Class Assignment 1. First, describe a biological engineering application or tool you want to develop and why.
I want to develop a membraneless organelle within plant cells that is able to detect breakage of the cell membrane by a foreign organism. This organelle, which is comprised of intrinsically disordered proteins (IDPs), would trigger immune system responses upon detection. The purpose of this organelle is to detect and shut down fungal plant pathogens that infect through breaching cell membranes. This novel application would lower yield loss in rice plants (primarily Oriza Sativa) from fungal diseases like Rice Blast (Magnaporthe grisea) which is responsible for 10%-30% yield losses every year for rice, preventing the possibility of feeding about 60 million people.
Part 1: Benchling & In-silico Gel Art Info This is a picture of the gel art I designed on Benchling. The bands in the 1-6 ladders create the word “Hi” on completion. The restriction enzymes used on the Lambda DNA are listed above the diagram.
Assignment: Python Script for Opentrons Artwork This is a link to the code for my Opentrons Artwork.
AI Contributions: I used AI to generate large portions of my code as I am largely unfamiliar with python programming. I used Gemini AI and asked it to integrate my coordinates for my artwork into the code in Colab.
Part A. Conceptual Questions How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Assuming that meat contains about 20% protein, 500 grams of meat translates to ~100 grams of protein. If an amino acid weighs 100 Daltons, this will translate to 100 grams per mole of amino acids. Knowing this, we calculate 500 grams of meat to have one mole of amino acids. Using Avogadro’s constant, we can calulate the number to be 6 x 1023 molecules of amino acids in 500 grams of meat.
Part A: SOD1 Binder Peptide Design (From Pranam) Part 1: Generate Binders with PepMLM This is the human SOD1 sequence containing the A4V mutation: MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Binder Pseudo Perplexity 0 WHYYATGARWGE 16.929015 1 WRYGAVALELKK 12.714672 2 WRSPAAAARWWK 9.155765 3 WRYPATAAALKX 4.843841 4 FLYRWLPSRRGG N/A Info The table generated by PepMLM detailing possible peptides to bind to mutant SOD1 along with their pseudo perplexity scores. Peptide 4 is an already known SOD1-binding peptide.
Assignment: DNA Assembly What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? The Phusion High-Fidelity PCR Master Mix consists of Phusion DNA Polymerase, deoxynucleotides, and a reaction buffer (including MgCl2). The Phusion DNA Polymerase is a high-fidelity enzyme that is used to synthesize new, complementary nucleotides to the 3’ end of a DNA strand. Deoxynucleotides are present within the master mix to be added to the cloned DNA strand. The reaction buffer facilitates enzymatic function and stabilizes the DNA polymerase, allowing the PCR reaction to proceed smoothly.
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? Intracellular Artificial Neural Networks (IANNs) have several advantages over traditional Boolean genetic circuits. Traditional genetic circuits usually operate in an ON/OFF manner where genes are either expressed or not expressed. In contrast, IANNs can process inputs in a continuous and graded manner similar to biological systems. This allows IANNs to respond to multiple inputs at once with varying strengths instead of simple binary outputs. IANNs are also more capable of pattern recognition, noise tolerance, and complex decision making. These systems can integrate many molecular signals simultaneously and produce more flexible cellular behaviors than standard Boolean logic gates.
General homework questions/answers Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell-free protein synthesis has several advantages over traditional in vivo protein expression methods. In cell-free systems, researchers have direct control over experimental conditions such as pH, ion concentrations, temperature, substrate availability, and DNA concentration without needing to maintain living cells. This allows for rapid optimization and easier manipulation of gene expression conditions. Cell-free systems also avoid problems associated with cellular toxicity, metabolic burden, and membrane transport limitations. In addition, proteins can be produced much more quickly because there is no need for cell growth or transformation. Cell-free expression is especially beneficial when producing toxic proteins that would kill living cells and when rapidly prototyping genetic circuits for synthetic biology applications.
Homework: Final Project Measurement Draft Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc. For the experimental validation of Project Aero-Sentry, several distinct biological and biochemical components must be measured to ensure the engineered probiotic nasal spray functions as intended. First, the transcriptional activity and expression level of the chimeric EnvZ receptor must be quantified to confirm successful membrane integration. Second, the binding affinity and kinetic interaction between this engineered receptor and the target allergen, Bet v 1, must be measured to verify the precision of the sensory mechanism. Third, the transcriptional output of the sense-and-respond genetic circuit must be monitored, specifically tracking the induction of the Nb32ILZ nanobody gene under the control of the osmolarity-responsive pOmpF promoter. Finally, the total concentration of the secreted Nb32ILZ fusion nanobody in the extracellular environment must be measured alongside its ultimate neutralization efficiency when bound to Bet v 1, confirming that the therapeutic countermeasure is produced in effective quantities.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork For the artwork, I contributed to four pixels on the DNA strand. I really liked how the whole community came together to work on this one project. It was nice to interact with all the other students in the HTGAA course.
1. First, describe a biological engineering application or tool you want to develop and why.
I want to develop a membraneless organelle within plant cells that is able to detect breakage of the cell membrane by a foreign organism. This organelle, which is comprised of intrinsically disordered proteins (IDPs), would trigger immune system responses upon detection. The purpose of this organelle is to detect and shut down fungal plant pathogens that infect through breaching cell membranes. This novel application would lower yield loss in rice plants (primarily Oriza Sativa) from fungal diseases like Rice Blast (Magnaporthe grisea) which is responsible for 10%-30% yield losses every year for rice, preventing the possibility of feeding about 60 million people.
2. Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
To ensure an ethical future, it is imperative to use preventative measures against ecological harm or unintended spread of this tool. One sub-goal to support this goal is to pursue the genetic containment of any engineered constructs within the plant cells. This could be done through designing constructs that can only function within plant cells. Another sub-goal is ensuring reversibility of the tool and monitoring altered organsism over time. Molecular constructs that are able to disable the tool along with monitoring programs can secure reversibility in case of adverse effects to the environment. Another policy goal to pursue is promoting equitable and responsible agricultural use. Promoting equitable use can be done through confirming affordable implementation for small farmers. Practicing transparency on the mechanics of the engineered system with farmers and consumers along with informed consent are vital for responsible agricultural uses.
3. Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”).
Option 1. Currently, engineered crops are evaluated for environmental risk without any requirement for active containment mechanisms beyond general biosafety assessment. Federal regulators like the USDA-APHIS should create a requirement where engineered disease-responsive crops must have genetic containment features within its design to reduce unintended spread. This requirement should be a condition for any field trials/commercialization of genetically modified plants. The developers of the engineered crops will document containment logic and submit documentation to regulators within the federal agency for approval. This design assumes that genetic containment strategies will effectively reduce ecological risk and that regulators can consistently evaluate various containment designs across differing technologies. This option carries the risk of mutations or environmental variability bypassing containment procedures in practice. In addition, this option could slow innovation and burden smaller developers.
Option 2. Novel pathogen resistance strategies often only evaluated for efficacy and not for how they can shape pathogen evolution. Research institutions and academic funders (e.g., NSF, USDA-NIFA) can create new incentives for researchers to create new design strategies for pathogen resistance that take evolution and ecological pressure into consideration. Academic funders can reward researchers for designing damage-based or multi-signal immune systems over single effector type targets. This can be enforced through giving priority funding or recognition to projects that demonstrate reduced selective pressure. This option assumes that researchers can accurately anticipate evolutionary dynamics upon design. There is still a risk of pathogens adapting in an unforeseen way. It is also important to note that pathogen resistance strategies can lead to overly sensitive immune responses in crops which could hinder crop yield.
Option 3. Many engineered crops today are owned and controlled by companies which makes it more difficult for farmers to use these technologies. By using regulatory or financial incentives, we can encourage developers to commit to equitable licensing and deployment models to ensure more accessibility for small farmers. Government agencies can offer incentives such as faster regulatory review or public grant eligibility so that developers will agree to low-cost licensing and clear communication to farmers on benefits and limitations. This policy assumes that broader access to engineered crops will improve food security and the incentives are enough to influence company behavior. A possible issue with this policy is that smaller crop developers may struggle to meet equity requirements, unintentionally favoring bigger companies.
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
2
1
N/A
• By helping respond
1
2
N/A
Foster Lab Safety
• By preventing incident
2
2
N/A
• By helping respond
1
2
N/A
Protect the environment
• By preventing incidents
2
1
N/A
• By helping respond
1
2
N/A
Other considerations
• Minimizing costs and burdens to stakeholders
3
2
1
• Feasibility?
2
2
2
• Promote Accessibility?
3
3
1
• Not impede research
3
2
2
• Promote constructive applications
2
1
1
4. Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
The scoring demonstrates that the best course of action would be to combine Option 2 (incentives for evolution-resilient design) and Option 3 (accessible deployment and licensing). Option 2 would be prioritized as it performs the strongest in prevention-focused categories (biosecurity and environmental protection) while also minimizing burdens on researchers and scientific discovery. This option is vital in preventing detrimental harm to the environment.
Option 3 complements Option 2 by strengthening its gaps found outside of prevention-based categories. Option 3 allows for accessibility while also minimizing costs to stakeholders. By combining these two options, my bioengineering tool maximizes accessibility and impact while minimizing risks to the environment.
While Option 1 is strong in response to risks, it scored poorly on cost, accessibility, and research freedom. Prioritizing this option would ultimately slow innovation and burden smaller research teams. With our chosen options, we assume that developers would prioritize equitable deployment incentives and that early design decisions are effective at preventing ecological/evolutionary risk. However, we are uncertain as to whether incentives may be enough to convince profit-driven companies and if preventative design measures are able to prevent pathogens from evolving in unforeseen ways.
Week 2 Lecture Preparation
Homework Questions from Professor Jacobson
1. Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
The error rate of polymerase is 1:106. Since the human genome is 3.2 billion base pairs long, this error rate would create about 32,000 errors per replication cycle. This would be catastrophic for genetic stability. Biology deals with this discrepancy by using error correcting polymerase, which actively corrects mistakes before they become permanent mutations in the DNA.
2. How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
The average human protein has about 1036 base pairs. With about 345 codons, there can easily be 10100 or more ways to code the average human protein. A possible reason for why all these different codes do not work to code for the protein of interest is that mRNA could have an altered structure which could affect translation initiation, elongation, or stability. Another possible reason is that some sequences can create splice sites, promoter elements, or miRNA binding sites.
Homework Questions from Dr. LeProust
1. What’s the most commonly used method for oligo synthesis currently?
The most commonly used method for oligo synthesis currently is phosphoramidite-based solid-phase synthesis.
2. Why is it difficult to make oligos longer than 200nt via direct synthesis?
The main reasons for why it is so difficult to make oligos longer than 200 nt via direct synthesis is because of various reasons. To start, each cycle has a small failure rate which dramatically drops yield after 200 cycles. Long synthesis also increases the risk of base damage and full-length product is difficult to separate from failure sequences of a similar length.
3. Why can’t you make a 2000bp gene via direct oligo synthesis?
You can’t make a 2000bp gene via direct oligo synthesis as the small failure rate for each cycle would make the yield essentially zero. Too many errors would accumulate for a 2000-mer sequence.
Homework Questions from George Church
1. What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The 10 essential amino acid in all animals are histidine, isoleucine, lysine, methionine, phenylalanine, threonine, typtophan, valine, and arginine. This makes me feel as if the Lysine Contingency is a very flawed method. While the dinosaurs did require lysine to survive, they could have eaten lysine from any animal in their diet. A more robust contingency would’ve involved multiple essential nutrients or synthetic auxotrophy.
2. What code would you suggest for AA:AA interactions?
For AA:AA interactions, I would suggest a “Lock-and-Key” chemical match based on the charge, hydrophobicity, size, shape, and special pairs (like Cysteine forming strong bonds).
3. Given the one paragraph abstracts for these real 2026 grant programs sketch a response to one of them or devise one of your own:
If our most advanced biological medicines were as easy to store and ship as aspirin, this would mean that more people could receieve life-saving gene and cell therapies. We would not have to worry about ultra-cold freezers which saves hundreds of thousands of dollars. We would be able to reach people in remote areas who would not have the resources to have frigid freezers to store medicines. This would revolutionize medicine and allow for cheap distribution to every corner of the world.
Week 2 HW: DNA Read, Write, & Edit
Part 1: Benchling & In-silico Gel Art
Info
This is a picture of the gel art I designed on Benchling. The bands in the 1-6 ladders create the word “Hi” on completion. The restriction enzymes used on the Lambda DNA are listed above the diagram.
Part 3: DNA Design Challenge
3.1. Choose your protein.
The protein that I chose is the human testis-determining factor. I chose this protein because I find it interesting how one gene plays such a big role in sex differentiation and is the largest factor in deciding how a human embryo will grow. It is interesting to think about how one type of protein encoded by one gene in the human genome can spark significant change in the entire development process of humans. This sequence was taken from the NCBI:
>NP_003131.1 sex-determining region Y protein [Homo sapiens]
MQSYASAMLSVFNSDDYSPAVQENIPALRRSSSFLCTESCNSKYQCETGENSKGNVQDRVKRPMNAFIVWSRDQRRKMALENPRMRNSEISKQLGYQWKMLTEAEKWPFFQEAQKLQAMHREKYPNYKYRPRRKAKMLPKNCSLLPADPASVLCSEVQLDNRLYRDDCTKATHSRMEHQLGHLPPINAASSPQQRDRYSHWTKL
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
> reverse translation of NP_003131.1 sex-determining region Y protein [Homo sapiens] to a 612 base sequence of most likely codons.
ATGCAGAGCTATGCGAGCGCGATGCTGAGCGTGTTTAACAGCGATGATTATAGCCCGGCGGTGCAGGAAAACATTCCGGCGCTGCGCCGCAGCAGCAGCTTTCTGTGCACCGAAAGCTGCAACAGCAAATATCAGTGCGAAACCGGCGAAAACAGCAAAGGCAACGTGCAGGATCGCGTGAAACGCCCGATGAACGCGTTTATTGTGTGGAGCCGCGATCAGCGCCGCAAAATGGCGCTGGAAAACCCGCGCATGCGCAACAGCGAAATTAGCAAACAGCTGGGCTATCAGTGGAAAATGCTGACCGAAGCGGAAAAATGGCCGTTTTTTCAGGAAGCGCAGAAACTGCAGGCGATGCATCGCGAAAAATATCCGAACTATAAATATCGCCCGCGCCGCAAAGCGAAAATGCTGCCGAAAAACTGCAGCCTGCTGCCGGCGGATCCGGCGAGCGTGCTGTGCAGCGAAGTGCAGCTGGATAACCGCCTGTATCGCGATGATTGCACCAAAGCGACCCATAGCCGCATGGAACATCAGCTGGGCCATCTGCCGCCGATTAACGCGGCGAGCAGCCCGCAGCAGCGCGATCGCTATAGCCATTGGACCAAACTG
3.3. Codon optimization.
Optimizing a codon sequence can have various impacts. Codon optimization replaces less-favored codons in a specific organism with more common codons. An optimized codon sequences has a higher efficiency in translation which then leads to higher levels of protein expression. In addition, an optimized codon improves the stability of the mRNA since they are more likely to be recognized by tRNAs. Overall, an optimized codon is more likely to have increased protein expression.
> Optimized codon sequence of NP_003131.1 sex-determining region Y protein to Humans (Homo sapiens).
ATGCAGTCCTATGCCTCCGCCATGCTGAGCGTGTTTAACAGTGATGACTACTCCCCAGCCGTGCAGGAGAACATCCCAGCCCTGAGACGCAGCAGCTCATTCCTGTGTACCGAGTCTTGCAACTCCAAGTACCAGTGCGAGACCGGCGAGAACAGTAAGGGAAACGTGCAGGATCGCGTGAAAAGGCCCATGAACGCTTTCATCGTGTGGAGCCGCGATCAGAGGAGGAAGATGGCCCTGGAGAATCCCAGGATGCGGAACAGCGAAATCTCCAAGCAGCTGGGCTACCAGTGGAAGATGCTGACCGAGGCCGAGAAGTGGCCATTTTTCCAGGAGGCACAGAAGCTGCAGGCCATGCACAGAGAGAAGTACCCCAATTACAAGTACAGACCCAGAAGAAAGGCCAAAATGCTGCCTAAGAACTGTTCCCTGCTGCCCGCCGACCCAGCCTCCGTGCTGTGCTCTGAAGTCCAGCTGGACAACCGCCTGTACAGAGACGACTGTACCAAGGCCACCCACTCCCGCATGGAACACCAGCTGGGGCACCTGCCCCCCATTAATGCCGCATCCTCCCCCCAGCAGCGCGACCGGTACAGCCACTGGACAAAGCTG
3.4. You have a sequence! Now what?
To produce this protein from my DNA, we can use a multitude of both cell-dependent and cell-free methods.
We can living cells as “factories” for our proteins. We can design a plasmid containing our TDF-encoding gene that also has a promoter, ribosome binding site, and antibiotic resistance marker. Through transforming a host cell (like Escherichia coli) with this DNA, we can then induce expression of the TDF protein within the cells. Upon harvesting and purifying the protein, we can then have a batch of TDF proteins.
If a cell-free system was preferable, we could combine ribosomes, tRNAs, polymerases, amino acids + nucleotides, and an energy system with our DNA to create our protein in a test tube. This method would be much faster.
Part 4: Prepare a Twist DNA Synthesis Order
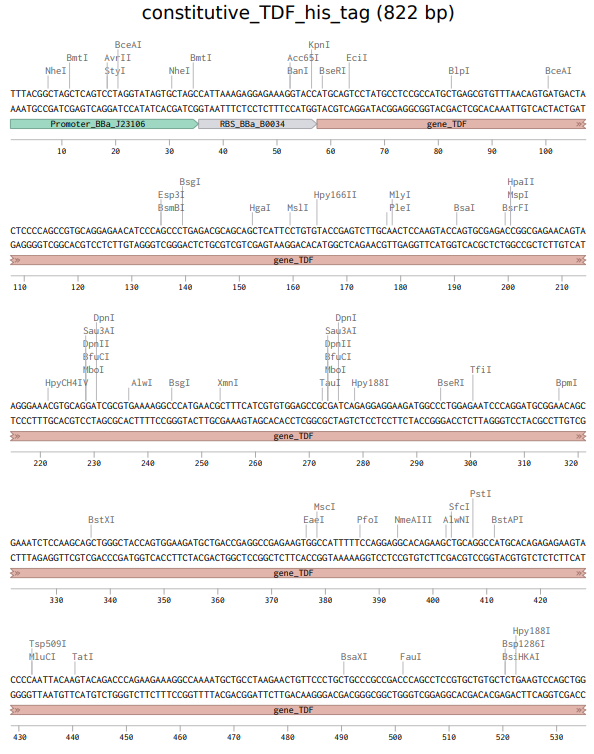
Info
The above photo is the sequence of the expression cassette to express TDF proteins. This photo was taken in Benchling.
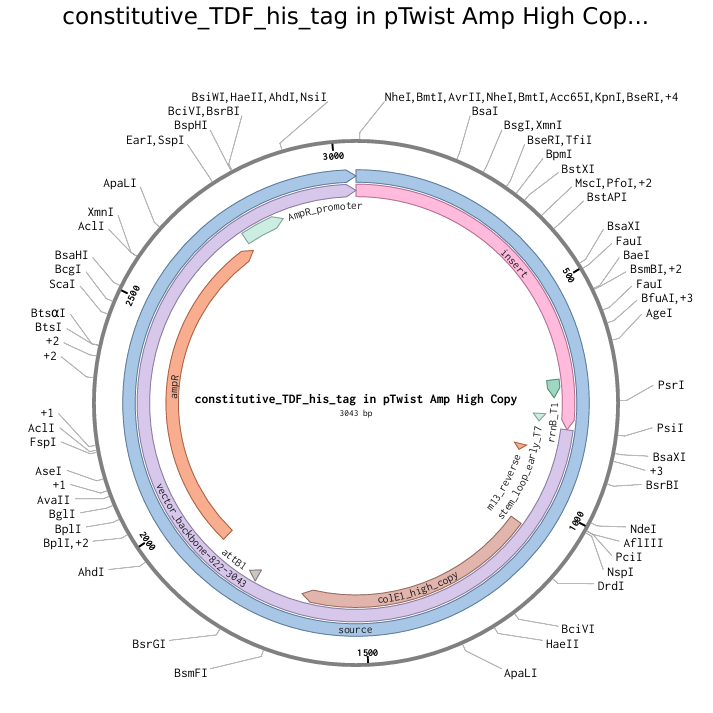
Info
The above photo is a pTwist Amp High Copy plasmid backbone with the TDF-expressing insert (shown by “insert”). This photo was taken in Twist Bioscience.
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
I would want to sequence the genome of a lactose intolerant person. I want to sequence this genome to better understand what genes are implicated in the reduced expression of lactase in lactose-intolerant patients.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
To sequence my DNA, I would use Illumina Whole-Genome Sequencing. I would use this method as it can give a complete, highly accurate view of someone’s entire DNA sequence. This is a second-generation sequencing method that can sequence millions of short fragments in parallel and uses PCR amplification. For our input, we must extract blood/saliva and purify the genomic DNA of these cells. Then, we shear the DNA into ~200-500 bp fragments using enzymes. Once we ligate synthetic adapters to both ends of the fragments, we can then PCR amplify the adapter-ligated fragments. The prepared DNA can be combined with complementary oligos from a flow cell to generate clusters. This method uses a sequencing-by-synthesis method. By adding fluorescently labled nucleotides, we can use a camera to record which color/nucleotide attached to the sequence. After removing the nucleotide chemically and repeating the process with each type of nucleotide, we can then generate a raw FASTQ file.
DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
I would like use synthesize a sequence of DNA that can work in a cell-free system and detect certain molecules present in a disease. This circuit will detect specific molecules from a pathogen like malaria. I want to create this biosensor to give impoverished areas a way to detect disease with cheap methods.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I will perform this DNA synthesis using Gibson Assembly and molecular cloning. I can order the parts of the genetic circuit and assemble them accordingly using Gibson Assembly. After assembling the plasmid, the plasmid can be amplified through transformation, cloning in a bacterial cell, and purification. The limitations of this method is that it can be slower due to colony screening and sequencing verification and could include failed ligations, wrong inserts, etc. It also requires that I have already synthesized DNA.
DNA Edit
(i) What DNA would you want to edit and why?
One application for editing human DNA is to cure lactose intolerance. By editing the genome of a lactose intolerant human to be able to produce lactase, we can cure his lactose intolerance and allow him to consume foods with lactose. This application of gene editing is just one example of how synthetic biology can be leveraged to solve common human disorders.
(ii) What technology or technologies would you use to perform these DNA edits and why?
Lactose intolerance is commonly caused due to reduced expression of the lactase enzyme. To fix this, one could modify regulatory variants near the LCT gene in the human genome. The best way of doing this uses base editing systems (a CRIPSR-derived technology) to convert one base into another at a targetted location. This process works by a guide RNA first directing a Cas protein to a specific DNA sequence. Upon finding this DNA sequence, the Cas protein binds to the target site and a fused enzyme will then chemically convert the base into another base. After the cell’s repair mechanisms fix the strand, there will be a single-letter DNA change.
To prepare to preform this DNA edit, you must first identify which regulatory variant is implicated in adult lactase expression. Then, you have to prepare a sequence-specific guide RNA, base editor protein, and delivery system (maybe viral?). As a note, this system will target human intestinal stem cells in vivo. This method, however, carries various limitations. Since it is in vivo, delivery will be extremely difficult and all cells will not be edited (mosaic editing). In addition, this editing method may cause unintended edits in other areas of the genome.
AI Contributions: I used AI to generate large portions of my code as I am largely unfamiliar with python programming. I used Gemini AI and asked it to integrate my coordinates for my artwork into the code in Colab.
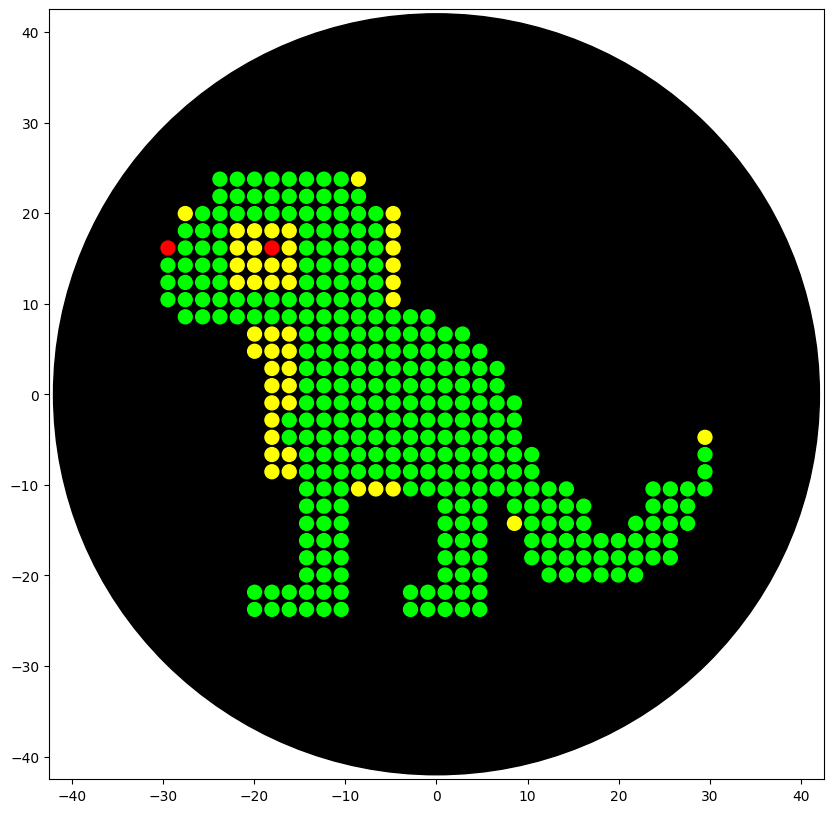
Info
This image is a simulation of the product of my code when ran on the Opentrons robot. It is pixel art of a lizard.
Info
An image of the result of my code being run on the Opentrons robot at William and Mary.
Post-Lab Questions
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
The published paper that I found is titled “Semiautomated Production of Cell-Free Biosensors” (Brown, 2025). In this publication, the researchers develop and demonstrate an automated pipeline using the Opentrons OT-2 liquid handling robot to (mostly) automate and scale the manufacturing of cell-free biosensors. In their pipeline, they used the Opentrons to produce a full 384-well plate of fluoride-sensing biosensors. The researchers had the objective of using the Opentrons OT-2 robot to develop a method capable of high-throughput manufacturing, reduced variability in sensor performance, and accessibility across global labs.
When compared to manually assembled biosensors, the biosensors created by the robot proved to have greater consistency among detection thresholds. This research suggested that facilities or field clinics could use Opentrons robots to assemble diagnostic tests on-demand rather than importing them from outside sources. In addition, the success of this study shows how the cheaper OT-2 robot can be used in replacement of industrial-grade liquid handlers so that the production of synthetic biology-based diagnostics can be done in resource-limited settings.
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
In my final project, I want to use automation tools to reliably produce biosensors and test them. I want to use the Opentrons robot to create multiple samples of protoplast cells altered by CRIPSR/Cas9 to produce a certain biosensor within the cell. These cells would be incubated and grown. Then, in an experiment, the automation tools would expose these cells to pathogens to see if the biosensor is able to reliably detect the presence of the pathogen. The fluoresence created by the biosensor can be measured by PHERAstar.
References
Brown, D. M., Phillips, D. A., Garcia, D. C., Arce, A., Lucci, T., Davies Jr., J. P., Mangini, J. T., Rhea, K. A., Bernhards, C. B., Biondo, J. R., Blurn, S. M., Cole, S. D., Lee, J. A., Lee, M. S., McDonald, N. D., Wang, B., Perdue, D. L., Bower, X. S., Thavarajah, W., … Lucks, J. S. (2025). Semiautomated Production of Cell-Free Biosensors. ACS Synthetic Biology, 14(3), 979-986. https://doi.org/10.1021/acssynbio.4c00703.s001
Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Assuming that meat contains about 20% protein, 500 grams of meat translates to ~100 grams of protein. If an amino acid weighs 100 Daltons, this will translate to 100 grams per mole of amino acids. Knowing this, we calculate 500 grams of meat to have one mole of amino acids. Using Avogadro’s constant, we can calulate the number to be 6 x 1023 molecules of amino acids in 500 grams of meat.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
When you eat beef or fish, you eat cells that contain various macromolecules like DNA, proteins, lipids, and carbohydrates. While these macromolecules would usually have an effect on a human’s cells, our bodies instead digest these molecules and break them down into smaller forms (Proteins into amino acids, DNA into nucleotides, etc.). So, by the time the macromolecules reach our bloodstream, there are no intact cow or fish genes/proteins. Our cells take up these digested molecules and uses them to rebuild our own human genome, not theirs.
Why are there only 20 natural amino acids?
There are only 20 natural amino acids because that number of different amino acids provides enough chemical diversity for protein function while also avoiding the drawbacks of having more amino acids. Adding any more amino acids would only supply new amino acids that would behave very similarly to one of the original 20 amino acids. In addition, since similar codons can encode chemically similar amino acids, adding an additional amino acid could increase the chance of a small genetic mutation causing a disruptive substitution in the protein structure.
Where did amino acids come from before enzymes that make them, and before life started?
Before life existed, amino acids were synthesized on Earth through ordinary chemistry. The Miller-Urey experiment demonstrated that early Earth’s atmosphere was capable of forming several types of amino acids without the help of cells or enzymes. Amino acids can form spontaneously through prebiotic chemical reactions that are driven by UV radiation, hydrothermal vents, lightning, etc.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
If you made an α-helix using D-amino acids, you would form a left-handed α-helix. However, if you used L-amino acids, you would create a right-handed α-helix. This is due to the peptide backbone’s fixed bond angles which make a D-amino acid-based α-helix energetically favorable in a left-handed form.
Can you discover additional helices in proteins?
Using synthetic biology, it is possible to design new helices in proteins. Researchers can use D-amino acids and noncanonical amino acids to modify backbone chemistry and engineer altered hydrogen bonding patterns. A new helice is unlikely to emerge naturally since the chemistry of protein backbones is heavily constrained.
Why are most molecular helices right-handed?
Most molecular helices are right-handed because of how the chiral L-amino acids and D-sugars act within the protein/DNA strand. Due to their stereochemistry, L-amino acids and D-sugars find right-handed helices more energetically favorable. Due to evolution, organisms tend to favor L-amino acids and D-sugars which causes most moelcular helices to instead be right-handed.
Why do β-sheets tend to aggregate?
β-sheets have hydrogen bond donors (N-H) and hydrogen bond acceptors (C=O). To satisy these untaken hydrogen bonds, β-sheets form hydrogen bonds with other strands. Unlike α-helices, β-sheets will depend on intermolecular hydrogen bonding. This causes them to become aggregation-prone.
What is the driving force for β-sheet aggregation?
The main driving force for β-sheet aggregation primarily includes backbone hydrogen bonding. Each added strand forms another array of H-bonds which increases stability. In addition, β-sheet-forming sequences usually have a hydrophobic effect where one face is hydrophobic while the other is polar. This causes water to be excluded from the packed-together hydrophobic faces.
Why do many amyloid diseases form β-sheets?
Amyloid fibrils share a universal Cross-β architecture motif. In this structure, β-strands run perpendicular to the fibril axis where sheets stack. In proteins that misfold, sequences are exposed that are capable of forming extended β-strands and are normally buried. This causes aggregation to become autocatalytic which results in additional monomers to attach to the growing fibril.
Can you use amyloid β-sheets as materials?
Amyloid β-sheets are strong, highly ordered, self-assembling, and chemically programmable. This makes them highly capable of being used as a material. Researchers are exploring them for use in nanofibers, biomaterials, drug delivery scaffolds, etc. Since they are sequence-programmable, researchers can engineer peptides that can be mechanically tunable and form controlled fibers.
Part B: Protein Analysis and Visualization
A 3D render of the structure of a homolog of lactase.
1. Briefly describe the protein you selected and why you selected it.
The protein that I chose was the lactase (LCT) enzyme (structure shown above). I chose this protein because this enzyme is heavily implicated in lactose intolerance. I have a large interest in using synthetic biology to be able to cure lactose intolerance.
2. Identify the amino acid sequence of your protein.
The most common amino acid is: L, which appears 170 times.
Using Uniprot’s BLAST tool, I found 250 protein sequence homologs for LCT. The result of the BLAST tool can be found here.
3. Identify the structure page of your protein in RCSB.
I was not able to find the same exact protein in RCSB. However, I found a close homolog in Escherichia coli. The PDB ID is pdb_00005e9a.
This structure was solved on 2016-10-26. The resolution of the structure is 2.56 Å. This is an acceptable/good quality structure.
In the structure, there are water molecules and typically bounder inhibitors or substrate analogs.
When put into SCOP, the PDB ID and Uniprot ID returns no results. I assume this is because the protein does not belong to any structure classification family.
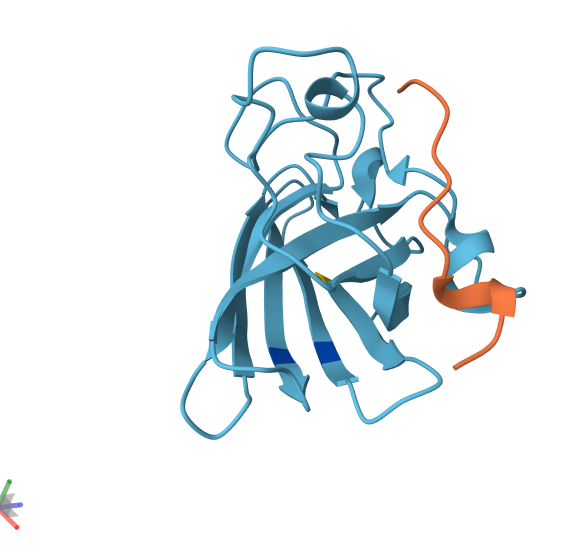
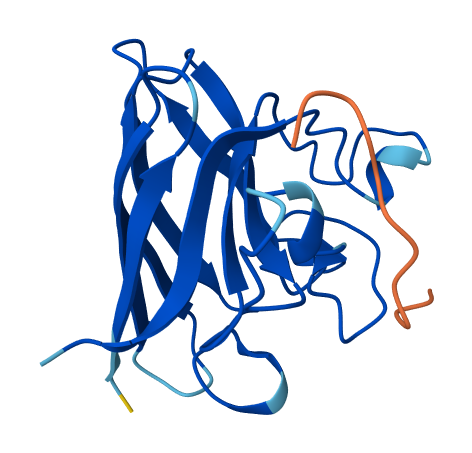
4. Open the structure of your protein in any 3D molecule visualization software.
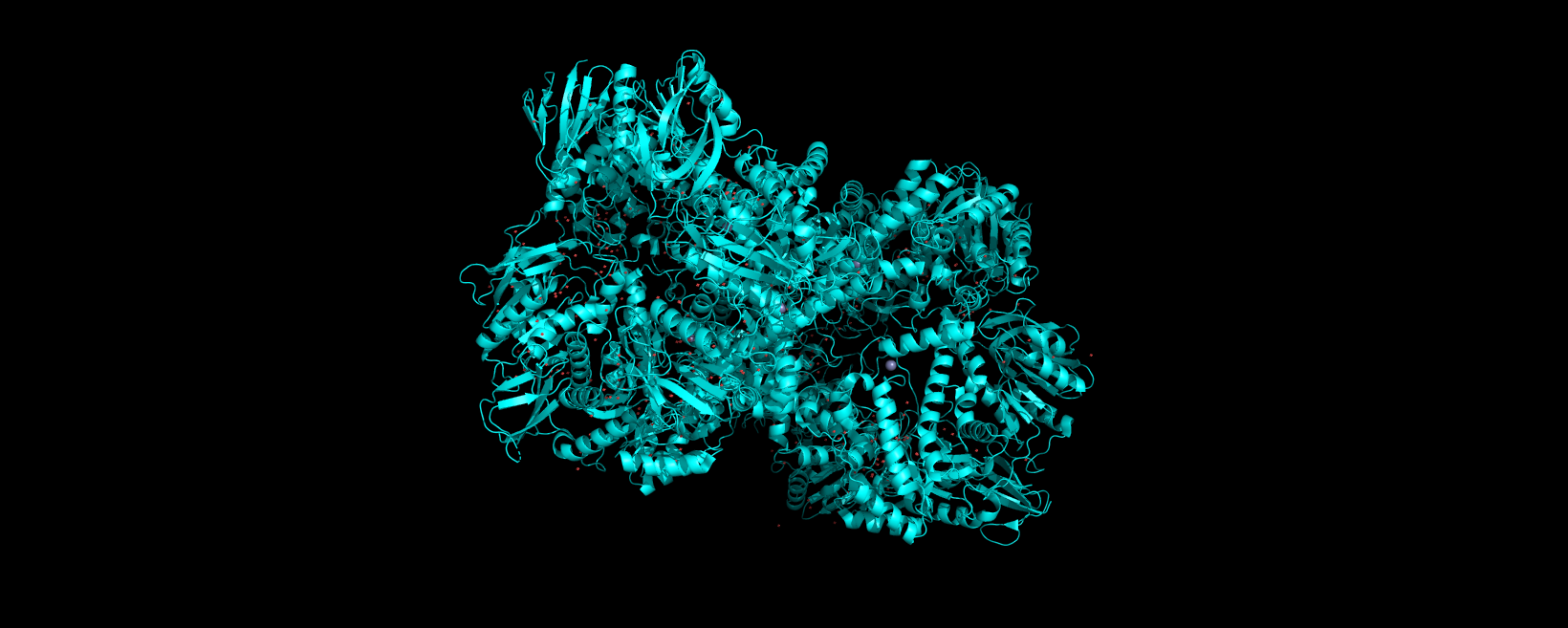
Info
3D visualization of “ball-and-stick” model of 5E9A protein.
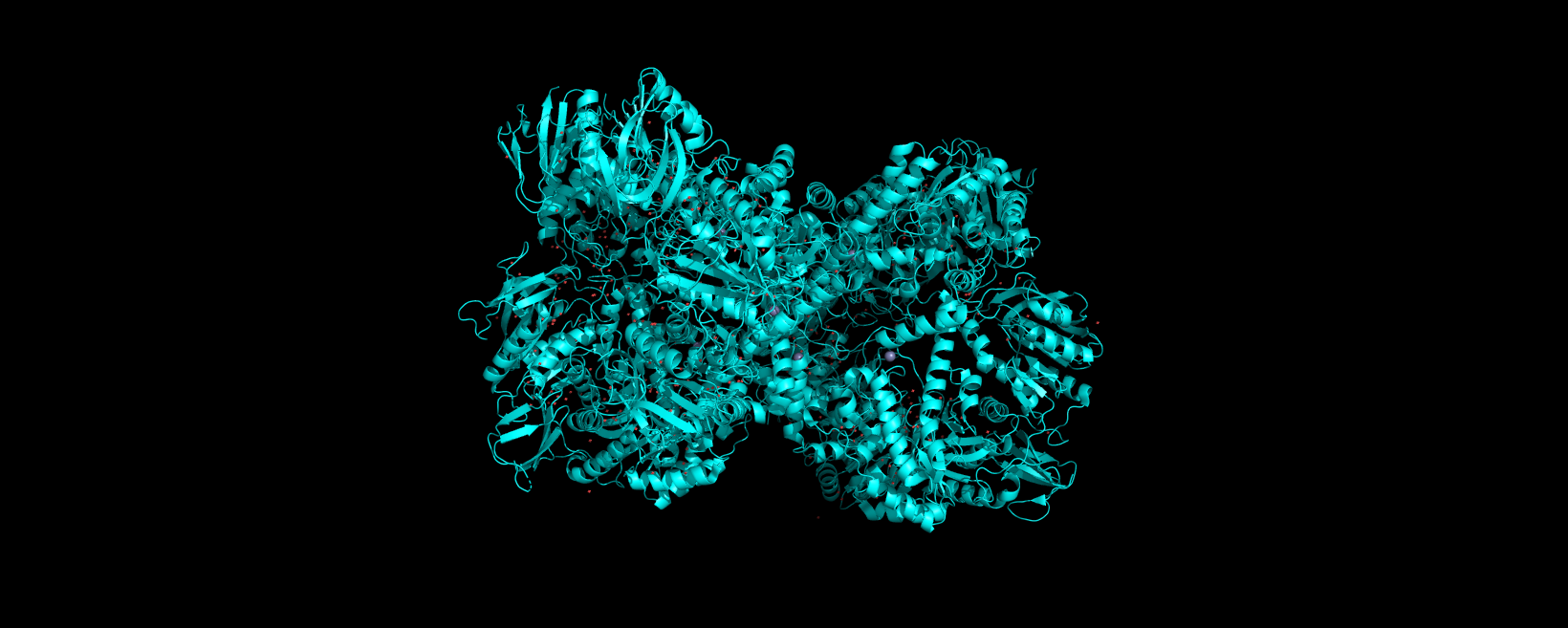
Info
3D visualization of “ribbon” model of 5E9A protein.
Info
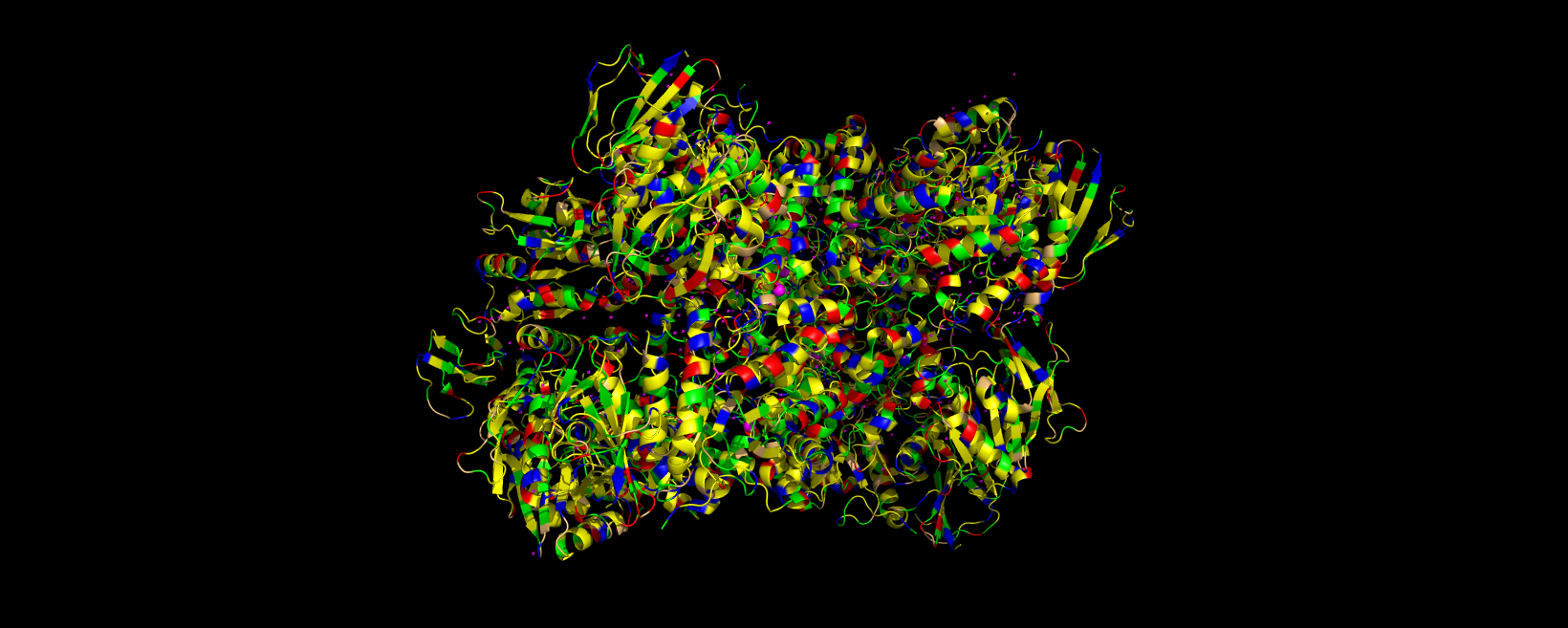
3D visualization of “cartoon” model of 5E9A protein.
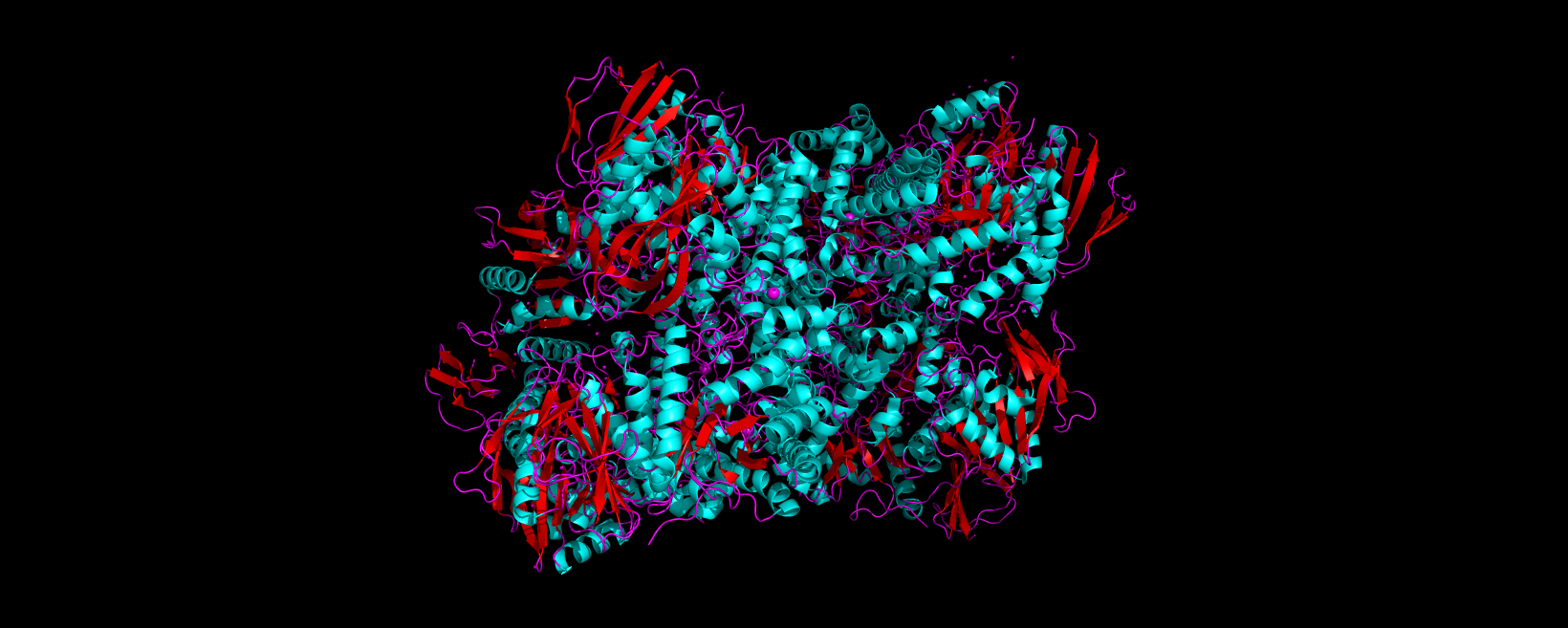
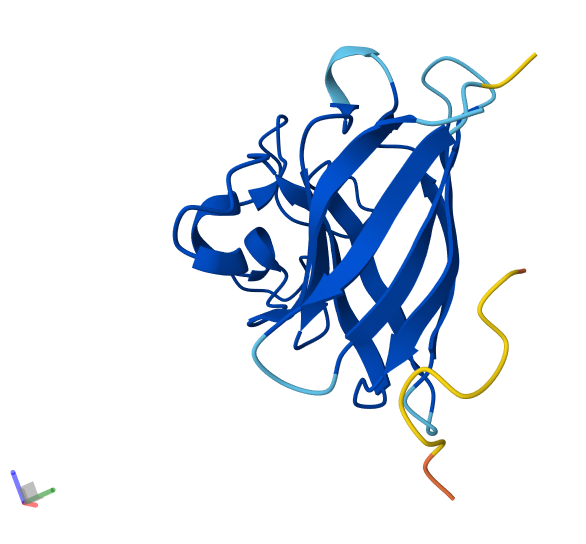
Info
3D visualization of 5E9A protein colored by secondary structure. Cyan is helix, red is sheet, and magenta is loop.
The above image of 5E9A colored by its secondary structure shows how the structure most likely contains more helices than sheets.
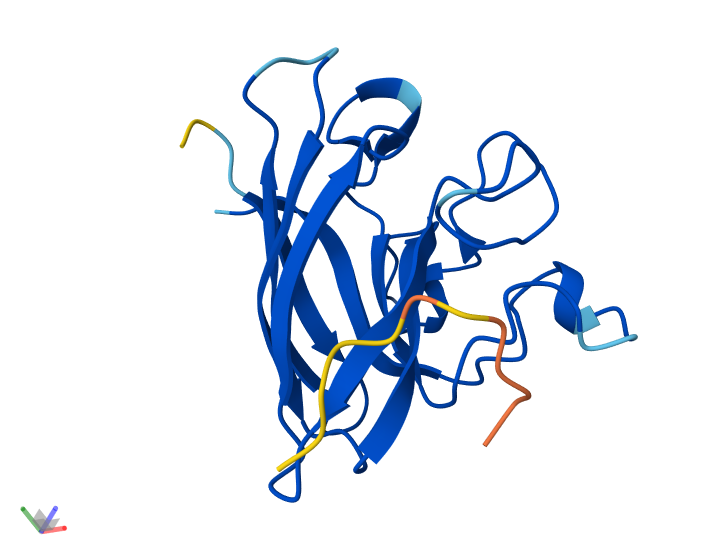
Info
3D visualization of 5E9A protein colored by residue type. Yellow is hydrophobic, green is polar (uncharged & hydrophilic), blue is positively charged (hydrophilic), red is negatively charged (hydrophilic), and light orange is Glycine (special case, both hydrophobic and hydrophilic).
The 3D visualization of the 5E9A protein colored by residue type shows how the protein is about half hydrophobic amino acids and half hydrophilic amino acids. The protein shows a somewhat alternating pattern between hydrophilic and hydrophobic residues. This pattern creates the helices, sheets, and loops shown in the model.
Info
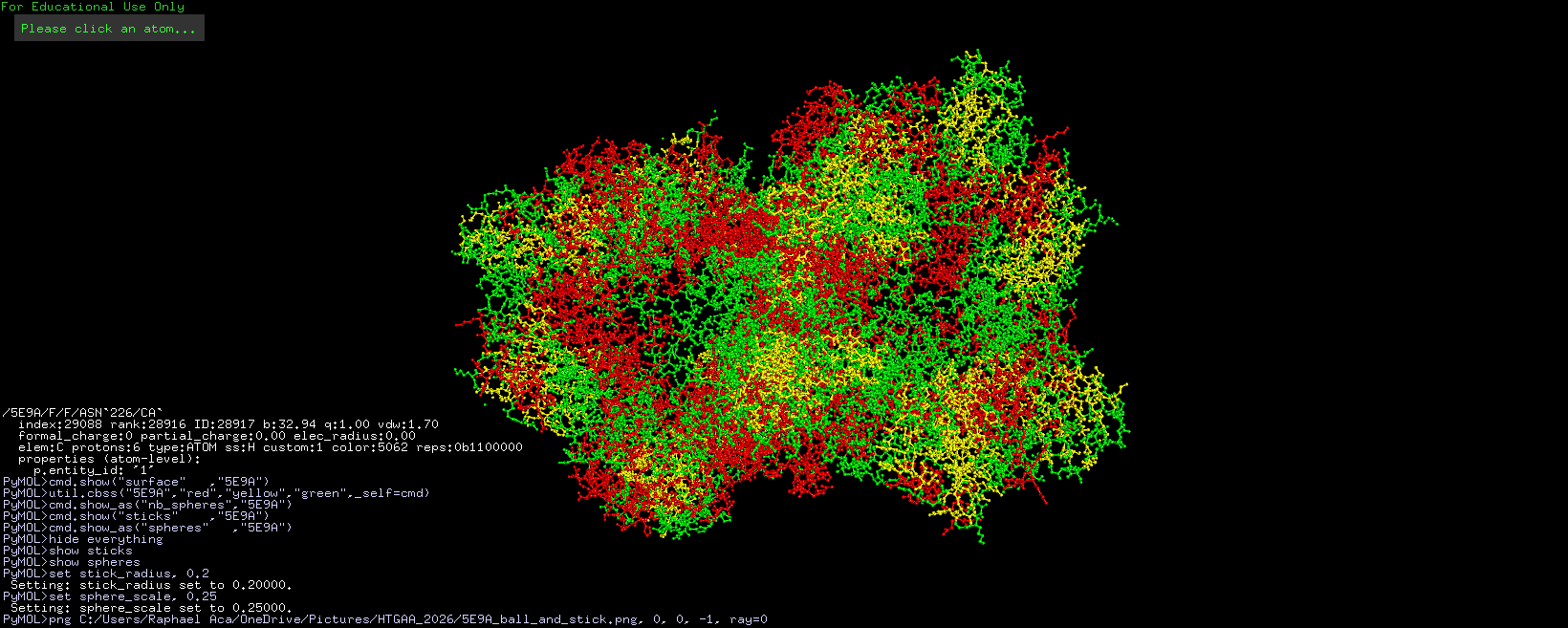
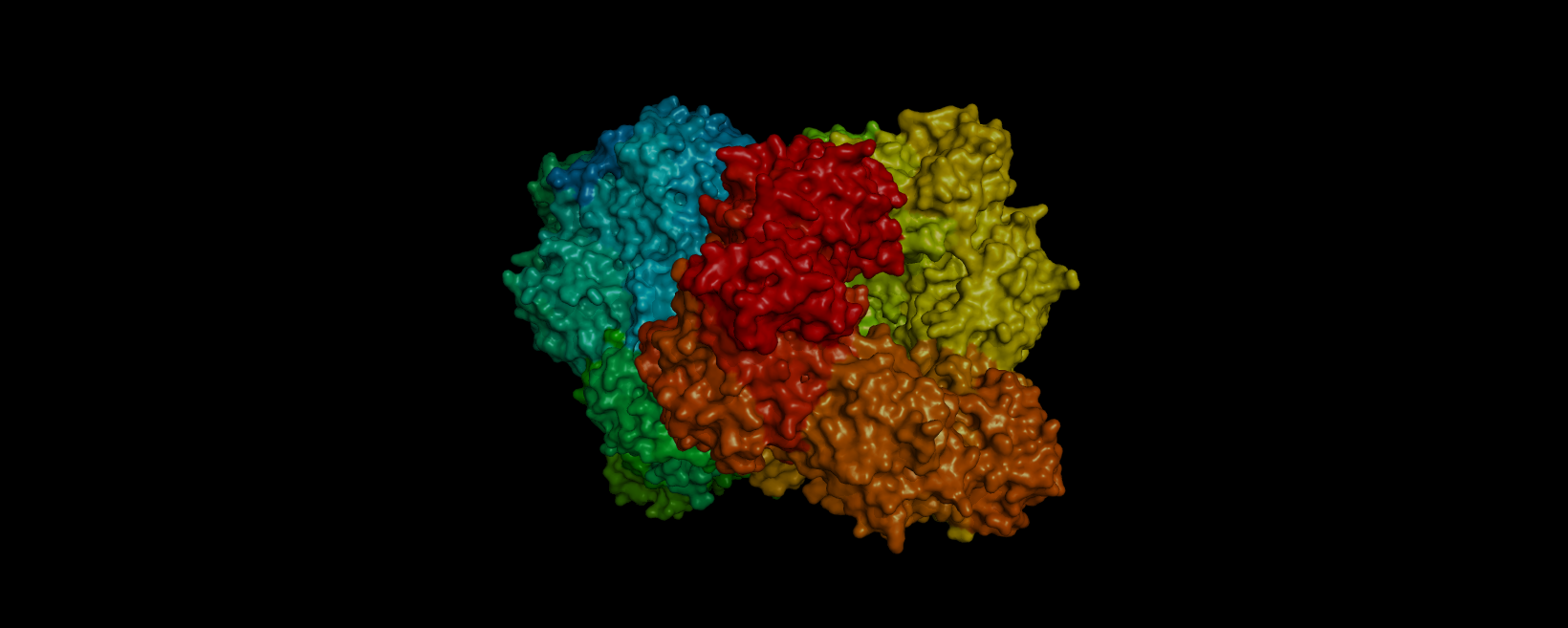
3D visualization of “surface” model of 5E9A protein. Colored rainbow by elem c.
The above model shows a “hole” in the enzyme which is its binding pocket. Not shown in the picture but on the other side of the protein there is another binding pocket. It is likely that these “holes” are where lactose binds.
Part C. Using ML-Based Protein Design Tools
For this part, I am continuing to use the lactase protein pdb_00005e9a.
C1. Protein Language Modeling
1. Deep Mutational Scans
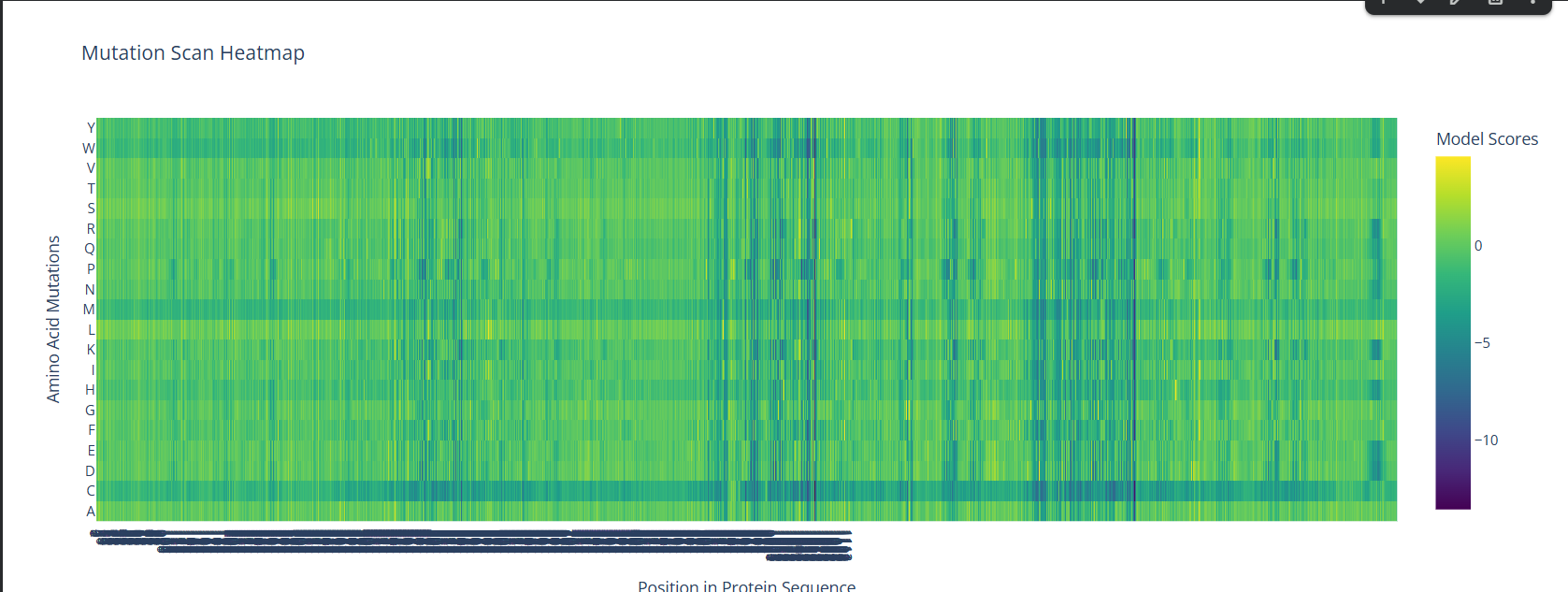
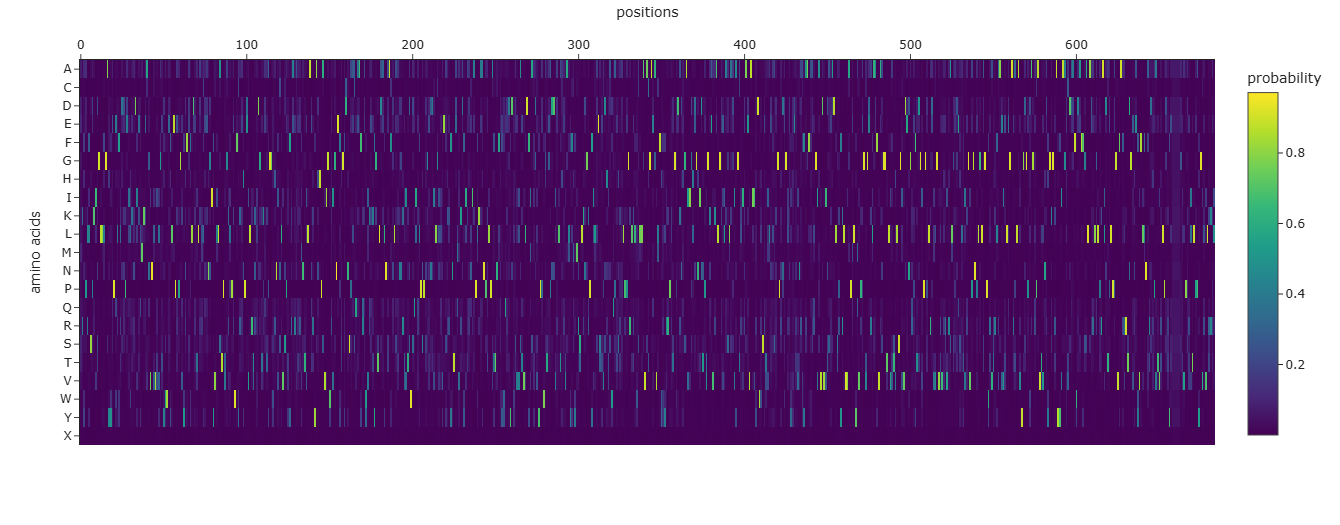
Info
A screenshot of the Mutation Scan Heatmap generated from the LCT protein sequence on ESM2.
The above mutation scan heatmap demonstrates stark patterns. For instance, the W, M, and C residues seem to have consistently negative values throughout the protein sequence. This means that these amino acid residues would most likely reduce fitness/function in the LCT protein if they were to replace nearly any amino acid in a mutation.
2. Latent Space Analysis
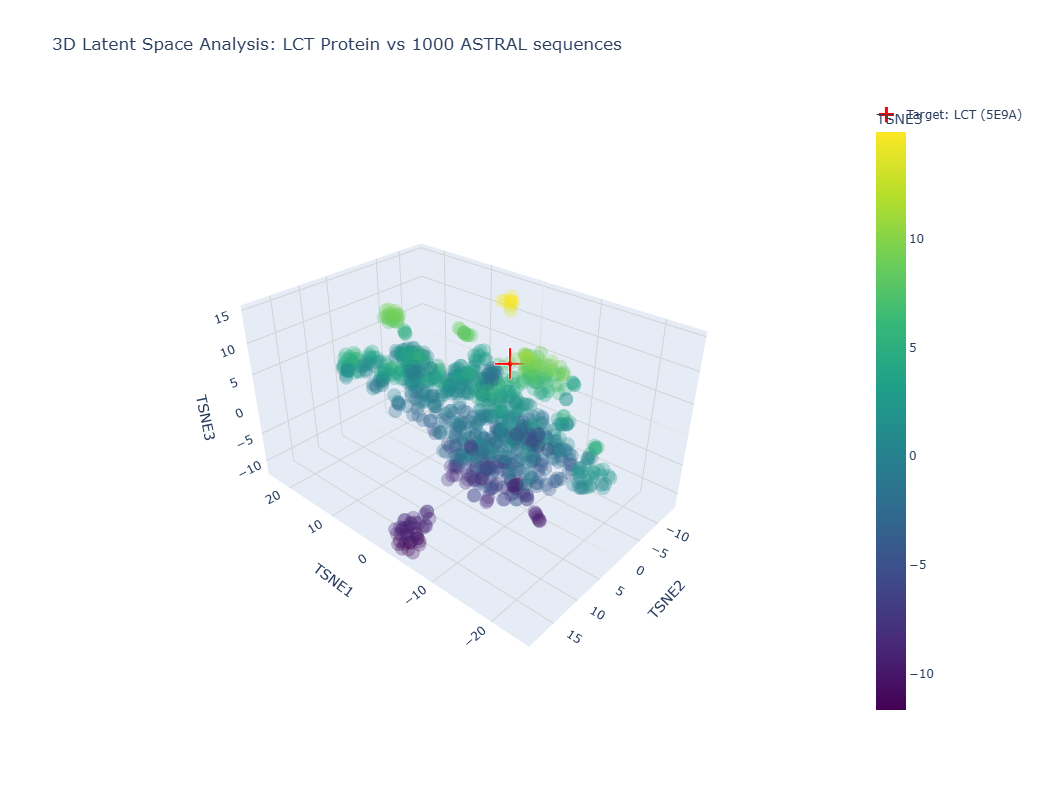
Info
Image showing a latent space analysis of various proteins. The red cross represents my chosen protein, LCT/E59A.
The different formed neighborhoods within the the latent space analysis which grouped proteins together mostly based on similar function and species. For example, HMG1 (High mobility group protein 1) of Rattus norvegicus (Norway rat) and HMG1 of Cricetulus griseus (Chinese hamster) were present in a cluster together.
My protein, E59A, had a higher TSNE3 (as shown above) and was grouped with various human (Homo sapien) proteins such as somatotropin, inferon-beta, and other automated matches. It seems to be similar to human proteins involved in human development. This is slightly expected as E59A is a human protein.
C2. Protein Folding
Info
A 3D representation of the truncated structure of 5E9A (560 residues) using ESMFold.
Due to the sequence of my protein being too long, I opted to truncate my protein sequence to 560 residues so that the ESMFold in Google Colab would be able to successfully run the sequence. The ESMFold prediction produced an average pLDDT score of 79.1, showing generally reliable structural confidence. The predicted TM-score (pTM = 0.583) shows moderate confidence in the overall global fold. These statistics imply that the individual domains are well predicted but their orientations may be less accurate. This is a plausible is structurally coherent fold.
The predicted coordinates do not exactly match my original structure as I had to truncate my protein in order for it to work with ESMFold. However, if I used my entire sequence, I predict that it would have matched well given the statistics of my fold.
After mutating the sequence a few times, I saw how my structure proved to be fairly resistant to mutations. After small mutations, the structure did not change significantly. However, large segments did produce significant change within the structure.
C3. Protein Generation
Info
Plot showing the probabilities of each amino acid in each index of the sequence based on the 3D structure of E59A.
This is the original sequence:
MNHKVHHHHHHIEGRHMELGTLEGSMTKFPLLSSKISGLLHGADYNPEQWLDHPDVLVRDVEMMKEARCNVMSVGIFSWSALEPEEGRYTFDWMDQVLNRLHENGISVFLATPSGARPAWMSQKYPQVLRVGRDRVPALHGGRHNHCMSSPVYREKVQLMNGQLAKRYAHHPAVIGWHISNEYGGECHCDTCQGQFRDWLKARYVTLDALNKAWWSTFWSHTYTDWSQLESPSPQGENGVHGLNLDWRRFNTDQVTRFCSEEIRPLKAENPALPATTNFMEYFNDYDYWKLAGVLDFISWDSYPMWHTRQDDIGLAAYTAMYHDLMRTLKQGKPFVLMESTPSFTNWQPTSKLKKPGMHILSSLQAVAHGADSVQYFQWRKSRGSCEKFHGAVVDHVGHIDTRVGREVAELGSILSALAPVAGSRVEAKVAIIFDWESRWAMDDAMGPRNAGLHYENTVADHYRALWAQGIAVDVINADCDLQGYDLVIAPMLYMVREGVGERISAFVQAGGRFVATYWSGIVNETDLCFLNGFPGPLRPVLGIWAEEIDSLTDEQHNSVAGVEGNALGLSGPYRASQLCEVIHLEGAAALATYGDDFYAGNPAVTVNLYGKGQAYYVASRNDQQFHADFFTALAKEMKLPRAINTPLPEGVTAARRTDGESEFIFLQNYNADNQTVALPQDYQDIVHGGNLPRKLTLPAFGCQILTRKITQ
This is the predicted sequence:
MKFPRLSPKIDGLLLGAEYYPELFLDDPELIERLIELMKEAKINVVRLGTHAWEYLEPEPGVYNFAWLDKTLDLLEKNGIYVLLATPTGKLPRWVYEEHPEVLRTKLDGTKEKYGGSHNICLVSPYFRALATEMNTKLASRYAHHPAVIGWEIGNEFSGYCNCPLCQEAFRKWVKEKFGTLDAFNKAGNLEKNNKVVTDWSQIKLPSPNGENDVLTLINLFKEYNTQLVKDFLTQLIEPLKKYNPNLPVTSNFDWWQEDYDYTELATVLDFIAYDSYPPWGTGPDDVRLAAEVAMYHDYMRSLKNGKPFILSETYVDHVNWQATSTSLPPGRLKLWCLLHVACGAEAVLFHYLRRPRTGWDKNHGAVIDHTGNIDTPVGKEVKALGDELASLKDIAGTKIEAEVAIVYDWKSRVILEASKGPLDAGLDYVGNVNRWFRAFWSQGIAVDVIDADADLSPFKLVVAPELYMVPAGVGDRLAAFVAAGGTFVATVLSGVVDEYGREFTDGRPGPLRDVLGVLVRRVRSLSADQTATVTGVAGNELGLSGPYTVTRLAAVVELRGAEALAVYGSGPDAGQPAVTVNRHGKGRAYYVAGRVDDAFWAAFFGSLAEKLKLKRAIDTPLPPGVFARRRTDGENEYVFLFNFRDTPVTVTLPGTYTXXXXXGTVPATITLPPYGVKVLTRKI
The predicted sequence is significantly distinct from the original sequence. The predicted sequence is longer than the original sequence and contains little similarities with the original sequence.
Info
A proposed 3D structure generated by ESMFold based on the predicted sequence generated by ProteinMPNN. The predicted strcture seems to be more clumped than the original structure generated by ESMFold.
Part D. Group Brainstorm on Bacteriophage Engineering
For this part of the homework, I joined a group of students consisting of Jason Ross, Jay Handfield, Nana Agyei Afrane-Asare, Xavier-Lewis Palmer, and myself. After going through the phage reading and reviewing the bacteriophage final project goals, we opted to increase the thermodynamic stability of the Lysis Protein.
Our group’s plan for engineering a bacteriophage includes multiple steps and various protein engineering tools. First, we will use BLAST and Clustal Omega to discover amino acid residues that are conserved between Lysis protein homologs. ESM2 will be used to score mutations for their evolutionary plausibility and ESM-Fold/ProteinMPNN/Boltz-1 will be used to refine the folded protein. EvolvePro will be used to computationally direct evolution in this protein structure. Lastly, we can use computational stress-testing under varying environmental conditions to test for destabilization.
Our group’s full one page proposal is linked here.
Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
This is the human SOD1 sequence containing the A4V mutation:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Binder
Pseudo Perplexity
0
WHYYATGARWGE
16.929015
1
WRYGAVALELKK
12.714672
2
WRSPAAAARWWK
9.155765
3
WRYPATAAALKX
4.843841
4
FLYRWLPSRRGG
N/A
Info
The table generated by PepMLM detailing possible peptides to bind to mutant SOD1 along with their pseudo perplexity scores. Peptide 4 is an already known SOD1-binding peptide.
Part 2: Evaluate Binders with AlphaFold3
Binder
ipTM
Peptide localizes near SOD1 N-terminus?
Engages β-barrel region?
Approaches dimer interface?
Surface-bound/partially buried peptide?
3D Model
0
WHYYATGARWGE
0.32
No
No
Yes
Yes
1
WRYGAVALELKK
0.18
No
No
Yes
Yes
2
WRSPAAAARWWK
0.42
No
Yes
No
Yes
3
WRYPATAAALKA
0.46
No
Yes
No
Yes
4
FLYRWLPSRRGG
0.29
No
No
Yes
Yes
Info
Table containing results from AlphaFold3 generations
Peptide generations 0, 1, and 4 contain ipTM scores lower than 0.4, demonstrating no meaningful binding between the peptide and the A4V mutated SOD1 protein. However, compared to the known binder 4, peptides 0, 2, and 3 have greater ipTM values. Peptides 2 and 3 contain ipTM scores significantly greater than the known binder 4 at 0.42 and 0.46, respectively.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
The results from PeptiVerse of the 5 peptides proved to be mostly consistent with the ipTM values in AlphaFold3. Peptides 0, 1, and 4 resulted in weak binding which is consistent with their low ipTM scores in AlphaFold3. However, peptide 2 also resulted in weak binding in PeptiVerse which was unexpected as it had an ipTM score of 0.42 in AlphaFold3. Peptide 3 resulted in medium binding which is consistent with its ipTM score of 0.46. All five peptides were soluable and non-hemolytic so they all had sufficient therapeutic properties. Only peptide 4 balanced predicted binding and therapeutic properties well.
Part 4: Generate Optimized Peptides with moPPIt
Peptide Sequence
Hemolysis ↓
Solubility ↑
Affinity ↑
Motif Match ↑
GGKKEYYYSRYP
0.9586
0.9167
7.21
0.1572
EKQYTCDTSTKM
0.9675
0.9167
6.18
0.8416
KKTTGYGECSYN
0.9639
1.0000
5.85
0.8290
GTYTCETTYTQW
0.9728
0.9167
6.68
0.8369
Info
Table of results from the moPPIt-v3 Colab using the A4V mutated SOD1 protein.
The peptides generated by the moPPIt-v3 Colab generated stronger binding and motif match results than the peptides generated by PepMLM. In addition, both groups of peptides had high solubility. However, the peptides generated by moPPIt-v3 demonstrated very high hemolysis results, insinuating high risks for red blood cell damage. I would evaluate these peptides to be unready for clinical studies since their hemolytic values are too high. These peptides would be too dangerous to use in therapeutic applications.
Part C: Final Project: L-Protein Mutants
I worked with Jason Ross, Xavier-Lewis Palmer, and Nana Agyei Afrane-Asare to generate mutated proteins to improve the stability of the L-Protein. Our results can be found in this Google Doc.
Week 6 HW: Genetic Circuits Part I: Assembly Technologies
Assignment: DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion High-Fidelity PCR Master Mix consists of Phusion DNA Polymerase, deoxynucleotides, and a reaction buffer (including MgCl2). The Phusion DNA Polymerase is a high-fidelity enzyme that is used to synthesize new, complementary nucleotides to the 3’ end of a DNA strand. Deoxynucleotides are present within the master mix to be added to the cloned DNA strand. The reaction buffer facilitates enzymatic function and stabilizes the DNA polymerase, allowing the PCR reaction to proceed smoothly.
What are some factors that determine primer annealing temperature during PCR?
The annealing temperature for a PCR reaction the primer base composition (proportions of A, T, G, and C), primer concentration, primer length, and ionic reaction environment.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR
Restriction Enzyme Digest
Purpose
Create millions of copies of a DNA segment (amplify the DNA segment).
Cut DNA at a specific site.
Active Enzymes
DNA Polymerase
Restriction Endonucleases
Targetting
Defined by synthetic primers.
Defined by palindromic recognition sequences.
Temperature
Alternates between denaturation, annealing, and extension temperatures (95°C, 55°C, 72°C, respectively).
Usually a steady incubation at 37°C.
Result
High-concentration amplicons of a single size
Different sized fragments of DNA based on the number of sites
Input DNA
Minimal amount of DNA template
High concentrations of DNA
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
We ensure that the DNA sequences that we have digested and PCR-ed are appropriate for Gibson cloning through preparing the correct conditions for Gibson cloning. The two DNA inserts we create must have identical ends that will overlap with one another. This is to ensure that they will stick together during Gibson assembly. In addition, the DNA used must be treated with DpnI and a cleanup kit (column purification) to remove the original template DNA and leftover salts/enzymes.
How does the plasmid DNA enter the E. coli cells during transformation?
The most common method for plasmid DNA to enter E. Coli cells during transformation is chemical transformation. In this method, cells are treated with calcium chloride which neutralizes the negative charges on the DNA and cell membrane to bring them closer to one another. Then, the cells are cooled down and suddenly heated to 42°C. This sudden change in temperature creates a pressure difference between the inside and outside of the cell, forming temporary pores in the membrane which allow the entry of the plasmid DNA into the cytoplasm.
Describe another assembly method in detail (such as Golden Gate Assembly)
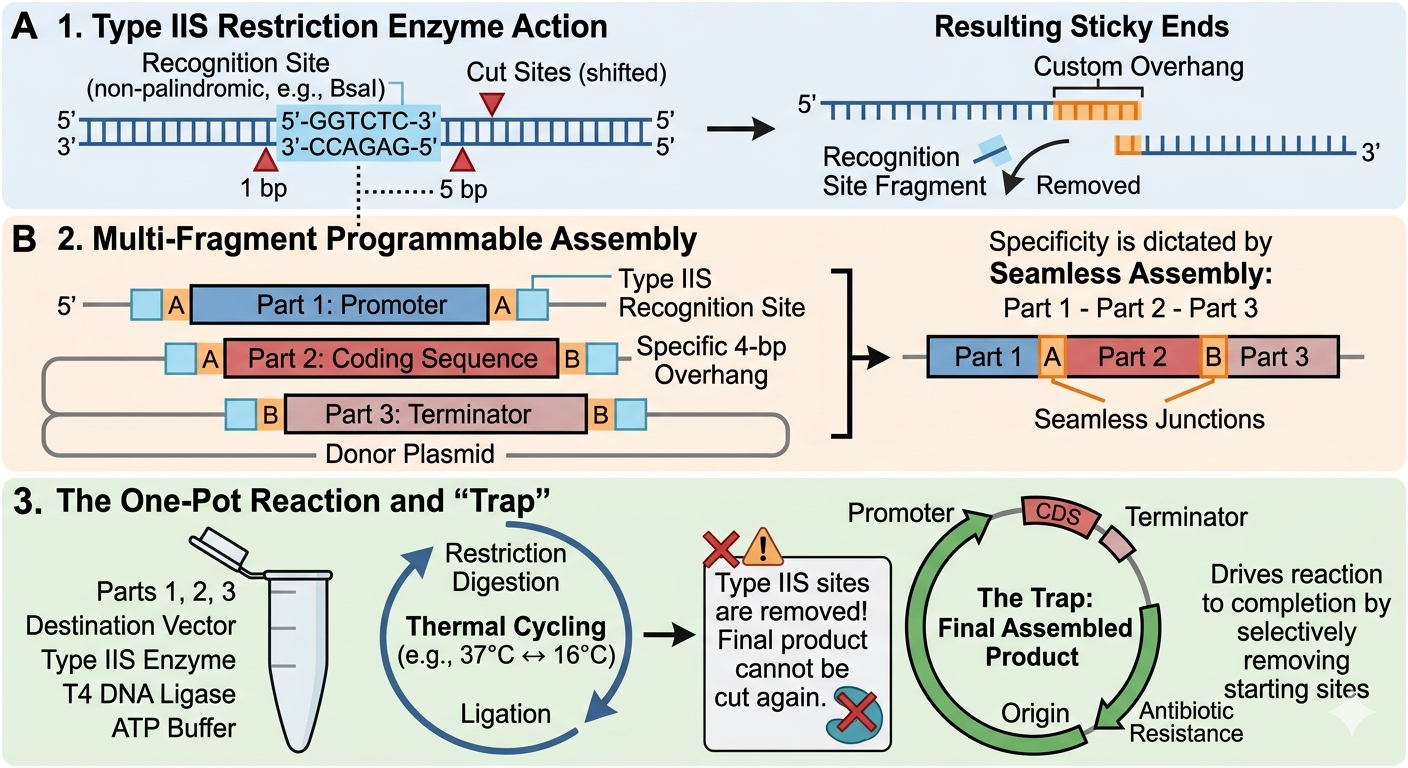
Golden Gate Assembly (GGA) is a highly efficient molecular cloning method that allows for the simultaneous assembly of multiple DNA fragments into a single piece. GGA relies on Type IIS restriction enzymes. These enzymes recognize specific DNA sequences but cut at a defined distance outside of that sequence. This allows the user to design complementary overhangs with any 4-base-pair sequence on different fragments so that they are forced to assemble in a specific order and orientation. Researchers can also design this process to cut off and discard the recognition site. By having the restriction digest and ligation happen at the same time, the DNA is sealed within its final, correct form without the possibility of being cut again. Golden Gate assembly allows for the restriction digest and ligation to happen all in one tube with the highly efficient, seamless and programmable arrangement of multiple segments of DNA.
Assignment: Asimov Kernel
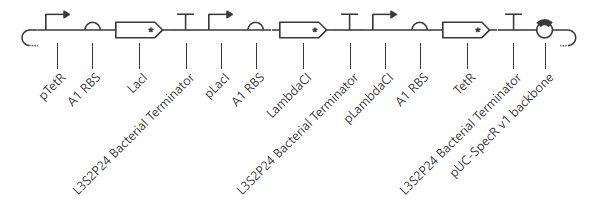
Under the RA_Repressilator_Construct tab, I recreated the repressilator found under the Bacterial Demos Repo and ran a simulation on it. The following is a schematic of the construct:
These are the settings used on the simulation:
Chassis:E. coli
Simulation duration: 72 hours
Simulation time step: 10 minutes
Transfection type: Transient transfection
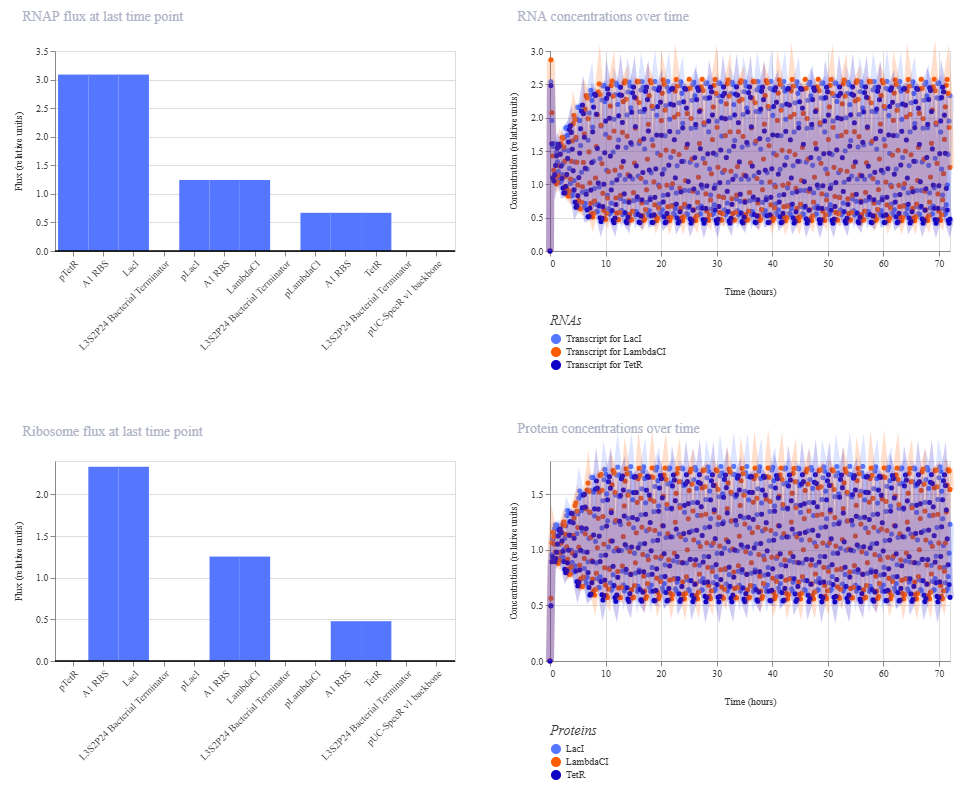
This is the results of the simulation:
Gene Circuit 1
I made a circuit that includes a GFP encoding gene under the GFP expressor tab. The promoter is activated by IPTG which enables transcription of A1 RBS and GFP.
These are the settings used on the simulation:
Chassis:E. coli
Simulation duration: 72 hours
Simulation time step: 10 minutes
Transfection type: Transient transfection
Ligand 1: IPTG added at hour 30
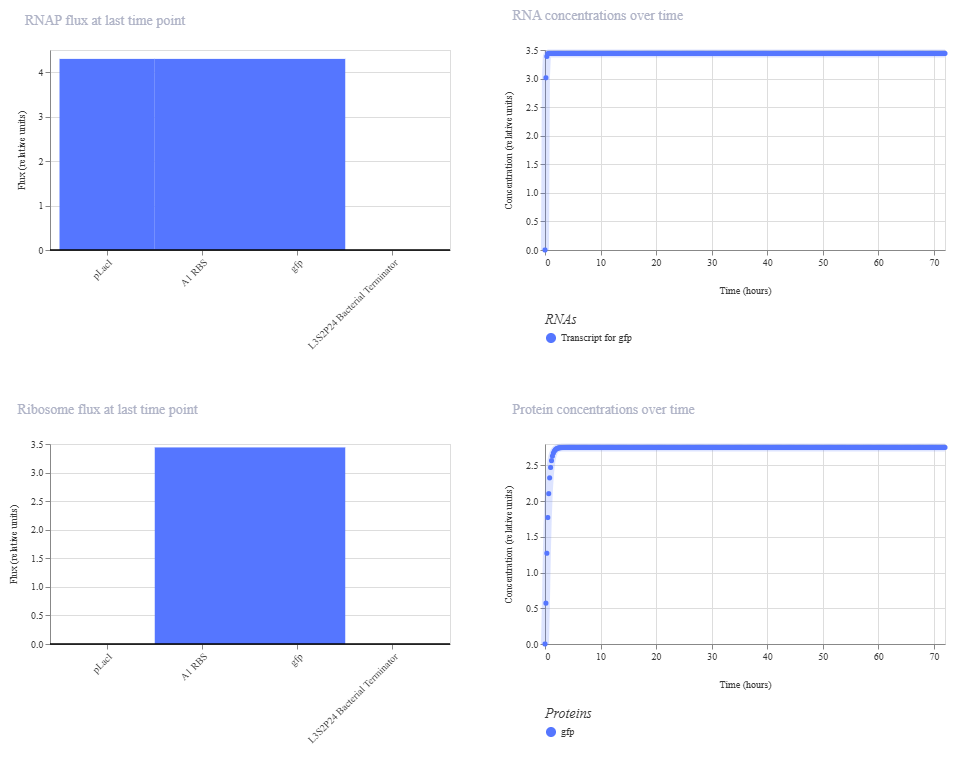
This is the results of the simulation:
Gene Circuit 2
I made the same gene construct, except I removed the A1 RBS sequence to see its effects under the GFP expressor without RBS tab. We expect for there to be a change in protein concentrations.
These are the settings used on the simulation:
Chassis:E. coli
Simulation duration: 72 hours
Simulation time step: 10 minutes
Transfection type: Transient transfection
Ligand 1: IPTG added at hour 30
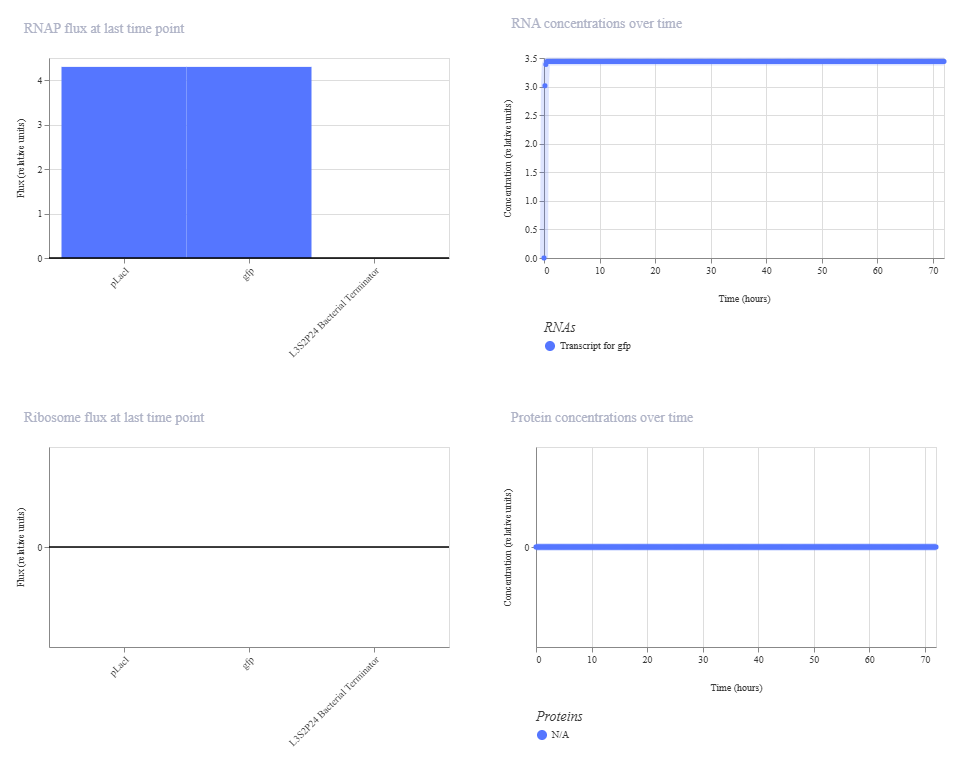
This is the results of the simulation:
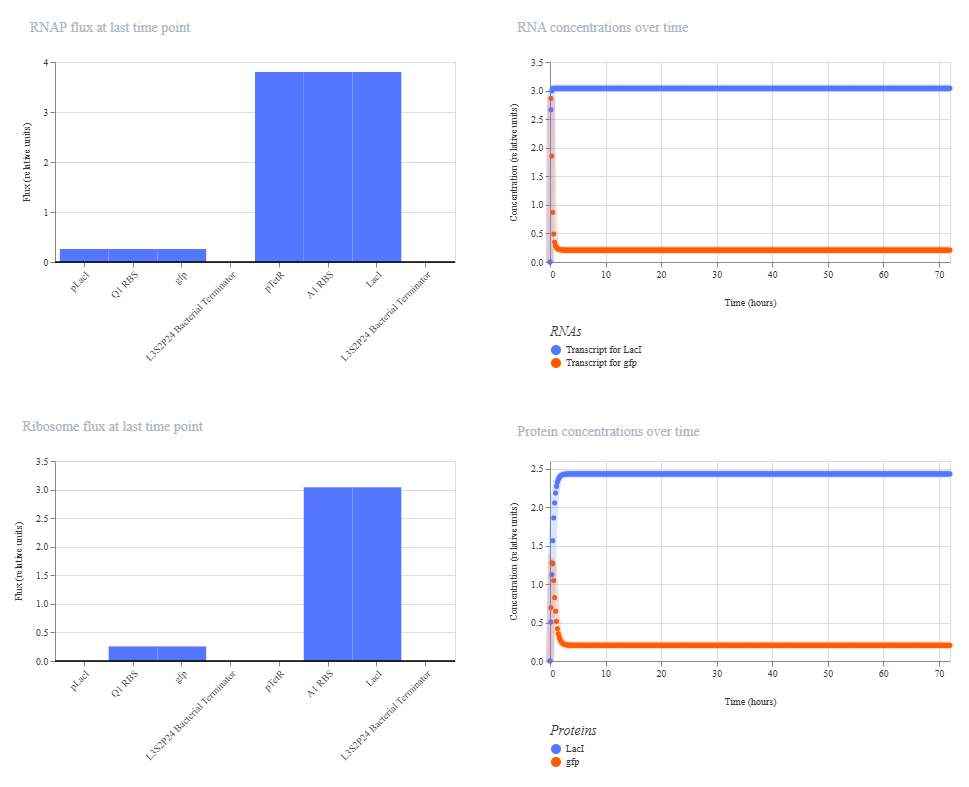
The change in ribosome flux and protein concentrations is most likely due to the fact that the RBS is required for translation.
Gene Circuit 3
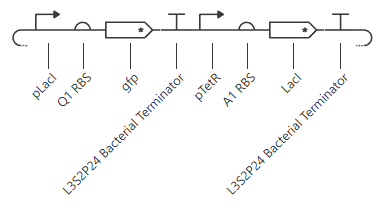
Under the LacI and TetR construct1 tab, I created a construct where transcription at the pLacI promoter will create GFP protein while transcription at the pTetR promoter will repress the pLacI promoter. Therefore, the pTetR promoter region will eventually remove all instance of GFP protein through the creation of LacI.
These are the settings used on the simulation:
Chassis:E. coli
Simulation duration: 72 hours
Simulation time step: 10 minutes
Transfection type: Transient transfection
This is the results of the simulation:
Week 7 HW: Genetic Circuits Part II
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Intracellular Artificial Neural Networks (IANNs) have several advantages over traditional Boolean genetic circuits. Traditional genetic circuits usually operate in an ON/OFF manner where genes are either expressed or not expressed. In contrast, IANNs can process inputs in a continuous and graded manner similar to biological systems. This allows IANNs to respond to multiple inputs at once with varying strengths instead of simple binary outputs. IANNs are also more capable of pattern recognition, noise tolerance, and complex decision making. These systems can integrate many molecular signals simultaneously and produce more flexible cellular behaviors than standard Boolean logic gates.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
One useful application for an IANN would be cancer detection and treatment within engineered immune cells. In this system, the inputs could be concentrations of different cancer biomarkers such as HER2, EGFR, and inflammatory cytokines. The IANN would process all of these molecular inputs simultaneously and determine whether the detected pattern matches that of a cancer cell. If the combined weighted inputs surpass a threshold value, the output would activate the expression of a therapeutic protein or induce apoptosis in the target cell. Unlike traditional Boolean systems which may require perfect combinations of biomarkers, the IANN could tolerate noisy or partial signals and still make accurate decisions. However, limitations include metabolic burden on the cell, unintended cross-talk between pathways, difficulty tuning regulatory weights, and slow response times caused by transcriptional and translational delays.
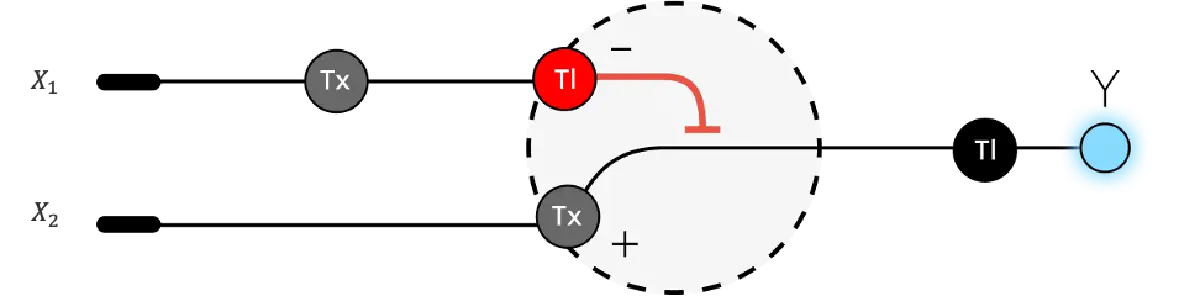
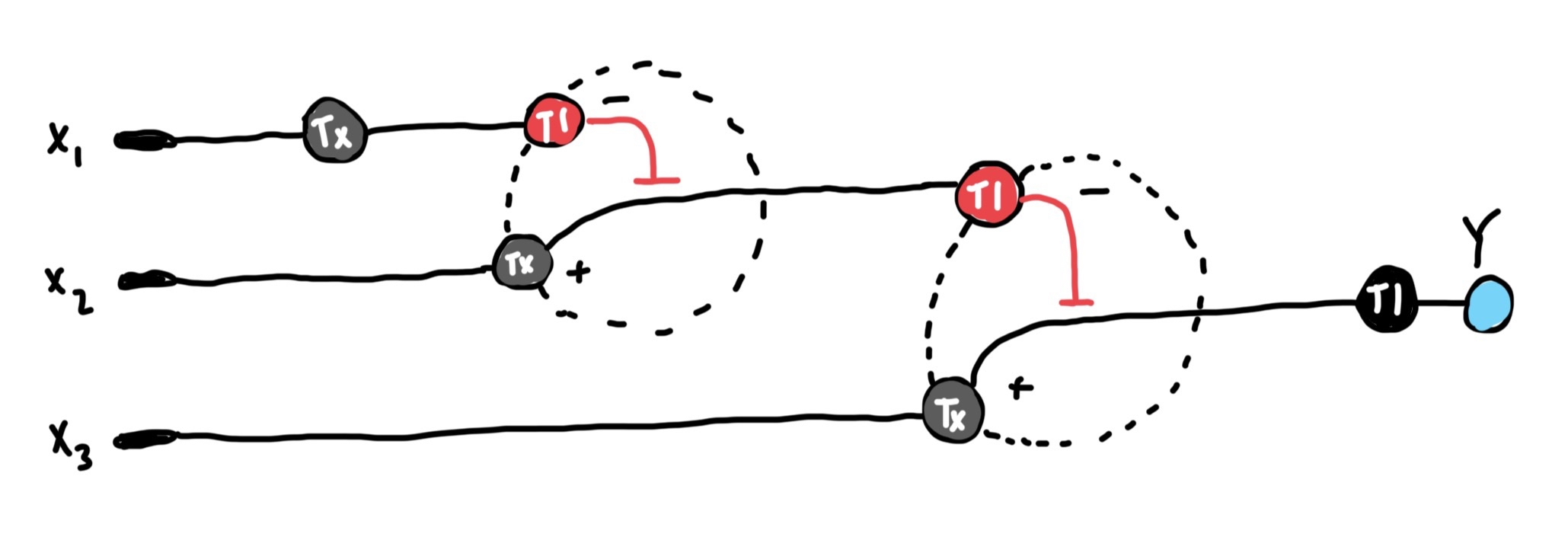
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation. Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Info
This is the multilayer perceptron diagram I created. The fluorescent protein is encoded in x3 which is regulated by the endoribonuclease encoded in the DNA of the x2 output. The x2 DNA’s transcribed mRNA is regulated by the endoribonuclease generated by x1’s DNA.
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Existing fungal materials include mycelium packaging materials, fungal leather, fungal insulation, and fungal construction materials. Mycelium packaging is used as a biodegradable replacement for polystyrene foam packaging. Fungal leather is used in clothing, shoes, and furniture as an alternative to animal leather. Mycelium insulation and building materials are being explored for sustainable construction due to their lightweight and fire-resistant properties. Advantages of fungal materials include biodegradability, sustainability, low energy production costs, and reduced environmental impact compared to plastics or animal-derived materials. However, disadvantages include lower durability, sensitivity to moisture, slower manufacturing times, and limited large-scale production infrastructure.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
One potential application of genetically engineered fungi would be to create self-healing construction materials. Fungi could be engineered to sense cracks or structural damage and produce adhesive biomaterials to repair the damaged region. Fungi could also be engineered for environmental remediation by degrading plastics, toxins, or oil contaminants in soil. Synthetic biology in fungi has several advantages over bacteria because fungi naturally grow as large multicellular networks called mycelia which can penetrate solid materials and survive in harsh environments. Fungi are also capable of secreting large amounts of enzymes and biomolecules into their surroundings. In addition, many fungi can process complex organic substrates that bacteria cannot efficiently metabolize. However, fungi generally grow more slowly and can be more difficult to genetically manipulate than bacteria.
Assignment Part 3: First DNA Twist Order
For this assignment, I reviewed the Individual Final Project documentation guidelines and submitted the Google Form with my draft Aim 1, final project summary, HTGAA industry council selections, and a shared folder for DNA designs.
Week 9 HW: Cell-Free Systems
General homework questions/answers
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis has several advantages over traditional in vivo protein expression methods. In cell-free systems, researchers have direct control over experimental conditions such as pH, ion concentrations, temperature, substrate availability, and DNA concentration without needing to maintain living cells. This allows for rapid optimization and easier manipulation of gene expression conditions. Cell-free systems also avoid problems associated with cellular toxicity, metabolic burden, and membrane transport limitations. In addition, proteins can be produced much more quickly because there is no need for cell growth or transformation. Cell-free expression is especially beneficial when producing toxic proteins that would kill living cells and when rapidly prototyping genetic circuits for synthetic biology applications.
Describe the main components of a cell-free expression system and explain the role of each component.
The main components of a cell-free expression system include a cell lysate, DNA template, amino acids, nucleotides, energy source, salts, and cofactors. The cell lysate contains ribosomes, tRNAs, transcription factors, RNA polymerase, and other enzymes required for transcription and translation. The DNA template contains the gene encoding the desired protein product. Amino acids are used as the building blocks for protein synthesis while nucleotides are required for transcription of mRNA. Energy sources such as ATP and phosphoenolpyruvate provide the energy needed for cellular reactions and protein synthesis. Salts and cofactors help stabilize enzymes and maintain proper reaction conditions for efficient protein production.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy regeneration is critical in cell-free systems because transcription and translation require large amounts of ATP and GTP. Without continuous energy regeneration, the reaction rapidly loses the ability to synthesize proteins and protein yield decreases significantly. One common method used to maintain ATP levels is the phosphoenolpyruvate (PEP) regeneration system. In this method, phosphoenolpyruvate serves as a high-energy phosphate donor which allows ATP to be continuously regenerated from ADP through enzymatic reactions. Another possible method is the use of creatine phosphate with creatine kinase to recycle ATP during the experiment.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic cell-free systems, such as E. coli lysates, are generally faster, cheaper, and produce high protein yields. However, they lack many post-translational modifications found in eukaryotic cells. Eukaryotic systems, such as wheat germ or rabbit reticulocyte lysates, are slower and more expensive but are capable of producing proteins requiring complex folding and post-translational modifications. A useful protein to produce in a prokaryotic system would be GFP because it folds efficiently and does not require glycosylation or other complex modifications. In contrast, a useful protein to produce in a eukaryotic system would be a monoclonal antibody because antibodies require proper disulfide bond formation and post-translational processing for functionality.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
To optimize membrane protein expression in a cell-free system, I would include lipid nanodiscs, detergents, or artificial liposomes within the reaction mixture to provide a membrane-like environment for proper protein folding and insertion. Membrane proteins are difficult to express because they are hydrophobic and tend to aggregate or misfold outside of membranes. I would also optimize temperature, magnesium concentration, and reaction duration to improve protein stability and translation efficiency. In addition, molecular chaperones could be added to assist with proper folding of the membrane protein during synthesis.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
One possible reason for low protein yield is degradation of the DNA template or mRNA by nucleases present in the lysate. This issue could be addressed by improving purification methods or adding nuclease inhibitors. A second possible reason is depletion of ATP or other energy substrates during the reaction. This could be solved by improving the energy regeneration system or increasing substrate concentrations. A third possible reason is poor protein folding or aggregation during translation. In this case, lowering the reaction temperature, adding molecular chaperones, or optimizing salt concentrations may improve protein stability and increase overall protein yield.
Homework questions from Kate Adamala
Pick a function and describe it.
The function of my synthetic minimal cell would be to detect peanut allergens in food samples and produce a fluorescent glow when peanut allergens are present. This system could be used as a rapid food safety sensor for people with severe peanut allergies.
a. What would your synthetic cell do? What is the input and what is the output?
The synthetic cell would sense peanut allergen proteins such as Ara h 1 or Ara h 2 and respond by expressing GFP as a fluorescent output signal. The input would be peanut allergen proteins diffusing into the synthetic cell environment while the output would be green fluorescence visible under blue or UV light.
b. Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Partially, but encapsulation would significantly improve the system. A purely cell-free system could detect allergens and produce GFP in solution, however encapsulation allows the synthetic cell to better control diffusion, protect internal reaction components, and more closely mimic cellular behavior. Encapsulation also improves stability and enables compartmentalized sensing.
c. Could this function be realized by genetically modified natural cell?
Yes. A genetically modified bacterium such as E. coli could potentially be engineered to detect peanut allergens and express GFP. However, synthetic minimal cells are safer because they are nonliving and cannot reproduce or spread in the environment. In addition, synthetic minimal cells avoid many regulatory and biosafety concerns associated with genetically modified organisms.
d. Describe the desired outcome of your synthetic cell operation.
The desired outcome is that the synthetic minimal cells remain inactive in the absence of peanut allergens but rapidly produce visible green fluorescence when peanut allergens are detected. This fluorescence would indicate contamination of the tested food sample with peanut proteins.
Design all components that would need to be part of your synthetic cell.
a. What would be the membrane made of?
The membrane would consist of phospholipids and cholesterol to create a stable lipid bilayer similar to biological membranes. Lipids such as POPC and cholesterol would help maintain membrane fluidity and structural integrity.
b. What would you encapsulate inside? Enzymes, small molecules.
Inside the synthetic cell I would encapsulate a bacterial cell-free transcription/translation system, ATP regeneration components, amino acids, nucleotides, ribosomes, RNA polymerase, and plasmid DNA encoding GFP under the control of a peanut allergen-responsive regulatory element. I would also include molecular chaperones to improve GFP folding efficiency.
c. Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason?
A bacterial cell-free expression system derived from E. coli would be sufficient for this application because GFP expression does not require complex mammalian post-translational modifications. In addition, bacterial systems are inexpensive, rapid, and produce high protein yields.
d. How will your synthetic cell communicate with the environment?
The synthetic cell membrane would contain membrane pores such as alpha-hemolysin channels to allow peanut allergen proteins or signaling molecules to diffuse into the synthetic cell. The membrane channels would allow environmental sensing while still maintaining compartmentalization of the internal Tx/Tl machinery.
Experimental Details
a. List all lipids and genes.
Lipids: POPC, cholesterol Cell-free system:E. coli lysate-based Tx/Tl system Genes: sfGFP reporter gene, alpha-hemolysin (aHL) membrane pore gene, allergen-responsive aptamer targeting Ara h 1/Ara h 2 peanut allergens
b. How will you measure the function of your system?
The function of the system would be measured by detecting GFP fluorescence using a fluorometer or fluorescence microscope. Increased GFP fluorescence would indicate the presence of peanut allergens within the tested sample. Fluorescence intensity could also be quantified over time to measure sensor sensitivity and response speed.
Homework question from Peter Nguyen
Write a one-sentence summary pitch sentence describing your concept.
My concept is a smart architectural paint containing freeze-dried cell-free systems that can detect carbon monoxide gas and produce a visible fluorescent glow to warn building occupants of dangerous conditions.
How will the idea work, in more detail?
The paint would contain embedded freeze-dried bacterial cell-free transcription/translation systems along with carbon monoxide-responsive genetic regulatory elements. When carbon monoxide diffuses into the paint layer, the cell-free system would become activated and induce the expression of a fluorescent reporter protein or luminescent enzyme. The glowing signal would provide a rapid visual warning that dangerous carbon monoxide levels are present within the environment. The paint could be applied near furnaces, garages, kitchens, industrial buildings, or enclosed spaces where carbon monoxide leaks are more likely to occur. Because the system is cell-free, there would be no living genetically modified organisms present within the material itself.
What societal challenge or market need will this address?
Carbon monoxide poisoning is a major public health issue because the gas is colorless and odorless, making leaks difficult to detect before harmful exposure occurs. This smart paint system could provide an inexpensive and highly visible method for early carbon monoxide detection in homes, schools, factories, and public buildings. In addition, the paint could improve safety in areas where electronic carbon monoxide detectors are unavailable, damaged, or lose power during emergencies.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
To improve stability, the freeze-dried cell-free components could be encapsulated within protective hydrogel microcapsules embedded throughout the paint. These capsules would protect the biological components from oxygen, UV radiation, and temperature fluctuations while remaining permeable to carbon monoxide gas. Small amounts of environmental humidity could help rehydrate the system when needed, while stabilizing molecules such as trehalose could increase the shelf-life of the biological components. In addition, modular paint layers or replaceable surface coatings could allow the sensing components to be periodically renewed after activation or degradation over time.
Homework question from Ally Huang
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting.
As humanity moves toward long-duration spaceflight and possible extraterrestrial colonization, understanding the effects of spaceflight conditions on fetal development becomes increasingly important. Microgravity and ionizing radiation have both been shown to alter gene expression and DNA stability, but their effects on epigenetic regulation during embryonic development remain poorly understood. Improper DNA methylation during development could permanently disrupt growth, organ formation, and viability of embryos in space. Understanding these mechanisms is essential for the future of human reproduction and multigenerational survival during deep-space missions.
Name the molecular or genetic target that you propose to study.
DNA methylation patterns and epigenetic regulation of the IGF2 and H19 genes during embryonic development under simulated spaceflight conditions.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses.
IGF2 and H19 are imprinted genes that play critical roles in fetal growth and embryonic development. Their expression is tightly regulated through DNA methylation patterns established early during development. Disruption of methylation at these loci can lead to developmental abnormalities and impaired fetal growth. Because microgravity and radiation are known to induce DNA damage and alter cellular regulation, studying methylation changes in IGF2 and H19 could reveal how spaceflight conditions interfere with embryonic development and reproductive success in space environments.
Clearly state your hypothesis or research goal and explain the reasoning behind it.
My hypothesis is that simulated spaceflight conditions, specifically microgravity and ionizing radiation, will disrupt normal DNA methylation patterns at the IGF2/H19 imprinting control region, leading to abnormal gene regulation during embryonic development. This hypothesis is based on previous studies demonstrating that radiation exposure and environmental stress can alter epigenetic regulation and DNA methylation stability. Because embryonic development relies heavily on tightly controlled epigenetic programming, even small methylation changes at growth-regulating genes such as IGF2 and H19 may significantly impair fetal development. Understanding these effects would provide insight into the biological limitations of long-term human reproduction in space and help guide future countermeasures for space colonization.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc.
I would expose cultured mammalian embryonic stem cells to simulated microgravity and radiation conditions while maintaining a parallel Earth-gravity control group. DNA samples would be collected and amplified using the miniPCR® thermal cycler. Methylation-sensitive analysis targeting the IGF2/H19 imprinting region would then be performed using BioBits® cell-free systems coupled to fluorescence-based reporters measured with the P51 Molecular Fluorescence Viewer. Fluorescence intensity differences between experimental and control groups would indicate changes in methylation-dependent gene regulation under simulated spaceflight conditions.
Homework Part B: Individual Final Project
I placed my final project slide in the slide deck and submitted the Final Project selection form. I did not end up making a Twist order as I have no lab access. Thus, all my work is done online.
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
For the experimental validation of Project Aero-Sentry, several distinct biological and biochemical components must be measured to ensure the engineered probiotic nasal spray functions as intended. First, the transcriptional activity and expression level of the chimeric EnvZ receptor must be quantified to confirm successful membrane integration. Second, the binding affinity and kinetic interaction between this engineered receptor and the target allergen, Bet v 1, must be measured to verify the precision of the sensory mechanism. Third, the transcriptional output of the sense-and-respond genetic circuit must be monitored, specifically tracking the induction of the Nb32ILZ nanobody gene under the control of the osmolarity-responsive pOmpF promoter. Finally, the total concentration of the secreted Nb32ILZ fusion nanobody in the extracellular environment must be measured alongside its ultimate neutralization efficiency when bound to Bet v 1, confirming that the therapeutic countermeasure is produced in effective quantities.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
Each target element within the genetic circuit requires a distinct biophysical or molecular assay to validate its performance. To measure the transcription of the chimeric receptor and the Nb32ILZ nanobody genes, cellular RNA will be isolated following allergen exposure and analyzed to determine relative fold-induction changes. To confirm that the physical proteins are properly translated, stable, and of the correct molecular weight, protein expression will be measured by evaluating cell lysates and supernatant fractions using molecular weight separation techniques paired with target-specific antibody probes. The physical binding kinetics of both the receptor-allergen interface and the nanobody-allergen interface will be measured by immobilizing the target allergen on a sensor surface and flowing the engineered binding domains over it to track molecular association and dissociation in real time. Lastly, the absolute quantity of the therapeutic nanobody secreted into the surrounding media will be determined using an antibody-coated plate assay that captures and quantifies the protein based on standard concentration curves.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
The primary technology used to measure gene transcription levels will be quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR), which will amplify cDNA reverse-transcribed from the bacterial mRNA to quantify expression changes relative to housekeeping controls. To confirm protein translation and size, sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) will be paired with Western blotting, allowing the visualization of the chimeric receptor and the secreted nanobody via anti-His tag antibodies directed at their engineered purification tags. Surface Plasmon Resonance (SPR) technology will be utilized to measure the precise binding kinetics, calculating the association rate (kon), dissociation rate (koff), and overall equilibrium dissociation constant (Kd) of Bet v 1 interaction with both the chimeric EnvZ receptor and the Nb32ILZ nanobody. Finally, an Enzyme-Linked Immunosorbent Assay (ELISA) will be implemented using a sandwich format to provide a highly sensitive, high-throughput colorimetric readout of the exact concentration of neutralizing nanobodies secreted by the engineered bacteria over time.
Homework: Waters Part I — Molecular Weight
Based on the predicted amino acid sequence of eGFP and any known modifications, what is the calculated molecular weight?
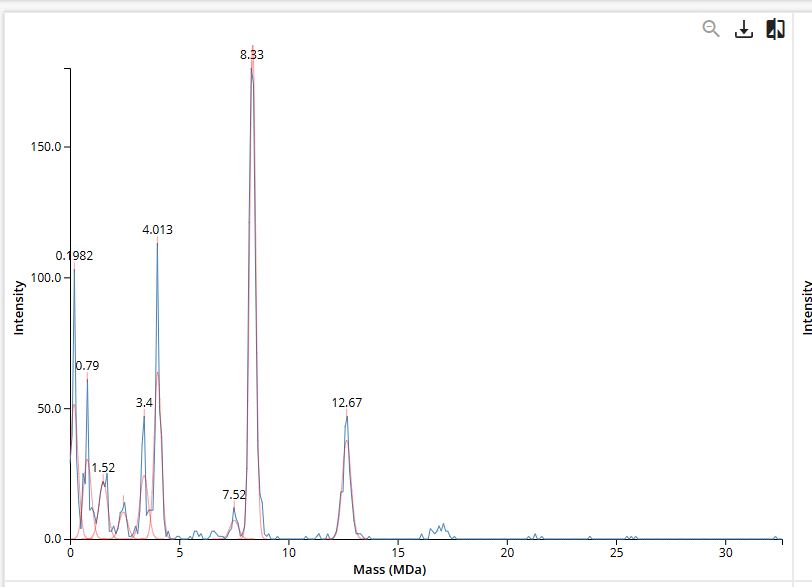
Using the provided eGFP amino acid sequence containing the LE linker and 6xHis tag, the ExPASy Compute pI/Mw tool yields an initial theoretical average molecular weight of 28,006.60 Da and a theoretical pI of 5.90. Because mature eGFP forms an internal chromophore structure during folding, it undergoes a post-translational modification resulting in a known mass loss of approximately 20 Da. Accounting for this maturation step (28,006.60 Da - 20 Da), the calculated theoretical molecular weight for the mature protein is determined to be 27,986.60 Da (or roughly 27.99 kDa).
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1).
Selecting two adjacent charge-state peaks from the intact LC-MS spectrum at 875.4421 and 848.9758 m/z, the charge state is calculated as z = 848.9758 / (875.4421 - 848.9758), which rounds to z = 32 for the 875.4421 peak and z = 33 for the 848.9758 peak. Deconvoluting the mass to account for proton ionization using the relationship MW = z(m/z - 1.0073) yields an experimental mass of 32 × (875.4421 - 1.0073) = 27,981.9 Da. Comparing this measured molecular weight of 27,982 Da against the mature theoretical mass yields a fractional error of |27,982 - 27,986.6| / 27,986.6, which equals approximately 0.016 percent error (or 160 ppm).
Does the zoomed-in peak around 1473.7 m/z still show a charge state? Determine the charge state and explain how it relates to the isotope spacing.
Analysis of the zoomed-in spectral envelope around 1473.7 m/z confirms a distinct multivalent charge state. Given that the calculated intact molecular weight of the eGFP sample is approximately 27,982 Da, dividing this mass by the local peak coordinate (27,982 / 1473.7) yields a value of roughly 19. This calculation identifies the region as representing the +19 charge state. This assignment is supported by the physical isotope spacing of the cluster, as the individual isotopic peaks for a +19 ion are separated by an expected interval of approximately 1/19 m/z.
Homework: Waters Part II — Secondary/Tertiary structure
This portion was optional and therefore skipped.
Homework: Waters Part III — Peptide Mapping - primary structure
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above.
Sequence analysis of the eGFP construct indicates the presence of 20 lysine (K) residues and 6 arginine (R) residues distributed across the polypeptide chain.
How many peptides will be generated from tryptic digestion of eGFP?
Running the primary sequence through the ExPASy PeptideMass tool under the specified cleavage parameters generates a predicted total of 19 distinct tryptic peptides with molecular weights above the 500 Da threshold.
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
The total ion chromatogram (TIC) exhibits approximately 20 distinct peptide peaks between the 0.5 and 6-minute retention time windows that register above the 10% relative abundance threshold.
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
The observed count of 20 peaks is closely aligned with the predicted 19 peptides, yielding a slight excess of chromatographic signals. This minor variance is typical in LC-MS maps due to factors like chemical noise, sample impurities, unexpected missed cleavages, or differential ionization efficiencies.
Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ([M + H]+) based on its m/z and z.
The primary monoisotopic signal centers at 525.76712 m/z. Because the fine isotope spacing within the spectral cluster is roughly 0.5 m/z, the predominant charge state is identified as z = 2. Applying the conversion formula adjusts for the ionizing protons, yielding a calculated singly charged monoisotopic mass of approximately 1050.5269 Da.
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
Cross-referencing the calculated 1050.5269 Da mass with the theoretical PeptideMass digestion matrix matches the fragment sequence FEGDTLVNR, which possesses an absolute theoretical mass of 1050.5214 Da. Comparing the measured value against this baseline reveals a tight mass accuracy variance of roughly 2.0 ppm.
What is the percentage of the sequence that is confirmed by peptide mapping?
According to the amino acid coverage map generated from the experimental BioAccord LC-MS run data, the overall confirmed primary sequence coverage stands at 88%.
Homework: Waters Part IV — Oligomers
General homework questions/answers
Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7): 7FU Decamer, 8FU Didecamer, 8FU 3-Decamer, and 8FU 4-Decamer.
The structural mass for each oligomeric state is calculated by multiplying the base subunit mass by the total number of associated subunits. Based on these calculations, the expected positions and observed peak correlations on the CDMS spectrum are resolved as follows:
Oligomeric Species
Calculation
Theoretical Mass
Observed Peak Position
7FU Decamer
10 × 340 kDa
3,400 kDa
~3.4 MDa (Strong signal)
8FU Didecamer
20 × 400 kDa
8,000 kDa
~8.33 MDa (Strong signal)
8FU 3-Decamer
30 × 400 kDa
12,000 kDa
~12.67 MDa (Strong signal)
8FU 4-Decamer
40 × 400 kDa
16,000 kDa
~16.00 MDa (Weak / low abundance)
In the experimental CDMS trace, the highest intensity distributions map directly to the 7FU decamer (~3.4 MDa), the 8FU didecamer (~8.33 MDa), and the 8FU 3-decamer (~12.67 MDa). The 8FU 4-decamer assembly aligns with the theoretical 16.00 MDa boundary but displays very weak relative abundance and lower resolution within the provided spectrum.
Homework: Waters Part V — Did I make GFP?
Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
Parameter
Theoretical
Observed/measured on the Intact LC-MS
PPM Mass Error
Molecular weight (kDa) of FEGDTLVNR
1050.5214
1050.511
9.90 ppm
Info
I was unable to attend Waters as a CL, so I obtained this data from other students.
Week 11 HW: Bioproduction & Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
For the artwork, I contributed to four pixels on the DNA strand. I really liked how the whole community came together to work on this one project. It was nice to interact with all the other students in the HTGAA course.
To make this collaborative art experiment better for next year, the art canvas could be made to be bigger.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate
BL21 (DE3) Star Lysate: Provides the core cellular translation machinery, including ribosomes, tRNAs, and aminoacyl-tRNA synthetases, alongside natively expressed T7 RNA Polymerase to drive robust transcription from T7 promoter-contained DNA templates.
Salts/Buffer
Potassium Glutamate: Serves as the primary intracellular monovalent salt to maintain optimal ionic strength and osmotic balance required for stable protein-nucleic acid interactions during translation.
HEPES-KOH pH 7.5: Functions as a zwitterionic biological buffer to maintain a stable, physiological pH profile throughout the duration of the metabolic incubation.
Magnesium Glutamate: Supplies critical divalent magnesium cations (Mg2+) necessary to stabilize ribosome structure, facilitate codon-anticodon base pairing, and serve as an enzymatic cofactor.
Potassium phosphate monobasic & Potassium phosphate dibasic: Forms a secondary buffering system that stabilizes pH while supplying an inorganic phosphate pool essential for continuous nucleotide recycling and energy regeneration.
Energy / Nucleotide System
Ribose: Acts as a fundamental sugar precursor used by the lysate’s salvage pathways to synthesize the ribose rings required for fresh nucleotide generation.
Glucose: Serves as the primary carbohydrate energy source, fueling endogenous glycolytic pathways within the lysate to regenerate ATP and GTP over extended incubation times.
AMP, CMP, GMP, UMP: Function as monophosphate nucleotide building blocks that are enzymatically phosphorylated by the system into active nucleoside triphosphates (NTPs) for mRNA transcription.
Guanine: Provides a supplementary purine nucleobase resource to prevent guanosine nucleotide depletion during long-term transcriptional operations.
Translation Mix (Amino Acids)
17 Amino Acid Mix: Supplies the foundational, balanced pool of standard amino acid building blocks required for ongoing polypeptide chain elongation.
Tyrosine: Added individually to circumvent solubility limits at neutral pH, ensuring sufficient availability of this hydrophobic residue for complete protein translation.
Cysteine: Supplemented separately to offset rapid chemical oxidation in cell-free systems, preserving the thiol groups necessary for proper disulfide bond formation and protein folding.
Additives
Nicotinamide: Functions as a precursor and stabilizer for nicotinamide adenine dinucleotide (NAD/NADH) cofactors, protecting the internal metabolic networks driving energy regeneration from degradation.
Backfill
Nuclease Free Water: Serves as the high-purity solvent matrix to adjust total reaction volumes without introducing foreign nucleases that could degrade DNA templates or mRNA transcripts.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above.
The 1-hour optimized master mix relies on pre-supplied nucleoside triphosphates (NTPs) and phosphoenolpyruvate (PEP) to provide immediate energy and building blocks for rapid transcription and translation. In contrast, the 20-hour optimized master mix switches to simpler, cost-effective precursors—nucleoside monophosphates (NMPs), ribose, and glucose—utilizing endogenous cellular enzymes in the lysate to sustainably regenerate energy and nucleotides over an extended incubation period. Additionally, the 20-hour mix adjusts salt concentrations (such as lower HEPES-KOH and slightly higher magnesium) and eliminates short-term additives like spermidine, cAMP, and folinic acid in favor of nicotinamide to support long-term metabolic stability.
Bonus question: How can transcription occur if GMP is not included but Guanine is?
Even without pre-added GMP, transcription can still occur because the E. coli lysate contains active, endogenous salvage pathway enzymes. The system utilizes hypoxanthine-guanine phosphoribosyltransferase (HGPRT) to attach a ribose-5-phosphate group from the available carbohydrate pool directly onto the free Guanine base, synthesizing GMP de novo. Once GMP is formed, native nucleotide kinases sequentially phosphorylate it into GDP and then into GTP, providing the necessary guanosine triphosphates required by the RNA polymerase to synthesize mRNA.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
Fluorescent Protein
Biophysical / Functional Property Affecting Expression or Readout
sfGFP
Superfolder GFP features enhanced folding kinetics and structural stability that allow it to mature extremely rapidly and express efficiently even when fused to poorly soluble target proteins.
mRFP1
This first-generation monomeric red fluorescent protein suffers from a relatively slow maturation time and lower quantum yield, often requiring longer incubation periods in cell-free systems to reach detectable signal thresholds.
mKO2
Monomeric Kusabira Orange 2 exhibits strict oxygen dependence for its chromophore maturation, meaning its orange readout can be severely limited in high-density or poorly aerated cell-free reaction drops.
mTurquoise2
This cyan variant possesses an exceptionally high quantum yield and high photostability, providing a very bright and reliable fluorescent readout even at lower protein expression levels.
mScarlet_I
Engineered for superior brightness, mScarlet_I features an accelerated maturation rate compared to older red variants, making it ideal for real-time tracking of transcription-translation kinetics.
Electra2
Optimized specifically for rapid-response assays, Electra2 undergoes remarkably fast protein folding and chromophore maturation, allowing almost immediate detection after translation begins.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
Hypothesis: Supplementing the master mix with a 1.5x higher concentration of nicotinamide and slightly increasing the dibasic potassium phosphate ratio to stabilize a higher pH will maximize mKO2 fluorescence over a 36-hour incubation. Nicotinamide will sustain the long-term metabolic energy pathways needed for continuous translation, while the optimized phosphate buffer will prevent reaction acidification. This stable environment ensures the translation machinery remains active long enough to support the slow, oxygen-dependent chromophore maturation required by mKO2.
The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
I composed master mix compositions in the given website as shown below.
Part D: Build-A-Cloud-Lab | (optional) Bonus Assignment
This was optional and skipped.
Week 12 HW: Building Genomes
Week 13 HW: AI, SynBio, and Scaling Health Innovation (ARPA-H)