Week 4 HW: Protein Design Part I
Protein Design
Part A: Conceptual Questions
1. Number of amino-acid molecules in 500 g meat
- Meat is roughly ~20% protein → ~100 g protein in 500 g meat (order-of-magnitude estimate).
- Average amino-acid residue ≈ 100 Da = 100 g/mol.
- Moles of amino acids ≈ 100 g ÷ 100 g/mol = 1 mol.
- Number of molecules ≈ (6x10^{23}) amino-acid residues. → Roughly 10^24 amino-acid molecules.
2. Why only 20 natural amino acids
Twenty amino acids provide a compromise between:
- Chemical diversity (charge, hydrophobicity, size, reactivity)
- Biosynthetic metabolic cost
- Translational accuracy and evolutionary robustness
This set is sufficient to build functional proteins.
3. Can we design non-natural amino acids
Yes. Non-natural amino acids are widely synthesized. Examples of designs:
- Fluorinated amino acids → increase stability
- Photocaged amino acids → allow light-controlled protein activation
- Azido- or alkyne-containing amino acids → enable click chemistry
- Conformationally restricted amino acids → stabilize protein folds
They can be incorporated using engineered tRNA–synthetase systems.
4. Origin of amino acids before life
Amino acids were likely produced by prebiotic chemistry:
- Atmospheric discharge reactions (e.g., Miller-Urey–type synthesis)
- Hydrothermal vent chemistry
- Organic molecules delivered by meteorites
- Photochemical synthesis in early oceans
These processes generated many amino acids before biological enzymes existed.
5. Handedness of α-helix made from D-amino acids
Proteins built from D-amino acids form left-handed α-helices, which are mirror images of the right-handed helices formed by L-amino acids.
6. Why most molecular helices are right-handed
Because natural proteins are composed mainly of L-amino acids. The stereochemistry of the peptide backbone energetically favors right-handed α-helices.
7. Why β-sheets tend to aggregate
β-strands expose backbone hydrogen-bond donors and acceptors, allowing intermolecular hydrogen bonding. Side chains may pack via hydrophobic interactions, promoting sheet-to-sheet association.
8. Why do many amyloid diseases form β-sheets?
Amyloid diseases are associated with protein misfolding. Normally folded proteins are stabilized by their native tertiary structure, but under pathological conditions partial unfolding can expose backbone hydrogen-bond donors and acceptors. These segments tend to re-associate into cross-β structures, where β-strands run perpendicular to the fibril axis and β-sheets stack along the fiber direction.
β-sheet conformations are favored because:
The peptide backbone can form extensive intermolecular hydrogen-bond networks, which provides high thermodynamic stability.
Side chains can pack tightly through hydrophobic interactions, reducing solvent exposure.
Once a nucleus forms, β-sheet aggregation becomes self-propagating, leading to fibril growth.
This aggregation tendency underlies diseases such as Alzheimer-type neurodegeneration and several systemic amyloidoses, where misfolded peptides accumulate into insoluble fibrils.
9. Can amyloid β-sheets be used as materials? Design of a β-sheet motif
Yes. Amyloid β-sheet assemblies are actively explored as biomaterials because they are:
- Mechanically strong due to dense hydrogen-bond networks
- Highly ordered at the nanoscale
- Capable of programmable self-assembly
- Biocompatible when designed carefully
A useful design strategy is to construct a short amphiphilic β-hairpin peptide motif that promotes controlled fibrillization.
Example β-sheet motif design
Where:
- H = hydrophobic residue (e.g., Val, Leu, Ile, Phe) to drive core packing
- X = charged or polar residue (e.g., Lys, Glu, Ser) to improve solubility and orientation
The turn region can use a Gly-Pro-Gly or similar flexible motif to stabilize the β-hairpin
Example specific sequence design:
- KLVFFAEGPGAEFVLK
Design principles:
- Include a nucleation core (often aromatic or hydrophobic residues) to trigger stacking.
- Balance solubility and aggregation tendency to avoid uncontrolled precipitation.
- Use end-capping charges to control fibril length if necessary.
Such motifs can form nanofibers, hydrogels, or scaffold materials for drug delivery, tissue engineering, and nanoelectronics.
Part B: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
The protein I selected is Superoxide dismutase 1 (SOD1). I selected it because it is a key antioxidant enzyme that protects cells from oxidative stress by catalyzing the conversion of superoxide radicals into oxygen and hydrogen peroxide. It is relevant to lung oxidative injury and can be integrated into redox-sensing diagnostic platforms.
2. Identify the amino acid sequence of your protein
The amino acid sequence of SOD1 is:
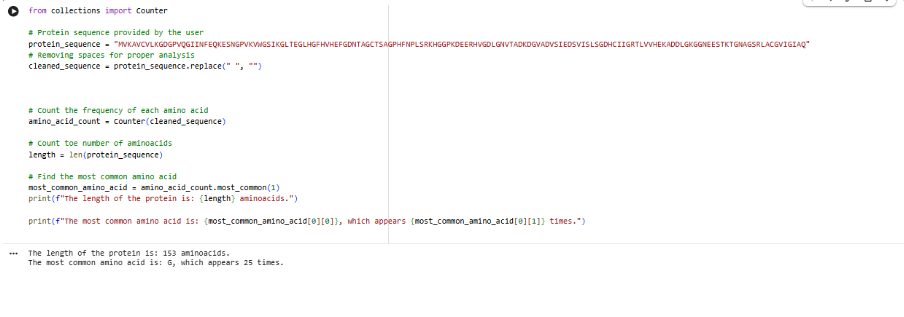
3. Sequence length and most frequent amino acid
- Length: 153 amino acids
- Most frequent amino acid: Glycine (G) is among the most frequent residues in this sequence.
4. How many protein sequence homologs are there for your protein?
Using UniProt BLAST search, SOD1 has hundreds of homologous sequences across different species. The enzyme is highly conserved because of its essential antioxidant function.
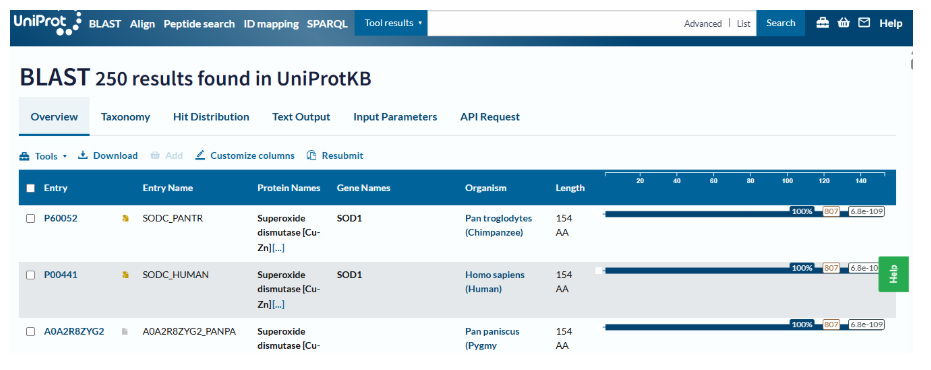
- BLAST search for SOD1 sequence returned approximately 250 homologous protein sequences, indicating that SOD1 is a highly conserved protein across many species.
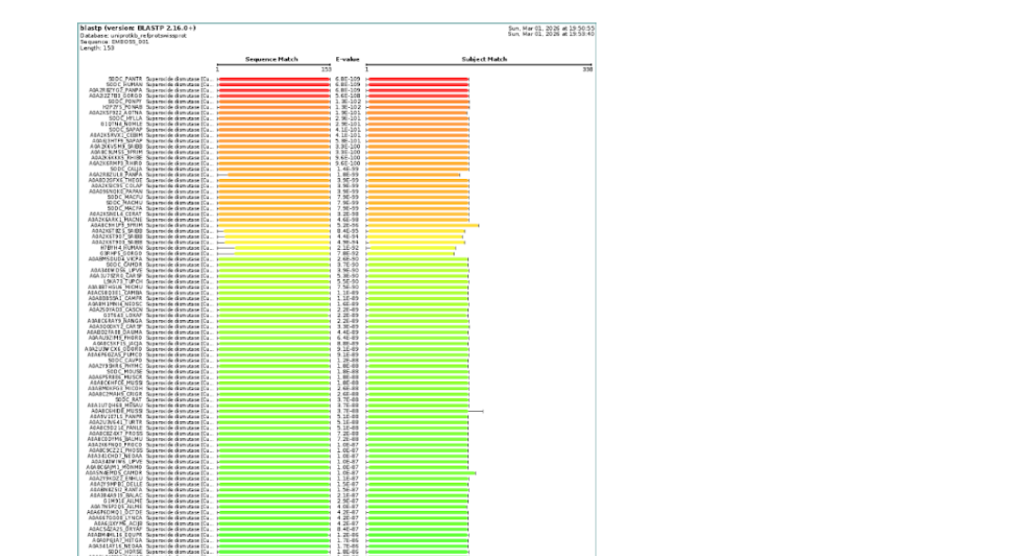
5. Protein family?
SOD1 belongs to the Cu/Zn superoxide dismutase family, which contains metalloenzymes that detoxify superoxide radicals.
6. Identify the structure page of your protein in RCSB. When was the structure solved? Is it a good quality structure? Are there any other molecules in the solved structure apart from protein?
Representative structure:
- Structure database: Human Cu,Zn Superoxide Dismutase 1 structure https://www.rcsb.org/structure/1HL5
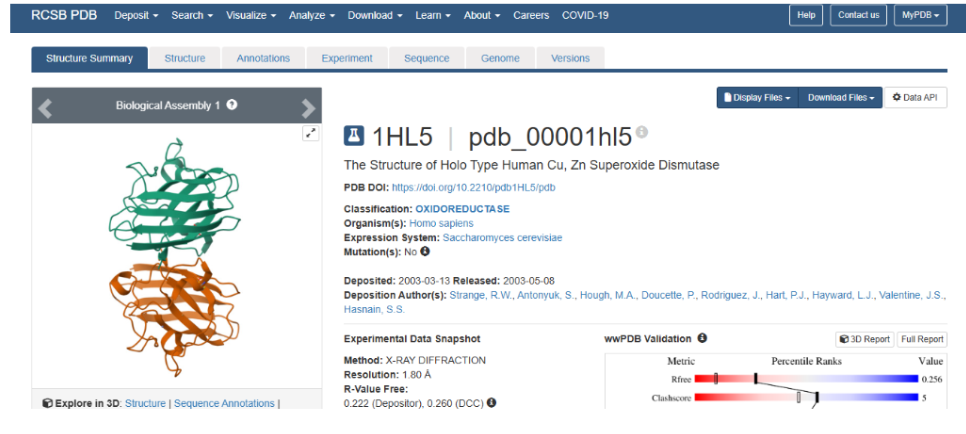
The structure of Human Cu,Zn Superoxide Dismutase 1 structure was deposited and released in 2003 (deposition date: 2003-03-13, release date: 2003-05-08). It is a good quality structure because the resolution is 1.80 Å, which is much smaller than the reference threshold of 2.70 Å. A resolution of 1.80 Å indicates high structural accuracy and reliable atomic detail.
Other molecules present in the structure:
- Copper (Cu²⁺)
- Zinc (Zn²⁺)
- Water molecules
7. Structure classification family
The protein belongs to the Cu,Zn superoxide dismutase family.
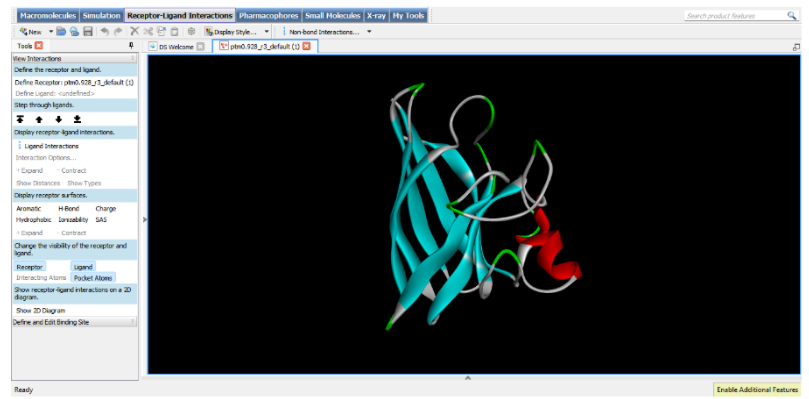
8. PyMoL Visualization
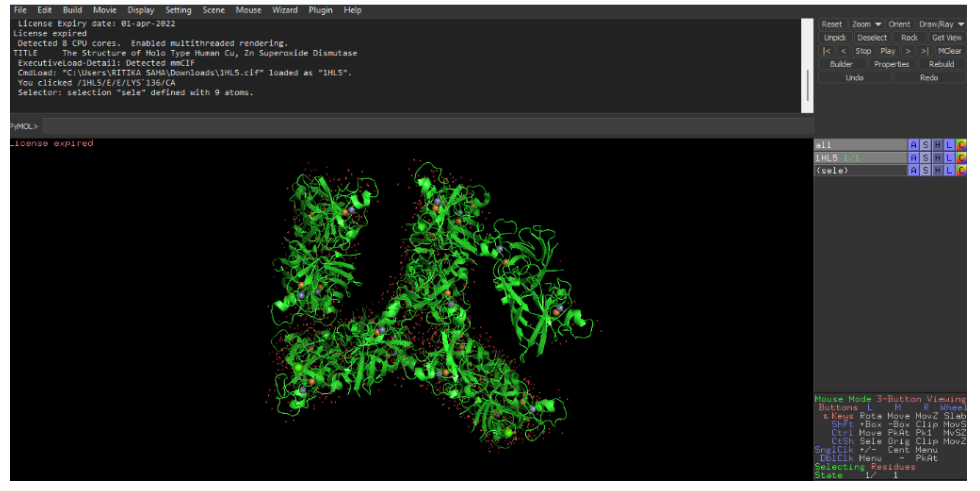
In PyMOL, you can visualize the same protein in different styles by creating multiple representations or by toggling representations in the same session.
Use the following commands:
Cartoon representation
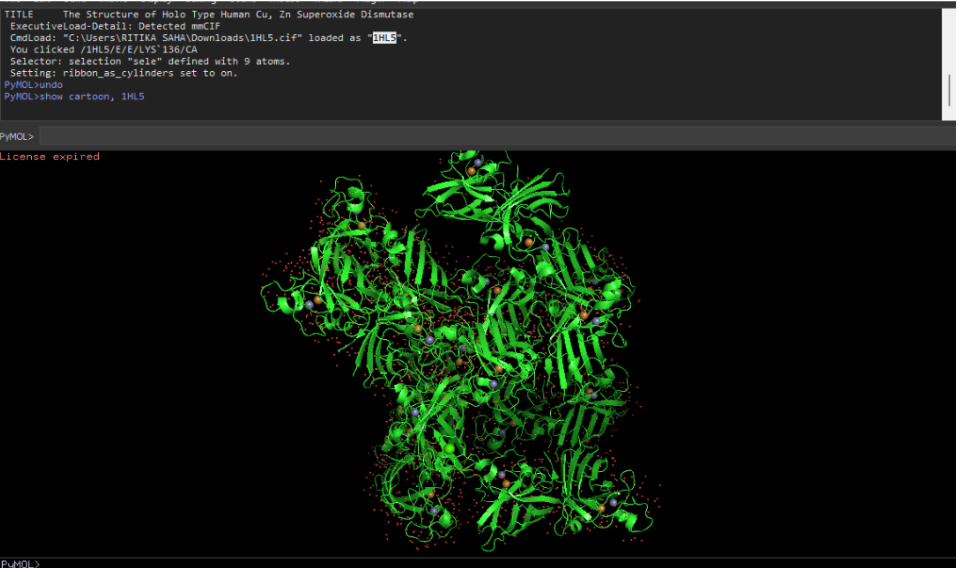
Ribbon representation
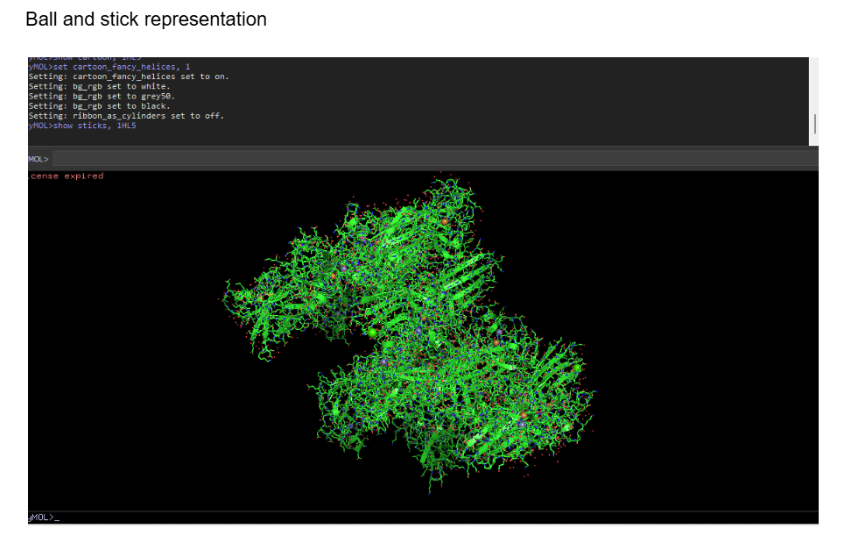
Ball and stick (sticks + spheres)
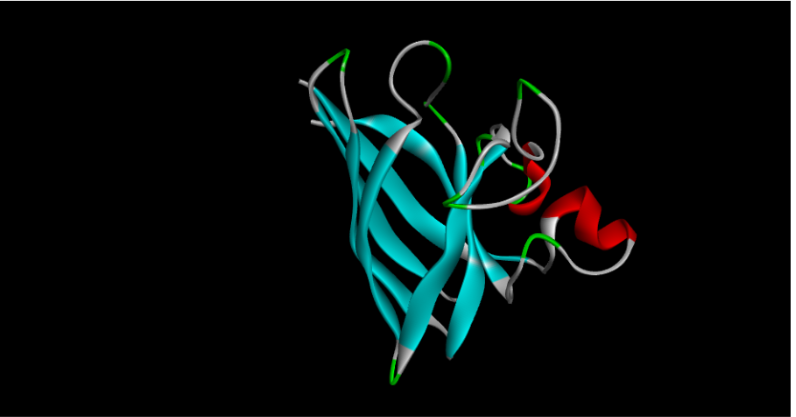
9. Secondary structure composition
SOD1 contains:
- More β-sheets than α-helices.
- The structure is mainly a β-barrel with several loop regions.
10. Hydrophobic vs hydrophilic residue distribution
- Hydrophobic residues are mainly buried inside the β-barrel core to stabilize the structure.
- Hydrophilic residues are exposed on the surface, facilitating solvent interaction and enzymatic activity.
Color protein by residue type
11. Surface pocket / binding cavity
- Yes, SOD1 has metal-binding pockets that coordinate copper and zinc ions. These pockets are essential for catalytic conversion of superoxide radicals.
Visualize protein surface and binding pockets
Part C: Using ML-Based Protein Design Tools
Part C1: Protein Language Modeling
a. Unsupervised deep mutational scan using ESM2
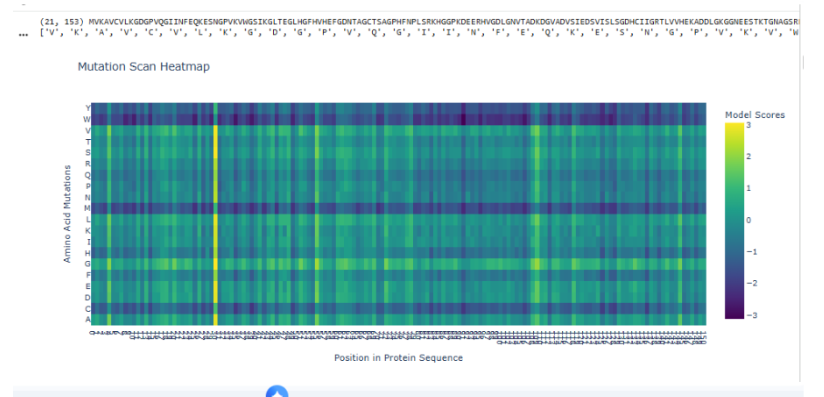
To generate an unsupervised deep mutational scan (DMS), the wild-type protein sequence is first passed through ESM2 to compute the log-likelihood of the native amino acid at each position. Then, for every residue position, all 19 possible single amino acid substitutions are introduced computationally, and the change in log-likelihood (Δ log P) relative to the wild type is calculated. These scores approximate how evolutionarily plausible each mutation is according to the language model. The resulting matrix (positions × 20 amino acids) is visualized as a heatmap, where strongly negative values indicate mutations that are highly disfavored and positive or near-zero values indicate tolerated substitutions. In this sequence, the scan reveals vertical dark bands at specific positions, suggesting strong evolutionary constraint, while other positions show a broader distribution of tolerated mutations, indicating structural or functional flexibility.
b. Interpretation of a specific pattern
One notable pattern appears around residue His45 within the motif GLHGFHVHEF. This region contains multiple histidines and glycines, suggesting structural or catalytic relevance. The heatmap shows that most substitutions at position 45 are strongly penalized, forming a pronounced vertical stripe. A particularly deleterious mutation is H45P (Histidine to Proline). Proline imposes rigid backbone constraints due to its cyclic structure and often disrupts helices or active-site conformations. If His45 participates in hydrogen bonding, catalysis, or metal coordination, replacing it with proline would disrupt both structural geometry and chemical functionality. ESM2 assigns a strongly negative likelihood change to this mutation, indicating that such substitutions are rarely observed across evolution. This pattern reflects evolutionary conservation and suggests that His45 is functionally important.

c. Latent Space Analysis
The given protein sequence
was embedded into a reduced-dimensional latent space together with the SCOP protein dataset using sequence-derived features and t-SNE dimensionality reduction. Each point in the latent space represents one protein domain, where spatial proximity indicates similarity in sequence-derived biochemical and structural properties.

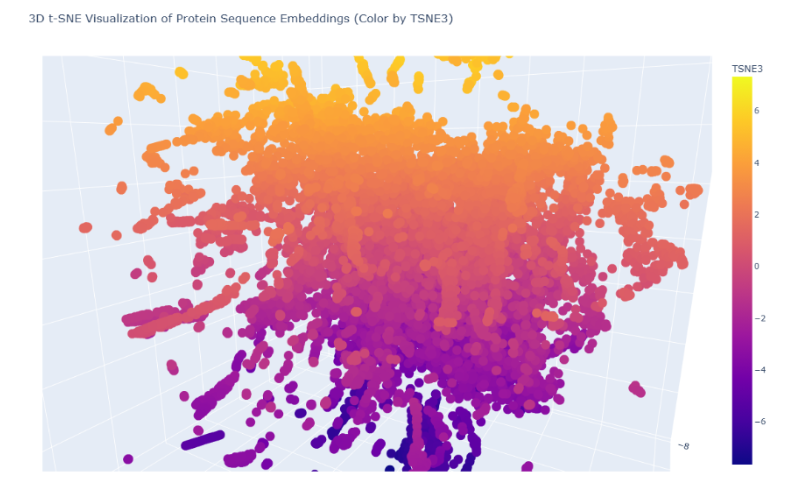
Neighborhood analysis: In the 3D t-SNE map, the protein lies within a dense central cluster of soluble proteins rather than in sparse peripheral branches. This indicates that its nearest neighbors share:
- similar amino-acid composition
- comparable length (~150 aa)
- soluble cytosolic nature
- globular enzyme-like fold
Thus, the latent space neighborhood approximates proteins with related structural class and biochemical characteristics.
Interpretation of protein properties from sequence
Position relative to neighbors
Do neighborhoods approximate similar proteins?
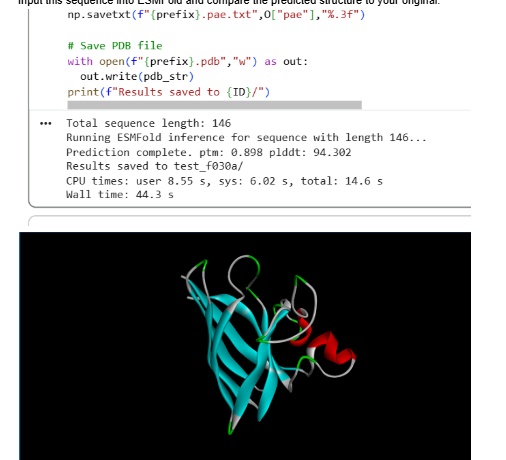
Part C2: Protein Folding
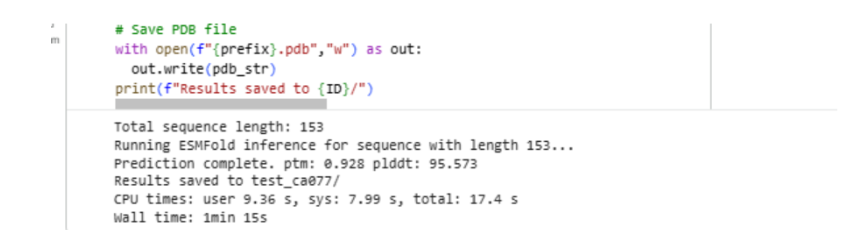

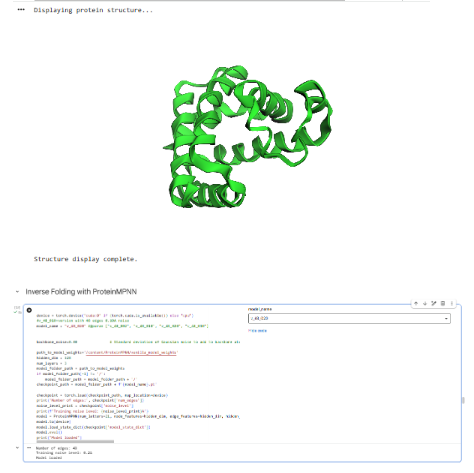
Part C3: Protein Generation

Part D: Group Project
- formed a group
- Group Project Link: https://docs.google.com/document/d/1ENvPHhRbBgtl0ERrfqmomJKxPg68nfvCugrPQrDdM7o/edit?usp=sharing
- Proposal: By: 2026a-nourelden-rihan, 2026a-ritika-saha, 2026a-rahul-yaji, 2026a-keerthana-gunaretnam
- We decided to focus on the main area of increasing the stability of the MS2 phage lysis protein L, with a possible secondary goal of reducing the dependency on host DnaJ, while still maintaining the lysis action.
- The tools AlphaFold, Clustal Omega, BLAST, ESM, and ESMFold were discussed.
- BLAST can pull out homologous lysis proteins from the databases.
- Clustal Omega can create MSAs to identify essential L48-S49 residues, and the pore-forming regions that must not be mutated.
- ESM can create mutation heatmaps, which can guide the use of ESMFold to obtain highest score foldings in mutatable regions.
- AlphaFold Multimer predicts whether the subunits of our protein can successfully create a pore in the host membrane, and also to check whether N-terminus can break the interaction with DnaJ.
- We also identified a few pitfalls, with majors ones dealing with limited training datasets, that may not be properly aligned towards creating a transmembrane lysis protein.
- Some other pitfalls include the lack of proper annotations for amurins; the possibility of an over-stable protein to form non-functional aggregates; and the vulnerability of modified protein to host proteases.