I’m Renzo Condori, a bioengineering student from Peru working at the intersection of synthetic biology, protein engineering, and computational modeling.
My goal is to design biological technologies that make healthcare more accessible in low-resource settings—especially diagnostics and protein-based therapeutics for neglected diseases in Latin America.
I’ve worked across wet lab, computation, and field settings: enzyme engineering, molecular dynamics simulations, and co-designing solutions with local communities. I enjoy translating biology into deployable tools that move beyond the lab.
At HTGAA, I want to prototype scalable biofabrication and synthetic biology systems that can become real-world products.
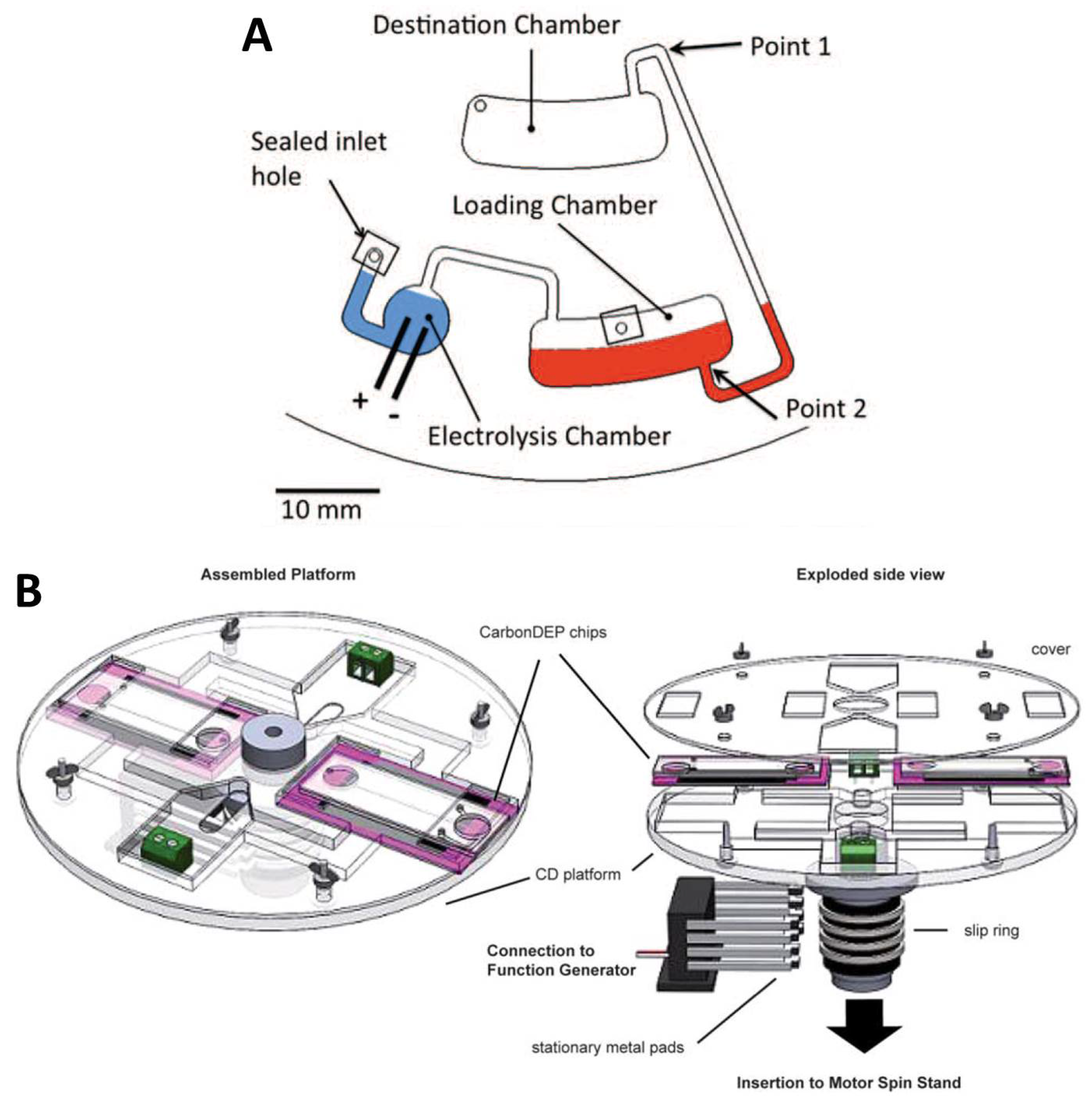
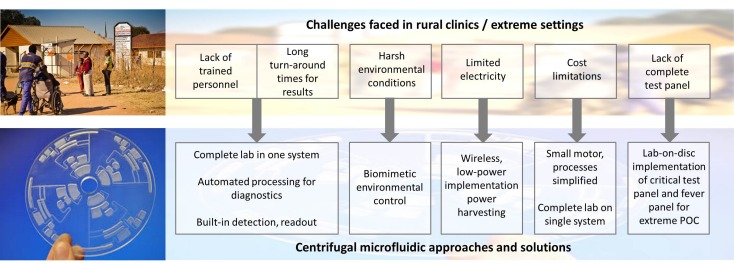
Early diagnostics trough microfluidics In current medical practice, prevention and early intervention are among the most effective strategies for improving health outcomes. However, in underserved regions such as the Peruvian Amazon, limited access to primary healthcare often leads to an accumulation of disease burden. Patients frequently seek medical attention only when symptoms have progressed to advanced or critical stages. In the region of Loreto, Perú, for example, widespread dependence on untreated river water exposes communities to persistent threats from pathogens, which can contribute to chronic malnutrition in children under five. Additionally, in remote and rural areas, the advanced laboratory infrastructure is scarce. In these contexts, delayed diagnosis is not just an inconvenience—it can be the difference between early treatment and a life-threatening outcome. Fast diagnostics save time, resources, and, most importantly, lives.
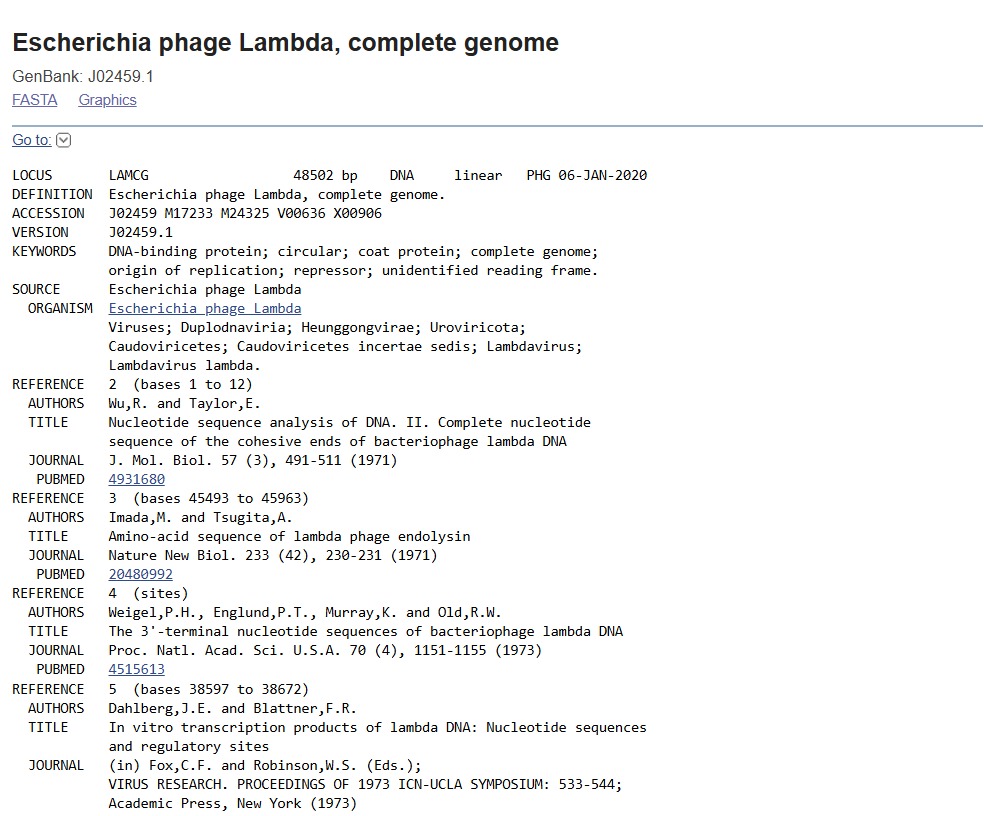
PART 1: Benchling & In-silico Gel Art First, I googled for the accession number of the Lambda phage full genome DNA sequence. I put “lambda dna sequence ncbi” in the google search box. The first result prompted this:
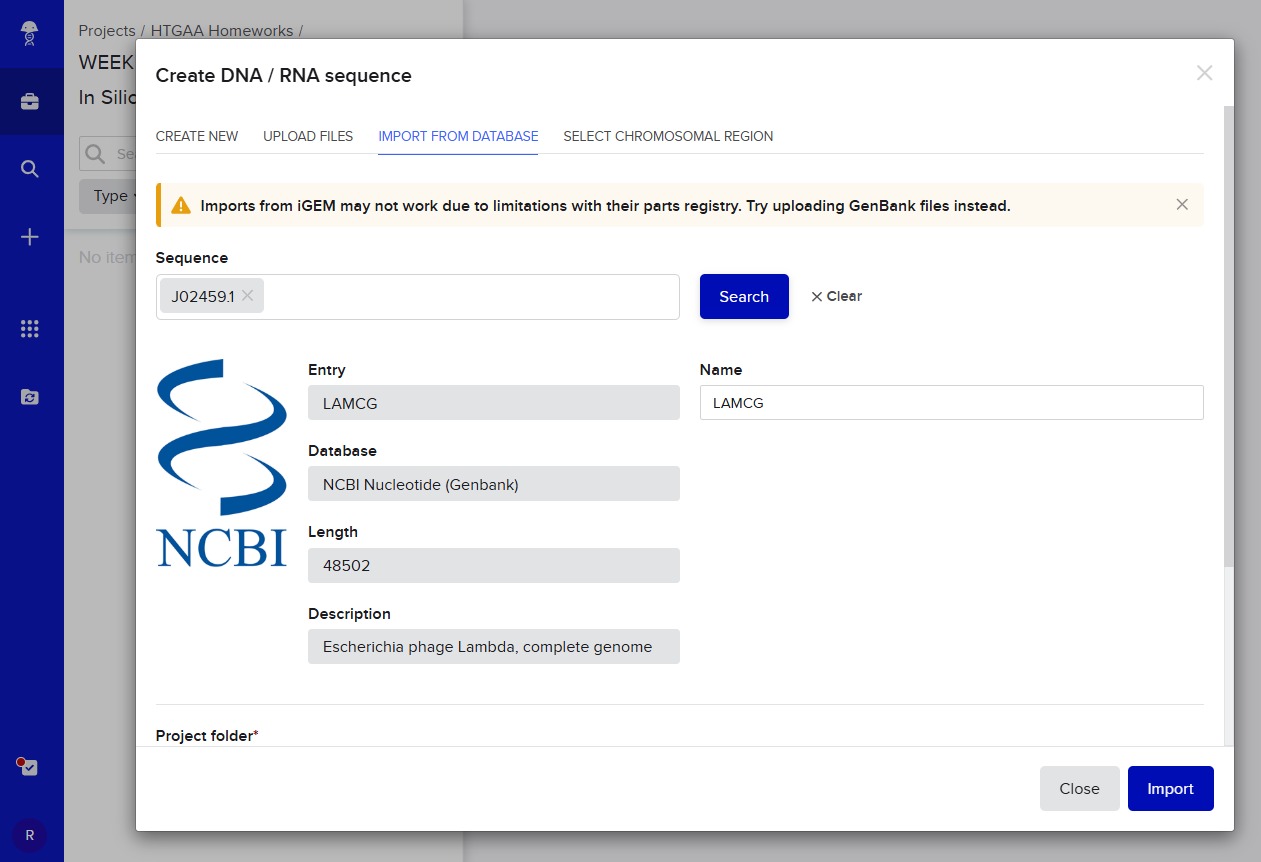
After this, I signed into Benchling and created a new project named “HTGAA HOMEWORKS/WEEK 2 - PART 1: Benchling & In Silico Gel Art”. There I imported Lambda DNA using the accession number: GenBank: J02459.1
Subsections of Homework
Week 1 HW: Principles and Practices
1. Early diagnostics trough microfluidics
In current medical practice, prevention and early intervention are among the most effective strategies for improving health outcomes. However, in underserved regions such as the Peruvian Amazon, limited access to primary healthcare often leads to an accumulation of disease burden. Patients frequently seek medical attention only when symptoms have progressed to advanced or critical stages. In the region of Loreto, Perú, for example, widespread dependence on untreated river water exposes communities to persistent threats from pathogens, which can contribute to chronic malnutrition in children under five. Additionally, in remote and rural areas, the advanced laboratory infrastructure is scarce. In these contexts, delayed diagnosis is not just an inconvenience—it can be the difference between early treatment and a life-threatening outcome. Fast diagnostics save time, resources, and, most importantly, lives.
Motivated by this reality, my project focuses on developing field-deployable diagnostic systems using centrifugal microfluidics, also known as Lab-on-a-Disc technology. This approach has the potential to bring essential diagnostic capabilities directly to communities that are far from centralized laboratories. A single disc can be designed to perform a critical panel of tests, including diagnostics for malaria, tuberculosis (TB), hepatitis, and waterborne pathogens, enabling earlier intervention for diseases that significantly impact maternal and child mortality.
To make diagnostics viable in extreme and resource-limited environments, this platform leverages centrifugal pseudo-forces to replace bulky and expensive laboratory equipment. By simply rotating a disc, the system can function as a centrifuge, mixer, and mechanical lyser within a compact format. Importantly, this design is well suited for regions with unreliable electricity, as it requires only a small spindle motor and can even be powered wirelessly using standards such as Qi induction charging.
For molecular detection, the platform can integrate Loop-Mediated Isothermal Amplification (LAMP), a technique capable of amplifying detectable DNA products in under 40 minutes at a constant temperature. This eliminates the need for complex thermal cycling and significantly reduces system complexity. Finally, when combined with smartphone-based imaging and analysis, the Lab-on-a-Disc platform offers a sensitive, specific, user-friendly, rapid, and practical pathway so that communities often left behind by modern healthcare systems are no longer excluded from timely and lifesaving diagnostics.
References
Smith, S., Mager, D., Perebikovsky, A., Shamloo, E., Kinahan, D., Mishra, R., Torres Delgado, S. M., Kido, H., Saha, S., Ducrée, J., Madou, M., Land, K., & Korvink, J. G. (2016). CD-Based Microfluidics for Primary Care in Extreme Point-of-Care Settings. Micromachines, 7(2), 22. https://doi.org/10.3390/mi7020022
Wu, C., Cheng, J., Zhang, Y., & Yao, P. (2025). A LAMP Detection System Based on a Microfluidic Chip for Pyricularia grisea. Sensors, 25(8), 2511. https://doi.org/10.3390/s25082511
Zhang, J., Fu, Y., Fong, C. Y., Hua, H., Li, W., & Khoo, B. L. (2025). Advancements in microfluidic technology for rapid bacterial detection and inflammation-driven diseases. Lab on a Chip, 25(16), 3348–3375. https://doi.org/10.1039/d4lc00795f
2. Governance and Policy Goals
The Lab-on-a-Disc diagnostic platform is designed for deployment in remote, high-biodiversity regions such as the Peruvian Amazon, where healthcare infrastructure is limited and ecological and cultural contexts differ significantly from those of centralized laboratories. Governance goals must therefore address not only safety and performance but also the ecological, social, and interpretive risks that may arise when advanced diagnostic technologies are introduced into sensitive environments.
To guide safe and responsible use, five refined governance goals are aligned with the four overarching principles: Goals 1 and 2 support Safety and Non-Malfeasance; Goal 3 supports Responsible Use; Goal 4 supports Trust and Transparency; and Goal 5 supports Equity and Access. This hierarchy shows how each goal translates broader principles into practical guidance.
Governance Goal 1: Prevent Clinical and Interpretive Harm in Low-Resource Settings
In environments where diagnostic results may directly influence treatment decisions without access to confirmatory testing, incorrect or misunderstood results can cause immediate harm.
Sub-goals:
Reduce the risk of false positives or negatives leading to inappropriate treatment, delayed care, or unnecessary referral.
Ensure results are interpretable by non-specialist users, minimizing reliance on implicit technical knowledge.
Governance Goal 2: Minimize Ecological and Biological Spillover Risks
Deploying molecular diagnostics in regions of high biodiversity raises concerns beyond human health, including accidental environmental exposure and biological contamination.
Sub-goals:
Prevent the release of amplified DNA, reagents, or biological samples into surrounding ecosystems.
Ensure safe containment and disposal of consumables in settings without formal biohazard waste infrastructure.
Avoid unintended ecological interactions, particularly in environments with poorly characterized microbial diversity.
Governance Goal 3: Prevent Misuse and Function Creep in Field Diagnostics
Even benign diagnostic tools can be repurposed or extended beyond their original intent, especially when deployed outside formal regulatory oversight.
Sub-goals:
Limit use of the platform to predefined diagnostic assays, preventing unauthorized adaptation for unvalidated testing.
Reduce the risk of informal surveillance, coercive testing, or testing without informed consent.
Prevent the normalization of diagnostic data collection without appropriate ethical or institutional oversight.
Governance Goal 4: Ensure Cultural Legitimacy and Trust in Local Contexts
Introducing unfamiliar technologies into marginalized communities can generate distrust, resistance, or misuse if local values and perceptions are not respected.
Sub-goals:
Avoid deployment models that frame the technology as an external or extractive intervention.
Ensure diagnostic practices align with local understandings of illness, care, and authority.
Support community engagement and transparency to prevent fear, stigma, or misinformation surrounding testing.
Governance Goal 5: Enable Equitable and Sustainable Access Without Dependency
Governance must ensure that access to diagnostics does not create long-term dependency on external suppliers or short-lived pilot programs.
Sub-goals:
Prevent reliance on proprietary consumables that cannot be locally sourced or substituted.
Support maintenance, repair, and adaptation at the local level.
Ensure that withdrawal of the technology does not leave communities worse off than before deployment.
References
National Academies of Sciences, Engineering, and Medicine. (2006). Globalization, biosecurity, and the future of the life sciences. National Academies Press.
United Nations Office for Disarmament Affairs. (2022). Global guidance framework for the responsible use of the life sciences: Mitigating biorisks and governing dual-use research. United Nations.
3. Governance Actions
Based on the governance goals outlined above, the following actions propose concrete interventions at different points in the development and deployment of the Lab-on-a-Disc diagnostic platform. Each action targets a specific failure mode while preserving the technology’s ability to deliver public health benefits in remote, high-biodiversity settings.
Governance Action 1: Assay Locking and Use-Restriction by Design
Purpose At present, microfluidic diagnostic platforms are often designed to be modular and adaptable. While this flexibility supports innovation, it also enables unvalidated or inappropriate use in the field. This action proposes restricting field-deployed devices to a predefined set of validated diagnostic assays.
Design
Embed physical or firmware-based constraints that limit the disc and reagents to specific, pre-approved assays (e.g., malaria, TB, hepatitis).
Require laboratory-level modification or authorization to expand assay capability.
Maintain an open design at the research stage, but enforce restrictions at the deployment stage, similar to how drones limit flight modes or altitudes for consumer users.
Assumptions
That misuse or function creep is more likely in the field than in controlled research environments.
That technical restrictions can meaningfully reduce misuse without preventing legitimate diagnostics.
Risks of Failure & Success
Restrictions may reduce adaptability to emerging pathogens.
Success could unintentionally slow emergency responses where rapid assay adaptation is needed.
Governance Action 2: Context-Specific Training and Certification for Field Use
Actors NGOs, public health agencies, academic partners, local health authorities
Purpose Currently, diagnostic tools are often deployed with minimal user training, assuming intuitive use. This action introduces a requirement for short, context-specific certification before field deployment.
Design
Develop localized training modules addressing sample handling, result interpretation, waste disposal, and cultural sensitivity.
Certification tied to device access, similar to operator licensing for drones or medical imaging equipment.
Training delivered in collaboration with local institutions to ensure cultural relevance.
Assumptions
That misuse and misinterpretation are more likely due to lack of contextual understanding than malicious intent.
That short, targeted training can significantly reduce error rates.
Risks of Failure & Success
Certification requirements could slow deployment in urgent situations.
Training programs may become symbolic rather than effective if poorly implemented.
Successful certification systems may unintentionally exclude informal healthcare providers who play key roles in remote regions.
Governance Action 3: Embedded Waste Containment and Disposal Protocols
Actors: Device designers, manufacturers, environmental health regulators
Purpose Standard biohazard disposal infrastructure is often absent in remote regions, increasing the risk of environmental contamination. This action proposes technical containment as a governance mechanism.
Design
Design disposable discs with sealed reaction chambers that remain closed after use.
Pair deployment with simple, low-tech disposal guidance suitable for rural settings, analogous to sharps containers in vaccination campaigns.
Assumptions
That ecological spillover risks are driven more by disposal practices than by the diagnostic process itself.
That technical containment can compensate for limited waste infrastructure.
Risks of Failure & Success
Increased manufacturing complexity and cost.
Improper disposal may still occur despite containment.
Widespread success could increase plastic waste if sustainability is not addressed in parallel.
Governance Action 4: Community Engagement and Consent-Based Deployment Models
Actors: NGOs, local governments, public health organizations, community leaders
Purpose Introducing unfamiliar diagnostic technologies without community engagement risks mistrust, resistance, or misuse. This action formalizes community involvement as a governance requirement rather than an optional add-on.
Design
Require community-level consultation before deployment.
Co-develop communication strategies explaining what the diagnostics can and cannot do.
Integrate informed consent practices adapted to local cultural norms, similar to participatory models used in environmental monitoring.
Assumptions
That cultural misalignment, rather than technical failure, is a major barrier to adoption.
That trust increases correct and ethical use.
Risks of Failure & Success
Engagement processes may slow deployment timelines.
Community leaders may not represent all stakeholders.
Successful engagement could create expectations of long-term support that exceed project resources.
A layered governance approach focused on Training and Certification, Embedded Waste Containment, and Community Engagement and Consent-Based Deployment should be prioritized, according to the above scoring. The most common failure modes in low-resource environments—misinterpretation of results, inappropriate handling of biological samples, and an excessive dependence on automated diagnostics—are directly addressed by training and certification. Deployment in high-biodiversity environments, where ecological spillover risks are significant and traditional biohazard disposal infrastructure is lacking, requires embedded waste containment. By ensuring that the technology is not viewed as an extractive or external intervention, community engagement promotes long-term sustainability, appropriate use, and trust. Despite offering robust protection against abuse, Assay Locking was not given priority as a primary mechanism because it may restrict adaptability in responding to emerging health threats. However, it remains a valuable complementary measure in high-risk or highly decentralized deployment contexts.
Uncertainties, Assumptions, and Trade-offs
Since training and community involvement may slow initial rollout, this approach accepts trade-offs between deployment speed and long-term legitimacy. Additionally, it makes the assumption that there is enough institutional capacity to support community consultation and certification programs. Political continuity, long-term funding, and scalability beyond pilot deployments are still uncertain, which emphasizes the need for adaptable, multi-layered governance as opposed to relying solely on one control mechanism.
In order to address health issues that stem from structural inequality and ecological vulnerability, this recommendation is mainly addressed to the Peruvian government, in particular public health and environmental authorities, as well as the United Nations, whose role in norm-setting, coordination, and support is crucial.
Reflection: Ethical Considerations and Governance Lessons from Week 1
Reflecting on this week’s lectures and discussions in How to Grow (Almost) Anything, one ethical concern that became particularly salient to me was the idea that engineering solutions can generate harm not through malicious intent but through misalignment with social, cultural, or ecological contexts. Prior to this week, I primarily associated ethical risk in biotechnology with dual-use or deliberate misuse. What was new to me was how easily harm can emerge from well-intentioned technologies when governance is treated as an afterthought rather than a design requirement.
In the context of my project, this raised concerns about how diagnostic technologies might alter decision-making in communities with limited access to follow-up care or how biological waste and amplified DNA could unintentionally interact with ecosystems characterized by high and poorly understood biodiversity. I also became more aware of the ethical implications of trust and authority: who is believed when a diagnostic result is produced and how that authority is perceived when the technology originates outside the community.
To address these concerns, I believe governance actions must extend beyond technical safeguards. Context-specific training, community engagement, and clear communication about the limitations of diagnostic tools are as important as analytical accuracy. Embedding governance into the lifecycle of the technology—from design to deployment—helps ensure that innovation supports, rather than overrides, local knowledge and autonomy.
This week reinforced for me that ethical biotechnology is not defined solely by what a technology can do, but by how, where, and for whom it is used, and that governance is a critical component of responsible bioengineering practice.
Week 2 HW: DNA Read, Write, and Edit
PART 1: Benchling & In-silico Gel Art
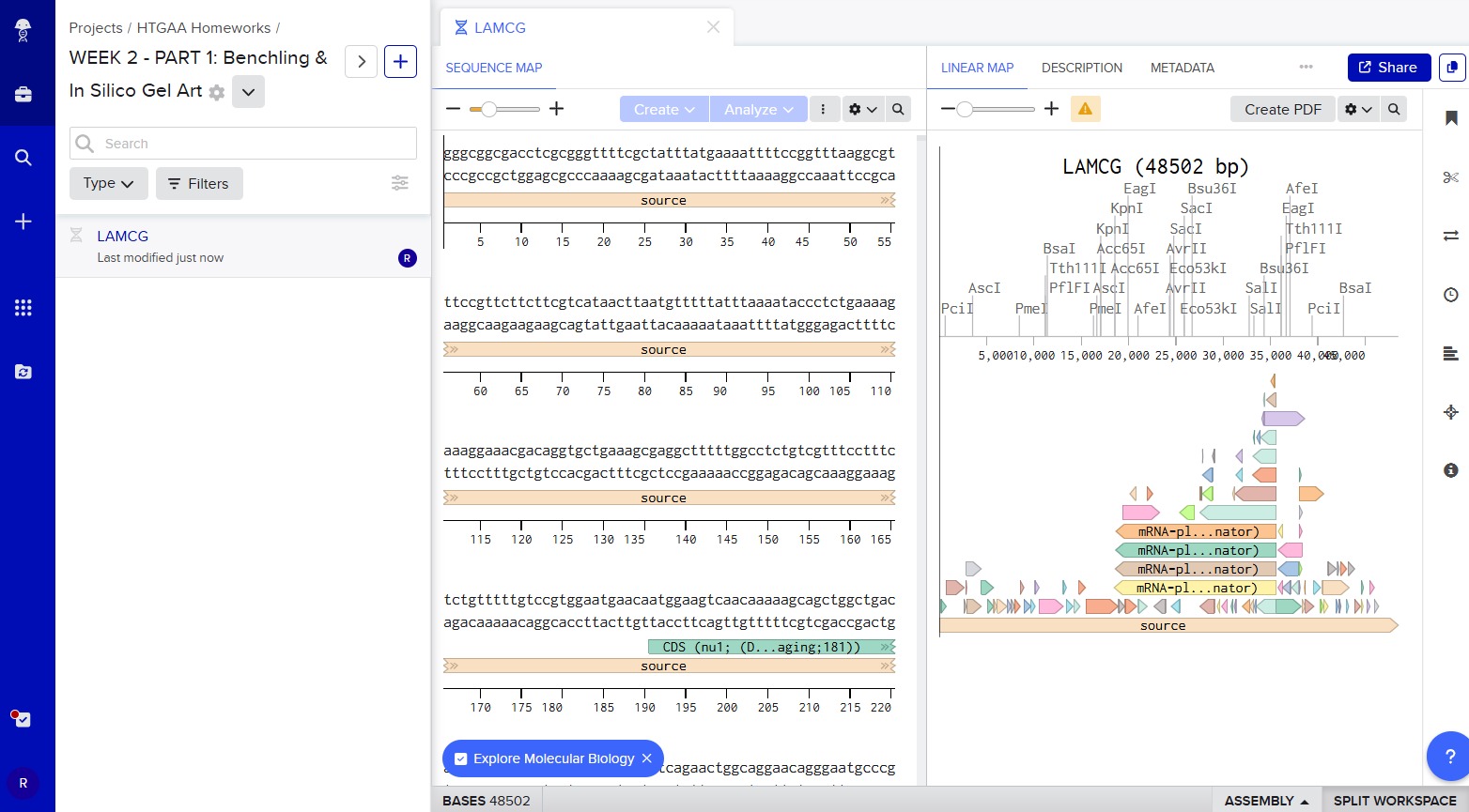
First, I googled for the accession number of the Lambda phage full genome DNA sequence. I put “lambda dna sequence ncbi” in the google search box. The first result prompted this:
After this, I signed into Benchling and created a new project named “HTGAA HOMEWORKS/WEEK 2 - PART 1: Benchling & In Silico Gel Art”. There I imported Lambda DNA using the accession number:
GenBank: J02459.1
Finally, we have everything to start with the task:
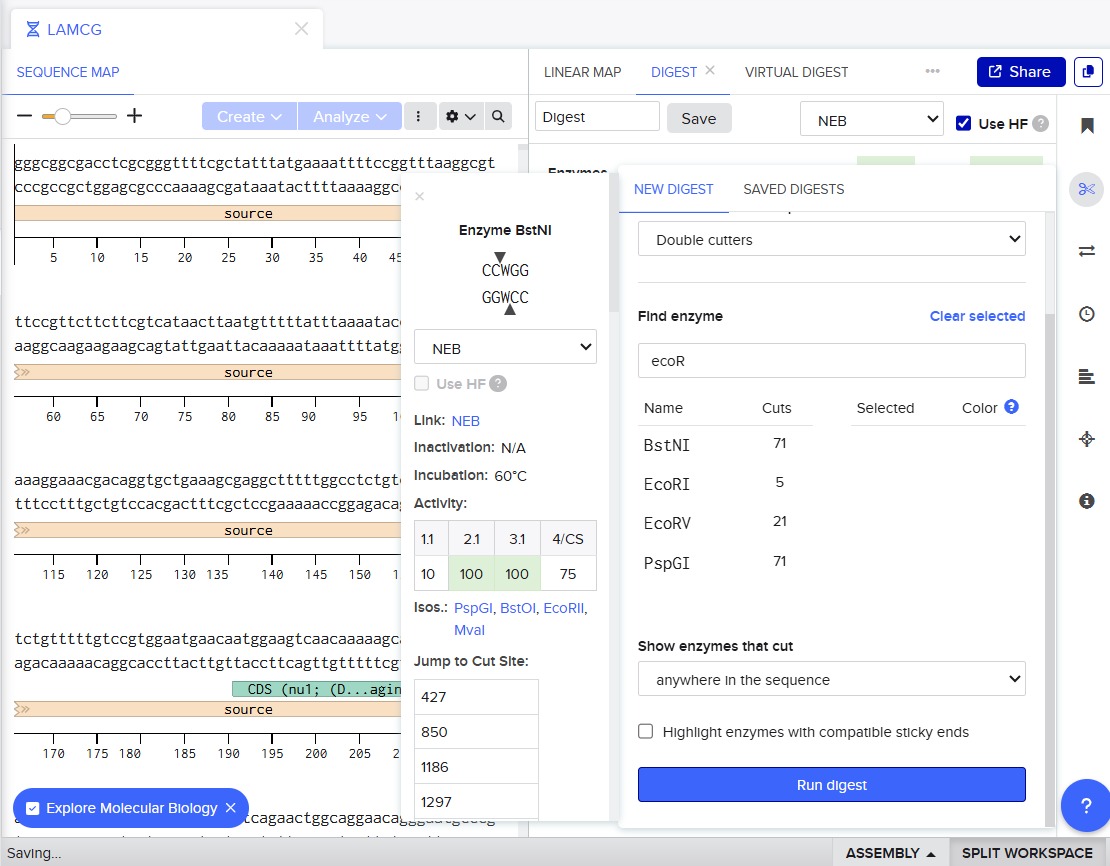
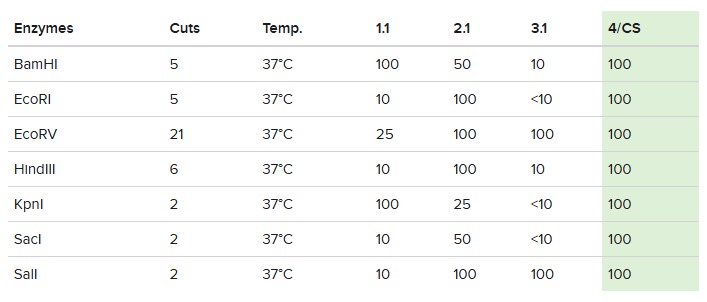
Simulate Restriction Enzyme Digestion with the following Enzymes:
EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, SalI
For this, we performed in-silico digestion by clicking on the scissors icon at the right vertical bar:
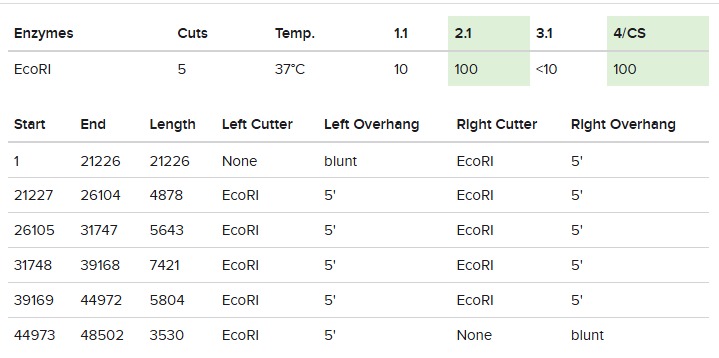
First we tested with only EcoRI:
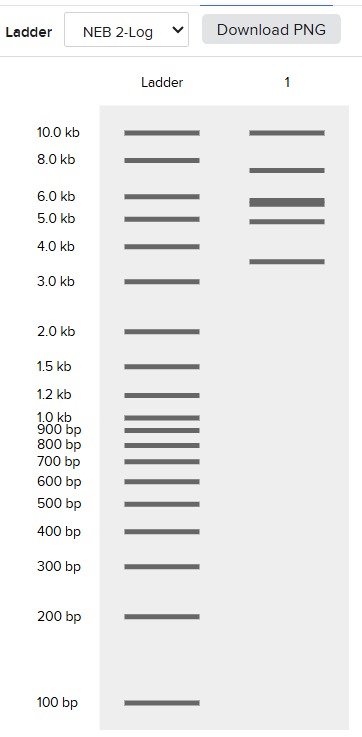
The final gel was this:
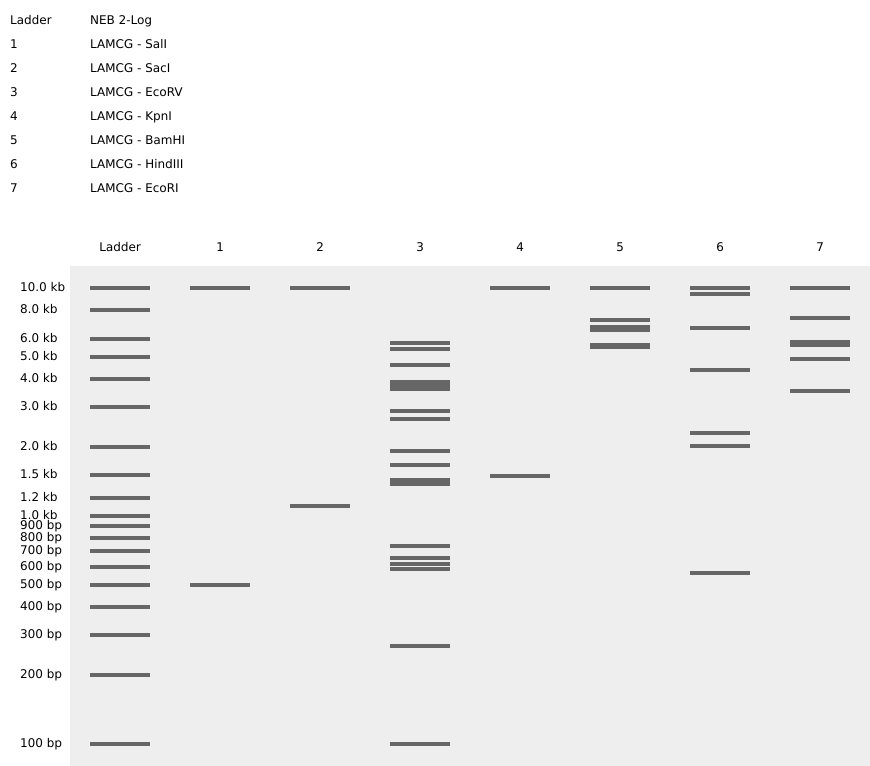
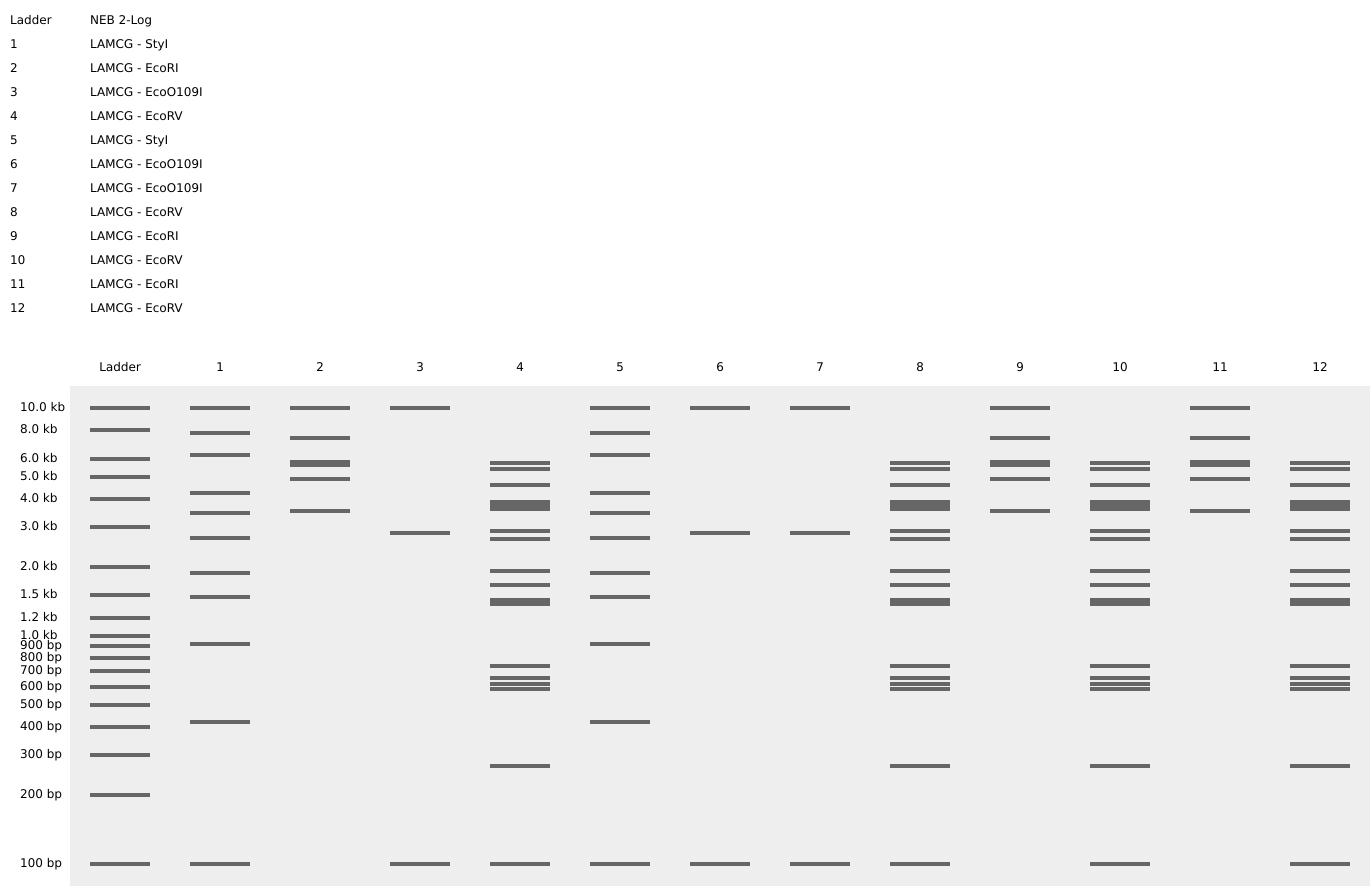
Following these steps, we obtained the final gel, including the rest of enzymes:
Each digestion product was ran on a separate lane in the virtual gel:
After some time testing different enzymes, I made the word “IgEm”.
PART 3: DNA Design Challenge
3.1. Choose your protein.
I chose the encapsulin shell protein from Thermotoga maritima because it is a self-assembling protein nanocage with strong potential in drug delivery and synthetic biology. Encapsulins form icosahedral structures composed of multiple identical subunits and can selectively encapsulate cargo proteins through short targeting peptides, making them highly programmable at the genetic level. Their robustness and modularity make them attractive platforms for engineering artificial nanocarriers.
However, despite their potential, encapsulins still present important limitations. Their high structural stability makes controlled cargo release difficult, which is a challenge for therapeutic applications. As bacterial proteins, they may also trigger immune responses in mammalian systems, and their long-term biocompatibility is not fully understood. In addition, modifying the shell surface for targeting or functionalization can disrupt assembly if not carefully designed. These constraints highlight that encapsulins are still an evolving technology rather than a fully optimized platform.
I was especially motivated to study encapsulins because they were first successfully obtained in Peru by a Peruvian research group last year. This encouraged me to explore their design and expression.
The sequence of the encapsulin shell protein (UniProt entry Q9WZP2) is:
Kwon, S., & Giessen, T. W. (2025). Engineering encapsulin nanocages for drug delivery. Materials Advances, 6, 6209–6220. https://doi.org/10.1039/D5MA00386E
Sutter, M., Boehringer, D., Gutmann, S., Günther, S., Prangishvili, D., Loessner, M. J., Stetter, K. O., Weber-Ban, E., & Ban, N. (2008). Structural basis of enzyme encapsulation into a bacterial nanocompartment. Nature Structural & Molecular Biology, 15(9), 939–947. https://doi.org/10.1038/nsmb.1473
Protein Data Bank. (2025). Cryo-EM structure of Thermotoga maritima encapsulin shell (PDB ID 9Y8P) [Data set]. RCSB PDB. https://doi.org/10.2210/pdb9Y8P/pdb :contentReference[oaicite:0]{index=0}
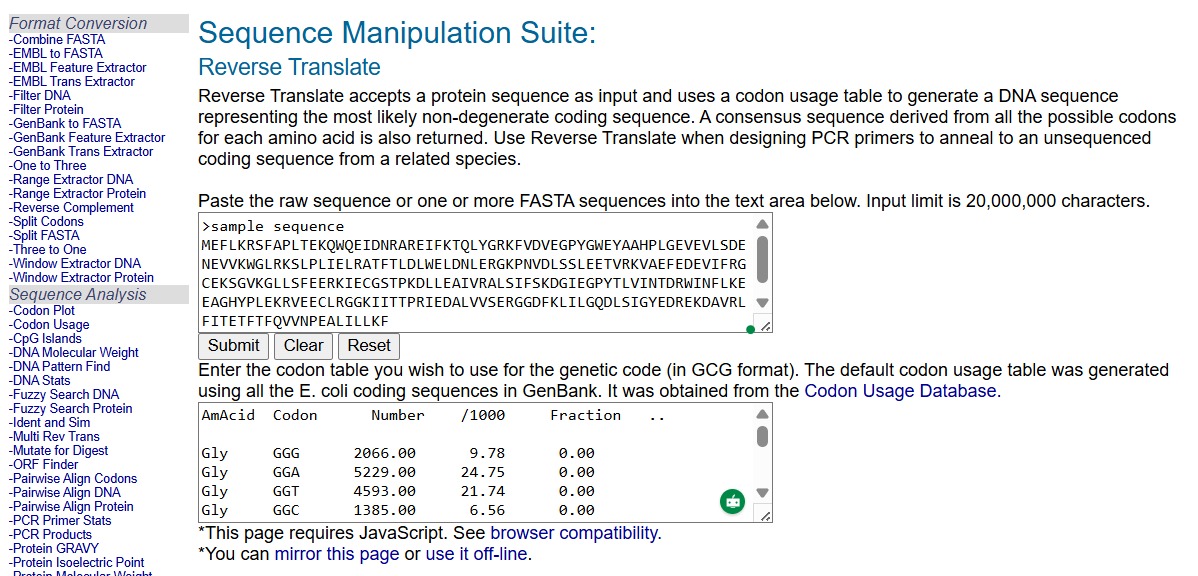
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
I reverse translated the aa sequence using the codon table from the Codon Usage Database ([https://www.kazusa.or.jp/codon/]).
I chose the first output, which is the predicted DNA sequence for the most likely codons for Bacillus Thurigiensis.
9Y8P_1|Type 1 encapsulin shell protein DNA sequence|Thermotoga maritima (2336)
atggaatttttaaaaagaagttttgcaccattaacagaaaaacaatggcaagaaattgataatagagcaagagaaatttttaaaacacaattatatggaagaaaatttgtagatgtagaaggaccatatggatgggaatatgcagcacatccattaggagaagtagaagtattaagtgatgaaaatgaagtagtaaaatggggattaagaaaaagtttaccattaattgaattaagagcaacatttacattagatttatgggaattagataatttagaaagaggaaaaccaaatgtagatttaagtagtttagaagaaacagtaagaaaagtagcagaatttgaagatgaagtaatttttagaggatgtgaaaaaagtggagtaaaaggattattaagttttgaagaaagaaaaattgaatgtggaagtacaccaaaagatttattagaagcaattgtaagagcattaagtatttttagtaaagatggaattgaaggaccatatacattagtaattaatacagatagatggattaattttttaaaagaagaagcaggacattatccattagaaaaaagagtagaagaatgtttaagaggaggaaaaattattacaacaccaagaattgaagatgcattagtagtaagtgaaagaggaggagattttaaattaattttaggacaagatttaagtattggatatgaagatagagaaaaagatgcagtaagattatttattacagaaacatttacatttcaagtagtaaatccagaagcattaattttattaaaattt
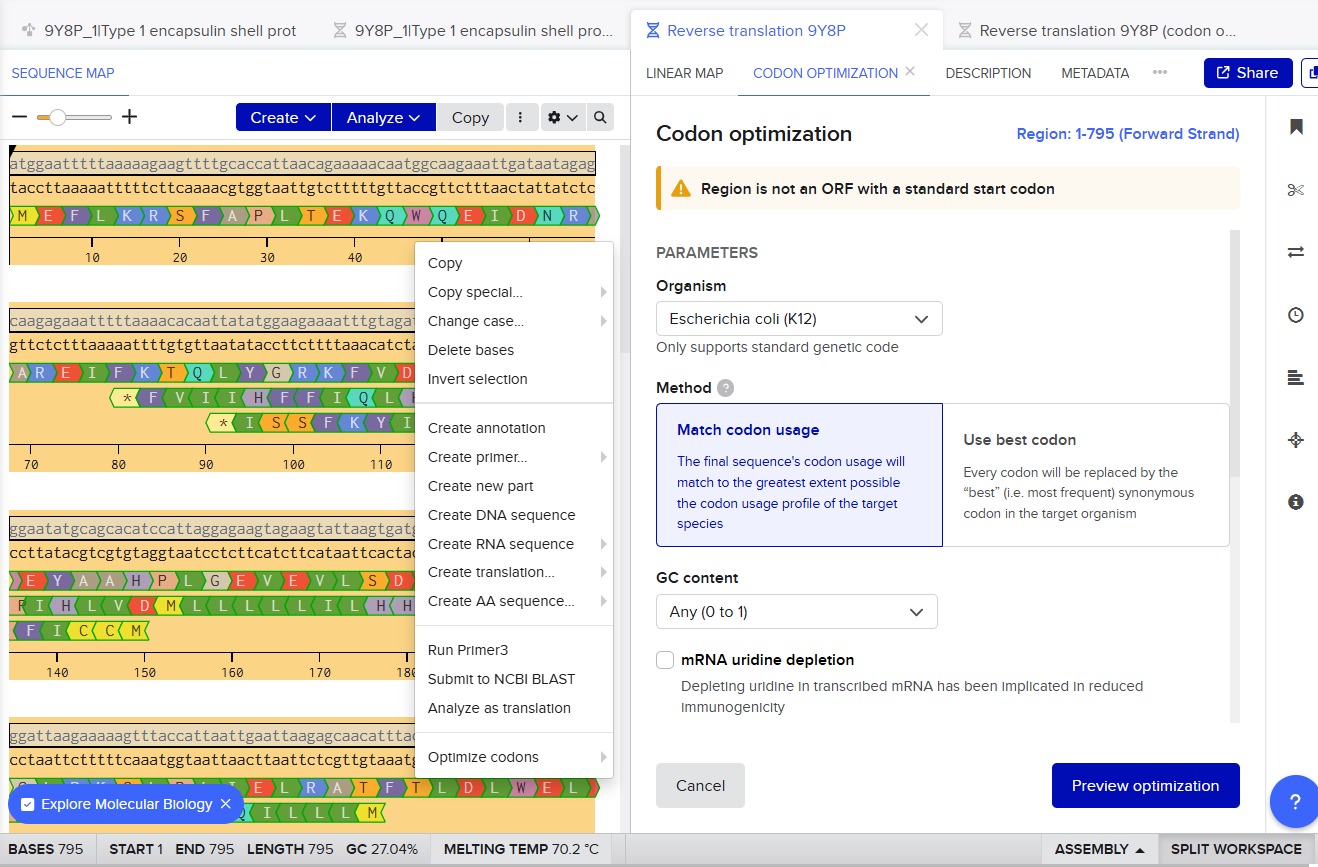
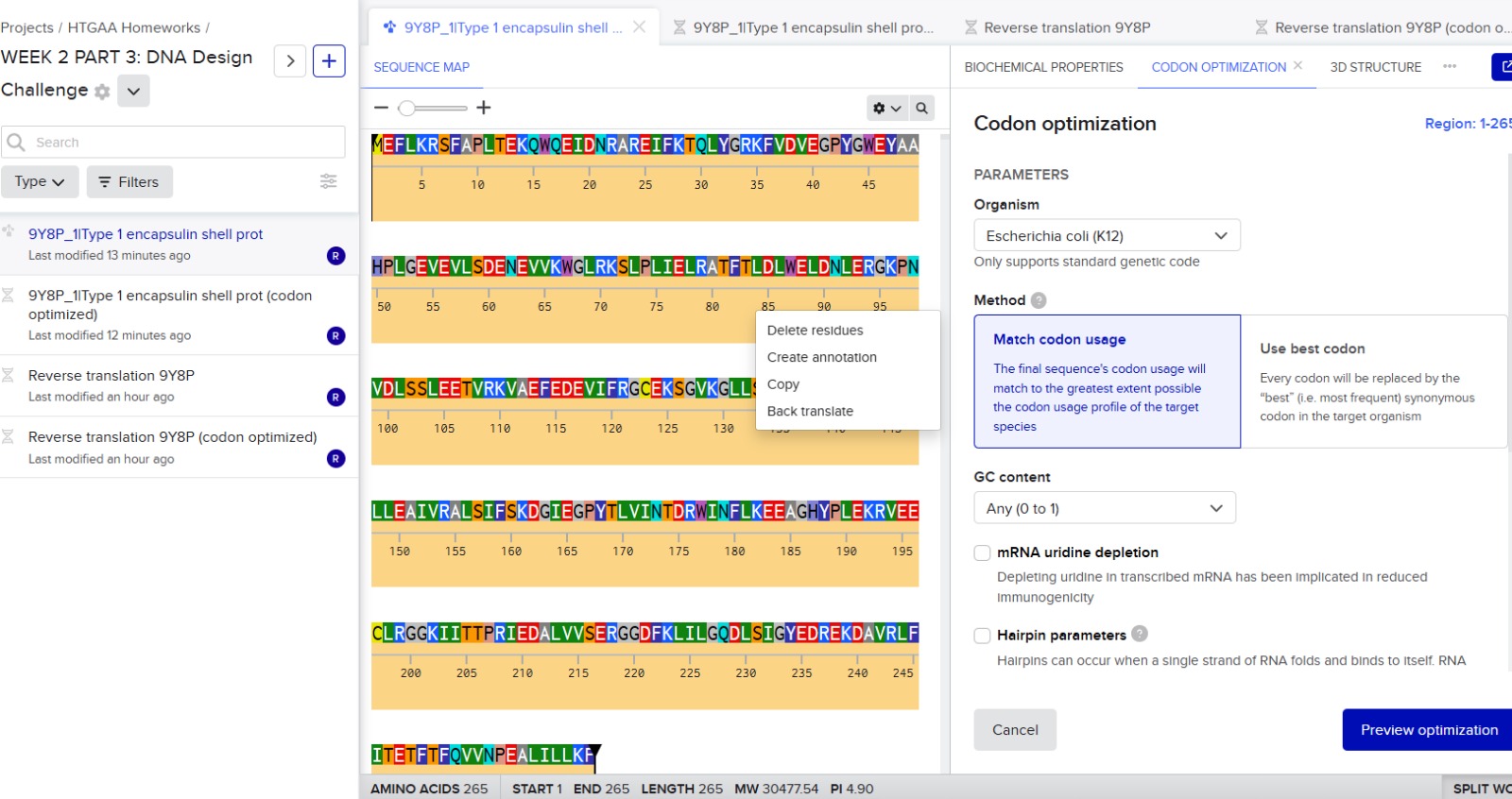
3.3. Codon optimization.
Codon optimization is necessary because different organisms prefer different codons to encode the same amino acid. Although the genetic code is universal, many amino acids are encoded by multiple codons, and each organism has a bias toward using certain ones more frequently. If a gene from Thermotoga maritima is expressed in another host without modification, the host’s ribosomes may translate it inefficiently due to rare codons, limited tRNA availability, or unfavorable mRNA secondary structures. This can reduce protein yield or even prevent proper expression.
For this project, I would optimize the encapsulin gene for Escherichia coli. E. coli is widely used for recombinant protein expression because it grows quickly, is inexpensive, and has well-established molecular biology tools. Encapsulins from T. maritima have previously been expressed successfully in E. coli, making it a practical and reliable host system.
9Y8P_1|Type 1 encapsulin shell protein DNA sequence|Thermotoga maritima (codon optimized)
ATGGAGTTCCTGAAGCGTAGTTTTGCACCATTGACTGAGAAGCAGTGGCAAGAGATTGATAACCGTGCGCGGGAAATTTTCAAAACCCAACTCTATGGCCGTAAATTTGTTGACGTCGAAGGTCCGTATGGGTGGGAATACGCAGCGCATCCTTTAGGTGAGGTTGAAGTGCTGTCGGATGAAAACGAAGTGGTCAAGTGGGGCCTGCGAAAAAGCTTACCCTTAATTGAACTCCGCGCGACCTTCACCCTGGATTTGTGGGAATTGGACAACCTTGAAAGGGGTAAGCCGAATGTAGATCTAAGCTCCCTGGAAGAAACCGTTCGTAAAGTAGCCGAGTTTGAAGATGAAGTCATATTTCGTGGGTGTGAGAAATCAGGCGTGAAAGGCCTGCTGAGCTTTGAAGAGCGCAAAATCGAATGCGGCTCGACGCCGAAAGACCTGCTGGAGGCCATTGTGCGTGCGTTAAGTATTTTTTCAAAAGACGGTATCGAAGGCCCGTATACCCTCGTTATTAATACGGATCGCTGGATTAACTTCTTGAAAGAAGAGGCGGGCCACTACCCGCTGGAAAAGCGCGTGGAAGAGTGCCTTCGCGGCGGTAAAATCATCACGACACCTCGCATCGAAGATGCTCTTGTAGTGAGCGAACGCGGCGGTGACTTCAAACTGATTCTGGGACAGGATCTGTCTATTGGATACGAAGATCGCGAGAAAGATGCCGTTCGTCTGTTTATTACAGAAACCTTTACTTTTCAGGTGGTGAATCCGGAAGCCTTAATCCTGCTTAAATTC
There is a second option in Benchling for reverse translating and optimizing the codons, which is by using the option Back translate.
The organism that I selected for the optimization was E.coli K12.
3.4. You have a sequence! What technologies could be used to produce this protein from your DNA?
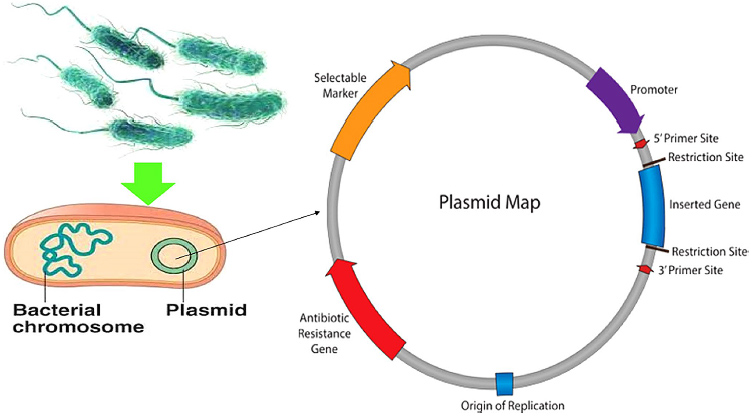
In a cell-dependent system, the optimized gene is inserted into a plasmid vector under the control of a promoter. The plasmid is introduced into E. coli through transformation. Inside the cell, RNA polymerase binds to the promoter and transcribes the DNA sequence into mRNA.
The mRNA is then translated by ribosomes. The ribosome reads the mRNA in triplets called codons, and each codon specifies one amino acid. Transfer RNAs (tRNAs) bring the correct amino acids to the ribosome, where they are linked together into a growing polypeptide chain. This process continues until a stop codon is reached. The resulting encapsulin monomers fold into their native structure and spontaneously self-assemble into a 60-subunit icosahedral nanocage.
In a cell-free system, purified transcription–translation machinery (often derived from E. coli lysates) is used outside living cells. The DNA template is added directly to the reaction mixture, and transcription and translation occur in vitro. This method allows faster prototyping and tighter experimental control, though it is typically more expensive than in vivo expression.
3.5. How does it work in nature/biological systems?
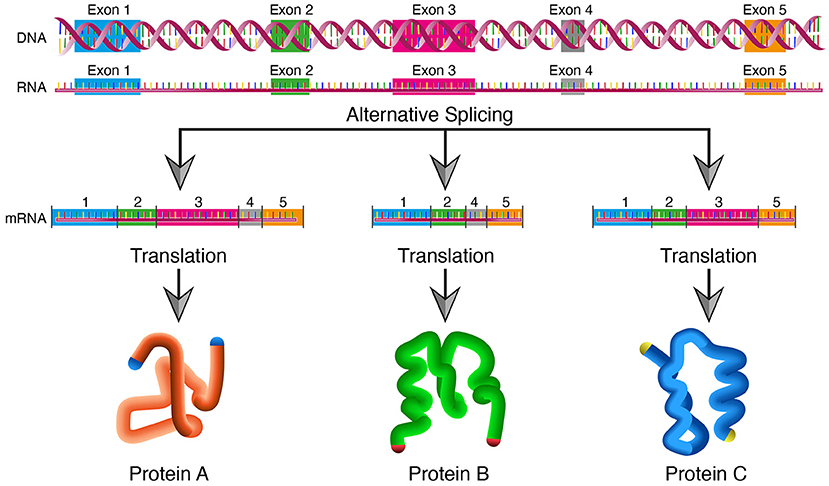
In biological systems, a single gene can produce multiple proteins at the transcriptional level through mechanisms that generate different RNA transcripts from the same DNA sequence.
In eukaryotes, the most common mechanism is alternative splicing. During transcription, a primary RNA transcript is produced that contains both exons (coding regions) and introns (non-coding regions). Different combinations of exons can be retained or removed before translation, producing distinct mRNA molecules. Each mRNA variant is translated into a different protein isoform with altered structure or function. Because the exon composition differs, the amino acid sequences of the resulting proteins differ.
Other transcriptional mechanisms include the use of alternative promoters or transcription start sites, which produce mRNAs with different 5′ ends, and alternative polyadenylation sites, which generate different 3′ ends. In some cases, different reading frames or alternative translation start sites can also produce distinct proteins from the same gene.
These mechanisms increase protein diversity without increasing genome size and allow organisms to regulate protein function in a context-dependent manner.
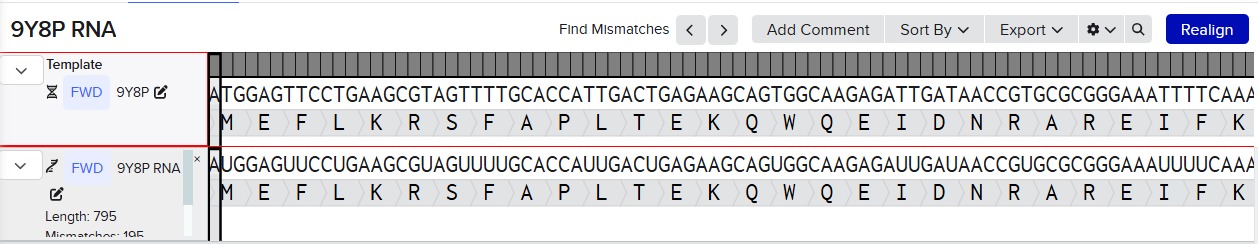
Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!! See example below.
Special note that all “T” were transcribed into “U” and that the 3-nt codon represents 1-AA.
Alignment RNA and DNA:
PART 4: Prepare a Twist DNA Synthesis Order
4.1. Create a Twist account and a Benchling account
Already created.
4.2. Build Your DNA Insert Sequence
The first step was to import the optimized DNA sequence for the encapsulin:
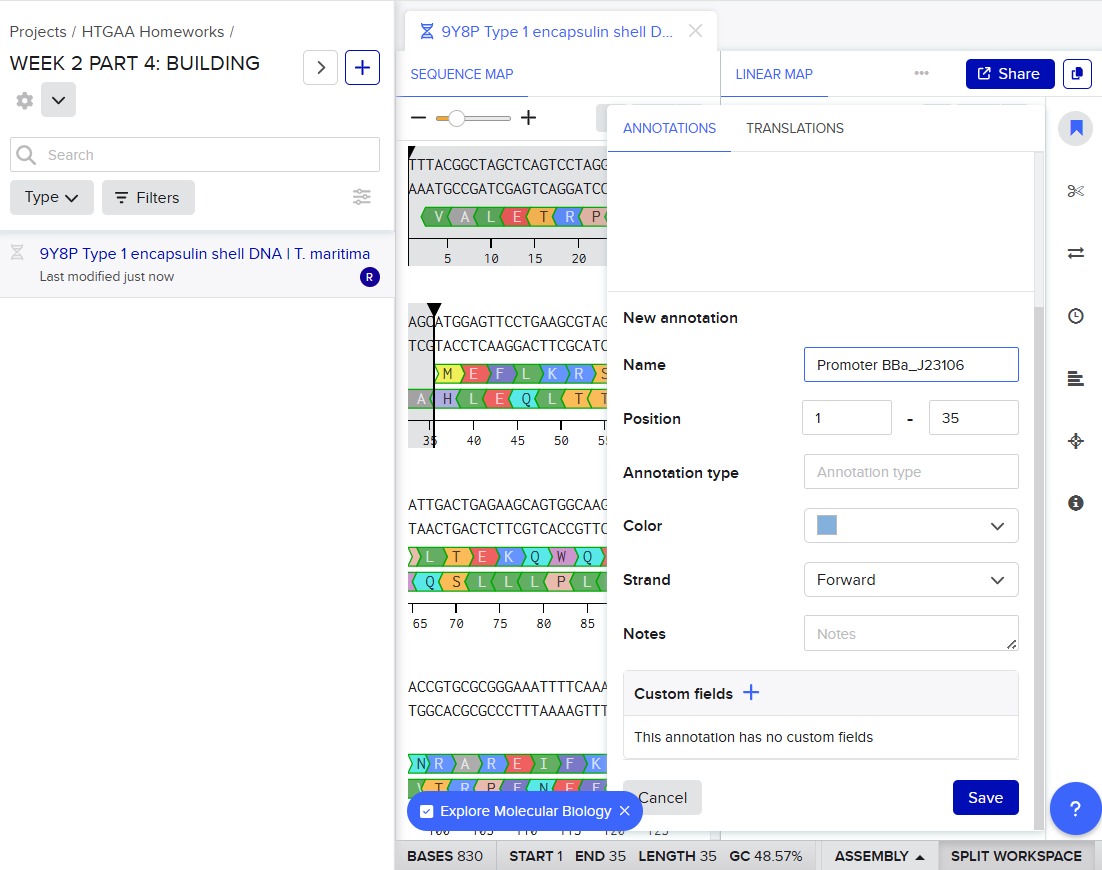
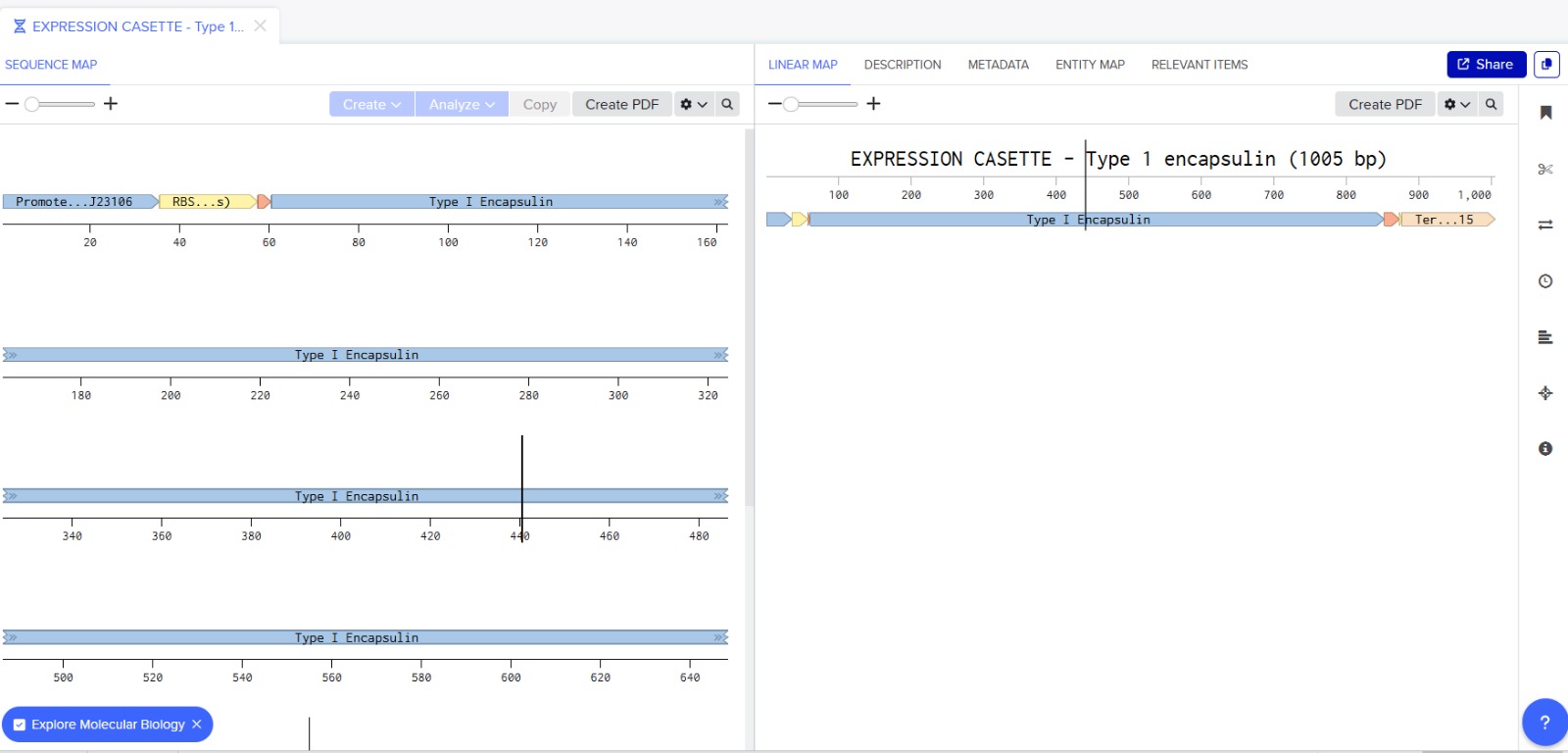
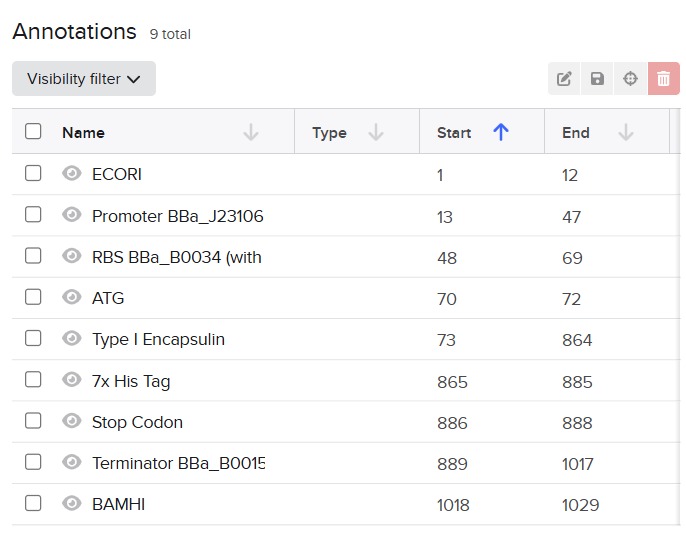
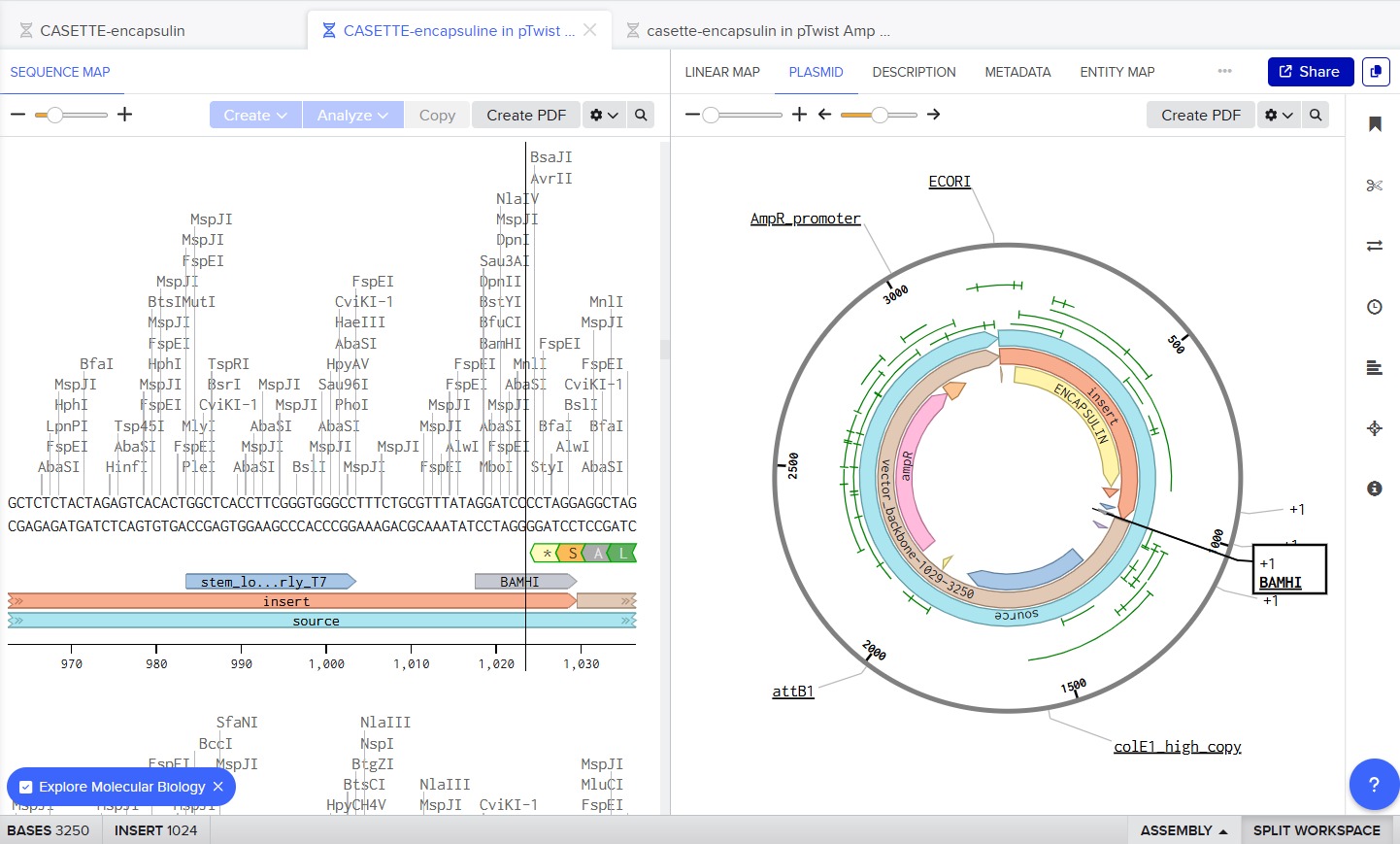
Next, I added the required genetic elements from the HGTAA website, including promoters, RBS, His-tags, and terminators. Each element was annotated with different colors for clarity:
Before finalizing the cassette, I added restriction enzyme sites at the ends of the sequence: EcoRI and BamHI. These enzymes do not cut any region of the plasmid used for cloning nor any part of the cassette.
The final expression cassette can be accessed via Benchling:
([https://benchling.com/s/seq-LwbFj7EEX7O47AzP8oMk?m=slm-kN07oTYhfDiiAiZL0IJa])
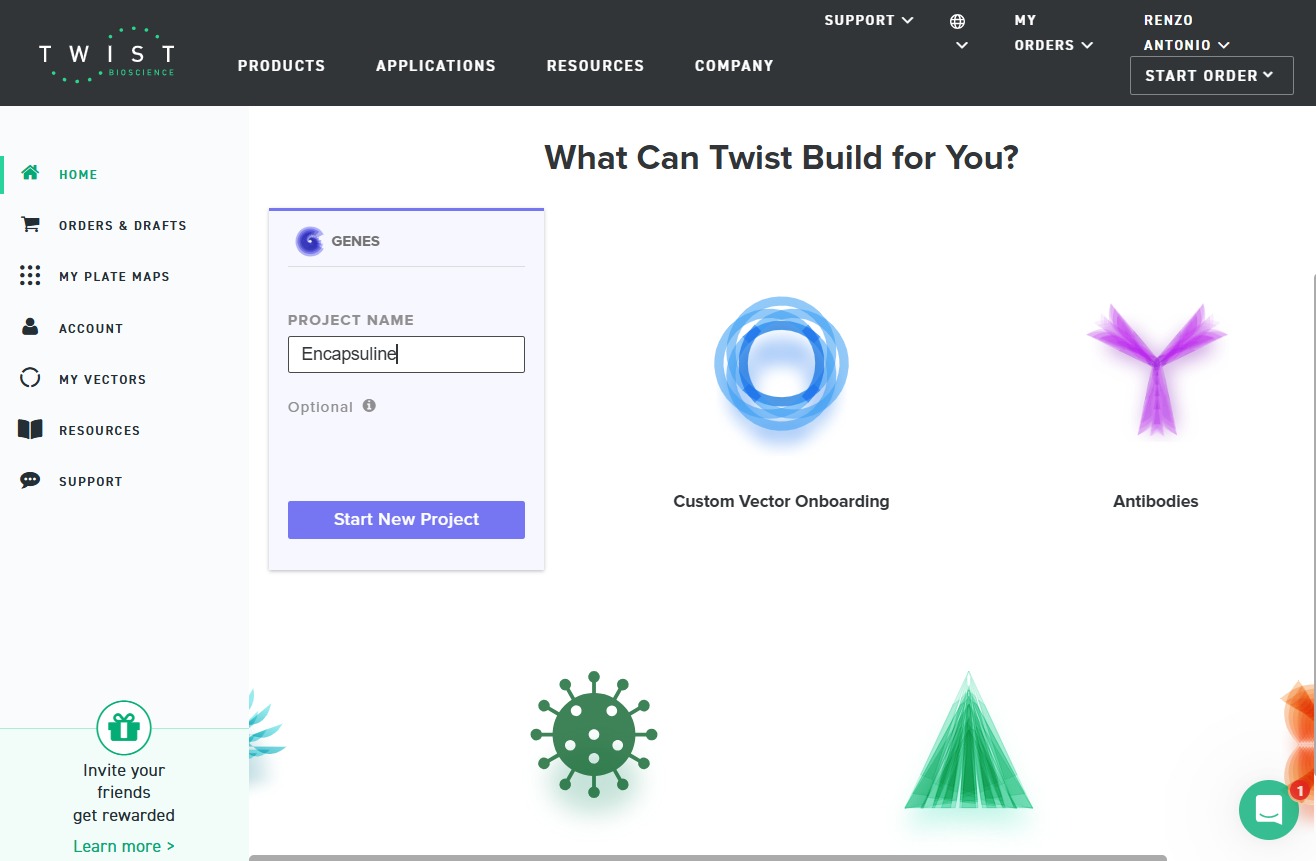
4.3. On Twist, Select The “Genes” Option
To begin the synthesis process, select the Genes option in Twist:
4.4. Select “Clonal Genes” option
The Clonal Genes option was selected to allow ordered synthesis and cloning.
4.5. Import your sequence
The sequence created in Benchling was successfully imported into Twist for ordering.
4.6. Choose Your Vector
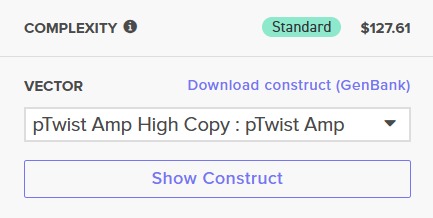
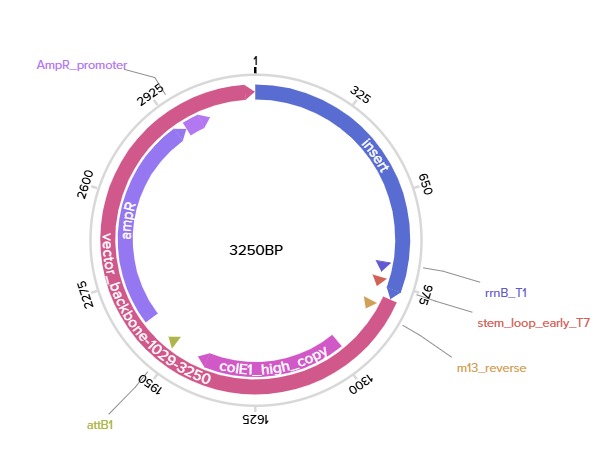
After selecting the desired cloning vector (pTwist Amp High Copy), Twist provides a visualization of the construct along with the price estimate:
Finally, the GenBank (.gb) file was downloaded and opened in Benchling for further review:
PART 5: DNA Read, Write, and Edit
5.1 DNA Read
(i) What DNA would you want to sequence and why?
I would like to sequence the genomes of new or previously uncharacterized organisms. The aim is to understand how life adapts to different environments and to uncover novel biological mechanisms. A clear example is sequencing the genomes of extremophiles living under high salinity conditions, which can reveal unique genetic adaptations for survival in harsh environments.
Sequencing these genomes can also identify new enzymes, proteins, and biomolecules with high potential for biotechnology, medicine, and industrial applications. Exploring previously unknown organisms expands our understanding of biodiversity and offers valuable resources for scientific and commercial use.
(ii) Sequencing Technology
Chosen Technologies:
Illumina (second-generation, short-read sequencing) for high-accuracy genome analysis.
Oxford Nanopore (third-generation, long-read sequencing) to capture complex genomic structures and long repetitive regions.
Fragment DNA into smaller pieces (for Illumina; optional for Nanopore).
Ligate adapters for sequencing.
PCR amplification (for Illumina; optional for Nanopore).
Essential Steps / Base Calling:
Illumina: DNA fragments are amplified on a flow cell, fluorescent nucleotides are incorporated one by one, and base identity is determined by imaging fluorescence signals.
Oxford Nanopore: Single DNA molecules pass through nanopores, changes in ionic current are measured, and base calling algorithms decode the sequence from the signal.
Output:
Raw nucleotide sequences (FASTQ files) with quality scores, which can be assembled into complete genomes and analyzed for novel genes or adaptations.
5.2 DNA Write
(i) What DNA would you want to synthesize and why?
I would synthesize an expression cassette encoding the enzymes responsible for siderophore production in bacteria.
Siderophores are small, high-affinity iron-chelating molecules secreted by many soil bacteria. They bind Fe³⁺ and increase iron bioavailability for both microbes and plants. Because iron is often limited in agricultural soils, enhancing siderophore production could:
Improve iron uptake in crops
Promote plant growth and yield
Reduce dependency on synthetic fertilizers
Support sustainable and environmentally friendly agriculture
The expression cassette would include:
A strong and regulated bacterial promoter
Ribosome binding site (RBS)
Coding sequences for siderophore biosynthesis enzymes (e.g., enzymes involved in nonribosomal peptide synthesis or other siderophore pathways)
The cassette could be cloned into a plasmid for high expression or integrated into the bacterial genome for stable, long-term expression. Codon optimization would be performed based on the selected bacterial host (E.coli).
(ii) DNA Synthesis Technology
Chosen Technology: Commercial gene synthesis combined with modular DNA assembly
To construct the expression cassette, I would use commercial gene synthesis services to synthesize either the complete construct or modular fragments.
Essential Steps:
In silico design
Design the full cassette using software (e.g., Benchling or SnapGene).
Optimize codon usage for the chosen bacterial host.
Remove unwanted restriction sites and secondary structures.
Commercial DNA synthesis
Order the optimized gene fragments.
DNA is synthesized using automated phosphoramidite chemistry.
Assembly of fragments
Gibson Assembly or Golden Gate cloning to assemble multi-gene constructs.
Insert into an expression plasmid backbone.
Transformation and validation
Transform into bacterial host cells.
Confirm sequence by Sanger sequencing.
Evaluate siderophore production using iron-chelation or colorimetric assays.
Limitations:
Large biosynthetic clusters may require multi-step assembly.
Expression may impose metabolic burden on host cells.
Regulatory tuning may be required to balance growth and production.
Cost increases with construct length and complexity.
5.3 DNA Edit
(i) What DNA would you want to edit and why?
I would edit the DNA sequence encoding the protein encapsulin to better understand how its structure influences adhesion to specific molecules.
Encapsulins are self-assembling protein nanocompartments found in bacteria and archaea. They form cage-like structures capable of encapsulating enzymes or interacting with target molecules. By modifying specific amino acids located on the surface, I could:
Alter surface charge properties
Modify binding affinity to selected molecules
Improve structural stability
Investigate structure–function relationships
Targeted mutations, such as single amino acid substitutions, would allow systematic evaluation of how structural features affect molecular adhesion.
(ii) What technology or technologies would you use and why?
If the encapsulin gene is cloned into an expression plasmid:
Clone encapsulin gene into an expression vector.
Design primers containing the desired nucleotide mutation.
Perform PCR amplification incorporating the mutation.
Remove template DNA using restriction digestion.
Transform bacteria.
Confirm mutation via DNA sequencing.
Advantages:
High precision
Efficient for point mutations
No genomic double-strand breaks
Suitable for structure–function studies
Option 2: CRISPR-Cas9 Genome Editing
If editing the native encapsulin gene in the bacterial genome:
Design guide RNA targeting the encapsulin gene.
Introduce Cas9 and guide RNA into the cells.
Provide a donor DNA template containing the desired mutation.
Repair occurs through homology-directed repair (HDR).
Required Components:
Guide RNA (for CRISPR-based editing)
Cas9 or base editor system
Donor DNA template (for HDR-based edits)
Delivery system (plasmid transformation or electroporation)
Sequencing-based validation
Limitations:
Off-target effects in CRISPR systems
Variable editing efficiency depending on organism
Mutations may disrupt protein folding
Functional assays required to evaluate adhesion changes
References
Hoose, A., Vellacott, R., Storch, M., et al. (2023). DNA synthesis technologies to close the gene writing gap. Nature Reviews Chemistry, 7(3), 145–161. https://doi.org/10.1038/s41570-022-00456-9
Shendure, J., Balasubramanian, S., Church, G., et al. (2017). DNA sequencing at 40: Past, present, and future. Nature, 550(7676), 345–353. https://doi.org/10.1038/nature24286
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
DNA polymerase has an intrinsic error rate of roughly one mistake per million nucleotides incorporated. Given that the human genome contains approximately 3.2 billion base pairs—equivalent to about 6.4 billion nucleotides in a diploid cell—this error rate would theoretically result in around 6,400 replication errors per cell division, or about half that number in a haploid genome. Such a frequency suggests a substantial risk for mutations that could potentially be inherited by offspring. However, biological systems have evolved multiple mechanisms to enhance the accuracy of DNA replication. Over the past century, these fidelity-enhancing processes have been extensively studied. One example is the MutS homolog 1 (MSH1/MutS-1) protein, which recognizes and binds to mismatched or improperly paired DNA sequences. This mismatch-repair mechanism serves as an additional layer of quality control, significantly increasing the fidelity of de novo DNA synthesis. (Carr et al., 2004)
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
An average human protein is approximately 300 amino acids long. Although there are (20^{300}) possible amino-acid sequences of this length, the number of DNA sequences that can encode a specific protein is determined by the degeneracy of the genetic code. Because most amino acids are encoded by multiple synonymous codons (about 3 on average), a single protein sequence can theoretically be encoded by roughly (3^{300}) different DNA sequences.
Despite this large number, most of these sequences do not function efficiently in cells. Factors such as codon usage bias, which affects translation speed, mRNA stability, translational accuracy, mRNA secondary structure, and regulatory or genomic constraints significantly restrict which nucleotide sequences can be successfully transcribed and translated. As a result, only a limited subset of possible DNA sequences are biologically functional.
Homework questions from Dr. LeProust
What’s the most commonly used method for oligo synthesis currently?
The most commonly used method for oligonucleotide synthesis is solid-phase chemical synthesis using phosphoramidite chemistry. In this approach, nucleotides are added one at a time to a growing DNA chain that is anchored to a solid support. Each synthesis cycle involves nucleotide coupling, capping of unreacted ends, and oxidation, allowing precise control over the sequence being synthesized (Kosuri & Chuch, 2014).
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Because the coupling efficiency in each synthesis cycle is less than 100%. As the number of cycles increases, small inefficiencies accumulate, resulting in a high proportion of truncated or incorrect sequences. Additionally, side reactions and chemical damage to the growing oligo further reduce yield and fidelity as length increases.
Why can’t you make a 2000bp gene via direct oligo synthesis?
A 2000 base-pair gene cannot be synthesized directly because the cumulative error rate and truncation probability become extremely high over thousands of synthesis cycles. This leads to very low yields of full-length, error-free DNA, making direct synthesis impractical. Instead, long genes are typically assembled from shorter, high-quality oligonucleotides using enzymatic methods such as PCR-based assembly or ligation.
Homework question from George Church
Using Google & Prof. Church’s slide #4, What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
In animals, essential amino acids are those that cannot be synthesized de novo and must be obtained from the diet. Across animals, the 10 commonly cited essential amino acids are:
Histidine
Isoleucine
Leucine
Lysine
Methionine
Phenylalanine
Threonine
Tryptophan
Valine
Arginine (especially during growth)
The lysine contingency refers to a bioengineered safety mechanism in which organisms are modified to depend on an external supply of lysine for survival. Because lysine is an essential amino acid in all animals, this dependency creates a strong biological containment strategy: the engineered organism cannot survive or proliferate outside controlled environments lacking lysine.
Understanding that animals depend on multiple essential amino acids reinforces the strength of the lysine contingency. It highlights how leveraging fundamental metabolic constraints can improve biosafety, particularly for in vivo applications, by reducing the risk of unintended survival or spread of engineered organisms.