with DnaJ.
Generating random mutations in the Lysis protein while avoiding the loss of function or non sense codons.The Python script was generated solely by the Google Gemini 2.5 Flash, that is in-built in Google Colab. The prompt was:
Develop a Python program in Google Colab that processes an amino acid sequence and generates mutated versions of it based on experimental data. The program should perform the following steps:
The data contains information about amino acid changes and their associated ‘Lysis’ activity.
Filter the mutation data to include only ‘active’ mutations (where ‘Lysis’ is not 0). Extract the ‘Original_Residue’, ‘Position’, and ‘Mutated_Residue’ from the relevant columns (e.g., ‘Amino Acid Change’ and ‘Amino Acid Position’ or a ‘Mutation’ column like ‘X###Y’).
Create a helper function to format amino acid sequences by inserting a space after every 5 amino acids for better readability.
Implement a function generate_random_mutation_combinations(sequence, mutation_df, num_mutations) that takes an original amino acid sequence, the filtered active mutations DataFrame, and the desired number of mutations as input.
This function should:
Identify all valid mutation sites where the original residue in the sequence matches an original residue in the mutation_df.
Ensure that the num_mutations are applied to unique positions in the sequence. If there are fewer available unique mutation positions than num_mutations, it should apply all available unique mutations.
Randomly select mutations from the available options for the chosen unique positions.
Return the new mutated sequence and print the applied mutations.
Generate Multiple Mutated Sequences: Prompt the user for the number of mutated sequences they wish to generate. For each requested sequence:
Call the generate_random_mutation_combinations function.
Display the generated sequence with a clear heading (e.g., ‘Sequence 1:’, ‘Sequence 2:’, etc.).
Print both the original and the mutated sequences, using the formatting function defined in step 5.
In a separate code block, display each generated mutated sequence individually using display() so that each sequence is easily copyable by the user.
AF2 Multimer was used to co-fold the mutant Lysis protein (METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAFFLSKFTNQLLLSLLEAVIRTVTTLQQLLT) and DnaJ:
The plDDT score indicates that the model is not confident about the folding of the input random mutated L protein. Overall, it suggests that the random mutation approach is very time consuming to obtain leads, and very computation-intensive. Due to limited computational resources, cofolding was not performed for other sequences.
Later, cofolding was performed using Alphafold server, and the results obtained are shown below:
Individual Final Project
Carbon Forge Red: Engineering a Photoautotrophic System for the Conversion of CO₂ into L-Lactic Acid as a Raw Material for Poly Lactic Acid on Mars.
HTGAA 2026: Individual Final Project Documentation
SECTION 1: ABSTRACT
Abstract:
Sustainable Mars settlement requires In-Situ Resource Utilization (ISRU) to reduce dependence on Earth-based supply chains. This project addresses the critical need for manufacturing materials on Mars by engineering a biological system to convert atmospheric $CO_2$ into Polylactic Acid (PLA), a versatile bioplastic for 3D printing. The broad objective is to create a photoautotrophic platform using Chlorella vulgaris for carbon fixation and polymer precursor production. We hypothesize that by redirecting metabolic flux from pyruvate to lactate via the introduction of $L$-lactate dehydrogenase ($Lldh$) and pyruvate kinase ($pk$), while knocking down phosphoenolpyruvate carboxylase ($ppc$), significant yields of $L$-lactic acid can be achieved. Specific aims include genetically modifying the algae, validating lactate accumulation, and refining extraction protocols. Methods involve CRISPR-based metabolic engineering, cell lysis, and chromatography for purification, followed by chemical polymerization. This system bridges the gap in Martian ISRU by providing a renewable source for construction and tool fabrication.
SECTION 2: PROJECT AIMS
Define three aims for your final project (minimum one sentence per aim).
Aim 1: Experimental Aim The first aim of my final project is to engineer Chlorella vulgaris to produce $L$-lactic acid from $CO_2$ by utilizing CRISPR-Cas9 gene editing to introduce $L$-lactate dehydrogenase ($Lldh$) and pyruvate kinase ($pk$) genes while knocking down the phosphoenolpyruvate carboxylase ($ppc$) gene.
I will use Benchling for genetic circuit design and codon optimization, Addgene plasmids for the CRISPR backbone, and Asimov Kernel for metabolic modeling. The experimental workflow involves algal transformation, selection via antibiotic resistance, and verification of lactate secretion using high-performance liquid chromatography (HPLC).
Aim 2: Development Aim The second aim is to scale the biological production into a functional manufacturing pipeline by optimizing the downstream purification and polymerization of $L$-lactic acid into 3D-printable Poly Lactic Acid (PLA) filament. Following a successful Aim 1, this phase involves developing efficient cell lysis protocols, utilizing ion-exchange chromatography for high-purity lactic acid recovery, and performing ring-opening polymerization to create a resin suitable for extrusion into 3D printing filaments.
Aim 3: Visionary Aim The long-term vision for this project is to establish a self-sustaining In-Situ Resource Utilization (ISRU) framework for Mars settlement, where atmospheric carbon is converted into essential structural materials without Earth-based feedstock. By validating these experiments under simulated Martian atmospheric conditions, this project aims to address the major barrier of high-mass transport costs in space exploration, enabling a new paradigm of “biological manufacturing” where settlers can grow their own tools, spare parts, and habitats from the air they cannot breathe.
SECTION 3: BACKGROUND
Background and Literature Context
Provide background research that explains the current state of knowledge and identifies the gap your project addresses.
1. Briefly summarize two peer-reviewed research citations relevant to your research:
This work highlights how metabolic pathway redirection and enzyme engineering can transform cyanobacteria into cell factories for biofuel and bioplastic precursors.
The study applies synthetic biology to reprogram Synechocystis sp. PCC6803 for sustainable lactic acid production directly from CO₂.
A heterologous lactate dehydrogenase (LDH) gene from Bacillus subtilis was stably integrated into the cyanobacterial genome, enabling light-driven conversion of carbon flux into L-lactic acid.
To boost yields, the team coexpressed a soluble transhydrogenase from Pseudomonas aeruginosa, which enhanced NADH regeneration and improved metabolic efficiency.
Researchers engineered Synechocystis sp. PCC 6803 to produce optically pure D-lactic acid directly from CO₂ using sunlight, introducing a mutated glycerol dehydrogenase (GlyDH*) as a novel D-lactate dehydrogenase.
Productivity was enhanced through codon optimization of the heterologous gene and by balancing intracellular cofactors via expression of a soluble transhydrogenase from Pseudomonas aeruginosa.
Supplementation with acetate further boosted yields, not by serving as a direct carbon source for lactate, but by supporting biomass synthesis and redirecting carbon flux from CO₂ into lactate.
The engineered strains achieved titers up to 2.17 g/L, demonstrating how synthetic biology can reprogram cyanobacteria into efficient cell factories for sustainable bioplastic precursors like polylactic acid.
Polylactic acid (PLA) is biodegradable, compostable, and derived from renewable resources like corn starch and sugarcane. It has good mechanical strength (~60 MPa tensile), thermal stability (melting point 150–160 °C), and can degrade up to 90% within six months under composting conditions, making it a strong candidate to replace petroleum plastics.
PLA is synthesized from lactic acid obtained mainly via microbial fermentation using Lactobacillus or Rhizopus species. Polymerization occurs through condensation (low molecular weight) or ring‑opening polymerization (ROP), the latter being preferred for high molecular weight PLA with tailored properties.
PLA is widely used in packaging (biodegradable bags, food containers), biomedical devices (sutures, drug delivery systems, tissue scaffolds), textiles (eco‑friendly fibers), and 3D printing. Its biocompatibility and biodegradability make it especially valuable in medical and sustainability contexts.
Despite its promise, PLA faces hurdles such as high production costs, brittleness, and limited flexibility. Research focuses on improving catalysts, blending with other polymers, and integrating nanotechnology to enhance properties and scalability for broader industrial adoption.
2. Explain how your project is novel or innovative (minimum three sentences).
I. Integration of Biological Systems into Martian ISRU Paradigms
Traditional In-Situ Resource Utilization (ISRU) frameworks for Mars primarily focus on thermochemical processes, such as the Sabatier reaction, to produce fuel and oxygen. This project challenges existing paradigms by proposing a biological ISRU platform. By leveraging Chlorella vulgaris as a self-replicating “cell factory,” the research demonstrates a novel application of synthetic biology to convert the Martian atmosphere ($CO_2$) directly into structural materials (PLA), thereby bypassing the need for high-mass feedstock transport from Earth.
II. Precision Metabolic Flux Redirection in Chlorella spp.
While lactic acid production has been demonstrated in other species of prokaryotic cyanobacteria, this project innovates through a tripartite genetic modification strategy in the eukaryotic microalgae Chlorella vulgaris. By utilizing CRISPR-Cas9 to introduce heterologous $L$-lactate dehydrogenase ($Lldh$) and pyruvate kinase ($pk$) while simultaneously implementing a knockdown of phosphoenolpyruvate carboxylase ($ppc$), the research expands the boundaries of algal metabolic engineering. This specific combination of “push” and “pull” genetic edits is designed to maximize carbon flux toward $L$-lactic acid, representing a sophisticated advancement over single-gene insertions.
III. End-to-End “Cell-to-Component” Manufacturing Pipeline
A significant technological innovation of this project is the development of a closed-loop manufacturing methodology that bridges the gap between synthetic biology and additive manufacturing. The workflow integrates computational genetic design (via Asimov Kernel) with downstream chemical processing—specifically the refinement of extraction protocols and ring-opening polymerization (ROP) for filament extrusion. This establishes a new methodology for autonomous, light-driven production where biological output is directly translated into 3D-printable hardware, providing a scalable model for sustainable extraterrestrial settlement.
Reference Summary of Core Innovations
Innovation Category
Traditional Approach
Carbon Forge Red Approach
Material Source
Earth-shipped polymers
Martian $CO_2$ conversion
ISRU Method
Mechanical/Chemical
Synthetic Biology (Photoautotrophic)
Production Cycle
Linear Supply Chain
Circular “Cell-to-Filament” Pipeline
3. Explain why your project matters and what impact it could have (minimum five sentences).
The Carbon Forge Red project addresses the critical bottleneck of logistics in long-duration space exploration: the prohibitive cost and extreme risk associated with Earth-dependent supply chains. Current Mars mission architectures are severely constrained by the mass and volume of raw materials required for construction and maintenance, making the establishment of a permanent settlement logistically fragile. By engineering a photoautotrophic system to convert atmospheric $CO_2$ into Polylactic Acid (PLA), this project provides a vital solution for In-Situ Resource Utilization (ISRU), allowing settlers to manufacture essential tools and structural components locally rather than waiting for terrestrial shipments.
This project also marks a significant advancement in synthetic biology by demonstrating that eukaryotic algae can be reprogrammed to maintain high metabolic flux toward polymer precursors even in resource-limited environments. By validating the extraction and polymerization process under Martian atmospheric parameters, the project shifts the field-level paradigm from “exploration via cargo” to “settlement via biological manufacturing.” The impact of this work is the creation of a self-sustaining infrastructure loop, enabling pioneers to grow their own habitats, tools, and replacement parts directly from the air they cannot breathe. Ultimately, this creates the foundation for true Martian autonomy, where the success of a colony is limited only by its biological efficiency rather than its proximity to Earth.
4. Describe the ethical implications associated with your project and identify relevant ethical principles (minimum two paragraphs).
The ethical implications of the Carbon Forge Red project primarily center on the introduction of genetically modified organisms (GMOs) into extraterrestrial environments, often referred to as planetary protection. By engineering Chlorella vulgaris for Mars-based manufacturing, we encounter the risk of forward contamination—the accidental transfer of Earth-based life to another planet—which could permanently obscure our ability to detect indigenous Martian life or irreversibly alter the Martian “wilderness.” Furthermore, the dual-use nature of metabolic engineering poses a concern; while the intent is to produce bioplastics for habitats, the same gene-editing techniques could theoretically be repurposed to create harmful biological byproducts or disrupt local ecological balances if a biosphere were ever to exist. This involves the ethical principle of Non-Maleficence, ensuring that our technological footprint does not cause unintended harm to a pristine planetary environment.
To ensure ethical conduct and societal responsibility, rigorous containment and “biocontainment” protocols must be implemented (one can refer my homework of week 1), such as the use of Kill Switches—genetic circuits that cause the algae to self-destruct if they escape the controlled bioreactor environment. These measures align with the principle of Responsibility, emphasizing that scientists must be stewards of the environments they manipulate. Additionally, a framework of Transparency and Justice should be adopted, ensuring that the genetic resources and technologies developed for Martian settlement are not monopolized by a single entity but are shared to benefit the broader scientific community. Societal responsibility also dictates that we conduct extensive terrestrial simulations and ethical peer reviews to assess the long-term impact of “biological manufacturing” before any actual deployment occurs in space. But designing a kill switch is beyond the scope of this project right now, and will be addressed on a future date.
SECTION 4: EXPERIMENTAL DESIGN, TECHNIQUES, TOOLS, AND TECHNOLOGY
Create a detailed experimental plan for your final project. Include a timeline for each part (minimum 15 lines/sentences).
Experimental Plan: Optimizing Photoautotrophic L-Lactic Acid Production in Engineered Synechocystis sp. PCC6803
Phase 1: Genetic Construct Design and Molecular Cloning
Methods & Tools
Design and construct integration and replicative plasmids using standard BioBrick assembly techniques.
Codon Optimization: Target genes ($ldh$ from Bacillus subtilis and Lactococcus lactis, $sth$ from Pseudomonas aeruginosa, and $pk$ from Enterococcus faecalis) will be optimized using the OPTIMIZER application.
Regulatory Elements: Genes will be placed under strong promoters ($Ptrc$ or $Ptrc2$) and equipped with the transcriptional terminator BBa_B0014.
Vector Construction:
Integration: pBluescript II SK(+) backbone with homologous flanking regions targeting the slr0168 docking site or the ppc (phosphoenolpyruvate carboxylase) locus.
Replication: Utilize the pDF_LDH replicative plasmid (RSF1010 origin) for high-copy expression.
Cloning: PCR amplification using high-fidelity Pfu or Pwo DNA polymerases; all constructs cloned into E. coli XL-1 blue host cells.
Expected Results
Successful isolation of sequence-verified plasmids with correctly oriented $ldh$, $sth$, and $pk$ gene cassettes.
Functional pairing with kanamycin or streptomycin antibiotic resistance markers.
Phase 2: Cyanobacterial Transformation and Segregation
Methods & Tools
Transformation: Genomic integration via natural transformation of glucose-tolerant Synechocystis sp. PCC6803 wild-type cells using 5–10 μg of plasmid DNA.
Conjugation: Triparental mating for replicative plasmids using E. coli J53 (pRP4) as the helper strain.
Selection: Plating on BG-11 agar supplemented with 5 mM glucose, 0.3% sodium thiosulfate, and selective antibiotics.
Segregation: Drive complete chromosome segregation via repeated, step-wise restreaking onto plates with increasing antibiotic concentrations (up to 20 μg/mL kanamycin).
Verification: Confirm segregation using colony PCR with Taq DNA polymerase.
Expected Results
Generation of fully segregated, viable mutant strains (e.g., SAA023, SAW039, and SAW041).
Partial segregation for the ppc knockdown strain (SAW033), as ppc is an essential gene.
Phase 3: Culturing, Phenotyping, and Lactic Acid Quantification
Methods & Tools
Culturing: Batch liquid BG-11 medium (50 mM $NaHCO_3$ or 10 mM TES-KOH, pH 8) in shaking incubators at 30°C under constant white-light illumination.
Growth Kinetics: Monitor optical density at 730 nm (OD730) using a spectrophotometer.
Lactic Acid Assay: Quantify extracellular L-lactic acid in cell-free supernatants using a Megazyme 96-well enzymatic assay.
Analytical Profiling: Precise organic acid quantification via HPLC using a Rezex ROA-Organic Acid H+ column and a refractive index (RI) detector.
Expected Results
The SAW041 strain (co-expressing $pk$) is expected to achieve the highest titers, reaching approximately 9.3 mM lactic acid.
Anticipated growth retardation in high-yield strains due to metabolic burden.
SAW033 (ppc knockdown) expected to show the highest carbon partitioning ratio (over 30%).
Phase 4: Protein Expression Profiling and Enzymatic Assays
Methods & Tools
Cell Harvesting: Late-exponential phase cultures harvested and disrupted using 100-μm glass beads at 4°C.
Quantification: Protein concentrations determined via BCA protein assay.
Expression Profiling:SDS-PAGE stained with Coomassie Brilliant Blue (CBB) G-250 to verify heterologous protein bands.
In Vitro Assays:
LDH Activity: Monitor NADH/NADPH oxidation at 340 nm upon addition of sodium pyruvate.
PK Activity: Coupled reaction with exogenous LDH, monitoring NADH consumption initiated by phosphoenolpyruvate (PEP).
Expected Results
Confirmation of prominent protein bands for LDH and PK on SDS-PAGE.
Verification of up to 10-fold higher LDH activity in multi-copy strains.
Measurable shifts toward NADPH co-utilization in strains utilizing the mutated B. subtilis V38R LDH.
Techniques Checklist
[x] Pipetting [x] Lab Safety [x]Bioproduction [x]Registry of Standard Biological Parts [x]Chassis Selection (e.g., DH5alpha) ☒ Bioethical Considerations [x] Plasmid Preparation [x] Bacterial Culturing [x] Quality Control/Analysis [x] Bacterial Processing (Centrifugation, Lysis, DNA Purification) [x] DNA Construct Design [x] Restriction Enzyme Digestion [x] Gel Electrophoresis [x] DNA Purification From Gel ☐ Cell-Free Systems ☐ Freeze-Dried Cell-Free Systems [x] Databases (GenBank, NCBI, Ensembl, UCSC Genome Browser) ☐ miniPCR Tools [x] Protein Purification
Lab Automation
☐ Creating Code for Laboratory Automation ☐ Using Liquid Handling Robots (e.g., Opentrons) ☐ Designing a Twist Order ☐ Creating a plan to use the Autonomous Lab at Ginkgo Bioworks
CRISPR
☐ CRISPR/Cas9 ☐ Designing Prime Editing gRNA
Protein Design
☐ Protein Design ☐ Use of Boltz or PepMLM [x] Use of Asimov Kernel [x] Use of Benchling [x] Models and Notebooks [x] Databases
1. Expand upon two techniques you checked above (minimum four sentences).
Bioproduction: The metabolic engineering of Chlorella vulgaris facilitates a direct photoautotrophic transition from atmospheric $CO_2$ to $L$-lactic acid, leveraging the organism’s high photosynthetic efficiency to drive carbon flux toward polymer precursors. Achieving industrial-scale titers requires the transition from laboratory-scale benchtop setups to high-density photobioreactors, where precise control of light attenuation, $pCO_2$ saturation, and nutrient replenishment optimizes cellular productivity. Downstream processing involves high-throughput cell disruption and ion-exchange chromatography to isolate high-purity monomeric lactic acid, followed by catalytic ring-opening polymerization (ROP) to synthesize high-molecular-weight Poly Lactic Acid (PLA) resins. This integrated bioproduction pipeline is specifically designed for modularity, allowing for autonomous manufacturing cycles that transform raw Martian atmospheric components into a consistent supply of 3D-printable filaments for structural applications.
Quality control and assurance: QA and QC for the production of Martian PLA necessitate a multi-stage validation framework to ensure structural integrity and biocompatibility of the synthesized filaments. Initial spectroscopic analysis via High-Performance Liquid Chromatography (HPLC) and Nuclear Magnetic Resonance (NMR) is required to verify the chemical purity and optical isomer ratio of the $L$-lactic acid monomers prior to polymerization. Following resin synthesis, the resulting Poly Lactic Acid must undergo rigorous thermomechanical profiling, including Differential Scanning Calorimetry (DSC) to determine glass transition temperatures and tensile strength testing to ensure compliance with aerospace structural standards. Finally, a closed-loop feedback system utilizing real-time sensor data from the extrusion process monitors filament diameter consistency and rheological properties, guaranteeing that every batch of biologically derived material meets the precision requirements for automated additive manufacturing in extraterrestrial environments.
Identify any HTGAA Industry Council companies associated with your project (optional): [x] Addgene [] Epibone [x] Ginkgo Bioworks [] Helix Nano [] Millipore Sigma [x] BioFabricate [] Biome Consortia [] Bolt [] Boltz.bio [x] Cultivarium [] DeepCure [] Mycoworks [] New England Biolabs [x] Opentrons [x] SecureDNA [] Takeda Pharmaceuticals [] Thermo Fisher Scientific [] Transfyr.ai [] Twist Biosciences [] Upside Foods [] Waters Corporation
SECTION 5: RESULTS & QUANTITATIVE EXPECTATIONS
You are required to validate at least one aspect of your final project aims.
DNA Design:
What aspect of your project did you choose to validate?
I chose to validate the gene expression level, as it could be done virtually, without lab access.
Write a detailed protocol of how you validated it.
The construct should contain: Promoter, RBS, Start Codon, CDS, Stop Codon, Terminator.
Repeat the same steps for the one more gene.
Use the simulate option to get the graphical representation of the gene expression levels
What synthetic biology techniques did you use?
Promoter construction
Codon optimization
Terminator construction
Chassis organism selection
Present data and analysis (experimental or simulated).
Describe challenges, limitations, and alternative strategies. Due to technical challenges, the DNA couldn’t be constructed in Benchling, and therefore Asimov kernel was selected. But here, only E. coli is available as the chassis organism. A eukaryotic expression model would have been better.