Week 2 HW: DNA Read, Write, and Edit
PART 1: Benchling & In-silico Gel Art
First, I googled for the accession number of the Lambda phage full genome DNA sequence. I put “lambda dna sequence ncbi” in the google search box. The first result prompted this:
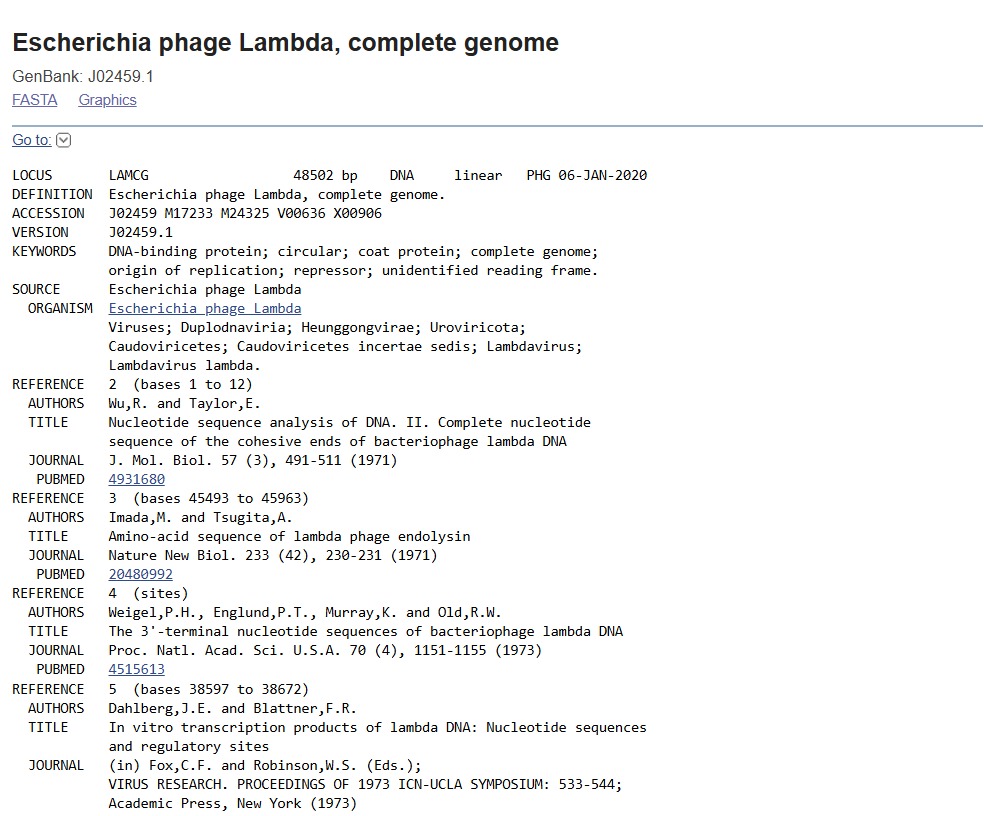
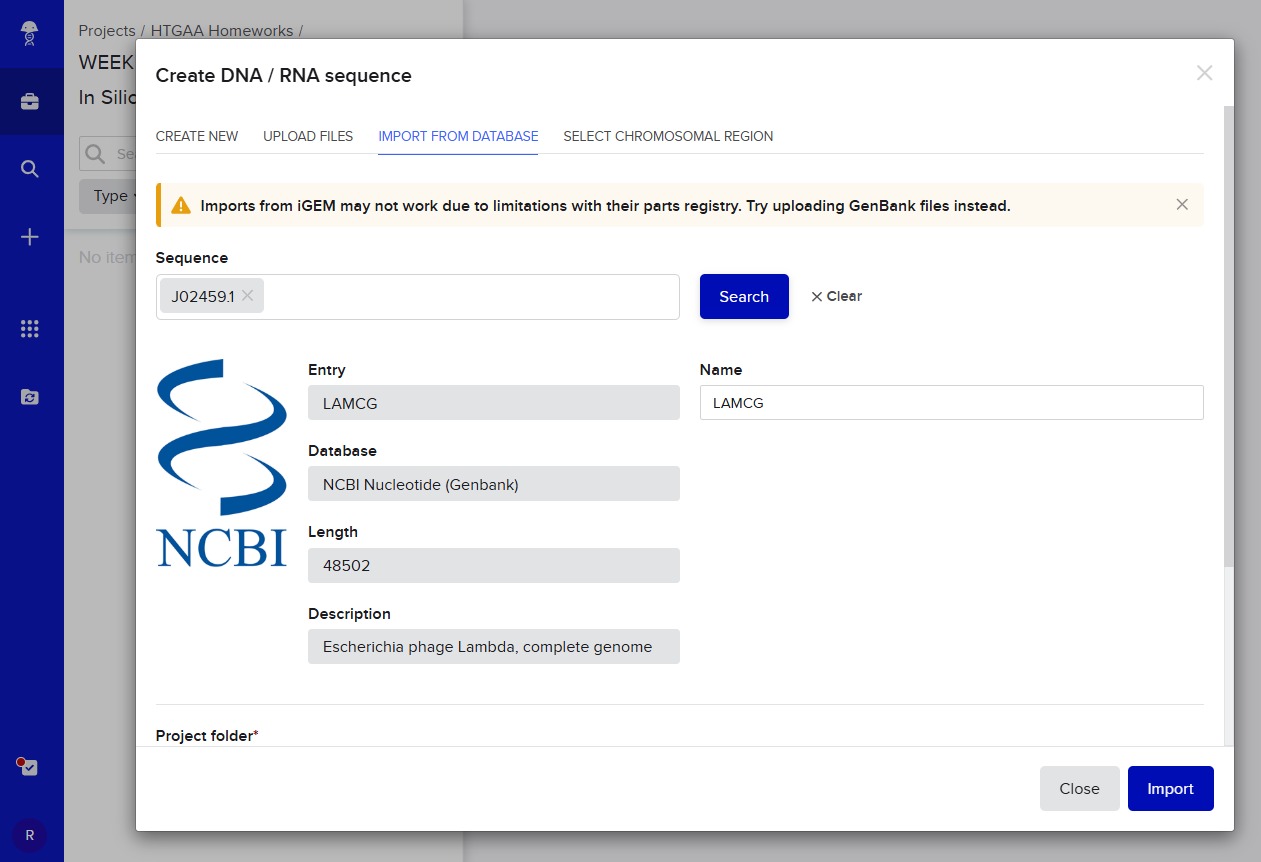
After this, I signed into Benchling and created a new project named “HTGAA HOMEWORKS/WEEK 2 - PART 1: Benchling & In Silico Gel Art”. There I imported Lambda DNA using the accession number: GenBank: J02459.1
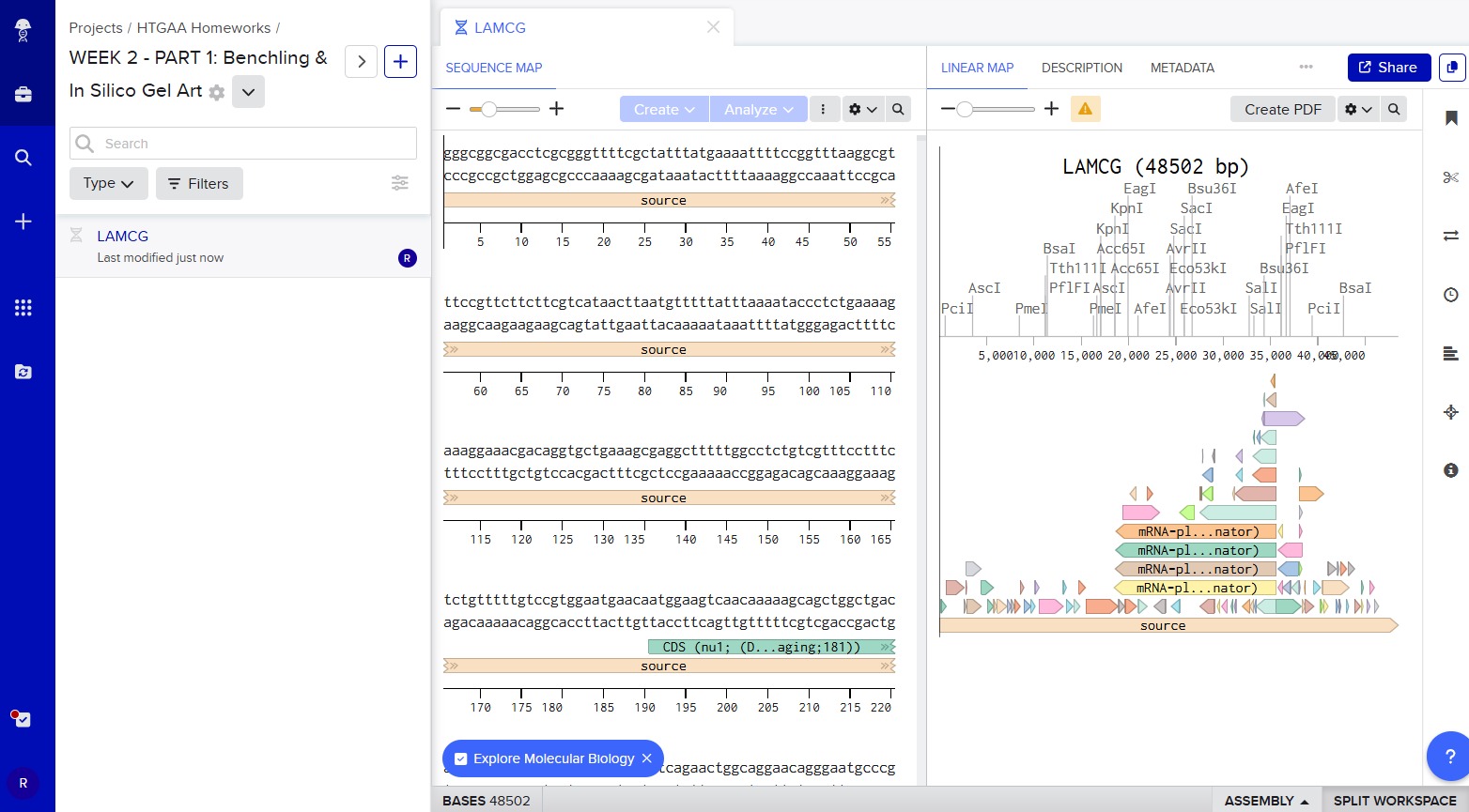
Finally, we have everything to start with the task:
Simulate Restriction Enzyme Digestion with the following Enzymes: EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, SalI
For this, we performed in-silico digestion by clicking on the scissors icon at the right vertical bar:
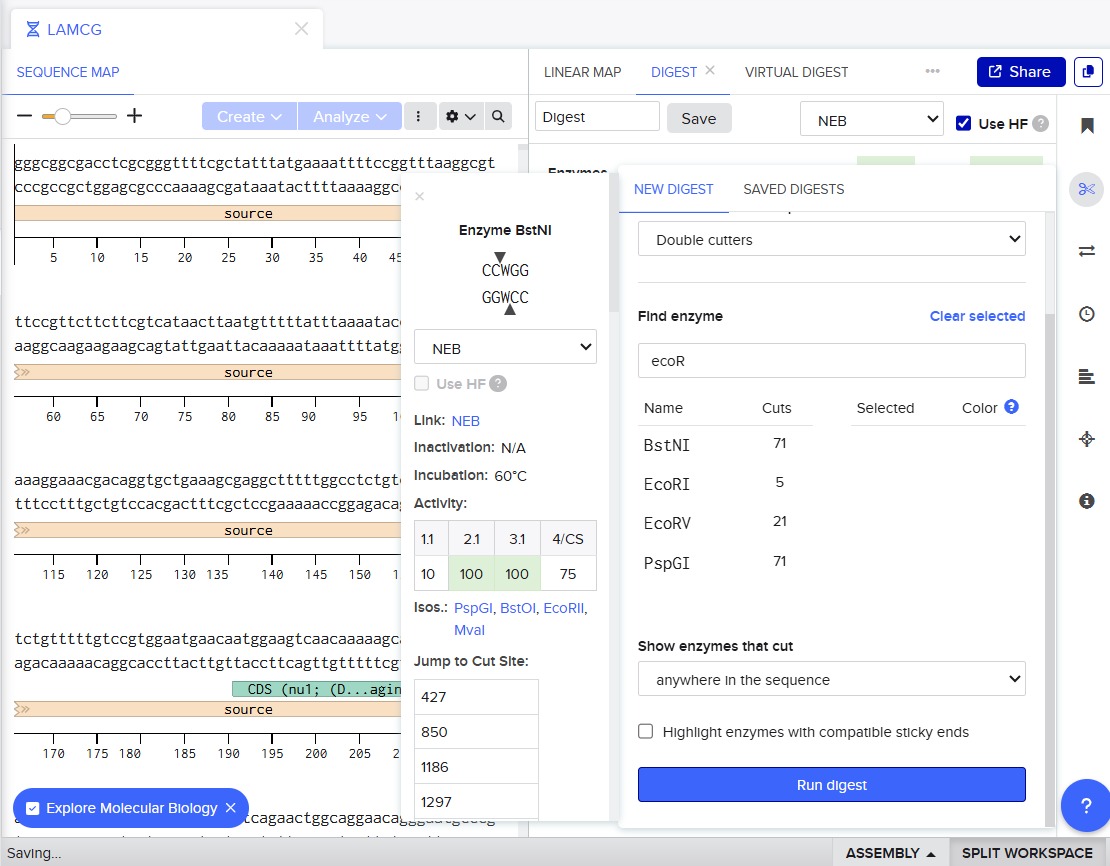
First we tested with only EcoRI:
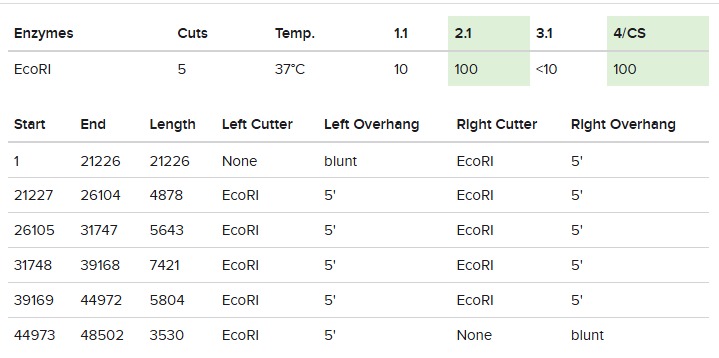
The final gel was this:
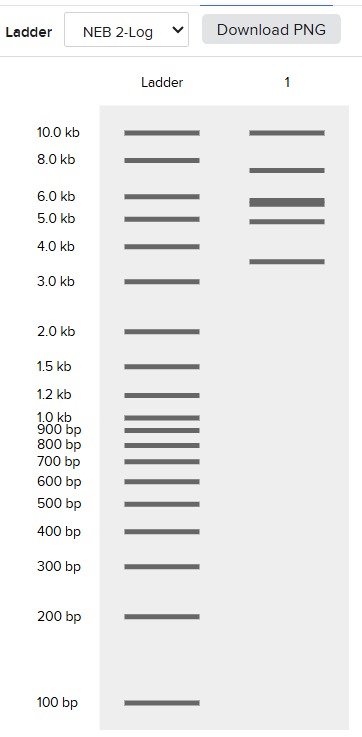
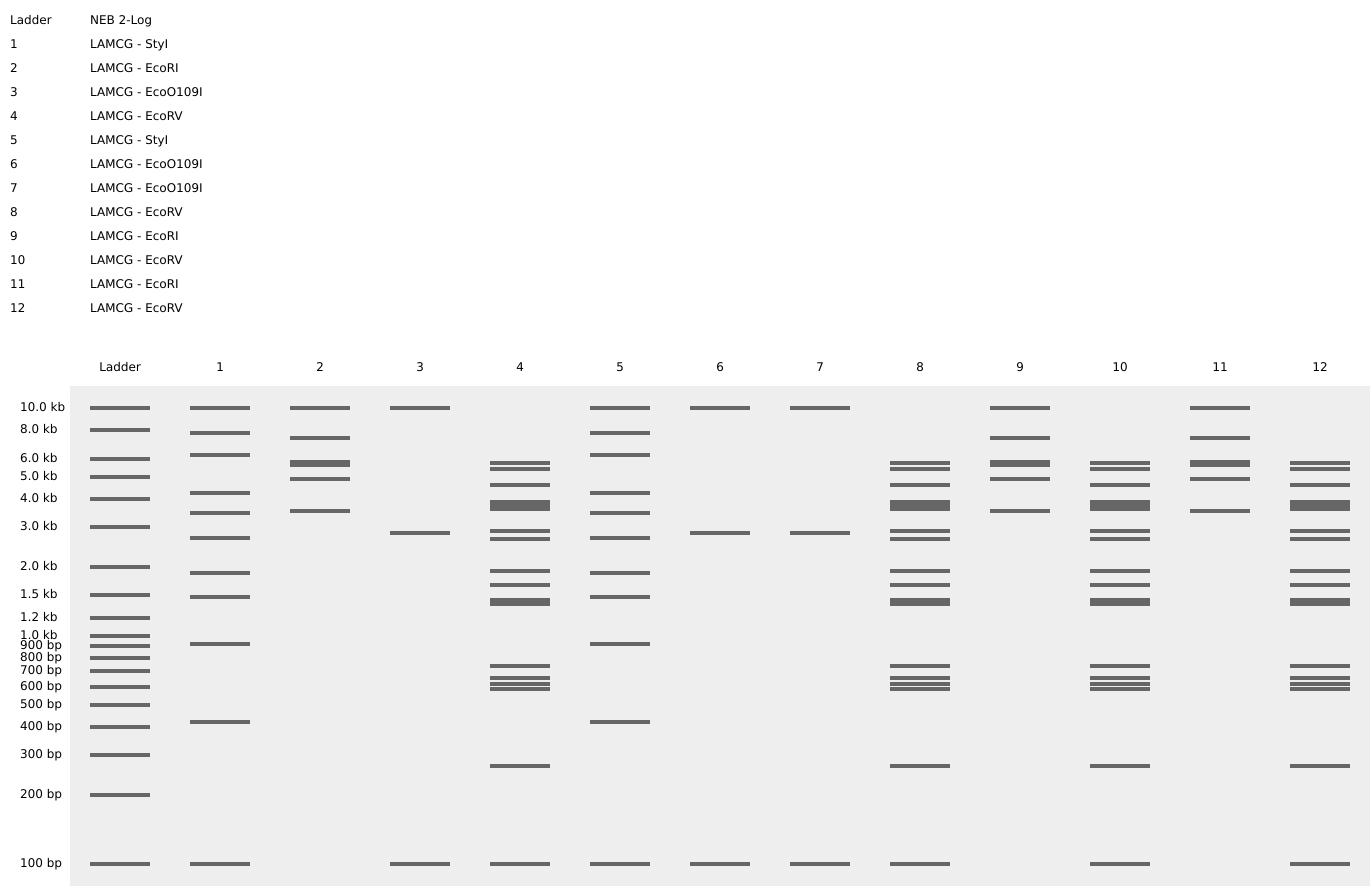
Following these steps, we obtained the final gel, including the rest of enzymes:
Each digestion product was ran on a separate lane in the virtual gel:
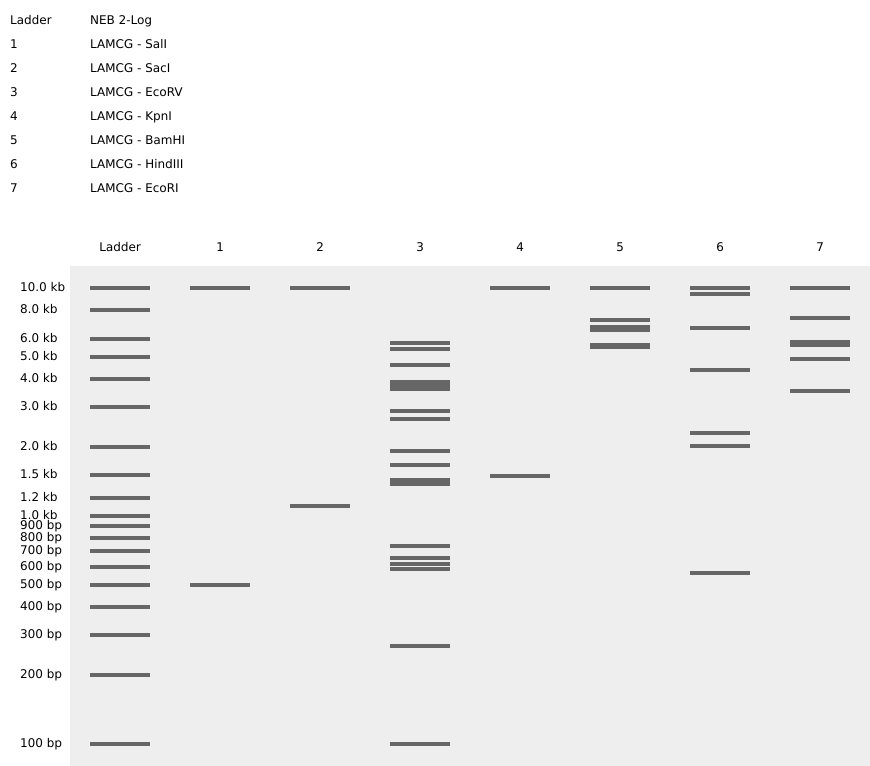
After some time testing different enzymes, I made the word “IgEm”.
PART 3: DNA Design Challenge
3.1. Choose your protein.
I chose the encapsulin shell protein from Thermotoga maritima because it is a self-assembling protein nanocage with strong potential in drug delivery and synthetic biology. Encapsulins form icosahedral structures composed of multiple identical subunits and can selectively encapsulate cargo proteins through short targeting peptides, making them highly programmable at the genetic level. Their robustness and modularity make them attractive platforms for engineering artificial nanocarriers.
However, despite their potential, encapsulins still present important limitations. Their high structural stability makes controlled cargo release difficult, which is a challenge for therapeutic applications. As bacterial proteins, they may also trigger immune responses in mammalian systems, and their long-term biocompatibility is not fully understood. In addition, modifying the shell surface for targeting or functionalization can disrupt assembly if not carefully designed. These constraints highlight that encapsulins are still an evolving technology rather than a fully optimized platform.
I was especially motivated to study encapsulins because they were first successfully obtained in Peru by a Peruvian research group last year. This encouraged me to explore their design and expression.
The sequence of the encapsulin shell protein (UniProt entry Q9WZP2) is:
9Y8P_1|Type 1 encapsulin shell protein|Thermotoga maritima (2336) MEFLKRSFAPLTEKQWQEIDNRAREIFKTQLYGRKFVDVEGPYGWEYAAHPLGEVEVLSDENEVVKWGLRKSLPLIELRATFTLDLWELDNLERGKPNVDLSSLEETVRKVAEFEDEVIFRGCEKSGVKGLLSFEERKIECGSTPKDLLEAIVRALSIFSKDGIEGPYTLVINTDRWINFLKEEAGHYPLEKRVEECLRGGKIITTPRIEDALVVSERGGDFKLILGQDLSIGYEDREKDAVRLFITETFTFQVVNPEALILLKF
References:
- Kwon, S., & Giessen, T. W. (2025). Engineering encapsulin nanocages for drug delivery. Materials Advances, 6, 6209–6220. https://doi.org/10.1039/D5MA00386E
- Sutter, M., Boehringer, D., Gutmann, S., Günther, S., Prangishvili, D., Loessner, M. J., Stetter, K. O., Weber-Ban, E., & Ban, N. (2008). Structural basis of enzyme encapsulation into a bacterial nanocompartment. Nature Structural & Molecular Biology, 15(9), 939–947. https://doi.org/10.1038/nsmb.1473
- Protein Data Bank. (2025). Cryo-EM structure of Thermotoga maritima encapsulin shell (PDB ID 9Y8P) [Data set]. RCSB PDB. https://doi.org/10.2210/pdb9Y8P/pdb :contentReference[oaicite:0]{index=0}
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
For Reverse Translating, there are 2 options. The first one is using the Reverse Translate tool online (https://www.bioinformatics.org/sms2/rev_trans.html).
I reverse translated the aa sequence using the codon table from the Codon Usage Database ([https://www.kazusa.or.jp/codon/]).
I chose the first output, which is the predicted DNA sequence for the most likely codons for Bacillus Thurigiensis.
9Y8P_1|Type 1 encapsulin shell protein DNA sequence|Thermotoga maritima (2336) atggaatttttaaaaagaagttttgcaccattaacagaaaaacaatggcaagaaattgataatagagcaagagaaatttttaaaacacaattatatggaagaaaatttgtagatgtagaaggaccatatggatgggaatatgcagcacatccattaggagaagtagaagtattaagtgatgaaaatgaagtagtaaaatggggattaagaaaaagtttaccattaattgaattaagagcaacatttacattagatttatgggaattagataatttagaaagaggaaaaccaaatgtagatttaagtagtttagaagaaacagtaagaaaagtagcagaatttgaagatgaagtaatttttagaggatgtgaaaaaagtggagtaaaaggattattaagttttgaagaaagaaaaattgaatgtggaagtacaccaaaagatttattagaagcaattgtaagagcattaagtatttttagtaaagatggaattgaaggaccatatacattagtaattaatacagatagatggattaattttttaaaagaagaagcaggacattatccattagaaaaaagagtagaagaatgtttaagaggaggaaaaattattacaacaccaagaattgaagatgcattagtagtaagtgaaagaggaggagattttaaattaattttaggacaagatttaagtattggatatgaagatagagaaaaagatgcagtaagattatttattacagaaacatttacatttcaagtagtaaatccagaagcattaattttattaaaattt
3.3. Codon optimization.
Codon optimization is necessary because different organisms prefer different codons to encode the same amino acid. Although the genetic code is universal, many amino acids are encoded by multiple codons, and each organism has a bias toward using certain ones more frequently. If a gene from Thermotoga maritima is expressed in another host without modification, the host’s ribosomes may translate it inefficiently due to rare codons, limited tRNA availability, or unfavorable mRNA secondary structures. This can reduce protein yield or even prevent proper expression.
For this project, I would optimize the encapsulin gene for Escherichia coli. E. coli is widely used for recombinant protein expression because it grows quickly, is inexpensive, and has well-established molecular biology tools. Encapsulins from T. maritima have previously been expressed successfully in E. coli, making it a practical and reliable host system.
9Y8P_1|Type 1 encapsulin shell protein DNA sequence|Thermotoga maritima (codon optimized) ATGGAGTTCCTGAAGCGTAGTTTTGCACCATTGACTGAGAAGCAGTGGCAAGAGATTGATAACCGTGCGCGGGAAATTTTCAAAACCCAACTCTATGGCCGTAAATTTGTTGACGTCGAAGGTCCGTATGGGTGGGAATACGCAGCGCATCCTTTAGGTGAGGTTGAAGTGCTGTCGGATGAAAACGAAGTGGTCAAGTGGGGCCTGCGAAAAAGCTTACCCTTAATTGAACTCCGCGCGACCTTCACCCTGGATTTGTGGGAATTGGACAACCTTGAAAGGGGTAAGCCGAATGTAGATCTAAGCTCCCTGGAAGAAACCGTTCGTAAAGTAGCCGAGTTTGAAGATGAAGTCATATTTCGTGGGTGTGAGAAATCAGGCGTGAAAGGCCTGCTGAGCTTTGAAGAGCGCAAAATCGAATGCGGCTCGACGCCGAAAGACCTGCTGGAGGCCATTGTGCGTGCGTTAAGTATTTTTTCAAAAGACGGTATCGAAGGCCCGTATACCCTCGTTATTAATACGGATCGCTGGATTAACTTCTTGAAAGAAGAGGCGGGCCACTACCCGCTGGAAAAGCGCGTGGAAGAGTGCCTTCGCGGCGGTAAAATCATCACGACACCTCGCATCGAAGATGCTCTTGTAGTGAGCGAACGCGGCGGTGACTTCAAACTGATTCTGGGACAGGATCTGTCTATTGGATACGAAGATCGCGAGAAAGATGCCGTTCGTCTGTTTATTACAGAAACCTTTACTTTTCAGGTGGTGAATCCGGAAGCCTTAATCCTGCTTAAATTC
There is a second option in Benchling for reverse translating and optimizing the codons, which is by using the option Back translate.
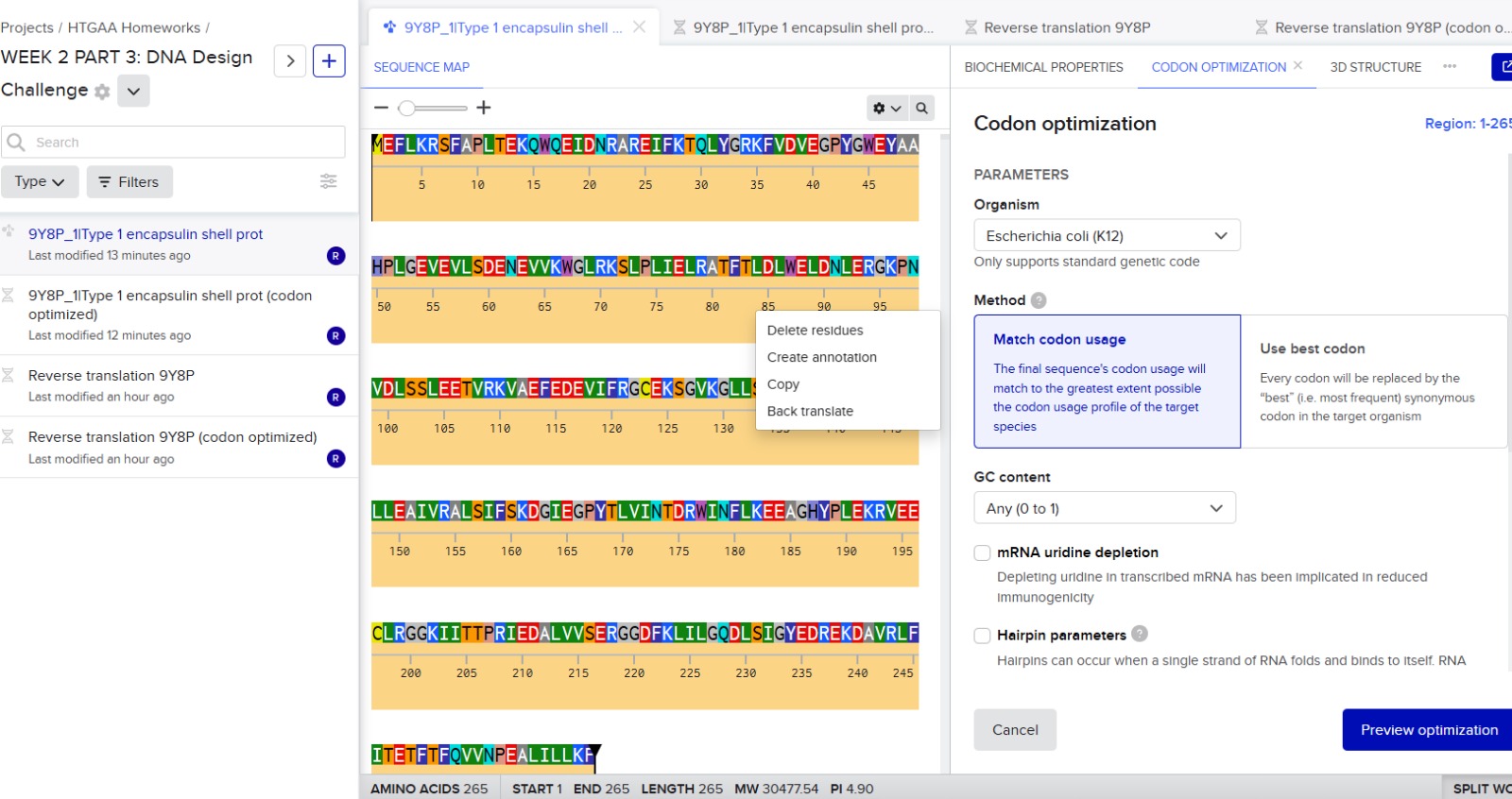
The organism that I selected for the optimization was E.coli K12.
3.4. You have a sequence! What technologies could be used to produce this protein from your DNA?
In a cell-dependent system, the optimized gene is inserted into a plasmid vector under the control of a promoter. The plasmid is introduced into E. coli through transformation. Inside the cell, RNA polymerase binds to the promoter and transcribes the DNA sequence into mRNA.
The mRNA is then translated by ribosomes. The ribosome reads the mRNA in triplets called codons, and each codon specifies one amino acid. Transfer RNAs (tRNAs) bring the correct amino acids to the ribosome, where they are linked together into a growing polypeptide chain. This process continues until a stop codon is reached. The resulting encapsulin monomers fold into their native structure and spontaneously self-assemble into a 60-subunit icosahedral nanocage.
In a cell-free system, purified transcription–translation machinery (often derived from E. coli lysates) is used outside living cells. The DNA template is added directly to the reaction mixture, and transcription and translation occur in vitro. This method allows faster prototyping and tighter experimental control, though it is typically more expensive than in vivo expression.
3.5. How does it work in nature/biological systems?
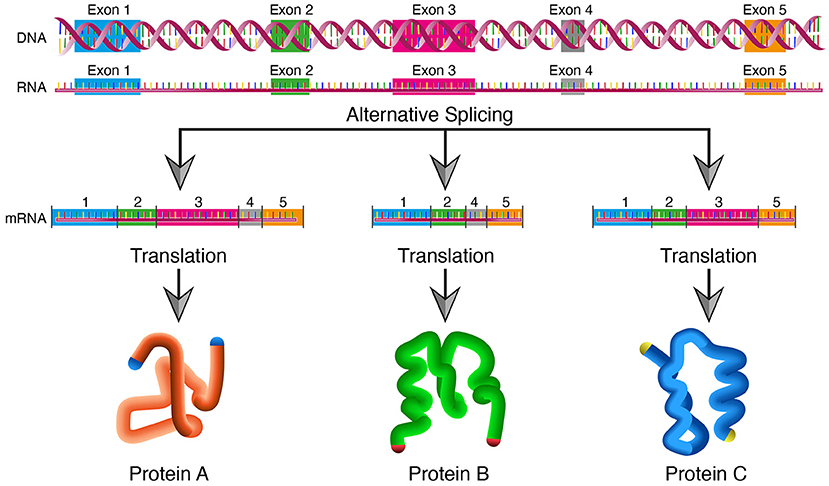
In biological systems, a single gene can produce multiple proteins at the transcriptional level through mechanisms that generate different RNA transcripts from the same DNA sequence.
In eukaryotes, the most common mechanism is alternative splicing. During transcription, a primary RNA transcript is produced that contains both exons (coding regions) and introns (non-coding regions). Different combinations of exons can be retained or removed before translation, producing distinct mRNA molecules. Each mRNA variant is translated into a different protein isoform with altered structure or function. Because the exon composition differs, the amino acid sequences of the resulting proteins differ.
Other transcriptional mechanisms include the use of alternative promoters or transcription start sites, which produce mRNAs with different 5′ ends, and alternative polyadenylation sites, which generate different 3′ ends. In some cases, different reading frames or alternative translation start sites can also produce distinct proteins from the same gene.
These mechanisms increase protein diversity without increasing genome size and allow organisms to regulate protein function in a context-dependent manner.
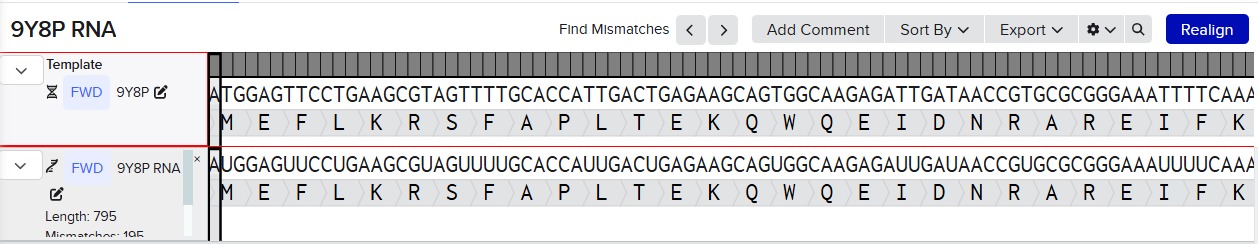
Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!! See example below. Special note that all “T” were transcribed into “U” and that the 3-nt codon represents 1-AA.
Alignment RNA and DNA:
PART 4: Prepare a Twist DNA Synthesis Order
4.1. Create a Twist account and a Benchling account
Already created.
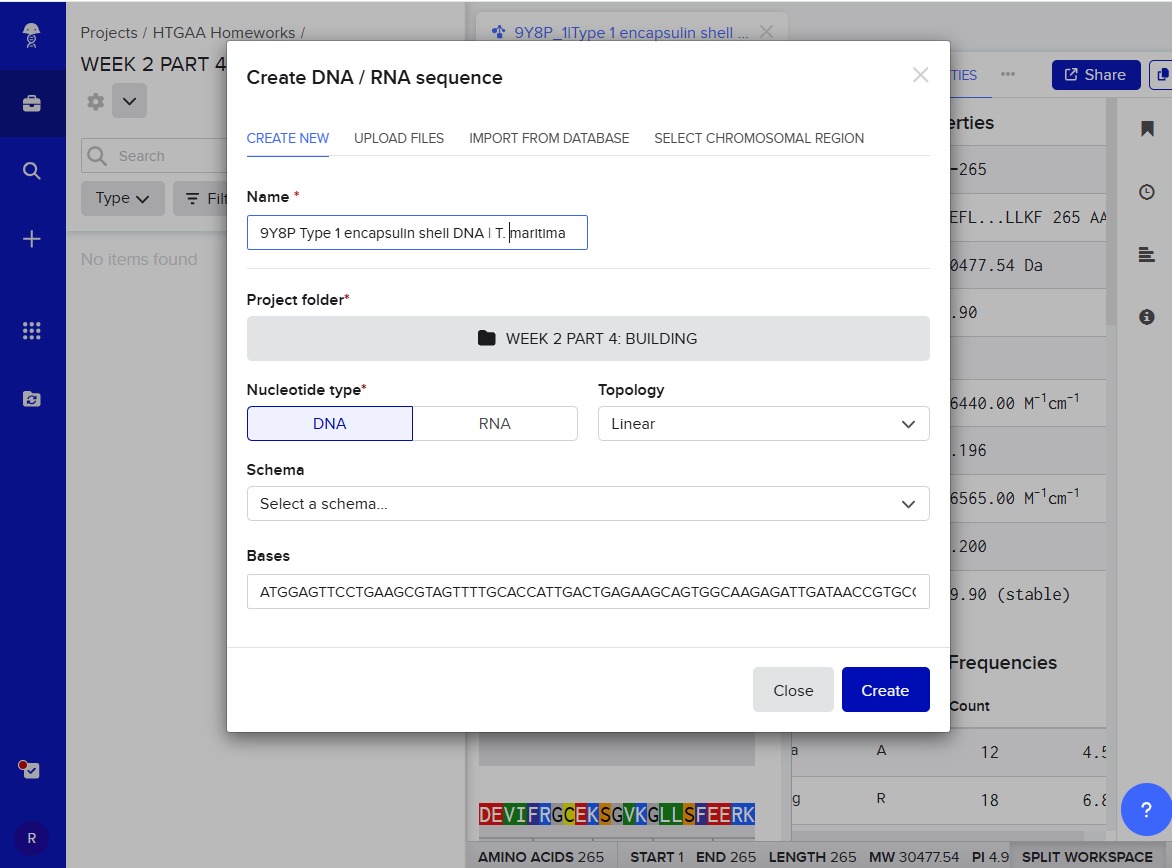
4.2. Build Your DNA Insert Sequence
The first step was to import the optimized DNA sequence for the encapsulin:
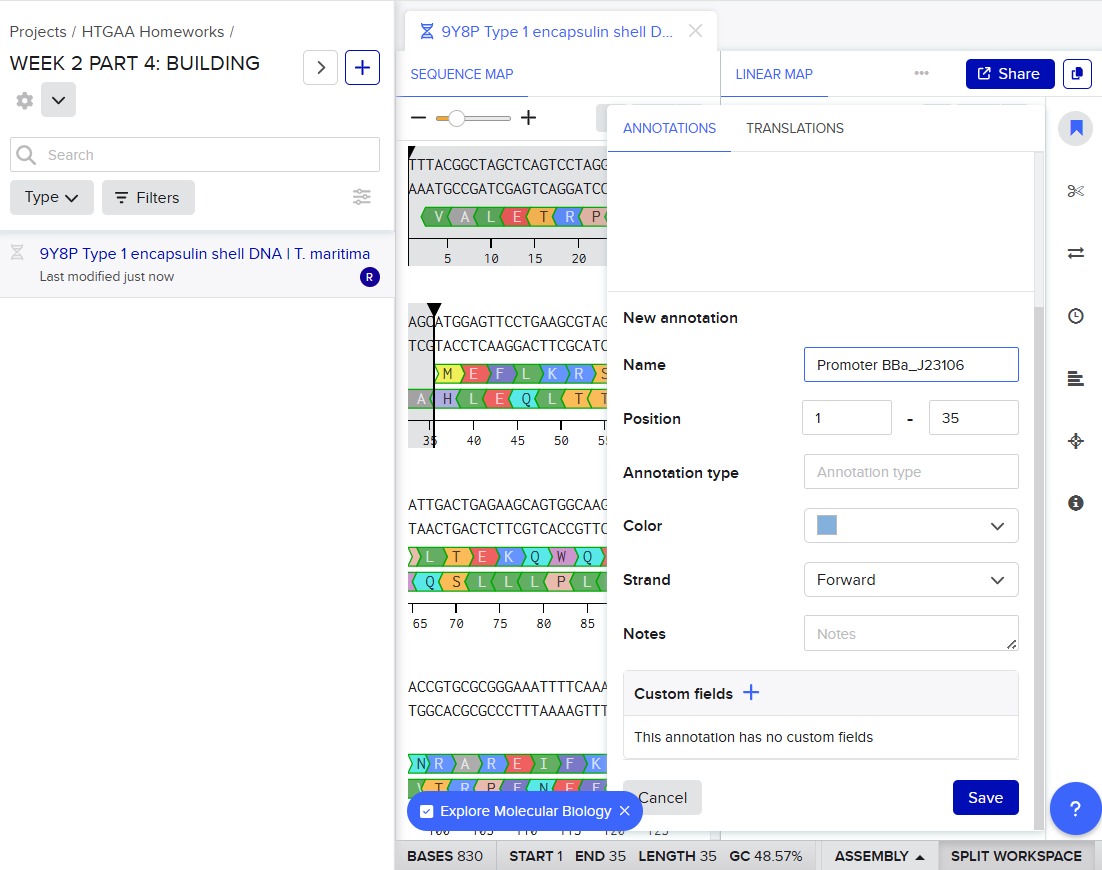
Next, I added the required genetic elements from the HGTAA website, including promoters, RBS, His-tags, and terminators. Each element was annotated with different colors for clarity:
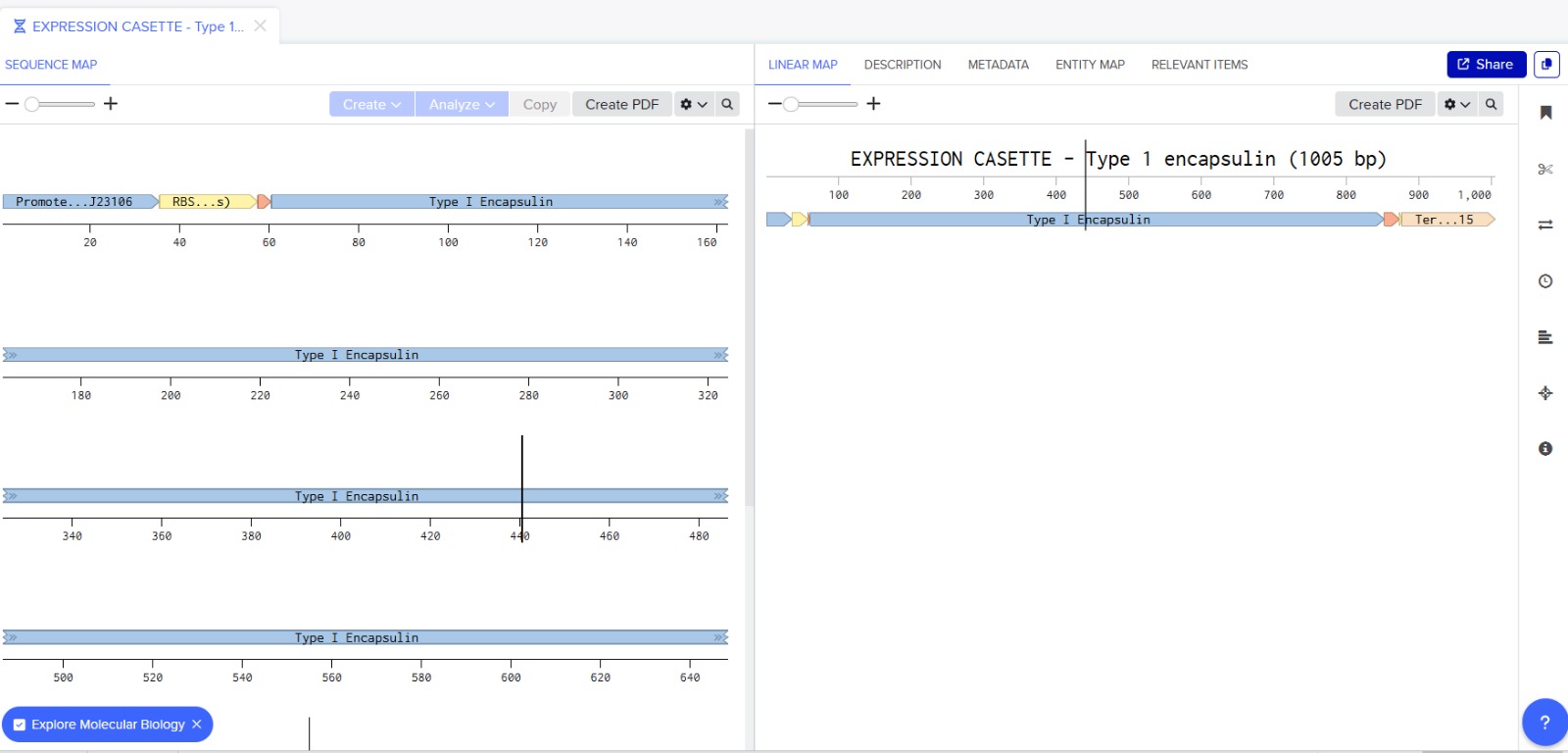
Before finalizing the cassette, I added restriction enzyme sites at the ends of the sequence: EcoRI and BamHI. These enzymes do not cut any region of the plasmid used for cloning nor any part of the cassette.
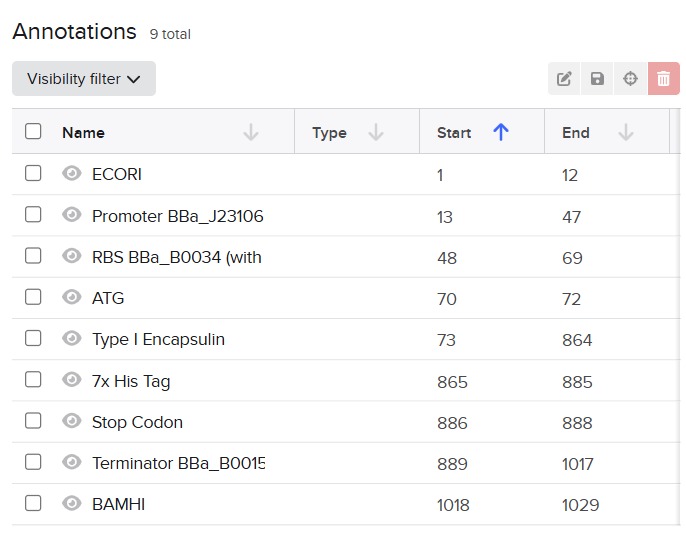
The final expression cassette can be accessed via Benchling: ([https://benchling.com/s/seq-LwbFj7EEX7O47AzP8oMk?m=slm-kN07oTYhfDiiAiZL0IJa])
4.3. On Twist, Select The “Genes” Option
To begin the synthesis process, select the Genes option in Twist:
4.4. Select “Clonal Genes” option
The Clonal Genes option was selected to allow ordered synthesis and cloning.
4.5. Import your sequence
The sequence created in Benchling was successfully imported into Twist for ordering.
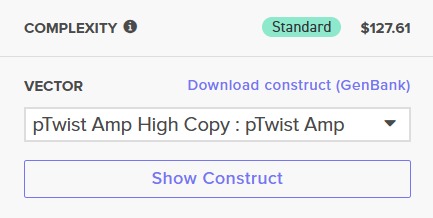
4.6. Choose Your Vector
After selecting the desired cloning vector (pTwist Amp High Copy), Twist provides a visualization of the construct along with the price estimate:
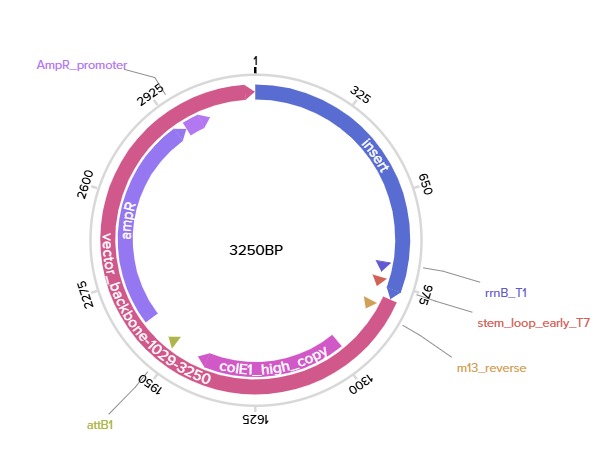
Finally, the GenBank (.gb) file was downloaded and opened in Benchling for further review:
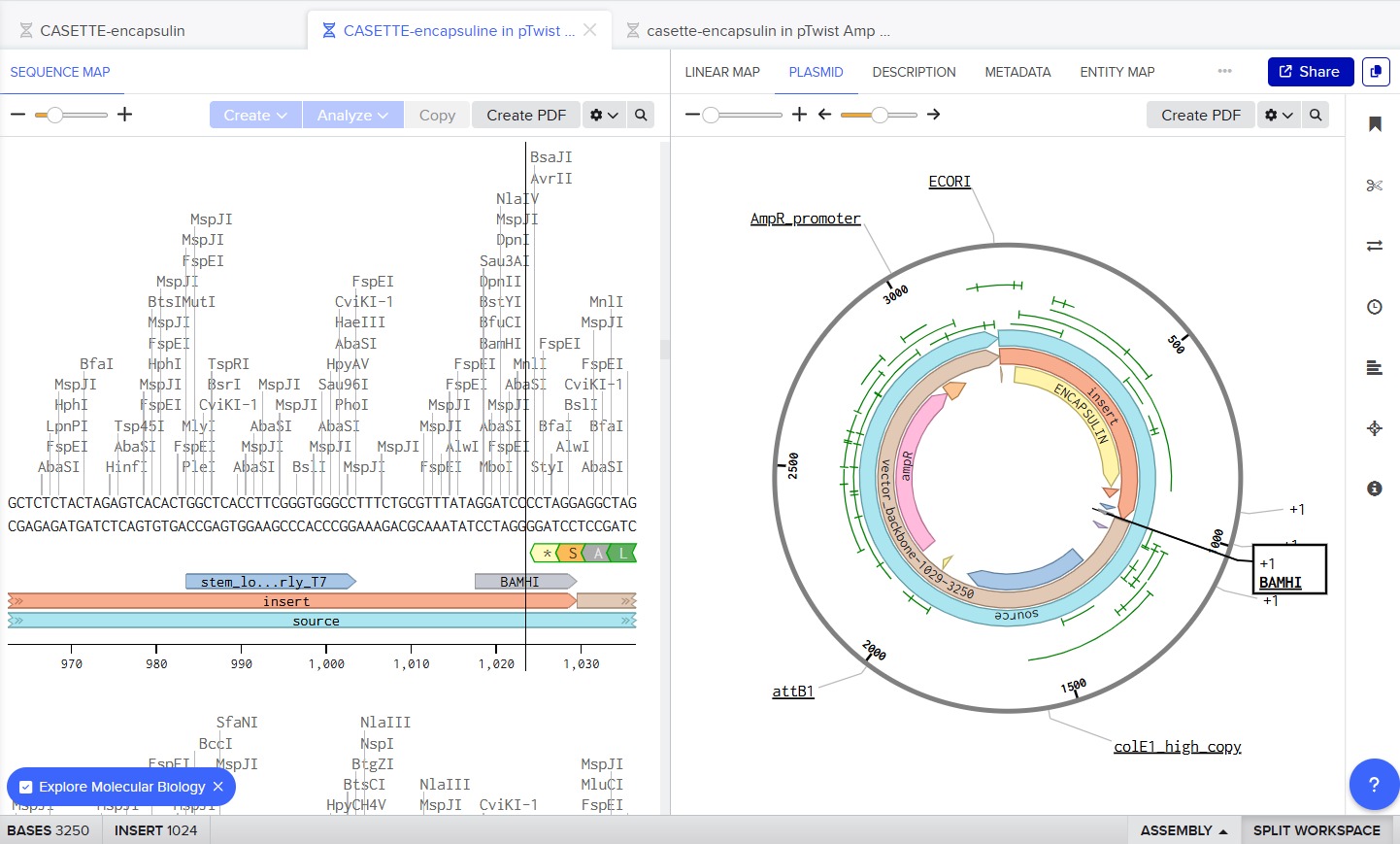
PART 5: DNA Read, Write, and Edit
5.1 DNA Read
(i) What DNA would you want to sequence and why?
I would like to sequence the genomes of new or previously uncharacterized organisms. The aim is to understand how life adapts to different environments and to uncover novel biological mechanisms. A clear example is sequencing the genomes of extremophiles living under high salinity conditions, which can reveal unique genetic adaptations for survival in harsh environments.
Sequencing these genomes can also identify new enzymes, proteins, and biomolecules with high potential for biotechnology, medicine, and industrial applications. Exploring previously unknown organisms expands our understanding of biodiversity and offers valuable resources for scientific and commercial use.
(ii) Sequencing Technology
Chosen Technologies:
- Illumina (second-generation, short-read sequencing) for high-accuracy genome analysis.
- Oxford Nanopore (third-generation, long-read sequencing) to capture complex genomic structures and long repetitive regions.
Generation:
- Illumina: second-generation, massively parallel short-read sequencing.
- Oxford Nanopore: third-generation, single-molecule long-read sequencing.
Input and Preparation:
- Extract genomic DNA from the organism.
- Fragment DNA into smaller pieces (for Illumina; optional for Nanopore).
- Ligate adapters for sequencing.
- PCR amplification (for Illumina; optional for Nanopore).
Essential Steps / Base Calling:
- Illumina: DNA fragments are amplified on a flow cell, fluorescent nucleotides are incorporated one by one, and base identity is determined by imaging fluorescence signals.
- Oxford Nanopore: Single DNA molecules pass through nanopores, changes in ionic current are measured, and base calling algorithms decode the sequence from the signal.
Output:
- Raw nucleotide sequences (FASTQ files) with quality scores, which can be assembled into complete genomes and analyzed for novel genes or adaptations.
5.2 DNA Write
(i) What DNA would you want to synthesize and why?
I would synthesize an expression cassette encoding the enzymes responsible for siderophore production in bacteria.
Siderophores are small, high-affinity iron-chelating molecules secreted by many soil bacteria. They bind Fe³⁺ and increase iron bioavailability for both microbes and plants. Because iron is often limited in agricultural soils, enhancing siderophore production could:
- Improve iron uptake in crops
- Promote plant growth and yield
- Reduce dependency on synthetic fertilizers
- Support sustainable and environmentally friendly agriculture
The expression cassette would include:
- A strong and regulated bacterial promoter
- Ribosome binding site (RBS)
- Coding sequences for siderophore biosynthesis enzymes (e.g., enzymes involved in nonribosomal peptide synthesis or other siderophore pathways)
- Transcriptional terminator
- Optional selectable marker
General structure:
Promoter – RBS – Siderophore biosynthesis genes – Terminator
The cassette could be cloned into a plasmid for high expression or integrated into the bacterial genome for stable, long-term expression. Codon optimization would be performed based on the selected bacterial host (E.coli).
(ii) DNA Synthesis Technology
Chosen Technology: Commercial gene synthesis combined with modular DNA assembly
To construct the expression cassette, I would use commercial gene synthesis services to synthesize either the complete construct or modular fragments.
Essential Steps:
In silico design
- Design the full cassette using software (e.g., Benchling or SnapGene).
- Optimize codon usage for the chosen bacterial host.
- Remove unwanted restriction sites and secondary structures.
Commercial DNA synthesis
- Order the optimized gene fragments.
- DNA is synthesized using automated phosphoramidite chemistry.
Assembly of fragments
- Gibson Assembly or Golden Gate cloning to assemble multi-gene constructs.
- Insert into an expression plasmid backbone.
Transformation and validation
- Transform into bacterial host cells.
- Confirm sequence by Sanger sequencing.
- Evaluate siderophore production using iron-chelation or colorimetric assays.
Limitations:
- Large biosynthetic clusters may require multi-step assembly.
- Expression may impose metabolic burden on host cells.
- Regulatory tuning may be required to balance growth and production.
- Cost increases with construct length and complexity.
5.3 DNA Edit
(i) What DNA would you want to edit and why?
I would edit the DNA sequence encoding the protein encapsulin to better understand how its structure influences adhesion to specific molecules.
Encapsulins are self-assembling protein nanocompartments found in bacteria and archaea. They form cage-like structures capable of encapsulating enzymes or interacting with target molecules. By modifying specific amino acids located on the surface, I could:
- Alter surface charge properties
- Modify binding affinity to selected molecules
- Improve structural stability
- Investigate structure–function relationships
Targeted mutations, such as single amino acid substitutions, would allow systematic evaluation of how structural features affect molecular adhesion.
(ii) What technology or technologies would you use and why?
Chosen Technologies:
- Site-directed mutagenesis (for plasmid-based expression systems)
- CRISPR-Cas9 or base editing (for genome-based editing)
Because the goal is precise modification of specific amino acids, high-accuracy editing methods are required.
Option 1: Site-Directed Mutagenesis (Plasmid-Based Editing)
If the encapsulin gene is cloned into an expression plasmid:
- Clone encapsulin gene into an expression vector.
- Design primers containing the desired nucleotide mutation.
- Perform PCR amplification incorporating the mutation.
- Remove template DNA using restriction digestion.
- Transform bacteria.
- Confirm mutation via DNA sequencing.
Advantages:
- High precision
- Efficient for point mutations
- No genomic double-strand breaks
- Suitable for structure–function studies
Option 2: CRISPR-Cas9 Genome Editing
If editing the native encapsulin gene in the bacterial genome:
- Design guide RNA targeting the encapsulin gene.
- Introduce Cas9 and guide RNA into the cells.
- Provide a donor DNA template containing the desired mutation.
- Repair occurs through homology-directed repair (HDR).
Required Components:
- Guide RNA (for CRISPR-based editing)
- Cas9 or base editor system
- Donor DNA template (for HDR-based edits)
- Delivery system (plasmid transformation or electroporation)
- Sequencing-based validation
Limitations:
- Off-target effects in CRISPR systems
- Variable editing efficiency depending on organism
- Mutations may disrupt protein folding
- Functional assays required to evaluate adhesion changes
References
Hoose, A., Vellacott, R., Storch, M., et al. (2023). DNA synthesis technologies to close the gene writing gap. Nature Reviews Chemistry, 7(3), 145–161. https://doi.org/10.1038/s41570-022-00456-9
Shendure, J., Balasubramanian, S., Church, G., et al. (2017). DNA sequencing at 40: Past, present, and future. Nature, 550(7676), 345–353. https://doi.org/10.1038/nature24286
Wannier, T., et al. (2021). Recombineering and MAGE. Nature Reviews Methods Primers, 1, 40. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9083505/
Wang, H., Doudna, J., et al. (2020). CRISPR technology: A decade of genome editing is only the beginning. Science, 367(6475), 1249–1250. https://www.science.org/doi/10.1126/science.add8643
HOMEWORK FROM WEEK 1
Homework questions from Prof. Jacobson
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
DNA polymerase has an intrinsic error rate of roughly one mistake per million nucleotides incorporated. Given that the human genome contains approximately 3.2 billion base pairs—equivalent to about 6.4 billion nucleotides in a diploid cell—this error rate would theoretically result in around 6,400 replication errors per cell division, or about half that number in a haploid genome. Such a frequency suggests a substantial risk for mutations that could potentially be inherited by offspring. However, biological systems have evolved multiple mechanisms to enhance the accuracy of DNA replication. Over the past century, these fidelity-enhancing processes have been extensively studied. One example is the MutS homolog 1 (MSH1/MutS-1) protein, which recognizes and binds to mismatched or improperly paired DNA sequences. This mismatch-repair mechanism serves as an additional layer of quality control, significantly increasing the fidelity of de novo DNA synthesis. (Carr et al., 2004)
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
An average human protein is approximately 300 amino acids long. Although there are (20^{300}) possible amino-acid sequences of this length, the number of DNA sequences that can encode a specific protein is determined by the degeneracy of the genetic code. Because most amino acids are encoded by multiple synonymous codons (about 3 on average), a single protein sequence can theoretically be encoded by roughly (3^{300}) different DNA sequences.
Despite this large number, most of these sequences do not function efficiently in cells. Factors such as codon usage bias, which affects translation speed, mRNA stability, translational accuracy, mRNA secondary structure, and regulatory or genomic constraints significantly restrict which nucleotide sequences can be successfully transcribed and translated. As a result, only a limited subset of possible DNA sequences are biologically functional.
Homework questions from Dr. LeProust
What’s the most commonly used method for oligo synthesis currently?
The most commonly used method for oligonucleotide synthesis is solid-phase chemical synthesis using phosphoramidite chemistry. In this approach, nucleotides are added one at a time to a growing DNA chain that is anchored to a solid support. Each synthesis cycle involves nucleotide coupling, capping of unreacted ends, and oxidation, allowing precise control over the sequence being synthesized (Kosuri & Chuch, 2014).
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Because the coupling efficiency in each synthesis cycle is less than 100%. As the number of cycles increases, small inefficiencies accumulate, resulting in a high proportion of truncated or incorrect sequences. Additionally, side reactions and chemical damage to the growing oligo further reduce yield and fidelity as length increases.
Why can’t you make a 2000bp gene via direct oligo synthesis?
A 2000 base-pair gene cannot be synthesized directly because the cumulative error rate and truncation probability become extremely high over thousands of synthesis cycles. This leads to very low yields of full-length, error-free DNA, making direct synthesis impractical. Instead, long genes are typically assembled from shorter, high-quality oligonucleotides using enzymatic methods such as PCR-based assembly or ligation.
Homework question from George Church
Using Google & Prof. Church’s slide #4, What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
In animals, essential amino acids are those that cannot be synthesized de novo and must be obtained from the diet. Across animals, the 10 commonly cited essential amino acids are:
- Histidine
- Isoleucine
- Leucine
- Lysine
- Methionine
- Phenylalanine
- Threonine
- Tryptophan
- Valine
- Arginine (especially during growth)
The lysine contingency refers to a bioengineered safety mechanism in which organisms are modified to depend on an external supply of lysine for survival. Because lysine is an essential amino acid in all animals, this dependency creates a strong biological containment strategy: the engineered organism cannot survive or proliferate outside controlled environments lacking lysine.
Understanding that animals depend on multiple essential amino acids reinforces the strength of the lysine contingency. It highlights how leveraging fundamental metabolic constraints can improve biosafety, particularly for in vivo applications, by reducing the risk of unintended survival or spread of engineered organisms.