Week 5
Execution of workflows:
@2026a-rahul-yaji
- Created a Python Script- L-Protein Mutagenisis to create random mutations at two distinct, non-conserved location L-protein, subject to the constraint that there should be no loss of lysis function due to the mutation.
- The data of the L-Protein Mutants document was used to avoid loss-of-lysis (Hereafter known as LoL) mutatioins.
- Key assumptions:
- All the LoL mutations occur in the conserved areas
- 0 indicates LoL, and 1 indicates intact lysis function
- If assumption 1 is true, MSA becomes redundant and therefore irrelavant
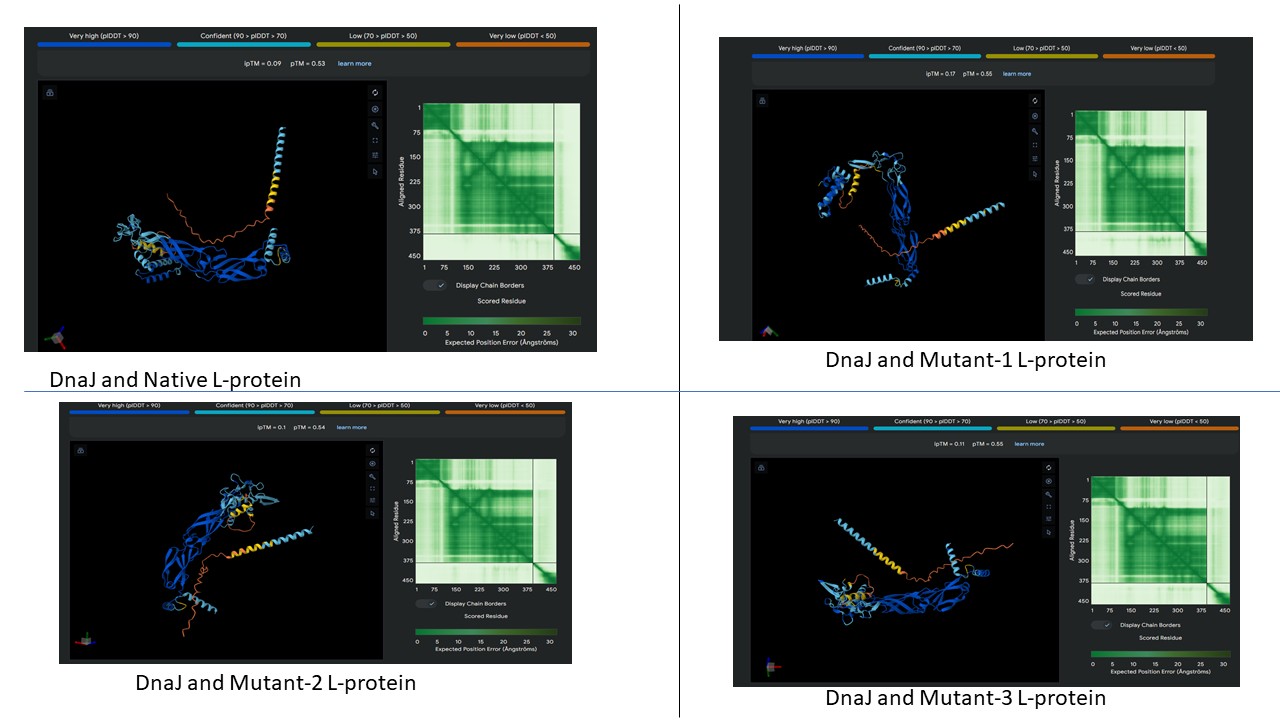
- The generated mutant sequences were cofolded with host DnaJ chaperone protein to analyze interactions
- So far, no mutant was found to have significant iPTM score, or interaction with DnaJ.
Generating random mutations in the Lysis protein while avoiding the loss of function or non sense codons.The Python script was generated solely by the Google Gemini 2.5 Flash, that is in-built in Google Colab. The prompt was:
Develop a Python program in Google Colab that processes an amino acid sequence and generates mutated versions of it based on experimental data. The program should perform the following steps:
Prompt the user to enter an amino acid sequence.
Load mutation data from a publicly accessible Google Sheet URL (https://docs.google.com/spreadsheets/d/11WzDDNkQDEiqbUSGV0ZCqITGctyNFpD7xnPlhsj2BhE/edit?gid=0#gid=0).
The data contains information about amino acid changes and their associated ‘Lysis’ activity.
Filter the mutation data to include only ‘active’ mutations (where ‘Lysis’ is not 0). Extract the ‘Original_Residue’, ‘Position’, and ‘Mutated_Residue’ from the relevant columns (e.g., ‘Amino Acid Change’ and ‘Amino Acid Position’ or a ‘Mutation’ column like ‘X###Y’).
Create a helper function to format amino acid sequences by inserting a space after every 5 amino acids for better readability.
Implement a function generate_random_mutation_combinations(sequence, mutation_df, num_mutations) that takes an original amino acid sequence, the filtered active mutations DataFrame, and the desired number of mutations as input.
This function should:
- Identify all valid mutation sites where the original residue in the sequence matches an original residue in the mutation_df.
- Ensure that the num_mutations are applied to unique positions in the sequence. If there are fewer available unique mutation positions than num_mutations, it should apply all available unique mutations.
Randomly select mutations from the available options for the chosen unique positions.
Return the new mutated sequence and print the applied mutations.
Generate Multiple Mutated Sequences: Prompt the user for the number of mutated sequences they wish to generate. For each requested sequence:
Call the generate_random_mutation_combinations function.
Display the generated sequence with a clear heading (e.g., ‘Sequence 1:’, ‘Sequence 2:’, etc.).
Print both the original and the mutated sequences, using the formatting function defined in step 5.
In a separate code block, display each generated mutated sequence individually using display() so that each sequence is easily copyable by the user.
The generated mutational sequences were:
0. METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT (Original)- METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAFFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
- METRFPQQSQQTLAATNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
- METRFPQQSQQTPASTNRRRPFKHGGYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
AF2 Multimer was used to co-fold the mutant Lysis protein (METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAFFLSKFTNQLLLSLLEAVIRTVTTLQQLLT) and DnaJ:
The plDDT score indicates that the model is not confident about the folding of the input random mutated L protein. Overall, it suggests that the random mutation approach is very time consuming to obtain leads, and very computation-intensive. Due to limited computational resources, cofolding was not performed for other sequences.
Later, cofolding was performed using Alphafold server, and the results obtained are shown below: