Carbon Forge Red: Engineering a Photoautotrophic System for the Conversion of CO₂ into L-Lactic Acid as a Raw Material for Poly Lactic Acid on Mars.
HTGAA 2026: Individual Final Project Documentation SECTION 1: ABSTRACT Abstract: Sustainable Mars settlement requires In-Situ Resource Utilization (ISRU) to reduce dependence on Earth-based supply chains. This project addresses the critical need for manufacturing materials on Mars by engineering a biological system to convert atmospheric $CO_2$ into Polylactic Acid (PLA), a versatile bioplastic for 3D printing. The broad objective is to create a photoautotrophic platform using Chlorella vulgaris for carbon fixation and polymer precursor production. We hypothesize that by redirecting metabolic flux from pyruvate to lactate via the introduction of $L$-lactate dehydrogenase ($Lldh$) and pyruvate kinase ($pk$), while knocking down phosphoenolpyruvate carboxylase ($ppc$), significant yields of $L$-lactic acid can be achieved. Specific aims include genetically modifying the algae, validating lactate accumulation, and refining extraction protocols. Methods involve CRISPR-based metabolic engineering, cell lysis, and chromatography for purification, followed by chemical polymerization. This system bridges the gap in Martian ISRU by providing a renewable source for construction and tool fabrication. SECTION 2: PROJECT AIMS Define three aims for your final project (minimum one sentence per aim).
Subsections of Projects
Group Final Project
Group Name: Phage Forge
Group Members:
@2026a-nourelden-rihan, @2026a-ritika-saha, @2026a-rahul-yaji, and @2026a-keerthana-gunaretnam
We decided to focus on the main area of increasing the stability of the MS2 phage lysis protein L, with a possible secondary goal of reducing the dependency on host DnaJ, while still maintaining the lysis action.
The tools AlphaFold, Clustal Omega, BLAST, ESM, and ESMFold were discussed.
BLAST can pull out homologous lysis proteins from the databases.
Clustal Omega can create MSAs to identify essential L48-S49 residues, and the pore-forming regions that must not be mutated.
ESM can create mutation heatmaps, which can guide the use of ESMFold to obtain highest score foldings in mutatable regions.
AlphaFold Multimer predicts whether the subunits of our protein can successfully create a pore in the host membrane, and also to check whether N-terminus can break the interaction with DnaJ.
We also identified a few pitfalls, with majors ones dealing with limited training datasets, that may not be properly aligned towards creating a transmembrane lysis protein.
Some other pitfalls include the lack of proper annotations for amurins; the possibility of an over-stable protein to form non-functional aggregates; and the vulnerability of modified protein to host proteases.
The study shows that the MS2 phage lysis protein L requires the host chaperone DnaJ for efficient host cell lysis. A missense mutation (P330Q) in the highly conserved C-terminal domain of DnaJ blocks MS2 L-mediated lysis at 30 °C and delays lysis at higher temperatures, without affecting overall L protein synthesis. The defect is specific to L-mediated lysis and does not affect lysis by other phage lysis proteins.
Genetic suppressor screening identified Lodj alleles of the L gene that bypass the DnaJ requirement. These alleles encode truncated L proteins lacking the highly basic N-terminal domain, indicating that this domain confers dependence on DnaJ. Biochemical assays demonstrated that wild-type L forms a membrane-associated complex with DnaJ, whereas the P330Q DnaJ variant cannot interact with L.
The authors propose that DnaJ functions as a chaperone that facilitates proper folding or conformational activation of full-length L, preventing steric interference from the N-terminal domain and allowing L to interact with its unknown cellular target. Removal of the dispensable N-terminal domain eliminates the need for chaperone assistance and accelerates lysis.
The work identifies DnaJ as a host factor regulating MS2 lysis timing and suggests that chaperone-dependent modulation of lysis may be an evolutionary strategy to optimize phage replication cycles.
This study performed comprehensive mutational and genetic analyses of the MS2 phage lysis protein L to identify residues and domains required for function. Random mutagenesis of the 75-aa L protein showed that most loss-of-function mutations cluster in the C-terminal half of the protein, especially around a conserved Leu-Ser (LS) dipeptide motif. Many inactivating mutations were conservative amino-acid substitutions and did not affect protein accumulation or membrane association, suggesting that L function depends on specific protein–protein interactions rather than nonspecific membrane disruption.
Functional studies demonstrated that L-mediated lysis requires interaction with the host chaperone DnaJ. The highly basic N-terminal domain of L is dispensable for lytic activity but mediates DnaJ dependence. Truncation of this domain or certain suppressor mutations bypassed the chaperone requirement and restored rapid lysis.
Biochemical and genetic data support a model in which L is an integral membrane protein whose essential domains (including the LS motif and neighboring regions) form a helical structure that likely engages a host membrane target protein. The interaction may occur near sites of membrane curvature associated with peptidoglycan biosynthesis rather than by forming nonspecific membrane lesions.
The work, supported in part by the Center for Phage Technology and associated laboratories including research by Ry Young, suggests that MS2 L functions through a specific heterotypic protein–protein interaction mechanism and that chaperone-dependent regulation helps control lysis timing during infection.
The study refines the mechanistic model of MS2 lysis, proposing that conserved structural motifs rather than general membrane disruption drive lytic activity.
This study provides detailed in vitro and in vivo characterization of the MS2 lysis protein MS2-L, focusing on its membrane insertion mechanism, oligomerization behavior, and interaction with the host chaperone DnaJ.
Key findings show that MS2-L is a 75-amino-acid phage toxin whose essential lytic activity resides in the C-terminal ~35 amino acids, which form a hydrophobic transmembrane region. The N-terminal soluble domain is not required for bacterial killing but modulates folding, membrane insertion efficiency, and chaperone interaction.
Biochemical assays demonstrate that MS2-L interacts directly with DnaJ, primarily through the soluble N-terminal domain. However, this interaction does not significantly affect membrane insertion, solubilization, or oligomerization of the toxin, suggesting that DnaJ functions more as a folding or stabilization partner rather than being essential for lytic activity.
Native mass spectrometry revealed that MS2-L assembles into high-order oligomeric complexes (≥10 monomers) after insertion into lipid nanodiscs, and oligomerization is driven mainly by the transmembrane domain. In detergent environments, oligomer formation is reduced, indicating that membrane lipid context is important for stable assembly.
Fluorescence microscopy and cryo-electron microscopy showed that MS2-L expression in bacteria leads to peripheral membrane clustering, followed by sequential lesion formation beginning in the outer membrane, then disruption of the peptidoglycan layer, and finally inner membrane disintegration with cytoplasmic leakage.
The data support a model in which MS2-L functions as a pore-forming phage toxin that kills cells through higher-order oligomerization within the bacterial membrane, rather than by directly inhibiting peptidoglycan biosynthesis. Chaperone DnaJ binds MS2-L but is not required for membrane insertion or pore assembly, suggesting its role is mainly in modulating toxin folding or stability.
These findings strengthen the concept that MS2-L belongs to the amurin/single-gene lysis protein family and may be useful for bioengineering applications such as bacterial ghost cell production and antimicrobial design.
This review from Elsevier surveys the biological mechanisms, clinical development, and future directions of phage therapy as a strategy to combat antimicrobial resistance. It explains that therapeutic phages should ideally be strictly lytic, highly host-specific, and thoroughly characterized to ensure safety and efficacy.
The article describes how phages kill bacteria through mechanisms such as inhibition of essential cellular processes, expression of lysis proteins, or disruption of bacterial membranes. It also discusses advances in phage engineering, including synthetic genome construction and modification of phage host range and virulence.
Clinical applications of phage therapy are highlighted, particularly for treating drug-resistant infections where antibiotics are ineffective. However, challenges remain, including bacterial resistance to phages, regulatory hurdles, manufacturing standardization, and the need to understand phage–host interactions.
Future directions include the use of genetically modified or synthetic phages, computational prediction of therapeutic candidates, and integration of phage therapy with conventional antimicrobial strategies. Overall, phage therapy is presented as a promising but still developing alternative to antibiotics in the fight against antimicrobial resistance.
This preprint reports the first experimental demonstration of generative design of complete bacteriophage genomes using genome language models (Evo 1 and Evo 2). The authors fine-tuned models on about 15,000 Microviridae phage genomes to enable autoregressive generation of full viral genomes guided by template-based prompts and biologically motivated design constraints.
The workflow involved computational generation followed by multi-tier filtering for sequence quality, host tropism specificity, and evolutionary diversity. Constraints included genome length (4–6 kb), GC content, absence of long homopolymers, preservation of phage-like gene architecture, and spike protein similarity to the template phage to maintain host targeting.
Experimental validation showed that about 285 of 302 synthesized genome candidates could be assembled, and 16 produced viable infectious phages that inhibited growth of the target host strain. These generated phages displayed substantial sequence novelty, containing hundreds of mutations relative to natural Microviridae genomes, while preserving functional genome organization.
Structural and functional analyses indicated that some generated phages possessed altered protein interfaces but maintained compatible capsid–protein interactions. Cryo-electron microscopy and structure prediction suggested context-dependent co-evolution of structural proteins such as capsid and packaging proteins.
Fitness assays showed that several AI-generated phages matched or exceeded the replication and lytic performance of the template phage, and phage cocktail experiments demonstrated rapid suppression of resistant bacterial strains through recombination and mutation-driven adaptation.
The study was conducted with biosafety considerations, including restricting model training to bacteriophage genomes and using well-characterized laboratory strains. The work was supported by researchers affiliated with institutions such as the Stanford University and the Arc Institute.
Overall, the paper proposes a framework for generative genome engineering, showing that AI models can design biologically viable and evolutionarily novel bacteriophages, potentially enabling future synthetic biology and phage-based therapeutic development.
Overview of the Project Proposal: Engineering the MS2 Phage Lysis Protein L
Our primary goal is to increase the structural stability of the MS2 bacteriophage lysis protein (L) while maintaining its ability to lyse bacterial cells.
Our secondary goal is to reduce the dependency of L on the host chaperone DnaJ, which normally assists the protein in folding or activation. Reducing this dependency could allow the lysis protein to function more efficiently and independently in engineered systems.
The MS2 L protein is a 75-amino-acid single-gene lysis toxin whose C-terminal region forms a hydrophobic transmembrane domain responsible for membrane disruption and pore formation, while the basic N-terminal domain interacts with host factors such as DnaJ. Previous studies show that truncation of the N-terminal region can bypass the DnaJ requirement while preserving lysis activity.
Therefore, our design strategy focuses on:
Stabilizing the transmembrane and oligomerization regions
Maintaining essential functional motifs such as the L48–S49 motif
Exploring modifications to the N-terminal region to reduce DnaJ dependence
2. Computational Tools and Approaches
We will use a multi-step computational protein engineering pipeline combining sequence analysis, machine-learning mutagenesis predictions, and structural modeling.
2.1 BLAST – Homolog Discovery
First, we will use BLAST to identify homologous lysis proteins from related bacteriophages.
Purpose:
Identify evolutionarily conserved residues
Discover natural sequence variations that maintain function
Build a dataset for multiple sequence alignment
This will help determine which regions are functionally constrained vs mutable.
Select Candidate Mutations (stability or N-terminal modifications)
Structure Prediction (ESMFold)
Complex/Oligomer Prediction (AlphaFold Multimer)
Final Mutant Candidates (stable + functional lysis protein)
3. Proposed Engineering Pipeline
Computational workflow we will follow.
4. Expected Outcomes
Our pipeline aims to produce engineered variants of the MS2 L protein with:
Increased structural stability
Reduced aggregation risk
Maintained transmembrane insertion
Potentially reduced dependency on host DnaJ
These optimized proteins could be useful in applications such as:
Synthetic phage engineering
Bacterial ghost cell production
Antimicrobial protein development
5. Potential Pitfalls
5.1 Limited Training Data
Most protein language models and structural predictors are trained primarily on globular proteins, not small transmembrane phage toxins.
This may reduce prediction accuracy for MS2 L.
5.2 Risk of Over-Stabilization
Mutations designed to increase stability may cause:
Protein aggregation
Improper membrane insertion
Loss of functional oligomerization
Thus stability must be balanced with function.
5.3 Poor Annotation of Amurin Proteins
Single-gene lysis proteins (also called amurins) are poorly annotated in sequence databases.
This may limit the quality of homologous sequences used for alignment and training.
5.4 Host Protease Sensitivity
Mutations may unintentionally expose protease cleavage sites, making the engineered protein less stable inside bacterial cells.
6. Future Work
If promising mutants are identified computationally, the next steps would include:
Experimental expression in E. coli
Measuring lysis timing
Measuring protein stability
Testing DnaJ independence
This would validate whether computational predictions translate into improved biological function.
Week 5
Execution of workflows:
@2026a-rahul-yaji
Created a Python Script- L-Protein Mutagenisis to create random mutations at two distinct, non-conserved location L-protein, subject to the constraint that there should be no loss of lysis function due to the mutation.
The data of the L-Protein Mutants document was used to avoid loss-of-lysis (Hereafter known as LoL) mutatioins.
Key assumptions:
All the LoL mutations occur in the conserved areas
0 indicates LoL, and 1 indicates intact lysis function
If assumption 1 is true, MSA becomes redundant and therefore irrelavant
The generated mutant sequences were cofolded with host DnaJ chaperone protein to analyze interactions
So far, no mutant was found to have significant iPTM score, or interaction with DnaJ.
Generating random mutations in the Lysis protein while avoiding the loss of function or non sense codons.The Python script was generated solely by the Google Gemini 2.5 Flash, that is in-built in Google Colab. The prompt was:
Develop a Python program in Google Colab that processes an amino acid sequence and generates mutated versions of it based on experimental data. The program should perform the following steps:
The data contains information about amino acid changes and their associated ‘Lysis’ activity.
Filter the mutation data to include only ‘active’ mutations (where ‘Lysis’ is not 0). Extract the ‘Original_Residue’, ‘Position’, and ‘Mutated_Residue’ from the relevant columns (e.g., ‘Amino Acid Change’ and ‘Amino Acid Position’ or a ‘Mutation’ column like ‘X###Y’).
Create a helper function to format amino acid sequences by inserting a space after every 5 amino acids for better readability.
Implement a function generate_random_mutation_combinations(sequence, mutation_df, num_mutations) that takes an original amino acid sequence, the filtered active mutations DataFrame, and the desired number of mutations as input.
This function should:
Identify all valid mutation sites where the original residue in the sequence matches an original residue in the mutation_df.
Ensure that the num_mutations are applied to unique positions in the sequence. If there are fewer available unique mutation positions than num_mutations, it should apply all available unique mutations.
Randomly select mutations from the available options for the chosen unique positions.
Return the new mutated sequence and print the applied mutations.
Generate Multiple Mutated Sequences: Prompt the user for the number of mutated sequences they wish to generate. For each requested sequence:
Call the generate_random_mutation_combinations function.
Display the generated sequence with a clear heading (e.g., ‘Sequence 1:’, ‘Sequence 2:’, etc.).
Print both the original and the mutated sequences, using the formatting function defined in step 5.
In a separate code block, display each generated mutated sequence individually using display() so that each sequence is easily copyable by the user.
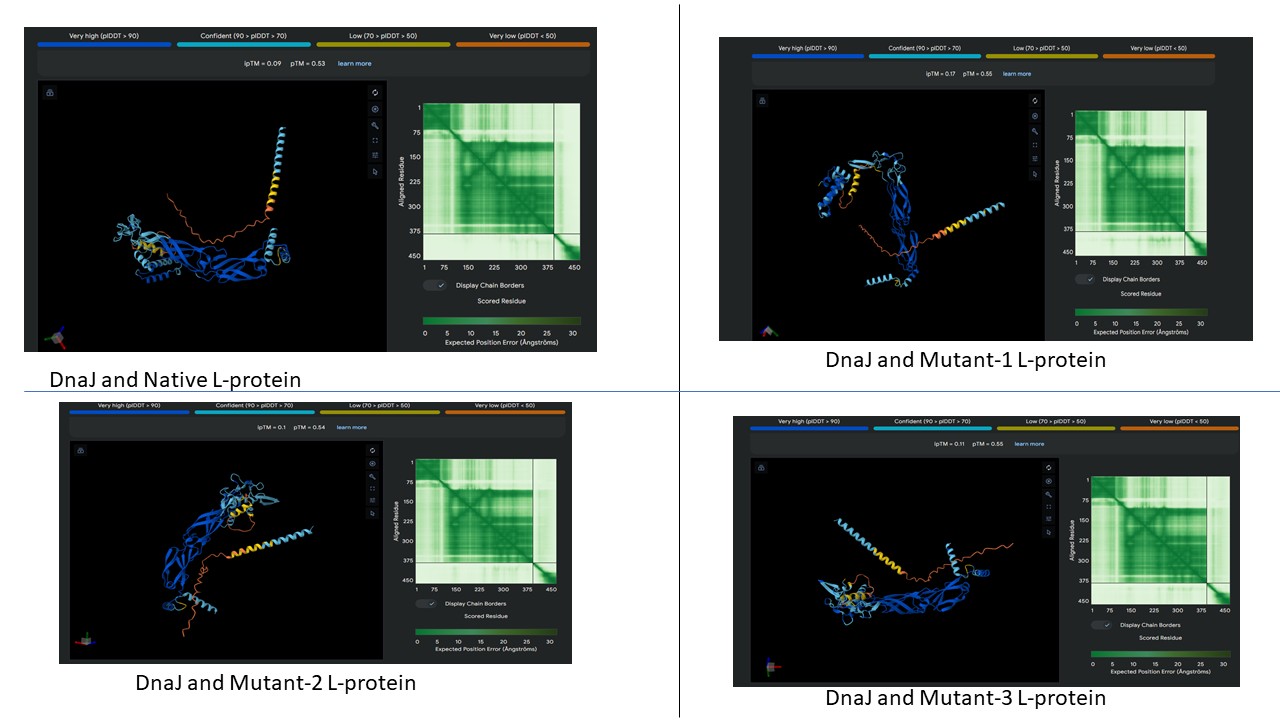
AF2 Multimer was used to co-fold the mutant Lysis protein (METRFPQQSQQTPASTNRRRPFKHEDYPCQRQQRSSTLYVLIFLAFFLSKFTNQLLLSLLEAVIRTVTTLQQLLT) and DnaJ:
The plDDT score indicates that the model is not confident about the folding of the input random mutated L protein. Overall, it suggests that the random mutation approach is very time consuming to obtain leads, and very computation-intensive. Due to limited computational resources, cofolding was not performed for other sequences.
Later, cofolding was performed using Alphafold server, and the results obtained are shown below:
Individual Final Project
Carbon Forge Red: Engineering a Photoautotrophic System for the Conversion of CO₂ into L-Lactic Acid as a Raw Material for Poly Lactic Acid on Mars.
HTGAA 2026: Individual Final Project Documentation
SECTION 1: ABSTRACT
Abstract:
Sustainable Mars settlement requires In-Situ Resource Utilization (ISRU) to reduce dependence on Earth-based supply chains. This project addresses the critical need for manufacturing materials on Mars by engineering a biological system to convert atmospheric $CO_2$ into Polylactic Acid (PLA), a versatile bioplastic for 3D printing. The broad objective is to create a photoautotrophic platform using Chlorella vulgaris for carbon fixation and polymer precursor production. We hypothesize that by redirecting metabolic flux from pyruvate to lactate via the introduction of $L$-lactate dehydrogenase ($Lldh$) and pyruvate kinase ($pk$), while knocking down phosphoenolpyruvate carboxylase ($ppc$), significant yields of $L$-lactic acid can be achieved. Specific aims include genetically modifying the algae, validating lactate accumulation, and refining extraction protocols. Methods involve CRISPR-based metabolic engineering, cell lysis, and chromatography for purification, followed by chemical polymerization. This system bridges the gap in Martian ISRU by providing a renewable source for construction and tool fabrication.
SECTION 2: PROJECT AIMS
Define three aims for your final project (minimum one sentence per aim).
Aim 1: Experimental Aim The first aim of my final project is to engineer Chlorella vulgaris to produce $L$-lactic acid from $CO_2$ by utilizing CRISPR-Cas9 gene editing to introduce $L$-lactate dehydrogenase ($Lldh$) and pyruvate kinase ($pk$) genes while knocking down the phosphoenolpyruvate carboxylase ($ppc$) gene.
I will use Benchling for genetic circuit design and codon optimization, Addgene plasmids for the CRISPR backbone, and Asimov Kernel for metabolic modeling. The experimental workflow involves algal transformation, selection via antibiotic resistance, and verification of lactate secretion using high-performance liquid chromatography (HPLC).
Aim 2: Development Aim The second aim is to scale the biological production into a functional manufacturing pipeline by optimizing the downstream purification and polymerization of $L$-lactic acid into 3D-printable Poly Lactic Acid (PLA) filament. Following a successful Aim 1, this phase involves developing efficient cell lysis protocols, utilizing ion-exchange chromatography for high-purity lactic acid recovery, and performing ring-opening polymerization to create a resin suitable for extrusion into 3D printing filaments.
Aim 3: Visionary Aim The long-term vision for this project is to establish a self-sustaining In-Situ Resource Utilization (ISRU) framework for Mars settlement, where atmospheric carbon is converted into essential structural materials without Earth-based feedstock. By validating these experiments under simulated Martian atmospheric conditions, this project aims to address the major barrier of high-mass transport costs in space exploration, enabling a new paradigm of “biological manufacturing” where settlers can grow their own tools, spare parts, and habitats from the air they cannot breathe.
SECTION 3: BACKGROUND
Background and Literature Context
Provide background research that explains the current state of knowledge and identifies the gap your project addresses.
1. Briefly summarize two peer-reviewed research citations relevant to your research:
This work highlights how metabolic pathway redirection and enzyme engineering can transform cyanobacteria into cell factories for biofuel and bioplastic precursors.
The study applies synthetic biology to reprogram Synechocystis sp. PCC6803 for sustainable lactic acid production directly from CO₂.
A heterologous lactate dehydrogenase (LDH) gene from Bacillus subtilis was stably integrated into the cyanobacterial genome, enabling light-driven conversion of carbon flux into L-lactic acid.
To boost yields, the team coexpressed a soluble transhydrogenase from Pseudomonas aeruginosa, which enhanced NADH regeneration and improved metabolic efficiency.
Researchers engineered Synechocystis sp. PCC 6803 to produce optically pure D-lactic acid directly from CO₂ using sunlight, introducing a mutated glycerol dehydrogenase (GlyDH*) as a novel D-lactate dehydrogenase.
Productivity was enhanced through codon optimization of the heterologous gene and by balancing intracellular cofactors via expression of a soluble transhydrogenase from Pseudomonas aeruginosa.
Supplementation with acetate further boosted yields, not by serving as a direct carbon source for lactate, but by supporting biomass synthesis and redirecting carbon flux from CO₂ into lactate.
The engineered strains achieved titers up to 2.17 g/L, demonstrating how synthetic biology can reprogram cyanobacteria into efficient cell factories for sustainable bioplastic precursors like polylactic acid.
Polylactic acid (PLA) is biodegradable, compostable, and derived from renewable resources like corn starch and sugarcane. It has good mechanical strength (~60 MPa tensile), thermal stability (melting point 150–160 °C), and can degrade up to 90% within six months under composting conditions, making it a strong candidate to replace petroleum plastics.
PLA is synthesized from lactic acid obtained mainly via microbial fermentation using Lactobacillus or Rhizopus species. Polymerization occurs through condensation (low molecular weight) or ring‑opening polymerization (ROP), the latter being preferred for high molecular weight PLA with tailored properties.
PLA is widely used in packaging (biodegradable bags, food containers), biomedical devices (sutures, drug delivery systems, tissue scaffolds), textiles (eco‑friendly fibers), and 3D printing. Its biocompatibility and biodegradability make it especially valuable in medical and sustainability contexts.
Despite its promise, PLA faces hurdles such as high production costs, brittleness, and limited flexibility. Research focuses on improving catalysts, blending with other polymers, and integrating nanotechnology to enhance properties and scalability for broader industrial adoption.
2. Explain how your project is novel or innovative (minimum three sentences).
I. Integration of Biological Systems into Martian ISRU Paradigms
Traditional In-Situ Resource Utilization (ISRU) frameworks for Mars primarily focus on thermochemical processes, such as the Sabatier reaction, to produce fuel and oxygen. This project challenges existing paradigms by proposing a biological ISRU platform. By leveraging Chlorella vulgaris as a self-replicating “cell factory,” the research demonstrates a novel application of synthetic biology to convert the Martian atmosphere ($CO_2$) directly into structural materials (PLA), thereby bypassing the need for high-mass feedstock transport from Earth.
II. Precision Metabolic Flux Redirection in Chlorella spp.
While lactic acid production has been demonstrated in other species of prokaryotic cyanobacteria, this project innovates through a tripartite genetic modification strategy in the eukaryotic microalgae Chlorella vulgaris. By utilizing CRISPR-Cas9 to introduce heterologous $L$-lactate dehydrogenase ($Lldh$) and pyruvate kinase ($pk$) while simultaneously implementing a knockdown of phosphoenolpyruvate carboxylase ($ppc$), the research expands the boundaries of algal metabolic engineering. This specific combination of “push” and “pull” genetic edits is designed to maximize carbon flux toward $L$-lactic acid, representing a sophisticated advancement over single-gene insertions.
III. End-to-End “Cell-to-Component” Manufacturing Pipeline
A significant technological innovation of this project is the development of a closed-loop manufacturing methodology that bridges the gap between synthetic biology and additive manufacturing. The workflow integrates computational genetic design (via Asimov Kernel) with downstream chemical processing—specifically the refinement of extraction protocols and ring-opening polymerization (ROP) for filament extrusion. This establishes a new methodology for autonomous, light-driven production where biological output is directly translated into 3D-printable hardware, providing a scalable model for sustainable extraterrestrial settlement.
Reference Summary of Core Innovations
Innovation Category
Traditional Approach
Carbon Forge Red Approach
Material Source
Earth-shipped polymers
Martian $CO_2$ conversion
ISRU Method
Mechanical/Chemical
Synthetic Biology (Photoautotrophic)
Production Cycle
Linear Supply Chain
Circular “Cell-to-Filament” Pipeline
3. Explain why your project matters and what impact it could have (minimum five sentences).
The Carbon Forge Red project addresses the critical bottleneck of logistics in long-duration space exploration: the prohibitive cost and extreme risk associated with Earth-dependent supply chains. Current Mars mission architectures are severely constrained by the mass and volume of raw materials required for construction and maintenance, making the establishment of a permanent settlement logistically fragile. By engineering a photoautotrophic system to convert atmospheric $CO_2$ into Polylactic Acid (PLA), this project provides a vital solution for In-Situ Resource Utilization (ISRU), allowing settlers to manufacture essential tools and structural components locally rather than waiting for terrestrial shipments.
This project also marks a significant advancement in synthetic biology by demonstrating that eukaryotic algae can be reprogrammed to maintain high metabolic flux toward polymer precursors even in resource-limited environments. By validating the extraction and polymerization process under Martian atmospheric parameters, the project shifts the field-level paradigm from “exploration via cargo” to “settlement via biological manufacturing.” The impact of this work is the creation of a self-sustaining infrastructure loop, enabling pioneers to grow their own habitats, tools, and replacement parts directly from the air they cannot breathe. Ultimately, this creates the foundation for true Martian autonomy, where the success of a colony is limited only by its biological efficiency rather than its proximity to Earth.
4. Describe the ethical implications associated with your project and identify relevant ethical principles (minimum two paragraphs).
The ethical implications of the Carbon Forge Red project primarily center on the introduction of genetically modified organisms (GMOs) into extraterrestrial environments, often referred to as planetary protection. By engineering Chlorella vulgaris for Mars-based manufacturing, we encounter the risk of forward contamination—the accidental transfer of Earth-based life to another planet—which could permanently obscure our ability to detect indigenous Martian life or irreversibly alter the Martian “wilderness.” Furthermore, the dual-use nature of metabolic engineering poses a concern; while the intent is to produce bioplastics for habitats, the same gene-editing techniques could theoretically be repurposed to create harmful biological byproducts or disrupt local ecological balances if a biosphere were ever to exist. This involves the ethical principle of Non-Maleficence, ensuring that our technological footprint does not cause unintended harm to a pristine planetary environment.
To ensure ethical conduct and societal responsibility, rigorous containment and “biocontainment” protocols must be implemented (one can refer my homework of week 1), such as the use of Kill Switches—genetic circuits that cause the algae to self-destruct if they escape the controlled bioreactor environment. These measures align with the principle of Responsibility, emphasizing that scientists must be stewards of the environments they manipulate. Additionally, a framework of Transparency and Justice should be adopted, ensuring that the genetic resources and technologies developed for Martian settlement are not monopolized by a single entity but are shared to benefit the broader scientific community. Societal responsibility also dictates that we conduct extensive terrestrial simulations and ethical peer reviews to assess the long-term impact of “biological manufacturing” before any actual deployment occurs in space. But designing a kill switch is beyond the scope of this project right now, and will be addressed on a future date.
SECTION 4: EXPERIMENTAL DESIGN, TECHNIQUES, TOOLS, AND TECHNOLOGY
Create a detailed experimental plan for your final project. Include a timeline for each part (minimum 15 lines/sentences).
Experimental Plan: Optimizing Photoautotrophic L-Lactic Acid Production in Engineered Synechocystis sp. PCC6803
Phase 1: Genetic Construct Design and Molecular Cloning
Methods & Tools
Design and construct integration and replicative plasmids using standard BioBrick assembly techniques.
Codon Optimization: Target genes ($ldh$ from Bacillus subtilis and Lactococcus lactis, $sth$ from Pseudomonas aeruginosa, and $pk$ from Enterococcus faecalis) will be optimized using the OPTIMIZER application.
Regulatory Elements: Genes will be placed under strong promoters ($Ptrc$ or $Ptrc2$) and equipped with the transcriptional terminator BBa_B0014.
Vector Construction:
Integration: pBluescript II SK(+) backbone with homologous flanking regions targeting the slr0168 docking site or the ppc (phosphoenolpyruvate carboxylase) locus.
Replication: Utilize the pDF_LDH replicative plasmid (RSF1010 origin) for high-copy expression.
Cloning: PCR amplification using high-fidelity Pfu or Pwo DNA polymerases; all constructs cloned into E. coli XL-1 blue host cells.
Expected Results
Successful isolation of sequence-verified plasmids with correctly oriented $ldh$, $sth$, and $pk$ gene cassettes.
Functional pairing with kanamycin or streptomycin antibiotic resistance markers.
Phase 2: Cyanobacterial Transformation and Segregation
Methods & Tools
Transformation: Genomic integration via natural transformation of glucose-tolerant Synechocystis sp. PCC6803 wild-type cells using 5–10 μg of plasmid DNA.
Conjugation: Triparental mating for replicative plasmids using E. coli J53 (pRP4) as the helper strain.
Selection: Plating on BG-11 agar supplemented with 5 mM glucose, 0.3% sodium thiosulfate, and selective antibiotics.
Segregation: Drive complete chromosome segregation via repeated, step-wise restreaking onto plates with increasing antibiotic concentrations (up to 20 μg/mL kanamycin).
Verification: Confirm segregation using colony PCR with Taq DNA polymerase.
Expected Results
Generation of fully segregated, viable mutant strains (e.g., SAA023, SAW039, and SAW041).
Partial segregation for the ppc knockdown strain (SAW033), as ppc is an essential gene.
Phase 3: Culturing, Phenotyping, and Lactic Acid Quantification
Methods & Tools
Culturing: Batch liquid BG-11 medium (50 mM $NaHCO_3$ or 10 mM TES-KOH, pH 8) in shaking incubators at 30°C under constant white-light illumination.
Growth Kinetics: Monitor optical density at 730 nm (OD730) using a spectrophotometer.
Lactic Acid Assay: Quantify extracellular L-lactic acid in cell-free supernatants using a Megazyme 96-well enzymatic assay.
Analytical Profiling: Precise organic acid quantification via HPLC using a Rezex ROA-Organic Acid H+ column and a refractive index (RI) detector.
Expected Results
The SAW041 strain (co-expressing $pk$) is expected to achieve the highest titers, reaching approximately 9.3 mM lactic acid.
Anticipated growth retardation in high-yield strains due to metabolic burden.
SAW033 (ppc knockdown) expected to show the highest carbon partitioning ratio (over 30%).
Phase 4: Protein Expression Profiling and Enzymatic Assays
Methods & Tools
Cell Harvesting: Late-exponential phase cultures harvested and disrupted using 100-μm glass beads at 4°C.
Quantification: Protein concentrations determined via BCA protein assay.
Expression Profiling:SDS-PAGE stained with Coomassie Brilliant Blue (CBB) G-250 to verify heterologous protein bands.
In Vitro Assays:
LDH Activity: Monitor NADH/NADPH oxidation at 340 nm upon addition of sodium pyruvate.
PK Activity: Coupled reaction with exogenous LDH, monitoring NADH consumption initiated by phosphoenolpyruvate (PEP).
Expected Results
Confirmation of prominent protein bands for LDH and PK on SDS-PAGE.
Verification of up to 10-fold higher LDH activity in multi-copy strains.
Measurable shifts toward NADPH co-utilization in strains utilizing the mutated B. subtilis V38R LDH.
Techniques Checklist
[x] Pipetting [x] Lab Safety [x]Bioproduction [x]Registry of Standard Biological Parts [x]Chassis Selection (e.g., DH5alpha) ☒ Bioethical Considerations [x] Plasmid Preparation [x] Bacterial Culturing [x] Quality Control/Analysis [x] Bacterial Processing (Centrifugation, Lysis, DNA Purification) [x] DNA Construct Design [x] Restriction Enzyme Digestion [x] Gel Electrophoresis [x] DNA Purification From Gel ☐ Cell-Free Systems ☐ Freeze-Dried Cell-Free Systems [x] Databases (GenBank, NCBI, Ensembl, UCSC Genome Browser) ☐ miniPCR Tools [x] Protein Purification
Lab Automation
☐ Creating Code for Laboratory Automation ☐ Using Liquid Handling Robots (e.g., Opentrons) ☐ Designing a Twist Order ☐ Creating a plan to use the Autonomous Lab at Ginkgo Bioworks
CRISPR
☐ CRISPR/Cas9 ☐ Designing Prime Editing gRNA
Protein Design
☐ Protein Design ☐ Use of Boltz or PepMLM [x] Use of Asimov Kernel [x] Use of Benchling [x] Models and Notebooks [x] Databases
1. Expand upon two techniques you checked above (minimum four sentences).
Bioproduction: The metabolic engineering of Chlorella vulgaris facilitates a direct photoautotrophic transition from atmospheric $CO_2$ to $L$-lactic acid, leveraging the organism’s high photosynthetic efficiency to drive carbon flux toward polymer precursors. Achieving industrial-scale titers requires the transition from laboratory-scale benchtop setups to high-density photobioreactors, where precise control of light attenuation, $pCO_2$ saturation, and nutrient replenishment optimizes cellular productivity. Downstream processing involves high-throughput cell disruption and ion-exchange chromatography to isolate high-purity monomeric lactic acid, followed by catalytic ring-opening polymerization (ROP) to synthesize high-molecular-weight Poly Lactic Acid (PLA) resins. This integrated bioproduction pipeline is specifically designed for modularity, allowing for autonomous manufacturing cycles that transform raw Martian atmospheric components into a consistent supply of 3D-printable filaments for structural applications.
Quality control and assurance: QA and QC for the production of Martian PLA necessitate a multi-stage validation framework to ensure structural integrity and biocompatibility of the synthesized filaments. Initial spectroscopic analysis via High-Performance Liquid Chromatography (HPLC) and Nuclear Magnetic Resonance (NMR) is required to verify the chemical purity and optical isomer ratio of the $L$-lactic acid monomers prior to polymerization. Following resin synthesis, the resulting Poly Lactic Acid must undergo rigorous thermomechanical profiling, including Differential Scanning Calorimetry (DSC) to determine glass transition temperatures and tensile strength testing to ensure compliance with aerospace structural standards. Finally, a closed-loop feedback system utilizing real-time sensor data from the extrusion process monitors filament diameter consistency and rheological properties, guaranteeing that every batch of biologically derived material meets the precision requirements for automated additive manufacturing in extraterrestrial environments.
Identify any HTGAA Industry Council companies associated with your project (optional): [x] Addgene [] Epibone [x] Ginkgo Bioworks [] Helix Nano [] Millipore Sigma [x] BioFabricate [] Biome Consortia [] Bolt [] Boltz.bio [x] Cultivarium [] DeepCure [] Mycoworks [] New England Biolabs [x] Opentrons [x] SecureDNA [] Takeda Pharmaceuticals [] Thermo Fisher Scientific [] Transfyr.ai [] Twist Biosciences [] Upside Foods [] Waters Corporation
SECTION 5: RESULTS & QUANTITATIVE EXPECTATIONS
You are required to validate at least one aspect of your final project aims.
DNA Design:
What aspect of your project did you choose to validate?
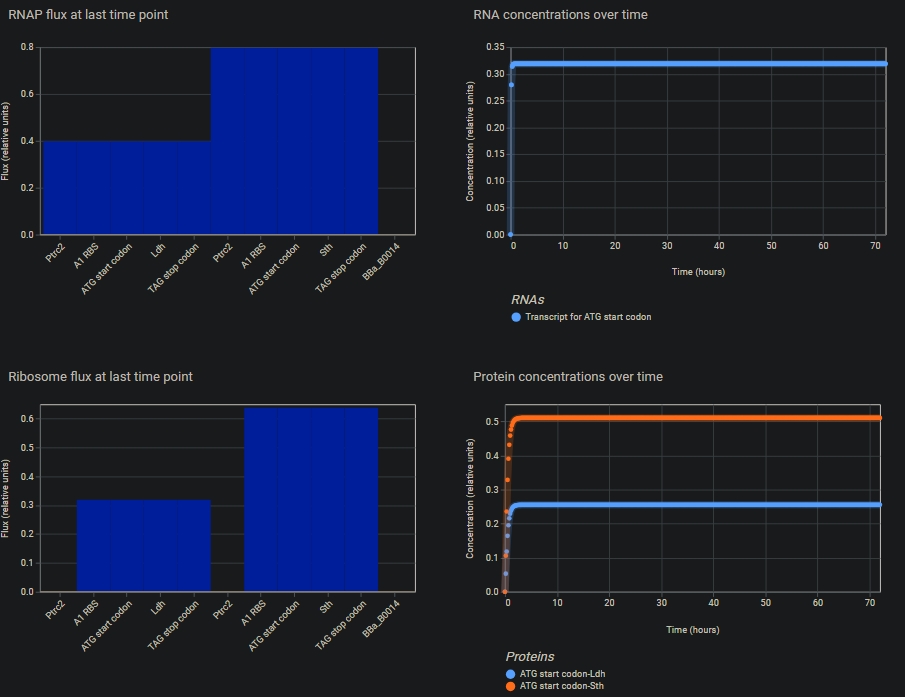
I chose to validate the gene expression level, as it could be done virtually, without lab access.
Write a detailed protocol of how you validated it.
The construct should contain: Promoter, RBS, Start Codon, CDS, Stop Codon, Terminator.
Repeat the same steps for the one more gene.
Use the simulate option to get the graphical representation of the gene expression levels
What synthetic biology techniques did you use?
Promoter construction
Codon optimization
Terminator construction
Chassis organism selection
Present data and analysis (experimental or simulated).
Describe challenges, limitations, and alternative strategies. Due to technical challenges, the DNA couldn’t be constructed in Benchling, and therefore Asimov kernel was selected. But here, only E. coli is available as the chassis organism. A eukaryotic expression model would have been better.