Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Assuming that meat contains about 20% protein, 500 grams of meat translates to ~100 grams of protein. If an amino acid weighs 100 Daltons, this will translate to 100 grams per mole of amino acids. Knowing this, we calculate 500 grams of meat to have one mole of amino acids. Using Avogadro’s constant, we can calulate the number to be 6 x 1023 molecules of amino acids in 500 grams of meat.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
When you eat beef or fish, you eat cells that contain various macromolecules like DNA, proteins, lipids, and carbohydrates. While these macromolecules would usually have an effect on a human’s cells, our bodies instead digest these molecules and break them down into smaller forms (Proteins into amino acids, DNA into nucleotides, etc.). So, by the time the macromolecules reach our bloodstream, there are no intact cow or fish genes/proteins. Our cells take up these digested molecules and uses them to rebuild our own human genome, not theirs.
Why are there only 20 natural amino acids?
There are only 20 natural amino acids because that number of different amino acids provides enough chemical diversity for protein function while also avoiding the drawbacks of having more amino acids. Adding any more amino acids would only supply new amino acids that would behave very similarly to one of the original 20 amino acids. In addition, since similar codons can encode chemically similar amino acids, adding an additional amino acid could increase the chance of a small genetic mutation causing a disruptive substitution in the protein structure.
Where did amino acids come from before enzymes that make them, and before life started?
Before life existed, amino acids were synthesized on Earth through ordinary chemistry. The Miller-Urey experiment demonstrated that early Earth’s atmosphere was capable of forming several types of amino acids without the help of cells or enzymes. Amino acids can form spontaneously through prebiotic chemical reactions that are driven by UV radiation, hydrothermal vents, lightning, etc.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
If you made an α-helix using D-amino acids, you would form a left-handed α-helix. However, if you used L-amino acids, you would create a right-handed α-helix. This is due to the peptide backbone’s fixed bond angles which make a D-amino acid-based α-helix energetically favorable in a left-handed form.
Can you discover additional helices in proteins?
Using synthetic biology, it is possible to design new helices in proteins. Researchers can use D-amino acids and noncanonical amino acids to modify backbone chemistry and engineer altered hydrogen bonding patterns. A new helice is unlikely to emerge naturally since the chemistry of protein backbones is heavily constrained.
Why are most molecular helices right-handed?
Most molecular helices are right-handed because of how the chiral L-amino acids and D-sugars act within the protein/DNA strand. Due to their stereochemistry, L-amino acids and D-sugars find right-handed helices more energetically favorable. Due to evolution, organisms tend to favor L-amino acids and D-sugars which causes most moelcular helices to instead be right-handed.
Why do β-sheets tend to aggregate?
β-sheets have hydrogen bond donors (N-H) and hydrogen bond acceptors (C=O). To satisy these untaken hydrogen bonds, β-sheets form hydrogen bonds with other strands. Unlike α-helices, β-sheets will depend on intermolecular hydrogen bonding. This causes them to become aggregation-prone.
What is the driving force for β-sheet aggregation?
The main driving force for β-sheet aggregation primarily includes backbone hydrogen bonding. Each added strand forms another array of H-bonds which increases stability. In addition, β-sheet-forming sequences usually have a hydrophobic effect where one face is hydrophobic while the other is polar. This causes water to be excluded from the packed-together hydrophobic faces.
Why do many amyloid diseases form β-sheets?
Amyloid fibrils share a universal Cross-β architecture motif. In this structure, β-strands run perpendicular to the fibril axis where sheets stack. In proteins that misfold, sequences are exposed that are capable of forming extended β-strands and are normally buried. This causes aggregation to become autocatalytic which results in additional monomers to attach to the growing fibril.
Can you use amyloid β-sheets as materials?
Amyloid β-sheets are strong, highly ordered, self-assembling, and chemically programmable. This makes them highly capable of being used as a material. Researchers are exploring them for use in nanofibers, biomaterials, drug delivery scaffolds, etc. Since they are sequence-programmable, researchers can engineer peptides that can be mechanically tunable and form controlled fibers.
Part B: Protein Analysis and Visualization
 A 3D render of the structure of a homolog of lactase.
A 3D render of the structure of a homolog of lactase.
1. Briefly describe the protein you selected and why you selected it.
The protein that I chose was the lactase (LCT) enzyme (structure shown above). I chose this protein because this enzyme is heavily implicated in lactose intolerance. I have a large interest in using synthetic biology to be able to cure lactose intolerance.
2. Identify the amino acid sequence of your protein.
This is the amino acid sequence of lactase:
>NP_002290.2 lactase/phlorizin hydrolase preproprotein [Homo sapiens]
MELSWHVVFIALLSFSCWGSDWESDRNFISTAGPLTNDLLHNLSGLLGDQSSNFVAGDKDMYVCHQPLPT FLPEYFSSLHASQITHYKVFLSWAQLLPAGSTQNPDEKTVQCYRRLLKALKTARLQPMVILHHQTLPAST LRRTEAFADLFADYATFAFHSFGDLVGIWFTFSDLEEVIKELPHQESRASQLQTLSDAHRKAYEIYHESY AFQGGKLSVVLRAEDIPELLLEPPISALAQDTVDFLSLDLSYECQNEASLRQKLSKLQTIEPKVKVFIFN LKLPDCPSTMKNPASLLFSLFEAINKDQVLTIGFDINEFLSCSSSSKKSMSCSLTGSLALQPDQQQDHET TDSSPASAYQRIWEAFANQSRAERDAFLQDTFPEGFLWGASTGAFNVEGGWAEGGRGVSIWDPRRPLNTT EGQATLEVASDSYHKVASDVALLCGLRAQVYKFSISWSRIFPMGHGSSPSLPGVAYYNKLIDRLQDAGIE PMATLFHWDLPQALQDHGGWQNESVVDAFLDYAAFCFSTFGDRVKLWVTFHEPWVMSYAGYGTGQHPPGI SDPGVASFKVAHLVLKAHARTWHHYNSHHRPQQQGHVGIVLNSDWAEPLSPERPEDLRASERFLHFMLGW FAHPVFVDGDYPATLRTQIQQMNRQCSHPVAQLPEFTEAEKQLLKGSADFLGLSHYTSRLISNAPQNTCI PSYDTIGGFSQHVNHVWPQTSSSWIRVVPWGIRRLLQFVSLEYTRGKVPIYLAGNGMPIGESENLFDDSL RVDYFNQYINEVLKAIKEDSVDVRSYIARSLIDGFEGPSGYSQRFGLHHVNFSDSSKSRTPRKSAYFFTS IIEKNGFLTKGAKRLLPPNTVNLPSKVRAFTFPSEVPSKAKVVWEKFSSQPKFERDLFYHGTFRDDFLWG VSSSAYQIEGAWDADGKGPSIWDNFTHTPGSNVKDNATGDIACDSYHQLDADLNMLRALKVKAYRFSISW SRIFPTGRNSSINSHGVDYYNRLINGLVASNIFPMVTLFHWDLPQALQDIGGWENPALIDLFDSYADFCF QTFGDRVKFWMTFNEPMYLAWLGYGSGEFPPGVKDPGWAPYRIAHAVIKAHARVYHTYDEKYRQEQKGVI SLSLSTHWAEPKSPGVPRDVEAADRMLQFSLGWFAHPIFRNGDYPDTMKWKVGNRSELQHLATSRLPSFT EEEKRFIRATADVFCLNTYYSRIVQHKTPRLNPPSYEDDQEMAEEEDPSWPSTAMNRAAPWGTRRLLNWI KEEYGDIPIYITENGVGLTNPNTEDTDRIFYHKTYINEALKAYRLDGIDLRGYVAWSLMDNFEWLNGYTV KFGLYHVDFNNTNRPRTARASARYYTEVITNNGMPLAREDEFLYGRFPEGFIWSAASAAYQIEGAWRADG KGLSIWDTFSHTPLRVENDAIGDVACDSYHKIAEDLVTLQNLGVSHYRFSISWSRILPDGTTRYINEAGL NYYVRLIDTLLAASIQPQVTIYHWDLPQTLQDVGGWENETIVQRFKEYADVLFQRLGDKVKFWITLNEPF VIAYQGYGYGTAAPGVSNRPGTAPYIVGHNLIKAHAEAWHLYNDVYRASQGGVISITISSDWAEPRDPSN QEDVEAARRYVQFMGGWFAHPIFKNGDYNEVMKTRIRDRSLAAGLNKSRLPEFTESEKRRINGTYDFFGF NHYTTVLAYNLNYATAISSFDADRGVASIADRSWPDSGSFWLKMTPFGFRRILNWLKEEYNDPPIYVTEN GVSQREETDLNDTARIYYLRTYINEALKAVQDKVDLRGYTVWSAMDNFEWATGFSERFGLHFVNYSDPSL PRIPKASAKFYASVVRCNGFPDPATGPHACLHQPDAGPTISPVRQEEVQFLGLMLGTTEAQTALYVLFSL VLLGVCGLAFLSYKYCKRSKQGKTQRSQQELSPVSSF
- The length of the protein is: 1927 aminoacids.
- The most common amino acid is: L, which appears 170 times.
- Using Uniprot’s BLAST tool, I found 250 protein sequence homologs for LCT. The result of the BLAST tool can be found here.
3. Identify the structure page of your protein in RCSB.
I was not able to find the same exact protein in RCSB. However, I found a close homolog in Escherichia coli. The PDB ID is pdb_00005e9a.
- This structure was solved on 2016-10-26. The resolution of the structure is 2.56 Å. This is an acceptable/good quality structure.
- In the structure, there are water molecules and typically bounder inhibitors or substrate analogs.
- When put into SCOP, the PDB ID and Uniprot ID returns no results. I assume this is because the protein does not belong to any structure classification family.
4. Open the structure of your protein in any 3D molecule visualization software.
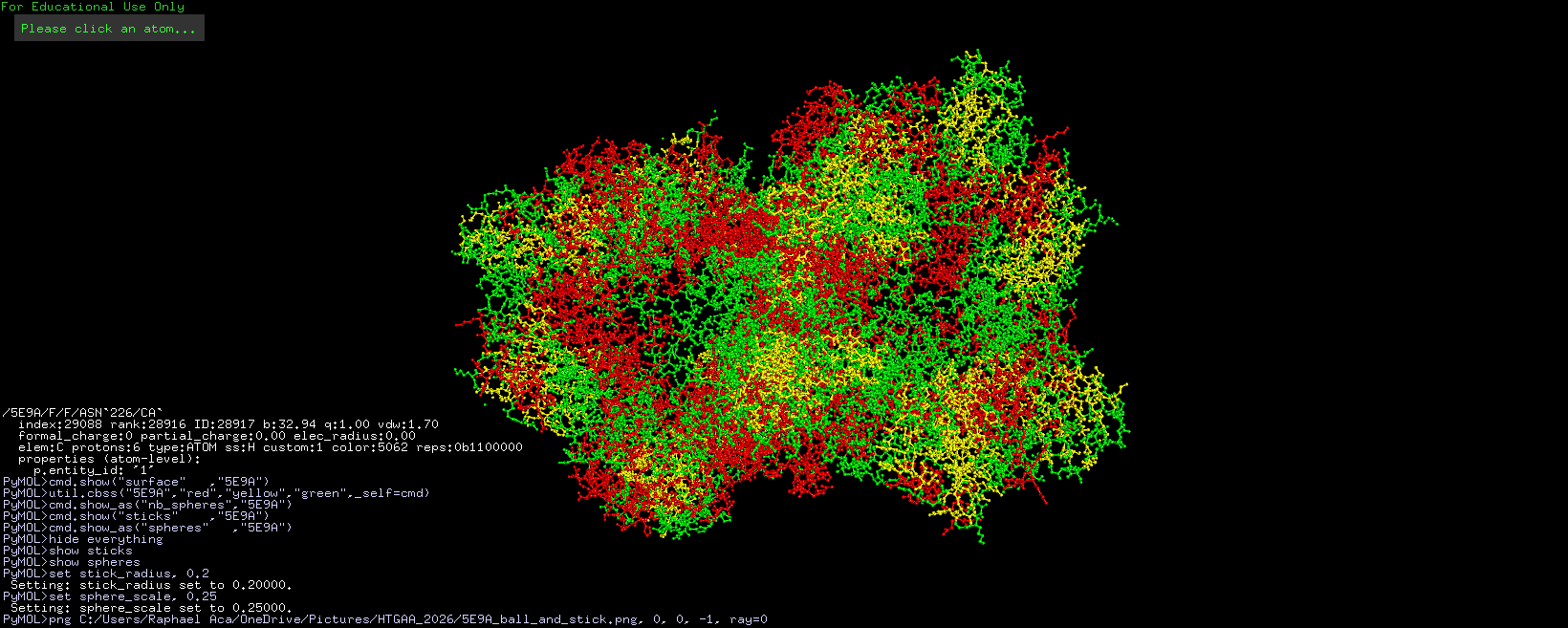
Info
3D visualization of “ball-and-stick” model of 5E9A protein.
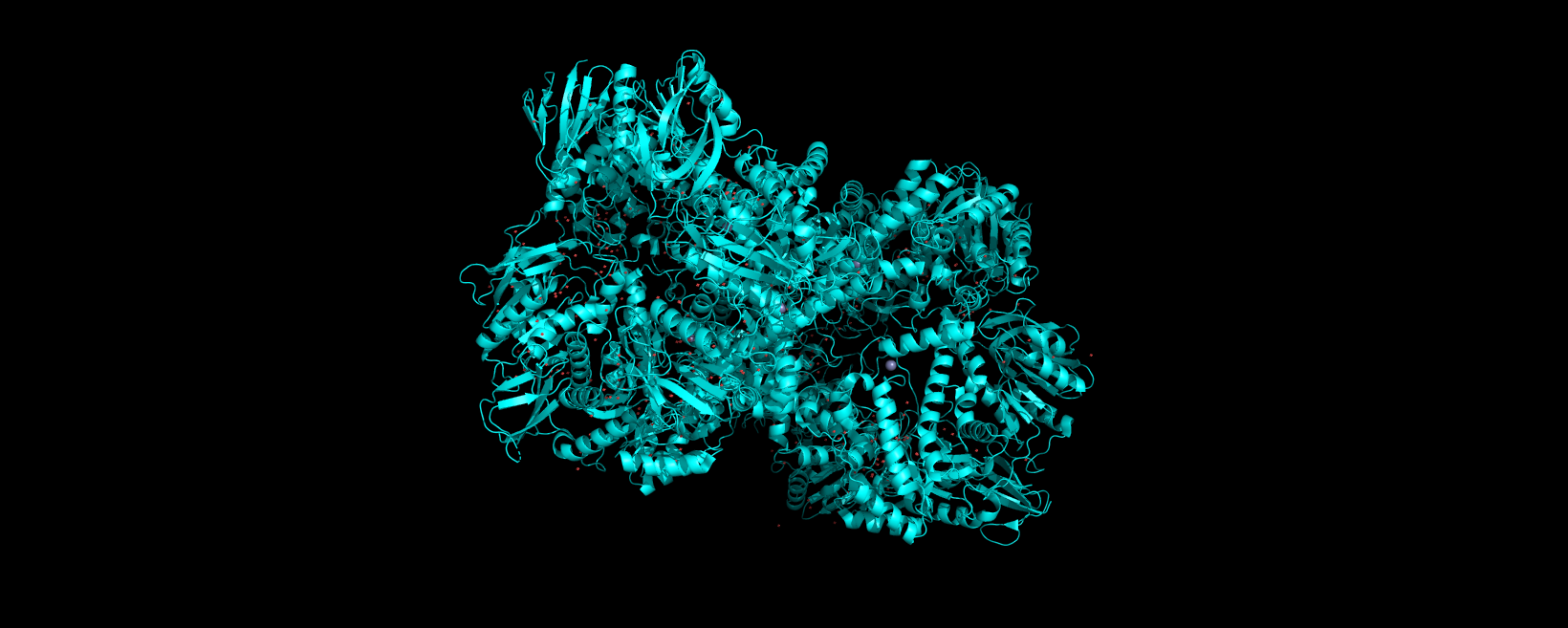
Info
3D visualization of “ribbon” model of 5E9A protein.
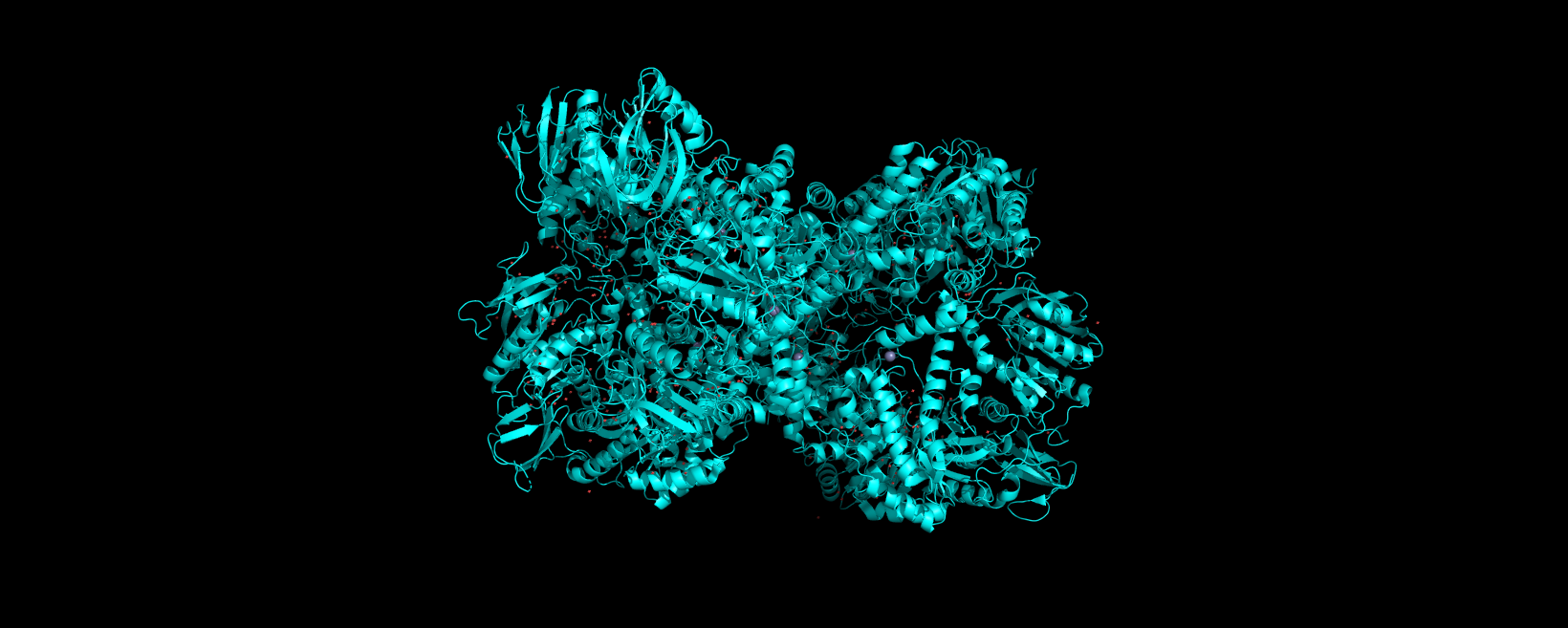
Info
3D visualization of “cartoon” model of 5E9A protein.
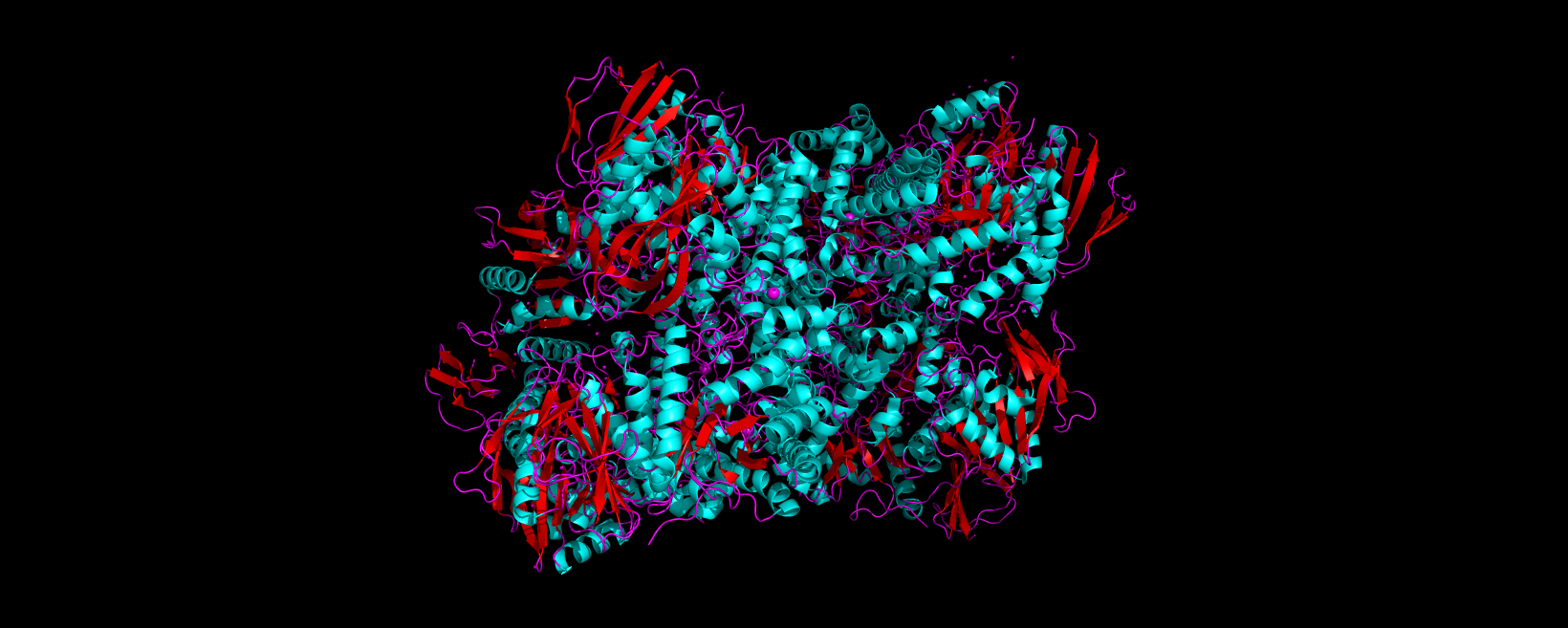
Info
3D visualization of 5E9A protein colored by secondary structure. Cyan is helix, red is sheet, and magenta is loop.
The above image of 5E9A colored by its secondary structure shows how the structure most likely contains more helices than sheets.
Info
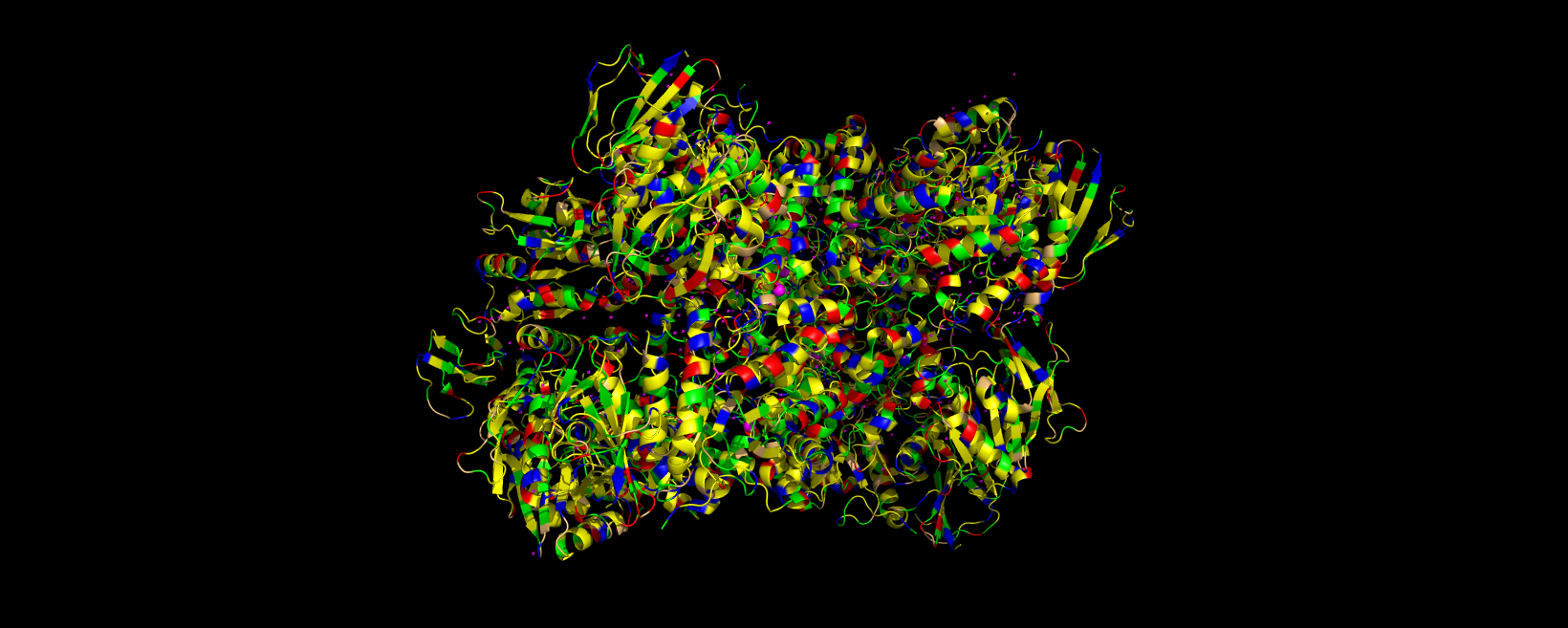
3D visualization of 5E9A protein colored by residue type. Yellow is hydrophobic, green is polar (uncharged & hydrophilic), blue is positively charged (hydrophilic), red is negatively charged (hydrophilic), and light orange is Glycine (special case, both hydrophobic and hydrophilic).
The 3D visualization of the 5E9A protein colored by residue type shows how the protein is about half hydrophobic amino acids and half hydrophilic amino acids. The protein shows a somewhat alternating pattern between hydrophilic and hydrophobic residues. This pattern creates the helices, sheets, and loops shown in the model.
Info
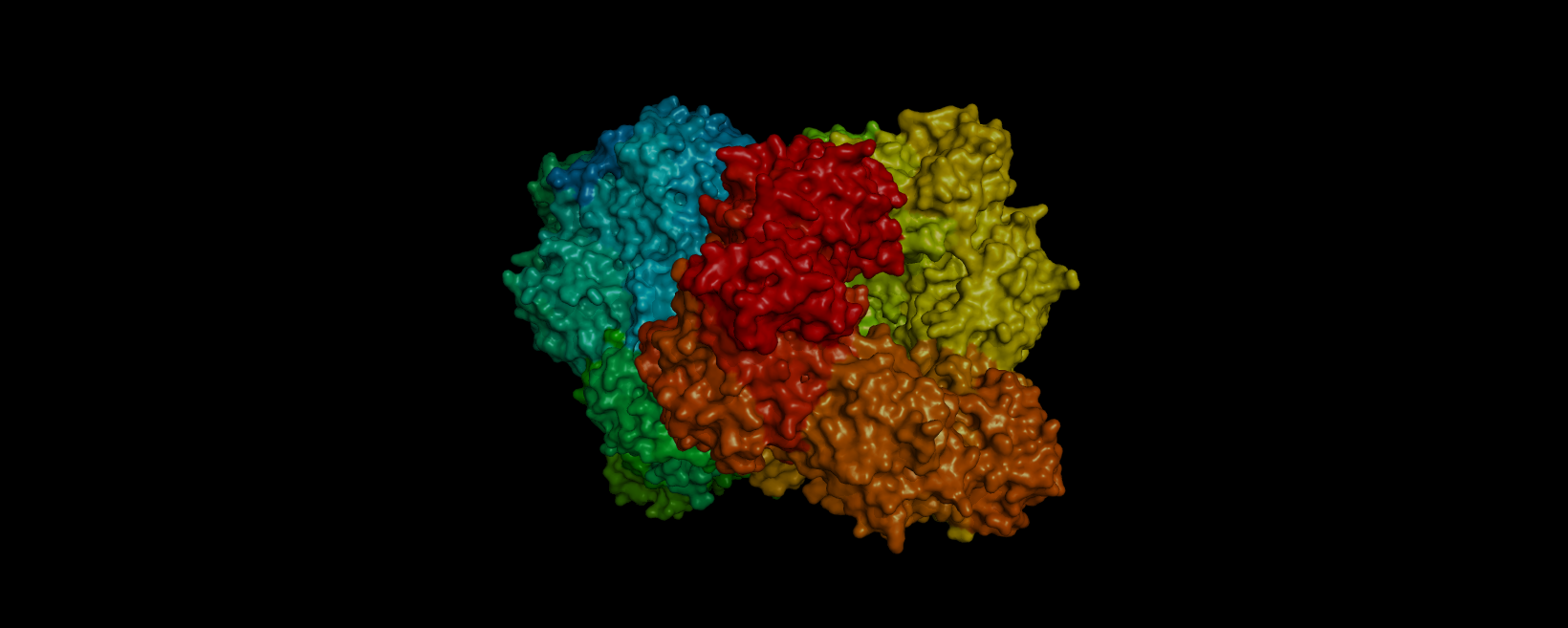
3D visualization of “surface” model of 5E9A protein. Colored rainbow by elem c.
The above model shows a “hole” in the enzyme which is its binding pocket. Not shown in the picture but on the other side of the protein there is another binding pocket. It is likely that these “holes” are where lactose binds.
Part C. Using ML-Based Protein Design Tools
For this part, I am continuing to use the lactase protein pdb_00005e9a.
C1. Protein Language Modeling
1. Deep Mutational Scans
Info
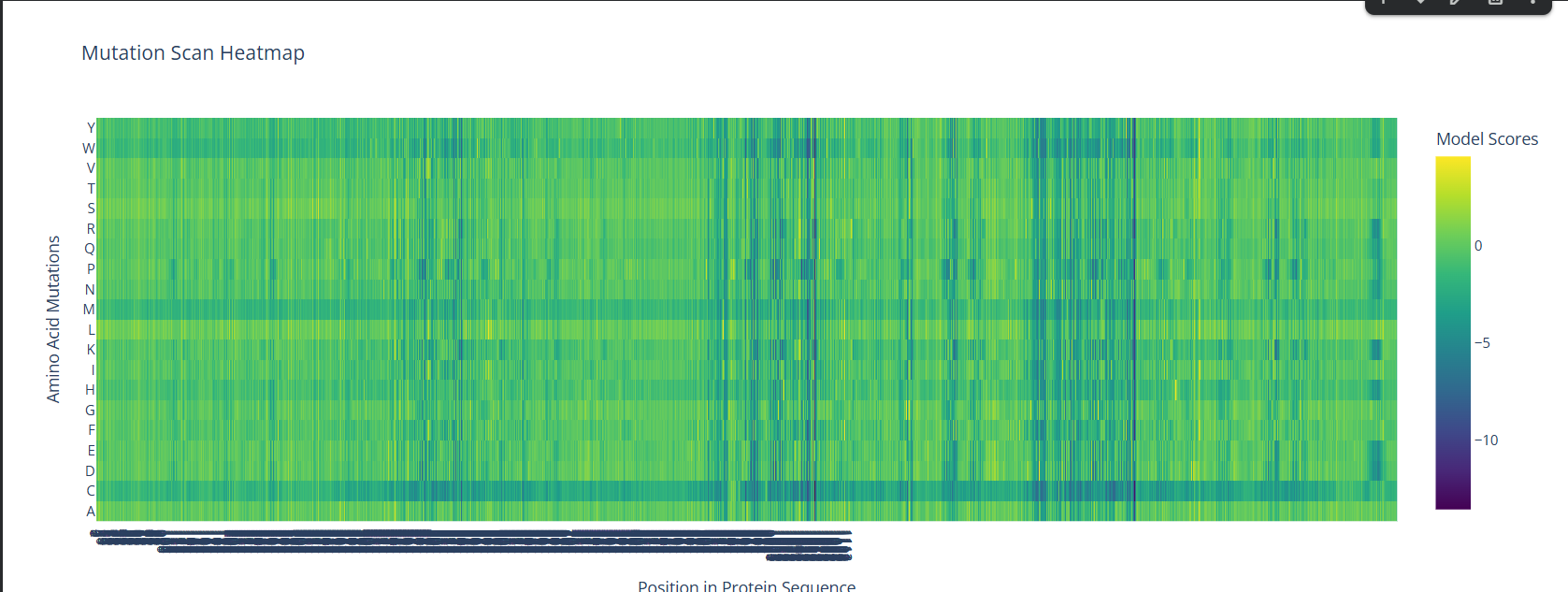
A screenshot of the Mutation Scan Heatmap generated from the LCT protein sequence on ESM2.
The above mutation scan heatmap demonstrates stark patterns. For instance, the W, M, and C residues seem to have consistently negative values throughout the protein sequence. This means that these amino acid residues would most likely reduce fitness/function in the LCT protein if they were to replace nearly any amino acid in a mutation.
2. Latent Space Analysis
Info
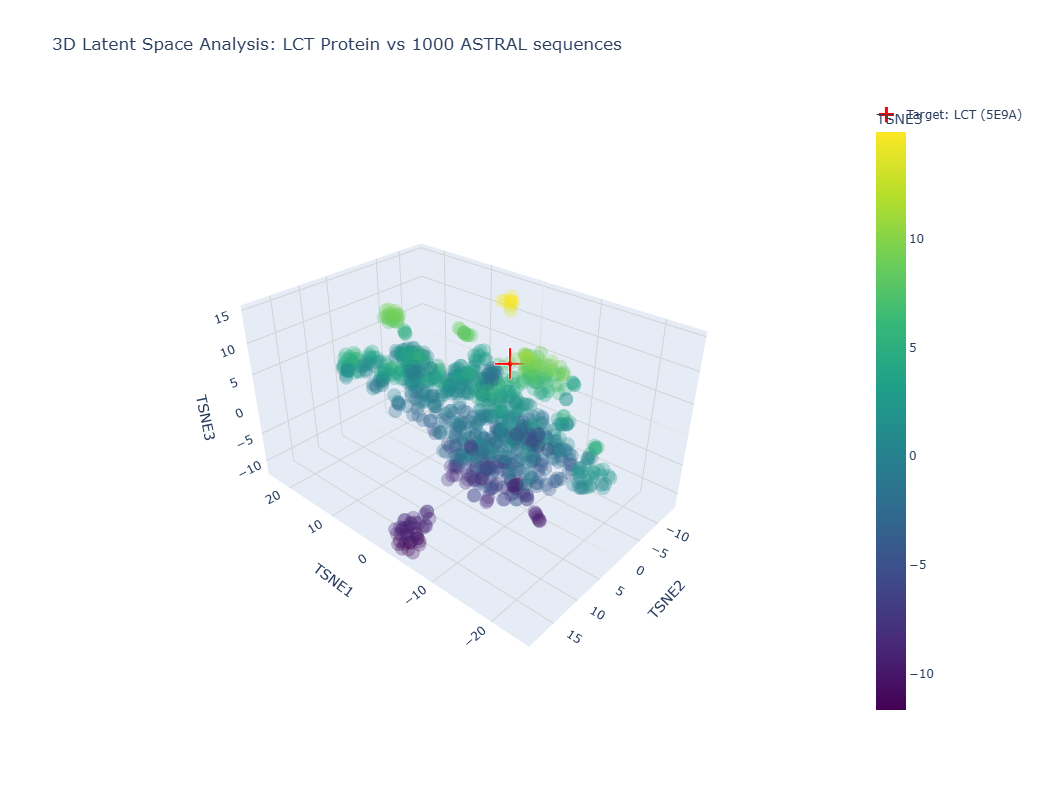
Image showing a latent space analysis of various proteins. The red cross represents my chosen protein, LCT/E59A.
The different formed neighborhoods within the the latent space analysis which grouped proteins together mostly based on similar function and species. For example, HMG1 (High mobility group protein 1) of Rattus norvegicus (Norway rat) and HMG1 of Cricetulus griseus (Chinese hamster) were present in a cluster together.
My protein, E59A, had a higher TSNE3 (as shown above) and was grouped with various human (Homo sapien) proteins such as somatotropin, inferon-beta, and other automated matches. It seems to be similar to human proteins involved in human development. This is slightly expected as E59A is a human protein.
C2. Protein Folding
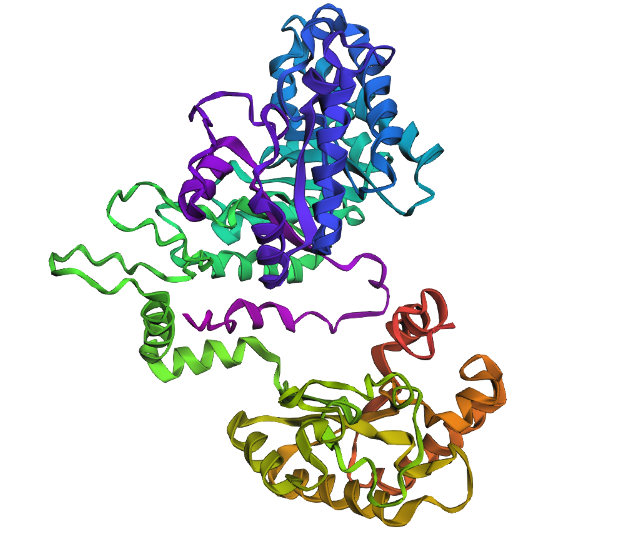
Info
A 3D representation of the truncated structure of 5E9A (560 residues) using ESMFold.
Due to the sequence of my protein being too long, I opted to truncate my protein sequence to 560 residues so that the ESMFold in Google Colab would be able to successfully run the sequence. The ESMFold prediction produced an average pLDDT score of 79.1, showing generally reliable structural confidence. The predicted TM-score (pTM = 0.583) shows moderate confidence in the overall global fold. These statistics imply that the individual domains are well predicted but their orientations may be less accurate. This is a plausible is structurally coherent fold.
The predicted coordinates do not exactly match my original structure as I had to truncate my protein in order for it to work with ESMFold. However, if I used my entire sequence, I predict that it would have matched well given the statistics of my fold.
After mutating the sequence a few times, I saw how my structure proved to be fairly resistant to mutations. After small mutations, the structure did not change significantly. However, large segments did produce significant change within the structure.
C3. Protein Generation
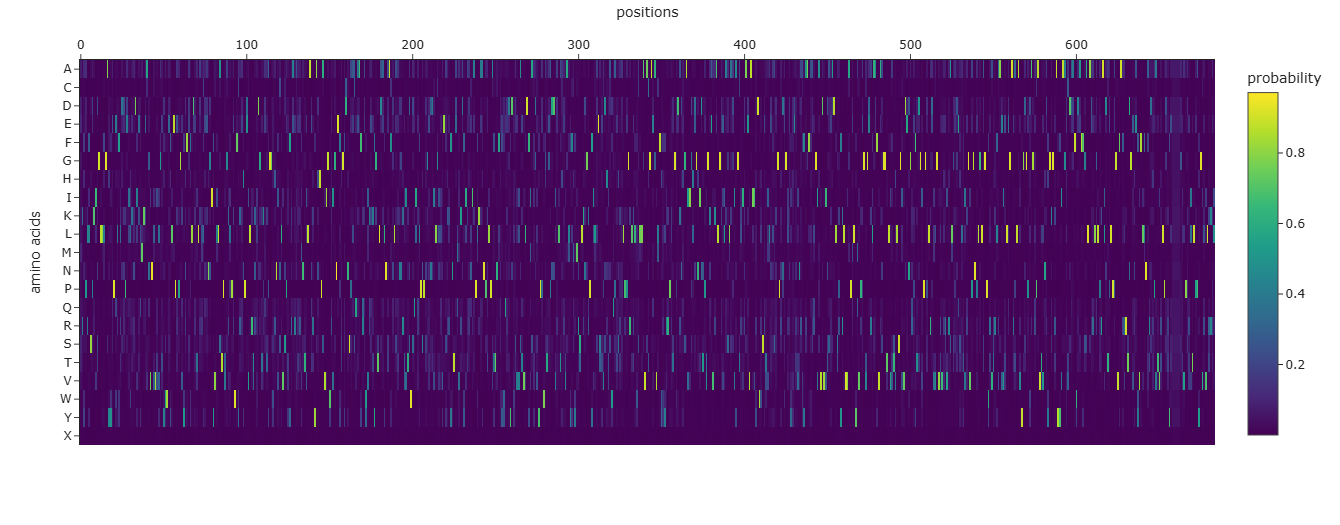
Info
Plot showing the probabilities of each amino acid in each index of the sequence based on the 3D structure of E59A.
This is the original sequence:
MNHKVHHHHHHIEGRHMELGTLEGSMTKFPLLSSKISGLLHGADYNPEQWLDHPDVLVRDVEMMKEARCNVMSVGIFSWSALEPEEGRYTFDWMDQVLNRLHENGISVFLATPSGARPAWMSQKYPQVLRVGRDRVPALHGGRHNHCMSSPVYREKVQLMNGQLAKRYAHHPAVIGWHISNEYGGECHCDTCQGQFRDWLKARYVTLDALNKAWWSTFWSHTYTDWSQLESPSPQGENGVHGLNLDWRRFNTDQVTRFCSEEIRPLKAENPALPATTNFMEYFNDYDYWKLAGVLDFISWDSYPMWHTRQDDIGLAAYTAMYHDLMRTLKQGKPFVLMESTPSFTNWQPTSKLKKPGMHILSSLQAVAHGADSVQYFQWRKSRGSCEKFHGAVVDHVGHIDTRVGREVAELGSILSALAPVAGSRVEAKVAIIFDWESRWAMDDAMGPRNAGLHYENTVADHYRALWAQGIAVDVINADCDLQGYDLVIAPMLYMVREGVGERISAFVQAGGRFVATYWSGIVNETDLCFLNGFPGPLRPVLGIWAEEIDSLTDEQHNSVAGVEGNALGLSGPYRASQLCEVIHLEGAAALATYGDDFYAGNPAVTVNLYGKGQAYYVASRNDQQFHADFFTALAKEMKLPRAINTPLPEGVTAARRTDGESEFIFLQNYNADNQTVALPQDYQDIVHGGNLPRKLTLPAFGCQILTRKITQ
This is the predicted sequence:
MKFPRLSPKIDGLLLGAEYYPELFLDDPELIERLIELMKEAKINVVRLGTHAWEYLEPEPGVYNFAWLDKTLDLLEKNGIYVLLATPTGKLPRWVYEEHPEVLRTKLDGTKEKYGGSHNICLVSPYFRALATEMNTKLASRYAHHPAVIGWEIGNEFSGYCNCPLCQEAFRKWVKEKFGTLDAFNKAGNLEKNNKVVTDWSQIKLPSPNGENDVLTLINLFKEYNTQLVKDFLTQLIEPLKKYNPNLPVTSNFDWWQEDYDYTELATVLDFIAYDSYPPWGTGPDDVRLAAEVAMYHDYMRSLKNGKPFILSETYVDHVNWQATSTSLPPGRLKLWCLLHVACGAEAVLFHYLRRPRTGWDKNHGAVIDHTGNIDTPVGKEVKALGDELASLKDIAGTKIEAEVAIVYDWKSRVILEASKGPLDAGLDYVGNVNRWFRAFWSQGIAVDVIDADADLSPFKLVVAPELYMVPAGVGDRLAAFVAAGGTFVATVLSGVVDEYGREFTDGRPGPLRDVLGVLVRRVRSLSADQTATVTGVAGNELGLSGPYTVTRLAAVVELRGAEALAVYGSGPDAGQPAVTVNRHGKGRAYYVAGRVDDAFWAAFFGSLAEKLKLKRAIDTPLPPGVFARRRTDGENEYVFLFNFRDTPVTVTLPGTYTXXXXXGTVPATITLPPYGVKVLTRKI
The predicted sequence is significantly distinct from the original sequence. The predicted sequence is longer than the original sequence and contains little similarities with the original sequence.
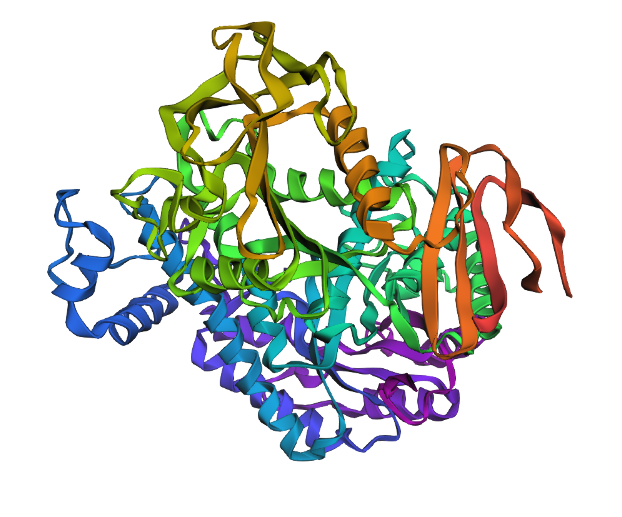
Info
A proposed 3D structure generated by ESMFold based on the predicted sequence generated by ProteinMPNN. The predicted strcture seems to be more clumped than the original structure generated by ESMFold.
Part D. Group Brainstorm on Bacteriophage Engineering
For this part of the homework, I joined a group of students consisting of Jason Ross, Jay Handfield, Nana Agyei Afrane-Asare, Xavier-Lewis Palmer, and myself. After going through the phage reading and reviewing the bacteriophage final project goals, we opted to increase the thermodynamic stability of the Lysis Protein.
Our group’s plan for engineering a bacteriophage includes multiple steps and various protein engineering tools. First, we will use BLAST and Clustal Omega to discover amino acid residues that are conserved between Lysis protein homologs. ESM2 will be used to score mutations for their evolutionary plausibility and ESM-Fold/ProteinMPNN/Boltz-1 will be used to refine the folded protein. EvolvePro will be used to computationally direct evolution in this protein structure. Lastly, we can use computational stress-testing under varying environmental conditions to test for destabilization.
Our group’s full one page proposal is linked here.