Week 5 HW: Protein Design Part II
Protein Design II
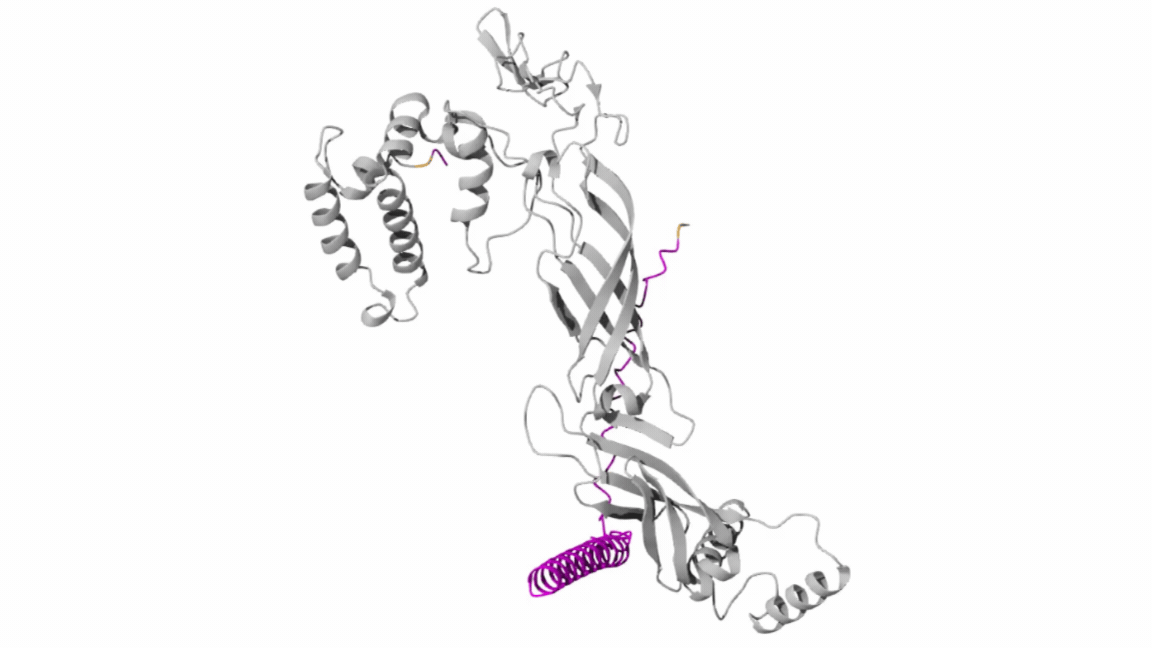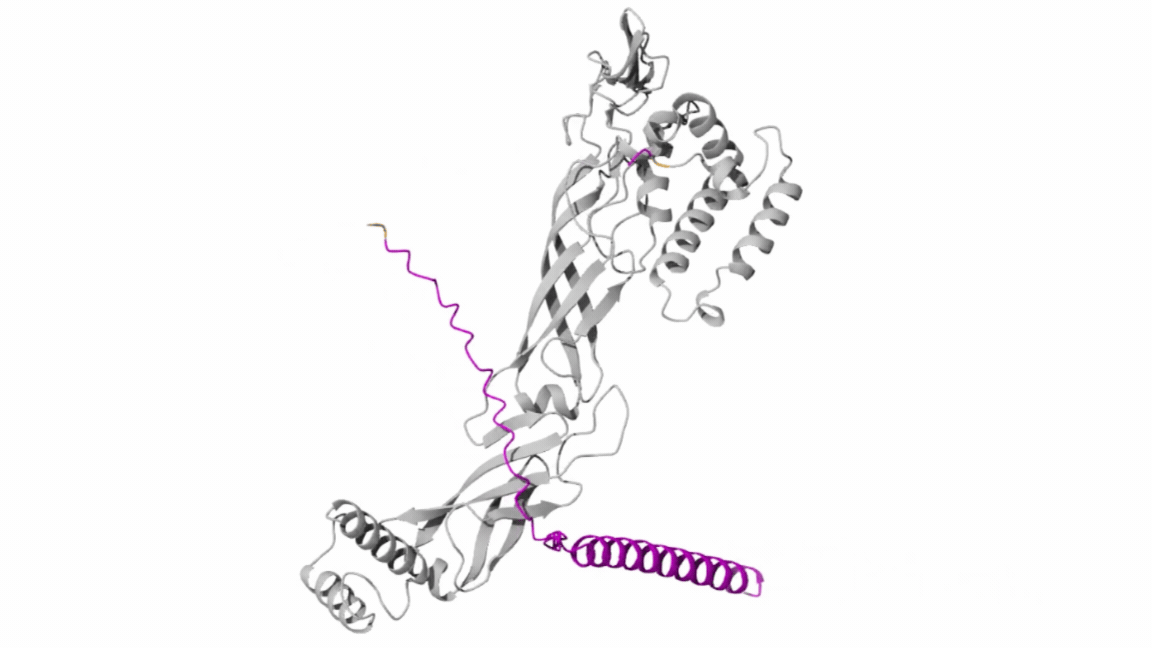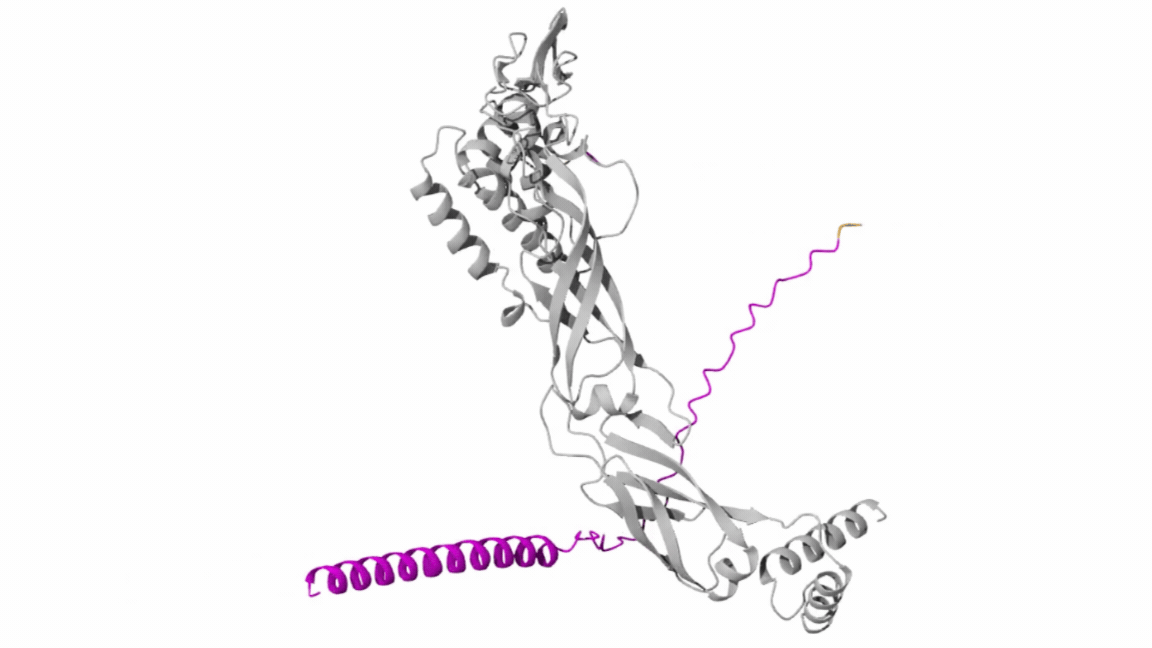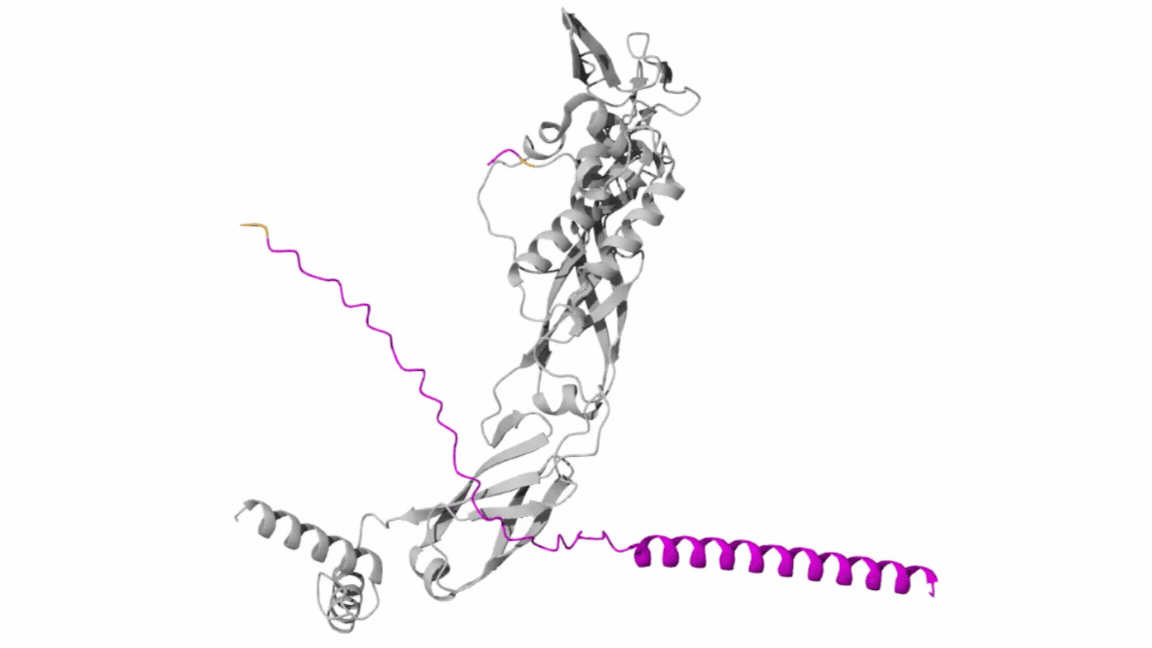
SOD1 Binder Peptide Design
Part 1: Generate Binders with PepMLM
- Retrieval of SOD1 Sequence
The human Superoxide Dismutase 1 (SOD1) protein sequence was retrieved from UniProt (Accession P00441).
Wild-type sequence (first region):
- Introduction of the A4V Mutation
The classical ALS mutation A4V replaces Alanine (A) with Valine (V) near the N-terminus.
However, examination of the provided sequence shows:
| Position | Residue |
|---|---|
| 1 | M |
| 2 | A |
| 3 | T |
| 4 | K |
| 5 | A |
| 6 | V |
Thus residue 4 is Lysine, not Alanine. The nearest Alanine occurs at position 5, so the mutation was applied there.
This substitution increases hydrophobicity near the N-terminus and is known to destabilize SOD1, promoting aggregation associated with aggressive familial ALS.
- Peptide Generation with PepMLM
Using the PepMLM-650M model Colab, the mutant SOD1 sequence was used as the conditioning context to generate four peptides of length 12 amino acids.
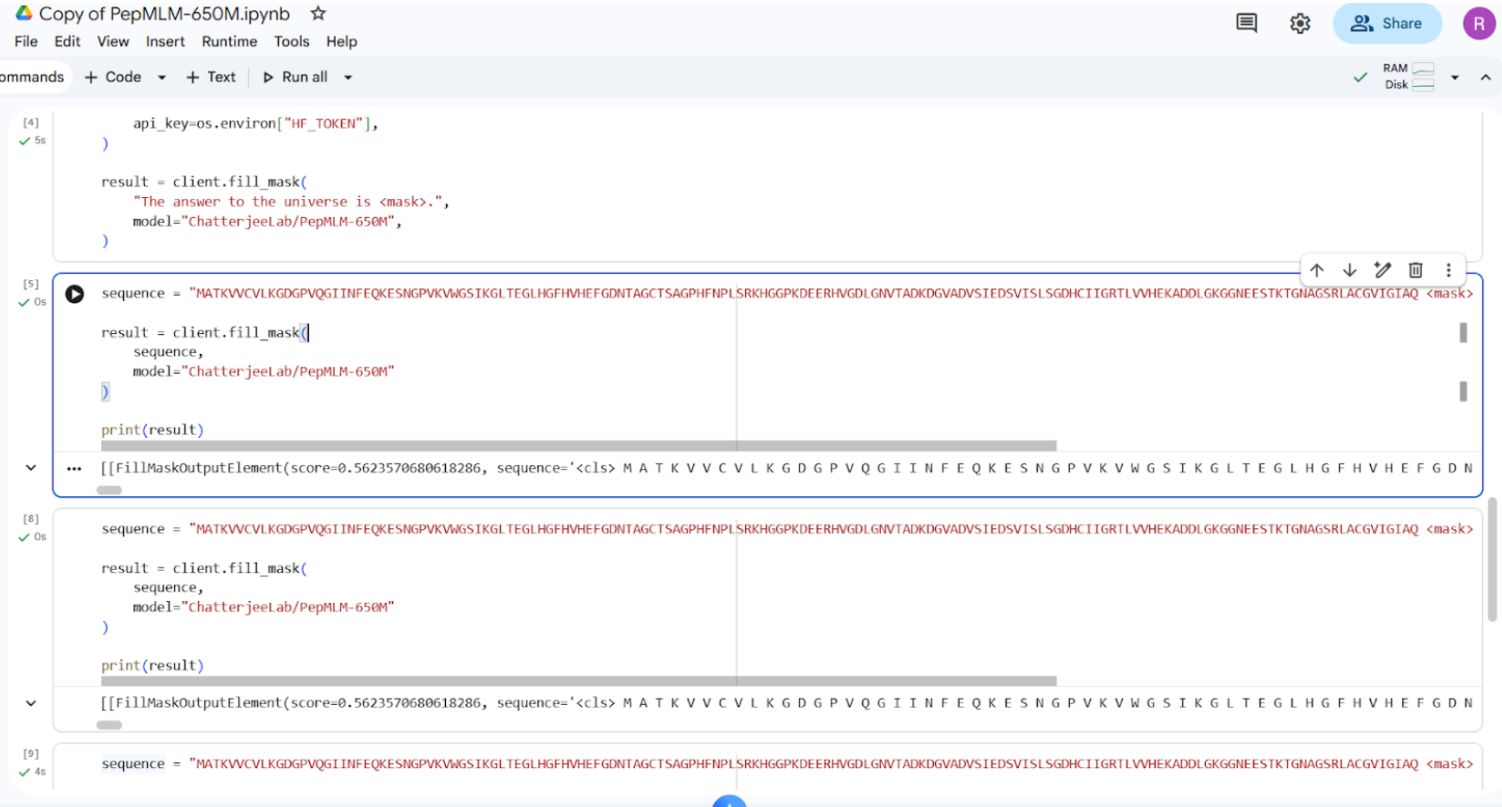
Snapshot of the output (of a particular section, not all)

Final generated peptides and control sequence is as follows:
| Peptide | Sequence |
|---|---|
| Pep1 | WRYYAVAAAHKX |
| Pep2 | WRYYAVAAAHKX |
| Pep3 | WRYYAVAAAHKX |
| Pep4 | WRYYAVAAAHKX |
| Control | FLYRWLPSRRGG |
PepMLM Token Prediction Scores:
| Position | Amino Acid | Score |
|---|---|---|
| 1 | W | 0.562357 |
| 2 | R | 0.230632 |
| 3 | Y | 0.458953 |
| 4 | Y | 0.257805 |
| 5 | A | 0.329096 |
| 6 | V | 0.214972 |
| 7 | A | 0.337871 |
| 8 | A | 0.136613 |
| 9 | A | 0.123724 |
| 10 | H | 0.186813 |
| 11 | K | 0.268938 |
| 12 | X | 0.243224 |
Part 2: Evaluate Binders with AlphaFold3
Submission to AlphaFold Server
The mutant A4V SOD1 FASTA sequence was submitted to the AlphaFold Server. For each test, the SOD1 mutant sequence was entered as the first chain, followed by the peptide sequence as the second chain to model the protein–peptide complex.
The following image shows the submission of SOD1 mutant sequence to the AlphaFold Server:
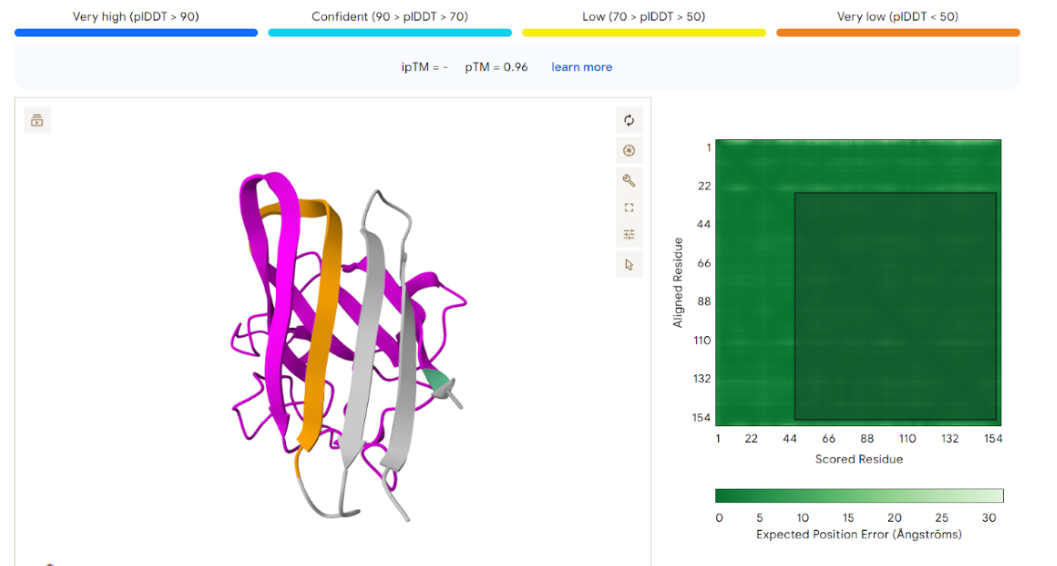
The result generated through this submission is as follows:
- Peptide 1 Evaluation
Original PepMLM Sequence
WRYYAVAAAHKX
Because X represents an unknown amino acid, it was replaced with E (Glutamic acid) before submission to AlphaFold:
Final peptide used:
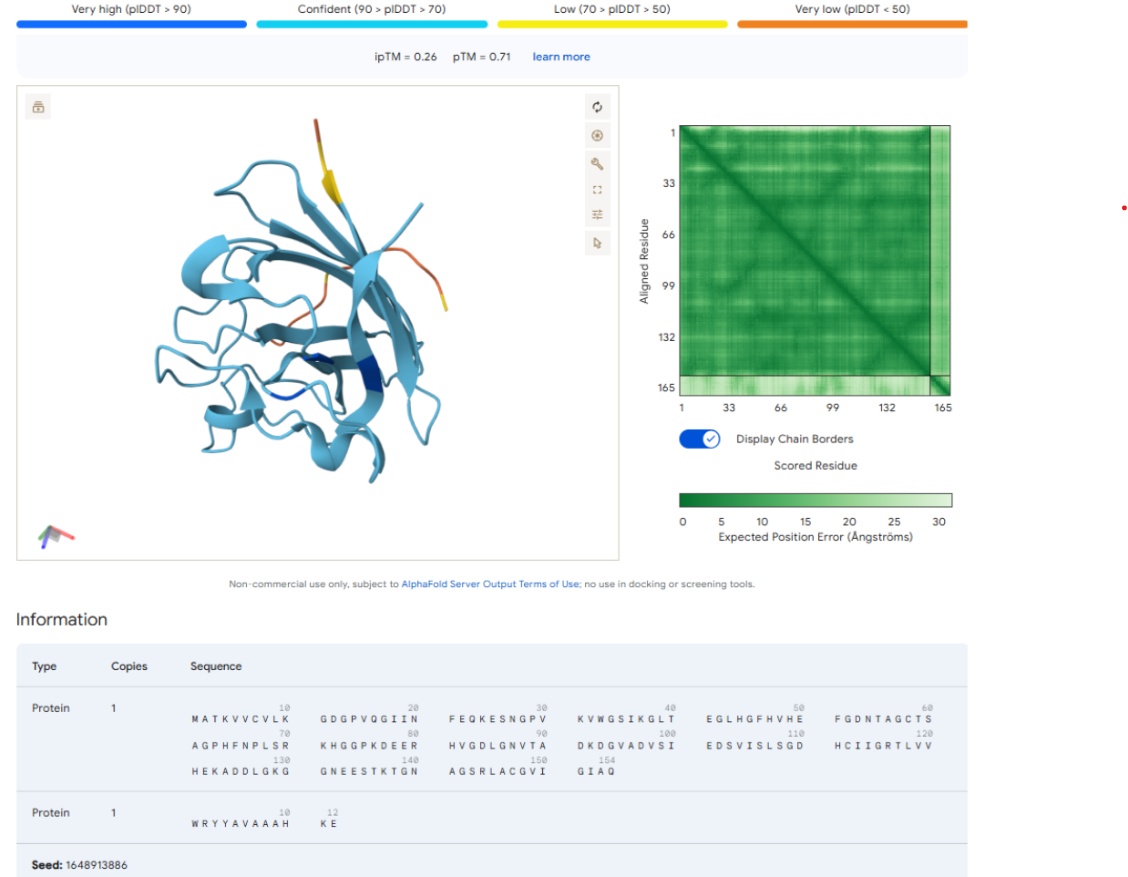
AlphaFold Scores
| Metric | Value |
|---|---|
| ipTM | 0.26 |
| pTM | 0.71 |
Structural Observation
The AlphaFold prediction produced an ipTM score of 0.26 and a pTM score of 0.71. The pTM value indicates that the overall SOD1 protein structure is predicted with reasonable confidence. However, the very low ipTM score suggests weak or negligible interaction between the peptide and SOD1.
Visualization of the predicted complex shows that the peptide is loosely positioned on the surface of the protein and does not form a clear binding interface. The peptide does not appear to localize near the N-terminal region where the A4V mutation occurs. Additionally, it does not penetrate the β-barrel core or interact with the dimer interface of the protein.
This result suggests that the PepMLM-generated peptide is unlikely to bind strongly to mutant SOD1.
- Control Peptide Evaluation
Control Sequence
FLYRWLPSRRGG
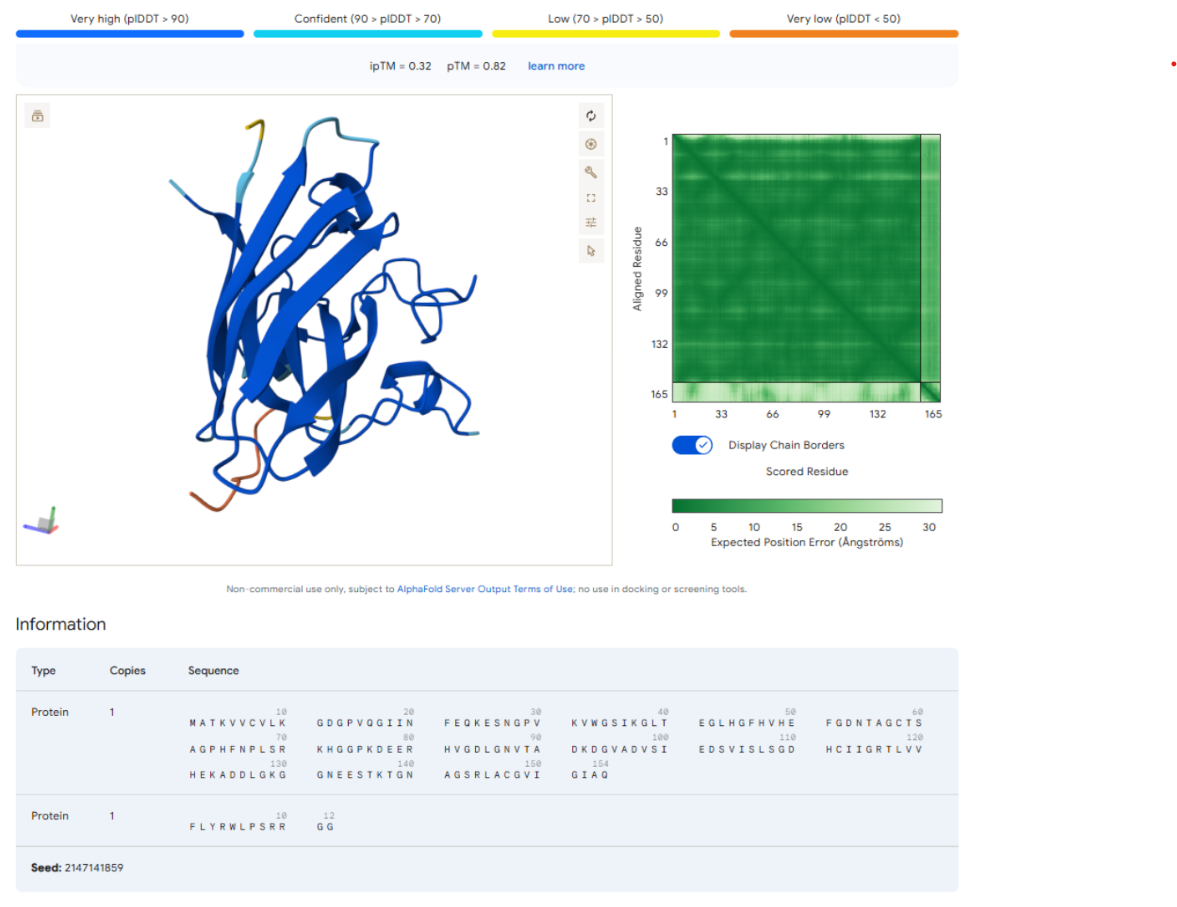
AlphaFold Scores
| Metric | Value |
|---|---|
| ipTM | 0.32 |
| pTM | 0.82 |
Structural Observation
The AlphaFold prediction for the control peptide produced an ipTM score of 0.32 and a pTM score of 0.82. The relatively high pTM value indicates that the overall SOD1 protein structure was predicted with high confidence, consistent with its known β-barrel fold.
However, the ipTM score remains relatively low, suggesting weak or unreliable interaction between the peptide and SOD1. Visualization of the predicted complex shows that the peptide is positioned along the outer surface of the protein rather than forming a well-defined binding pocket.
The peptide does not localize near the N-terminal region containing the A4V mutation and does not strongly engage the β-barrel core or the dimer interface. Instead, the peptide remains largely surface-bound, suggesting that the interaction may be nonspecific or transient.
- Summary of AlphaFold Results
| Peptide | Sequence | ipTM | Binding Observation |
|---|---|---|---|
| PepMLM peptide | WRYYAVAAAHKE | 0.26 | Peptide appears loosely positioned on the surface of SOD1 and does not form a well-defined binding interface. It does not localize near the A4V mutation site. |
| Control peptide | FLYRWLPSRRGG | 0.32 | Peptide remains surface-bound and does not strongly interact with the β-barrel core or dimer interface. |
- Binding Site Analysis
| Region | Observation |
|---|---|
| N-terminus (A4V site) | Peptide does not bind near this region |
| β-barrel core | Peptide does not penetrate the barrel |
| Dimer interface | Peptide does not appear positioned between monomers |
| Protein surface | Peptide appears loosely surface-bound |
- Final Interpretation
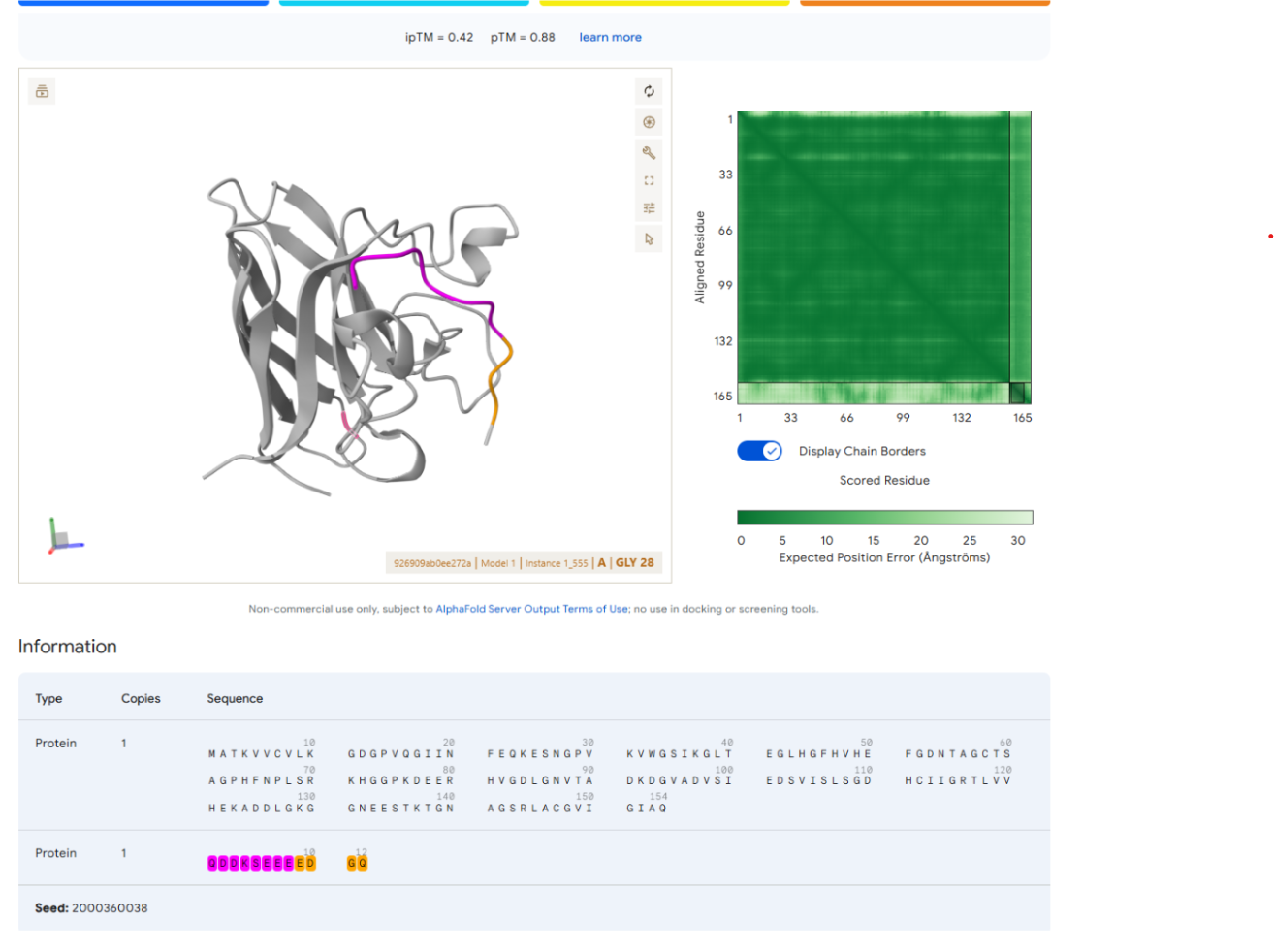
The AlphaFold predictions produced relatively low ipTM scores for both peptides, indicating weak predicted interactions with the SOD1 protein. The PepMLM-generated peptide (WRYYAVAAAHKE) showed an ipTM value of 0.26, suggesting very little confidence in a stable binding interface. The control peptide (FLYRWLPSRRGG) produced a slightly higher ipTM value of 0.32, but this value is still below the threshold typically associated with reliable protein–peptide interactions.
Visualization of the predicted complexes shows that both peptides remain largely surface-bound and do not interact strongly with the N-terminal A4V mutation site, the β-barrel core, or the dimer interface. None of the PepMLM-generated peptides matched or exceeded the predicted binding strength of the control peptide, and both peptides appear to form weak and nonspecific interactions with SOD1.
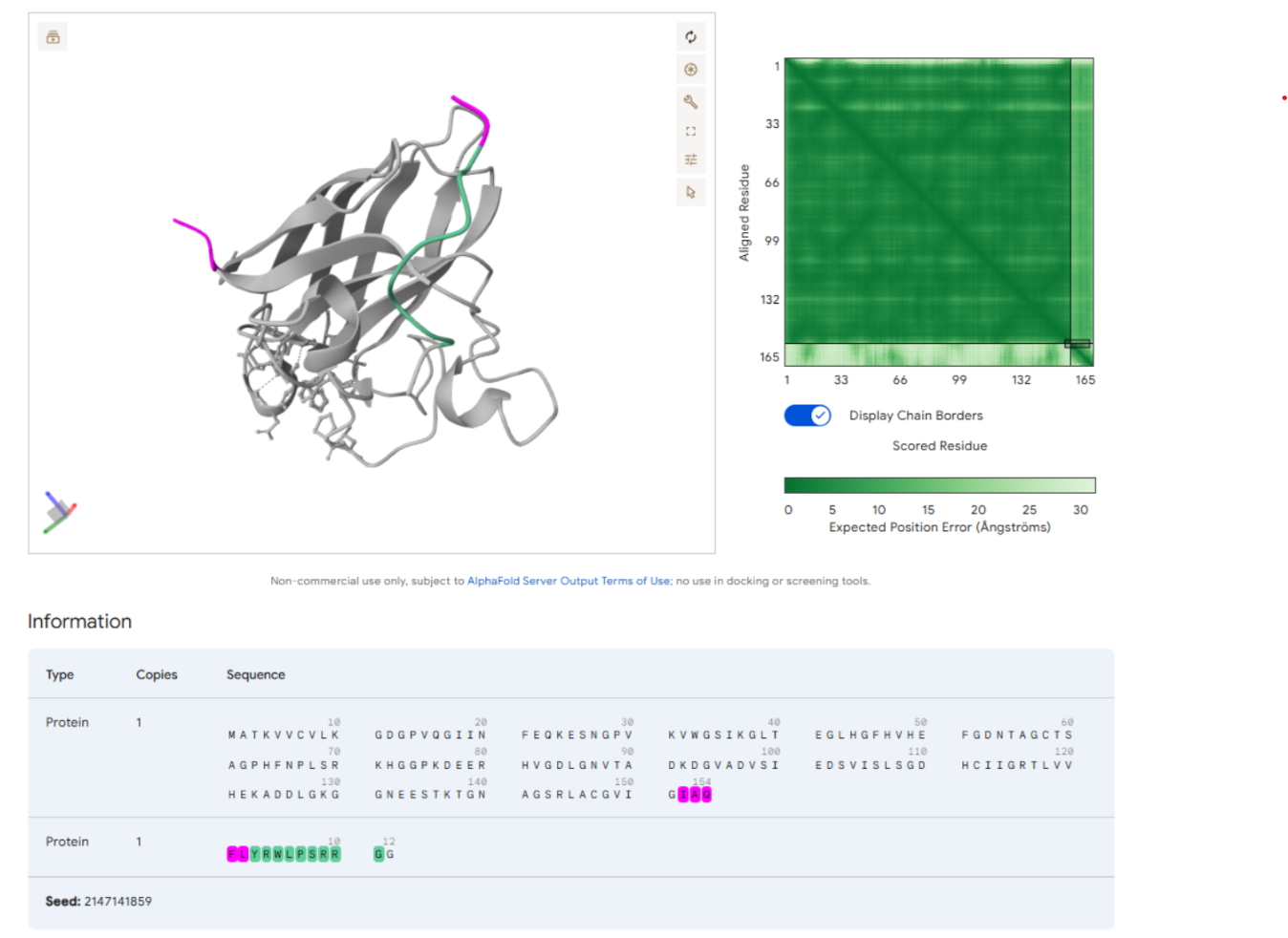
Highlighting the N-terminus Region
To further examine the predicted binding location, the N-terminal region of the SOD1 protein, which contains the A4V mutation, was highlighted in the AlphaFold structure. This visualization allowed for direct observation of whether the peptide interacts with or binds near this mutation site.
Upon inspection of the predicted complex, the peptide does not localize near the N-terminal region and does not appear to form interactions with residues surrounding the A4V mutation. Instead, the peptide remains positioned on the outer surface of the protein, away from the mutation site. This observation suggests that the peptide is unlikely to specifically target the A4V region of the mutant SOD1 protein.
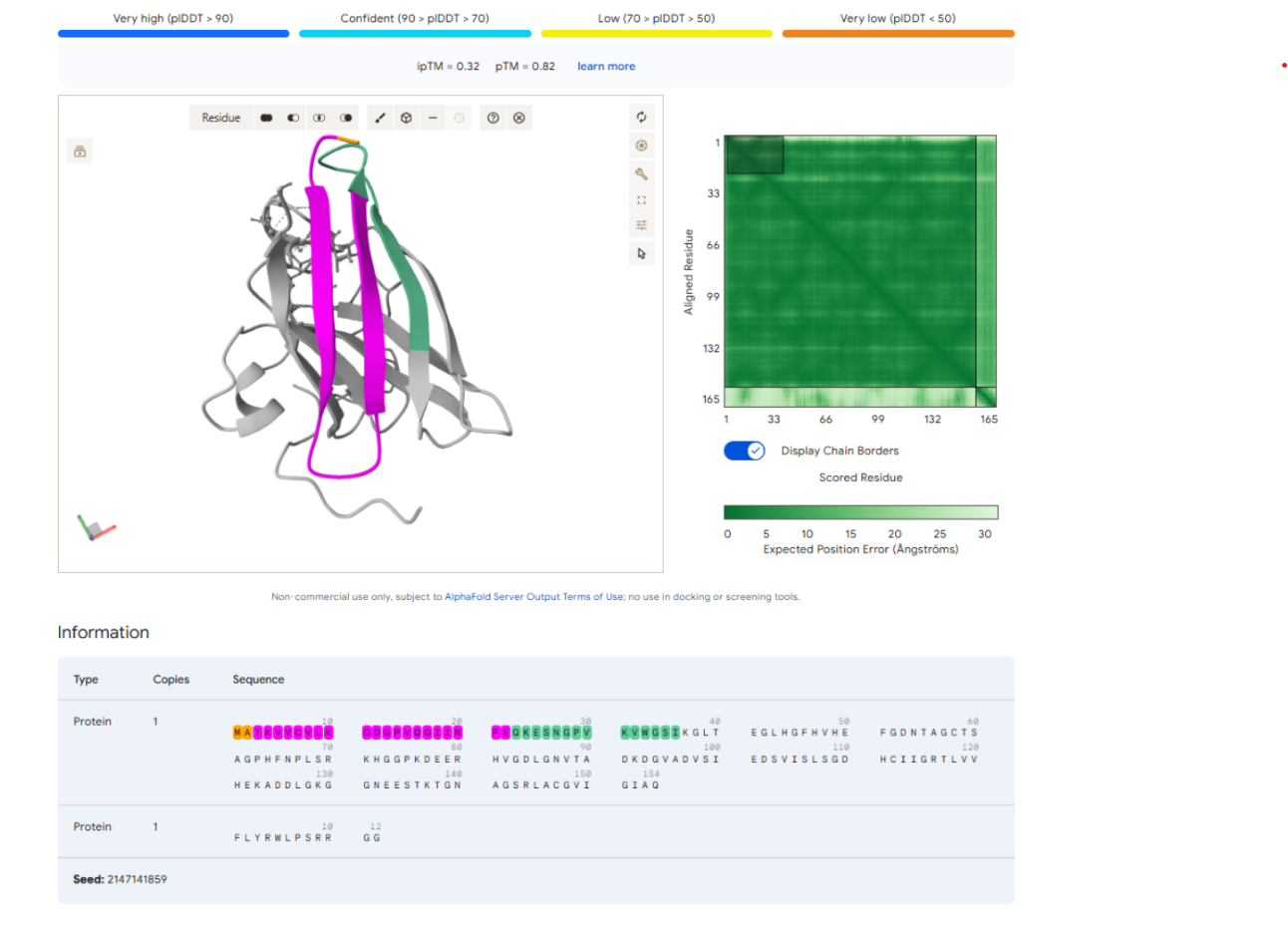
Highlighting the Control Peptide Sequence
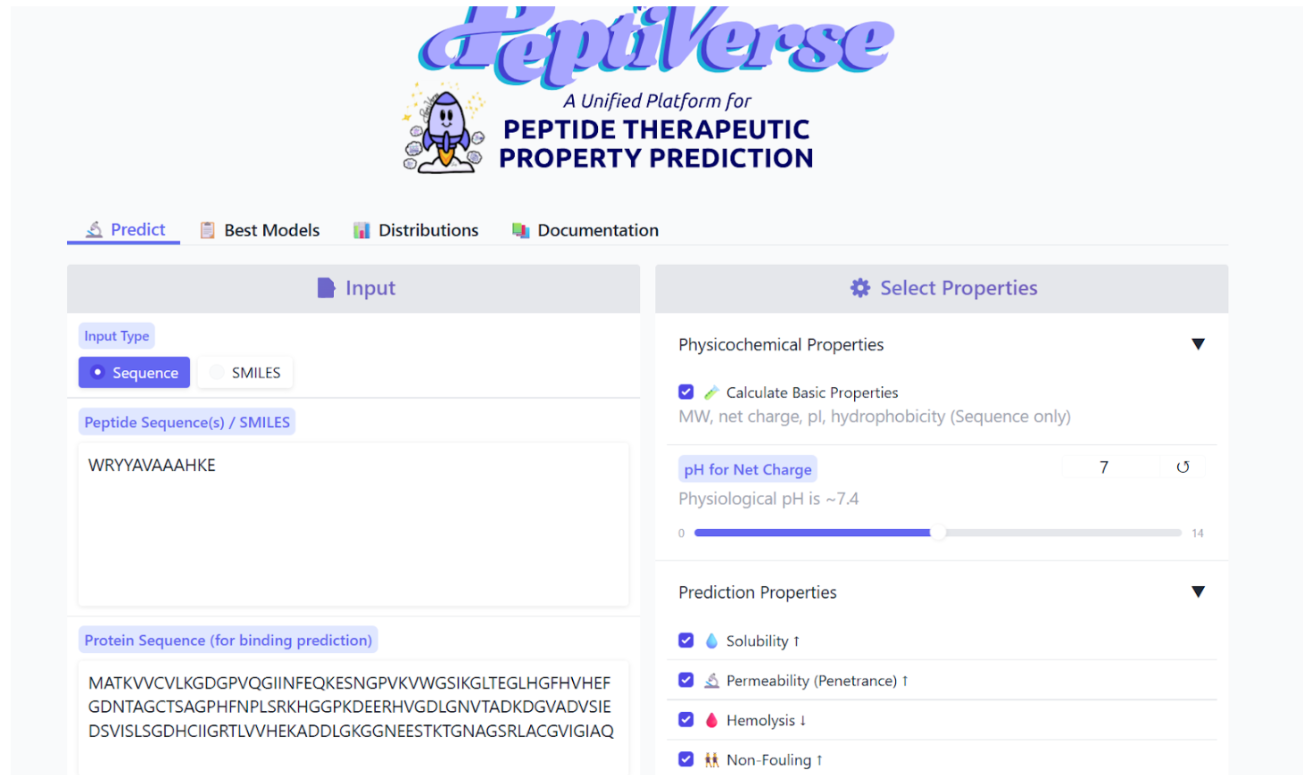
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
The therapeutic properties of the generated peptides were analyzed using the PeptiVerse platform.
Results obtained:
Therapeutic Property Evaluation Using PeptiVerse
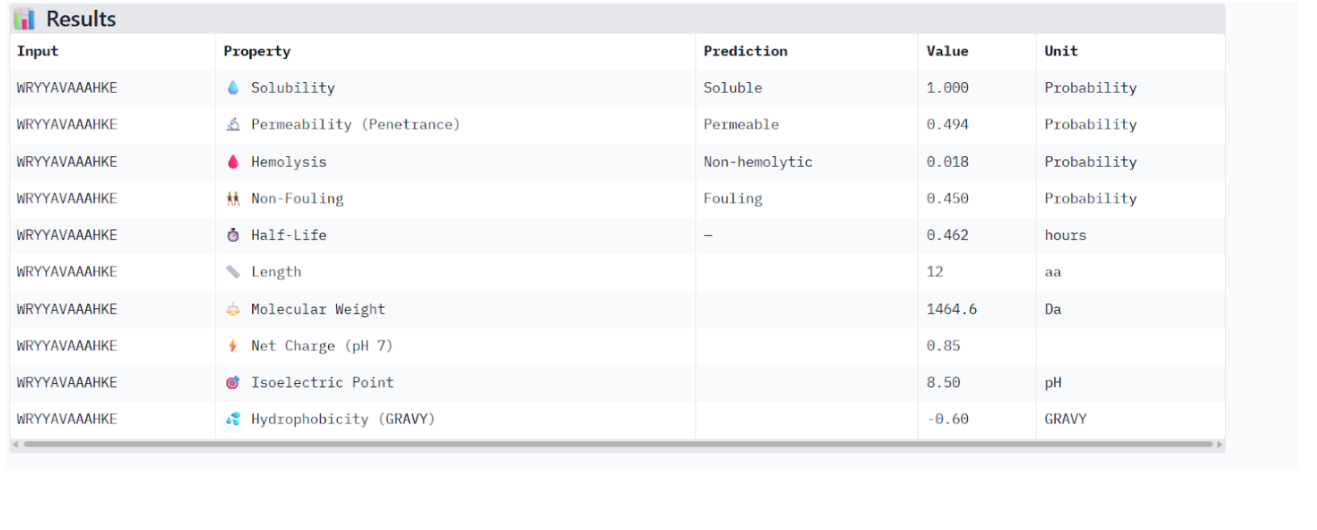
The peptide WRYYAVAAAHKE was further analyzed using PeptiVerse to evaluate its potential therapeutic properties. The peptide sequence and the A4V mutant SOD1 sequence were provided as inputs, and several relevant properties were predicted.
Predicted Peptide Properties
| Property | Predicted Value |
|---|---|
| Solubility Probability | 1.00 |
| Hemolysis Probability | 0.018 |
| Net Charge (pH 7) | 0.85 |
| Molecular Weight | 1464.6 Da |
| GRAVY Hydrophobicity | −0.60 |
| Cell Permeability | 0.494 |
| Estimated Half-Life | ~0.46 hours |
The peptide is predicted to be highly soluble, which is a desirable property for therapeutic peptides. It also shows a very low hemolysis probability, suggesting that it is unlikely to damage red blood cells. The moderate molecular weight and near-neutral net charge may support reasonable biological compatibility.
The GRAVY hydrophobicity score of −0.60 indicates that the peptide is relatively hydrophilic, which aligns with the predicted high solubility. However, the predicted cell permeability is moderate, and the estimated half-life of approximately 0.46 hours suggests limited stability in biological environments.
Comparison of Structural and Therapeutic Predictions
When comparing the structural predictions with the therapeutic property analysis, the results appear consistent. The low ipTM value from AlphaFold3 indicates weak predicted binding between the peptide and SOD1, and the structural visualization supports this by showing a surface-bound peptide without a well-defined binding interface.
Although the peptide does not demonstrate strong predicted binding affinity, it does not exhibit problematic therapeutic properties, such as high hemolysis risk or poor solubility, which are common limitations in peptide drug candidates.
Peptide Selection for Advancement
WRYYAVAAAHKE represents a reasonable peptide candidate to advance for further study. While its predicted binding strength to SOD1 is relatively weak, it demonstrates favorable therapeutic characteristics, including high solubility, low hemolysis probability, and acceptable physicochemical properties.
Future optimization approaches, such as targeted peptide redesign or guided peptide generation methods, could potentially improve binding affinity while preserving these favorable therapeutic traits.
Part 4: Generate Optimized Peptides with moPPIt
The given mutant sequence was used to generate the optimized peptide:
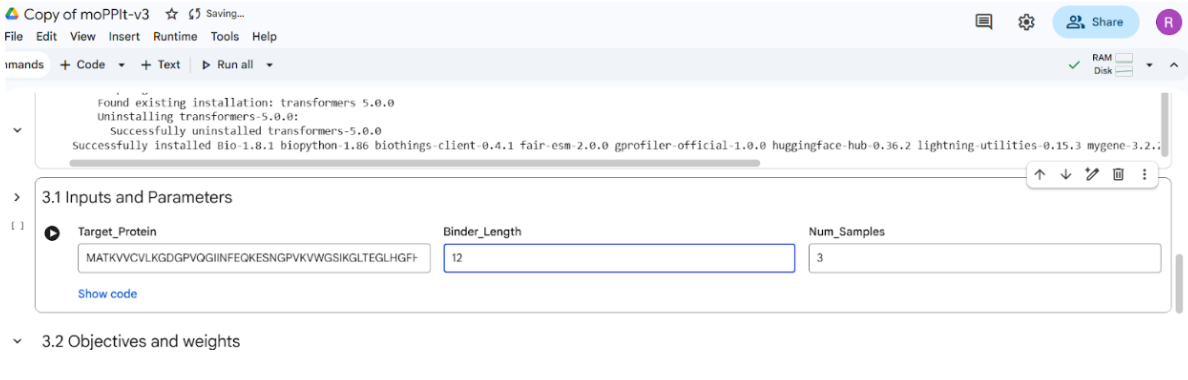
The motif positions were set to residues 1–10 during peptide generation. Additionally, only three optimization properties were selected in the notebook because the computation was performed on a T4 GPU in Google Colab, which has limited computational resources. Reducing the number of selected properties helped ensure that the notebook ran efficiently within the available GPU memory and runtime constraints.
It took >40 mins to implement the code

moPPIt Generated Peptides
The model generated three candidate peptides with predicted values for solubility, binding affinity, and motif score.
| Binder | Solubility | Predicted Affinity | Motif Score |
|---|---|---|---|
| YNQKYSQCKYAC | 0.9167 | 6.42 | 0.68 |
| IKYINQKLKELR | 0.6667 | 7.18 | 0.75 |
| QDDKSEEEEDGQ | 1.00 | 4.70 | 0.34 |
Comparison of moPPIt Peptides vs PepMLM Peptide
The moPPIt binder predictions produced three peptide candidates with varying physicochemical and predicted binding properties.
| Peptide | Solubility | Predicted Affinity | Motif Score |
|---|---|---|---|
| YNQKYSQCKYAC | 0.9167 | 6.42 | 0.68 |
| IKYINQKLKELR | 0.6667 | 7.18 | 0.75 |
| QDDKSEEEEDGQ | 1.00 | 4.70 | 0.34 |
For comparison, the PepMLM-generated peptide (WRYYAVAAAHKE) evaluated earlier showed:
Excellent solubility (1.0)
Very low hemolysis probability (0.018), indicating favorable therapeutic safety
However, AlphaFold3 predicted weak structural binding with an ipTM ≈ 0.26, suggesting low confidence in stable interaction with the SOD1 A4V protein.
In contrast, the moPPIt peptides show higher predicted binding affinity scores (4.7–7.18), suggesting stronger potential interaction with the target protein compared to the PepMLM peptide. However, the moPPIt peptides vary more in solubility. For example, IKYINQKLKELR shows only moderate solubility (0.67), which could potentially impact therapeutic delivery.
The moPPIt peptides appear optimized for binding affinity, whereas the PepMLM peptide appears optimized for favorable therapeutic properties, such as solubility and safety.
Evaluation Before Clinical Advancement
Before advancing any of these peptides to clinical studies, several additional evaluations would be necessary.
- Structural Validation
Further structural analysis should be performed using tools such as AlphaFold3 or molecular docking to confirm the predicted binding interface with the A4V mutant SOD1 protein. This would help determine whether the peptide binds near the N-terminal A4V mutation site, the β-barrel region, or the dimer interface.
- Binding Affinity Testing
Experimental assays such as surface plasmon resonance (SPR) or isothermal titration calorimetry (ITC) should be performed to measure the actual binding strength between the peptide and the SOD1 protein.
- Stability and Pharmacokinetics
Peptides should be evaluated for serum stability and biological half-life. Additional studies should assess protease resistance and degradation rates to determine whether the peptide remains stable in physiological conditions.
- Toxicity and Safety
Safety evaluation is essential before clinical use. Experiments should test hemolysis, cytotoxicity, and potential immunogenic responses in relevant cell culture models.
- Functional Assays
Functional assays should determine whether the peptide can reduce aggregation or toxicity of mutant SOD1, which is an important mechanism in ALS therapeutic development.
Interpretation The moPPIt peptides demonstrate stronger predicted binding affinity, particularly IKYINQKLKELR, which shows the highest affinity and motif score among the generated candidates. However, the PepMLM peptide shows superior solubility and safety predictions.
An ideal therapeutic peptide would balance strong binding affinity with favorable physicochemical and safety properties. Therefore, further computational validation and experimental testing would be required to determine which peptide candidate provides the best overall balance of binding performance, stability, and therapeutic safety.
Visualization of moPPIt Peptides
- YNQKYSQCKYAC
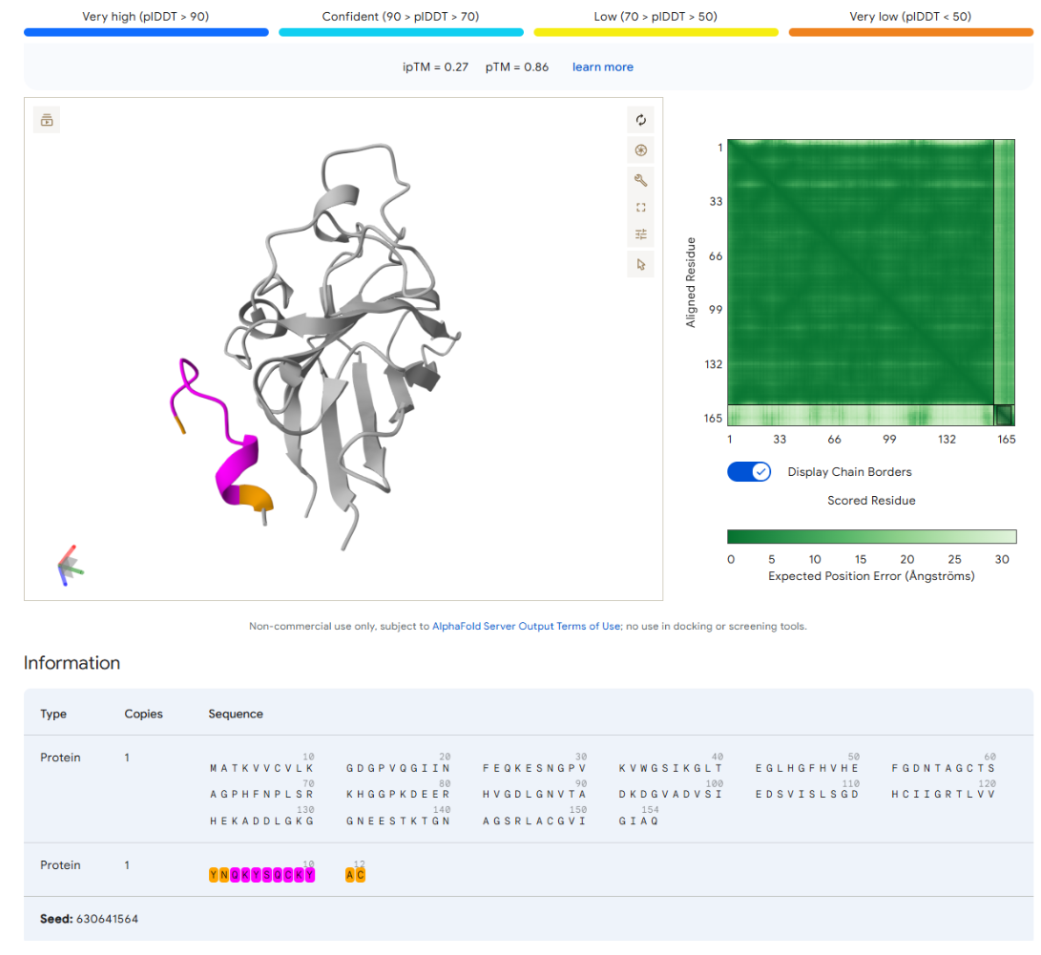
- IKYINQKLKELR
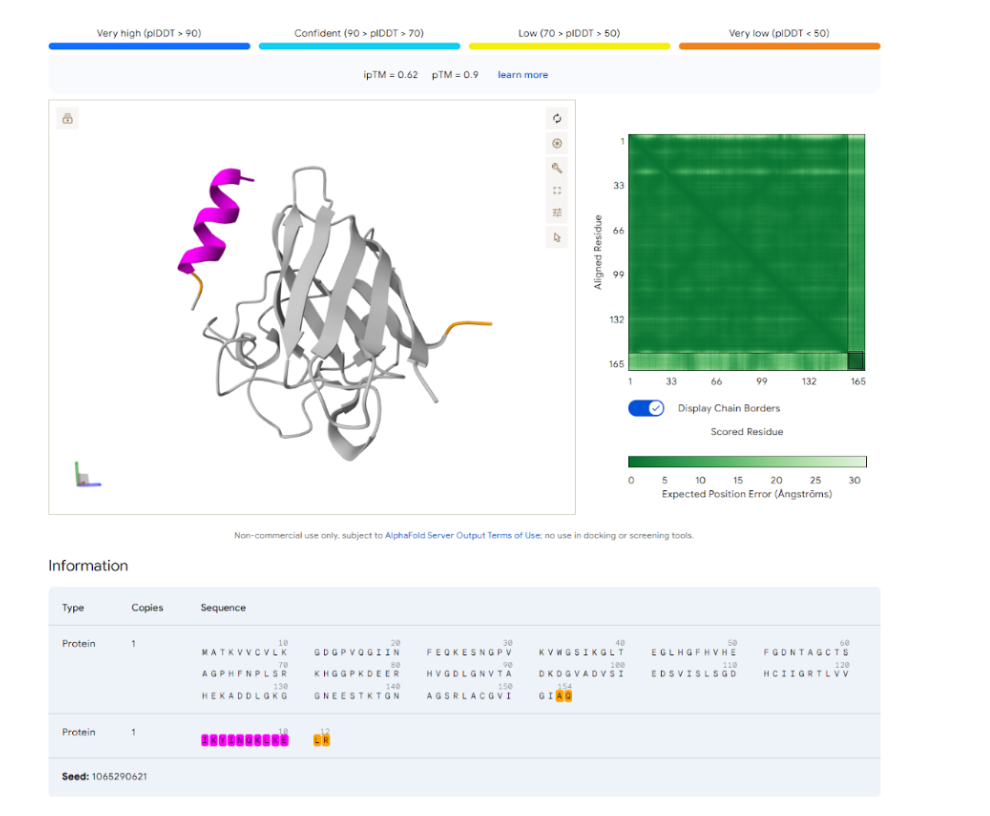
- QDDKSEEEEDGQ
FINAL GROUP PROJECT Phage Lysis Protein Design Challenge
- Introduction
Bacteriophage lysis proteins are responsible for disrupting the host bacterial membrane during phage infection, allowing the release of viral particles. The MS2 lysis protein is a small membrane-associated protein composed of 75 amino acids and contains two major functional regions:
| Domain | Residues | Function |
|---|---|---|
| Soluble domain | 1–40 | Interaction with host chaperone protein DnaJ |
| Transmembrane helix | 41–75 | Membrane insertion and pore formation |
Lysis Protein Sequence
Design Objective
Design five mutations in the lysis protein:
2 mutations in the soluble region
2 mutations in the transmembrane region
1 mutation anywhere in the sequence
These mutations should preserve protein function while potentially improving stability or membrane activity.
- Evolutionary Analysis
2.1 Protein BLAST
Homologous sequences for the MS2 lysis protein were obtained using Protein BLAST.
The sequences were downloaded in FASTA format and used for multiple sequence alignment.
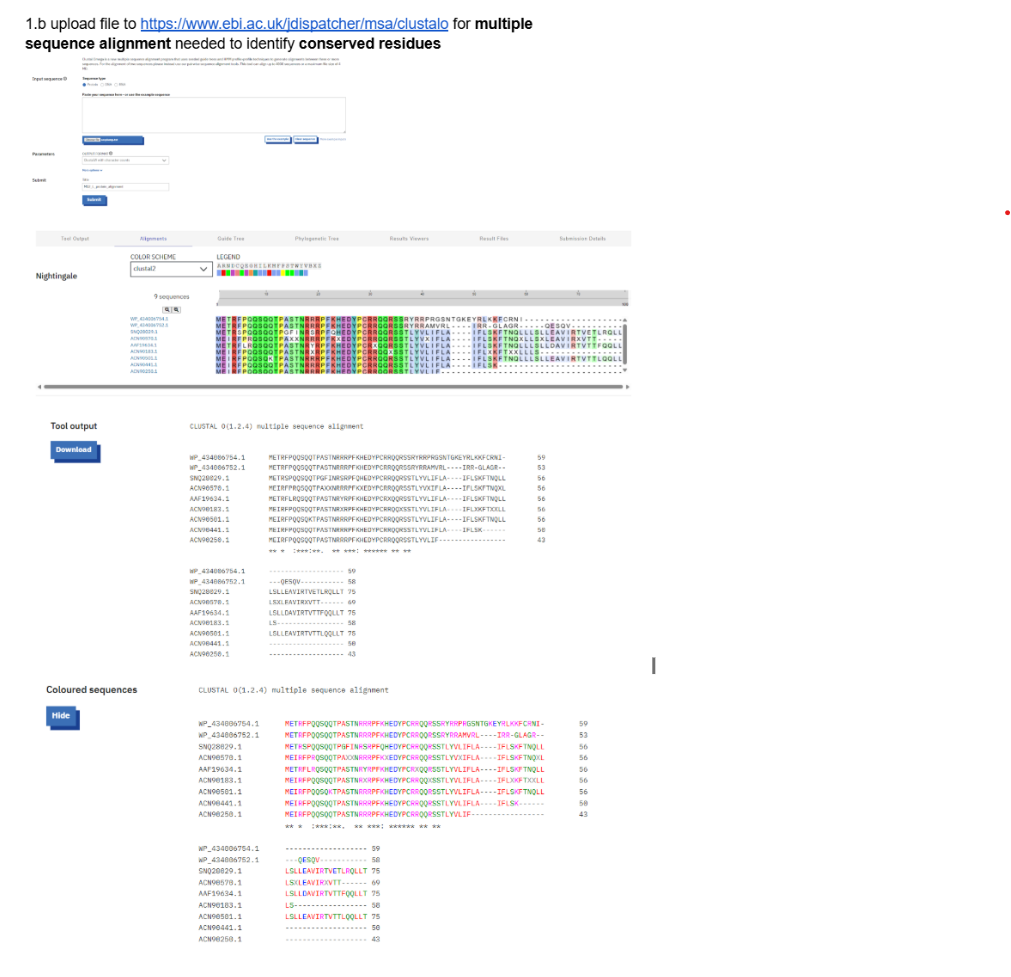
2.2 Multiple Sequence Alignment
Multiple sequence alignment was performed using Clustal Omega.
Tool used:
Homologous sequences used
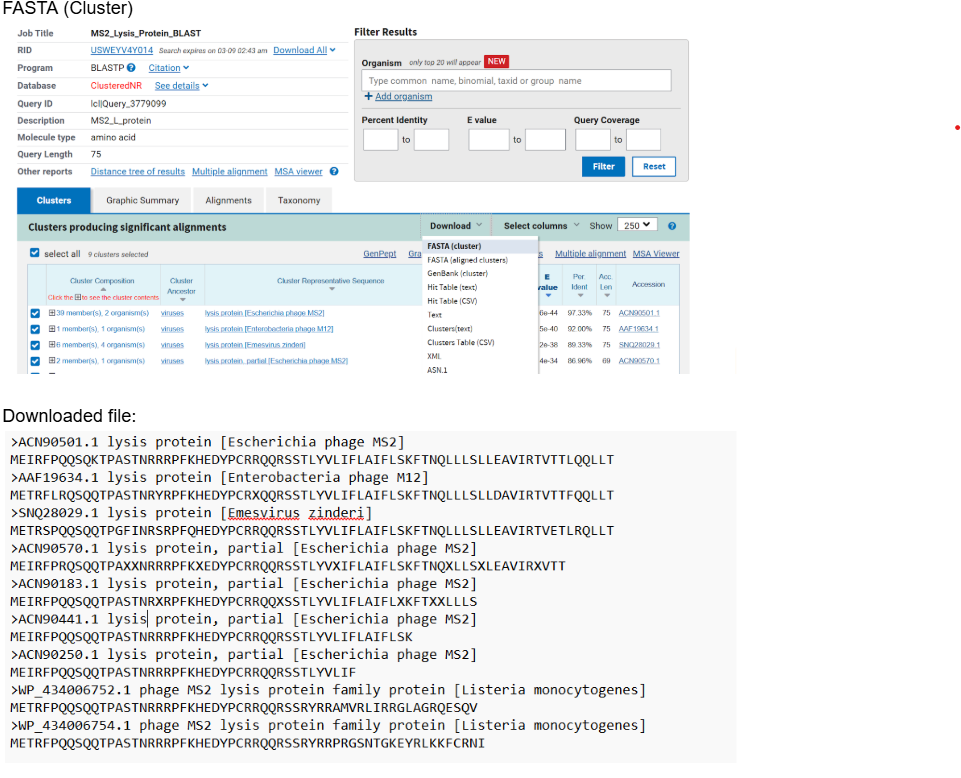
WP_434006754.1
WP_434006752.1
SNQ28029.1
ACN90570.1
AAF19634.1
ACN90183.1
ACN90501.1
ACN90441.1
ACN90250.1
These sequences represent related phage lysis proteins.
After, running the BLAST- downloaded the FASTA(CLUSTER) FILE:
- Conservation Analysis
Clustal Omega indicates conservation using the following symbols:
| Symbol | Meaning |
|---|---|
| * | Fully conserved residue |
| : | Strongly conserved |
| . | Weakly conserved |
Example conservation pattern:
Key Conserved Motifs
Highly conserved motifs observed in the alignment include:
These residues are likely essential for structural stability or host protein interaction, particularly with DnaJ.
Therefore, fully conserved residues should not be mutated.
- Variable Regions
Regions showing substitutions or alignment gaps indicate evolutionary variability.
Example variable region:
Variation is also observed in the C-terminal region, where some sequences contain truncations or insertions.
Implication
Variable regions are better candidates for mutational engineering because they are less likely to disrupt protein function.
- Domain Analysis
The MS2 lysis protein contains two main structural regions:
| Region | Residues | Function |
|---|---|---|
| Soluble domain | 1–40 | Interaction with DnaJ |
| Transmembrane domain | 41–75 | Membrane insertion and pore formation |
- Soluble Region Conservation
The N-terminal soluble domain shows high conservation across homologous sequences.
Example conserved sequence:
Mutations in this region must therefore be chosen carefully.
Candidate mutation sites
| Position | Residue | Reason |
|---|---|---|
| 12 | Q | Weakly conserved |
| 17 | N | Variable among homologs |
| 26 | Y | Moderate variability |
These positions may tolerate substitutions without disrupting protein folding.
- Transmembrane Region Conservation
The C-terminal region forms a transmembrane helix.
Example sequence:
This region is highly hydrophobic, which is required for membrane insertion.
However, conservative substitutions between hydrophobic residues may be tolerated.
Candidate mutation sites
| Position | Residue | Reason |
|---|---|---|
| 52 | L | Hydrophobic substitution possible |
| 55 | I | Minor hydrophobic change |
| 59 | V | Frequently mutated experimentally |
- Key Observations from Alignment
The N-terminal region is highly conserved, indicating functional importance in host interaction.
Some residues in the soluble domain show moderate variability.
The transmembrane region remains hydrophobic but allows conservative substitutions.
Some homologous proteins exhibit C-terminal truncations, suggesting structural flexibility in this region.
- Mutation Design Strategy
- The mutation design followed several biological constraints:
Rules applied
Avoid fully conserved residues
Prefer weakly conserved or variable residues
Maintain hydrophobicity in transmembrane helices
Preserve overall protein folding and stability
- Mutational Scoring Using Protein Language Models
Mutation effects are predicted using protein language models, such as:
ESM-1b
MSA Transformer
ProteinBERT
Mutation scoring used log-likelihood ratio (LLR) values.
LLR Interpretation
| Score | Interpretation |
|---|---|
| > 2 | Very favorable |
| 1–2 | Moderately favorable |
| 0–1 | Weakly favorable |
| < 0 | Unfavorable |
Following image indicates results obtained using Protein Language Models (ESM).ipynb
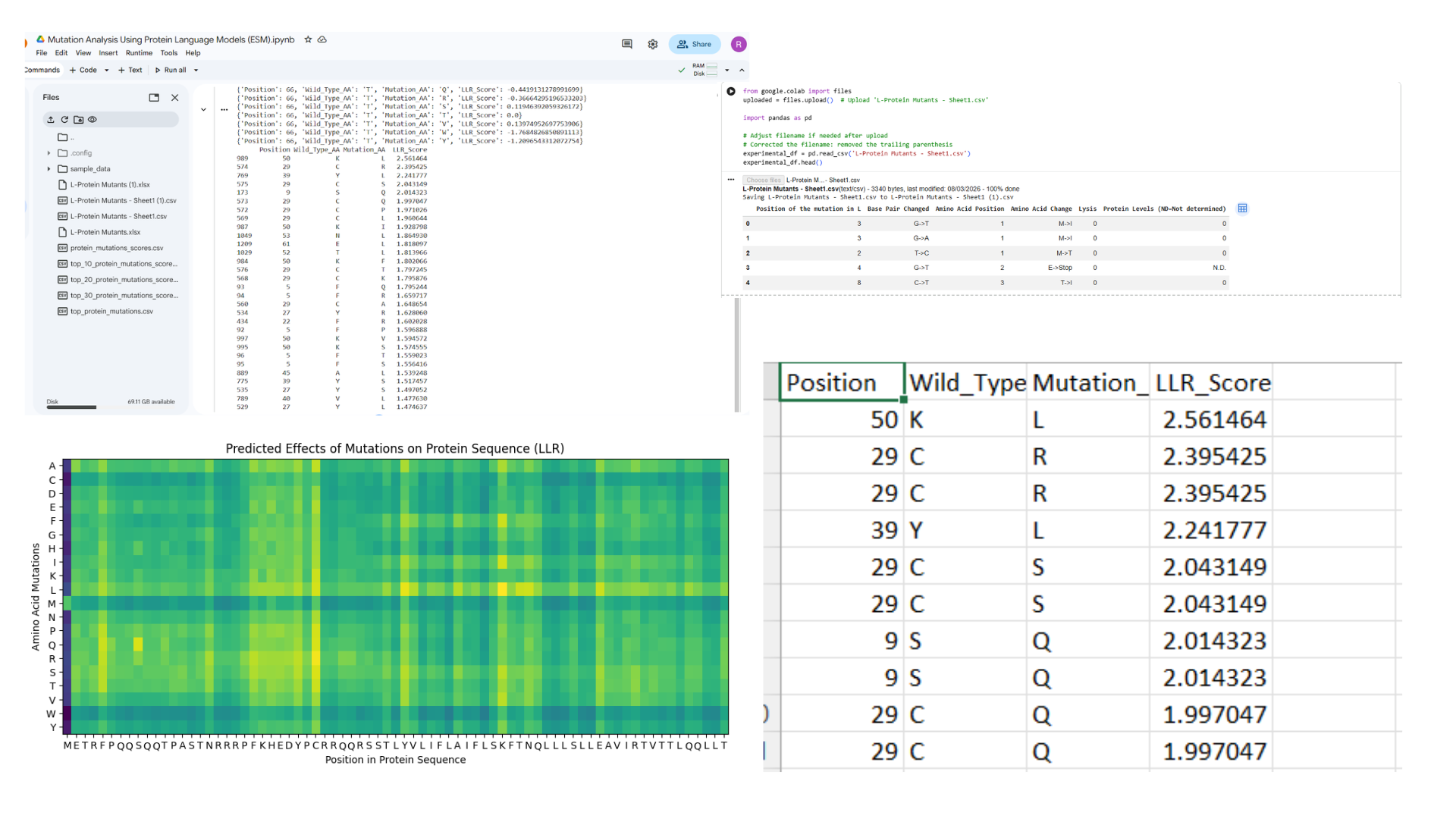
- Top Ranked Mutations
| Position | WT | Mutation | LLR Score |
|---|---|---|---|
| 50 | K | L | 2.56 |
| 29 | C | R | 2.39 |
| 39 | Y | L | 2.24 |
| 29 | C | S | 2.04 |
| 9 | S | Q | 2.01 |
| 53 | N | L | 1.86 |
| 52 | T | L | 1.81 |
| 61 | E | L | 1.81 |
Many favorable mutations convert residues to Leucine (L) because leucine stabilizes membrane helices due to its strong hydrophobicity.
- Mapping Mutations to Protein Regions
Soluble Region (1–40)
| Mutation | Score |
|---|---|
| C29R | 2.39 |
| C29S | 2.04 |
| S9Q | 2.01 |
| Y39L | 2.24 |
| F5Q | 1.79 |
Transmembrane Region (41–75)
| Mutation | Score |
|---|---|
| K50L | 2.56 |
| T52L | 1.81 |
| N53L | 1.86 |
| E61L | 1.81 |
| A45L | 1.53 |
- Biological Filtering
Risky mutations were removed using biological constraints.
Mutations excluded
- C29R
- C29S
Reason: cysteine residues may form structural interactions.
Safer alternatives
- Y39L
- S9Q
- F5Q
- Final Selected Mutations
| Mutation | Region | LLR Score |
|---|---|---|
| S9Q | Soluble | 2.01 |
| Y39L | Soluble | 2.24 |
| K50L | Transmembrane | 2.56 |
| T52L | Transmembrane | 1.81 |
| N53L | Anywhere | 1.86 |
- Mutated Protein Sequence
Original Sequence
Mutated Sequence
Mutations applied:
- S9Q
- Y39L
- K50L
- T52L
- N53L
- Comparison with Experimental Data
Experimental data supports mutational tolerance at several selected positions.
| Mutation | Position | Evidence | Interpretation |
|---|---|---|---|
| S9Q | 9 | No experimental mutation reported | Likely tolerant |
| Y39L | 39 | Y→H mutation reported | Position mutable |
| K50L | 50 | Multiple substitutions observed | Highly tolerant |
| T52L | 52 | Mutation recorded | Mutation tolerated |
| N53L | 53 | Several variants reported | Flexible boundary residue |
These results support the predicted soluble and membrane domain boundaries.
- Structural Prediction Using AlphaFold
The mutated sequence was modeled using AlphaFold Multimer
It required several attempts to successfully obtain a PDB file. Initially, an 8-sequence oligomer model was submitted for prediction; however, the system crashed during the run due to the high computational load. After adjusting the input and rerunning the analysis, a successful prediction was eventually completed and the resulting outputs were documented as follows.
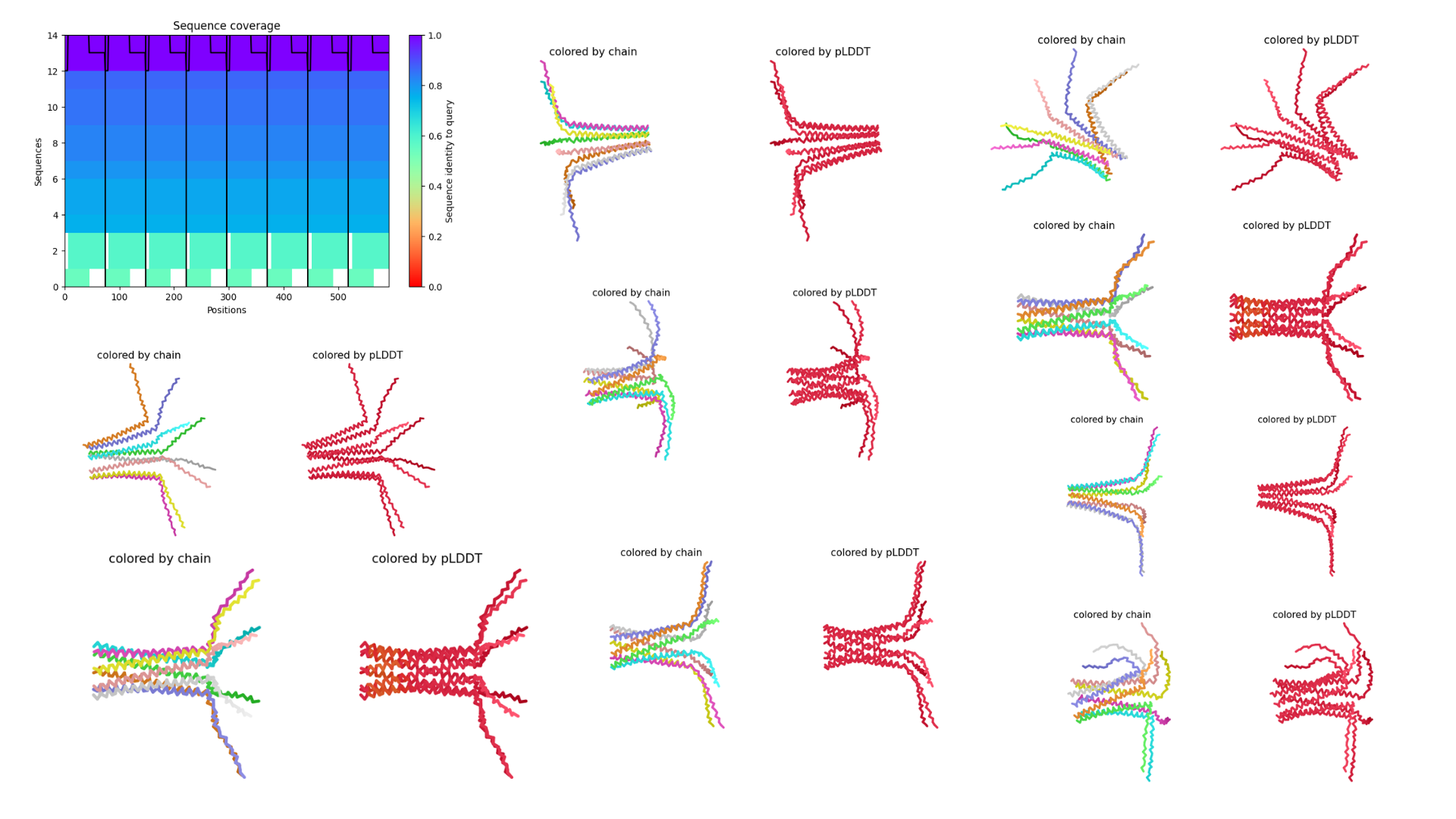
Interpretation
To improve the prediction results, the analysis was repeated using a different input configuration. Instead of running an eight-sequence oligomer model, which previously caused the system to crash, a four-oligomer sequence setup was used. This reduced computational complexity and allowed the prediction to run successfully, enabling the generation of structural outputs for further analysis.
Results obtained:
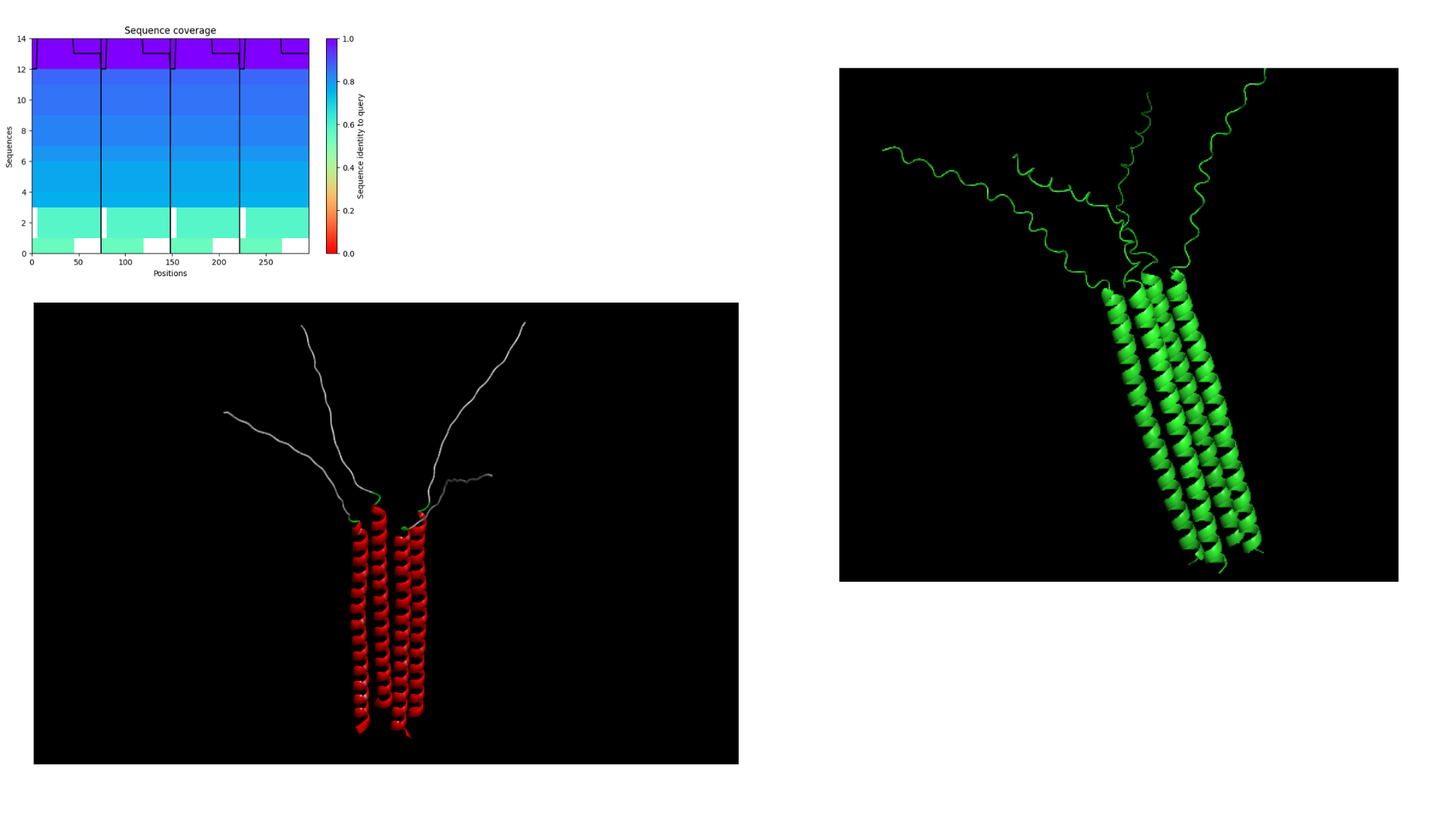
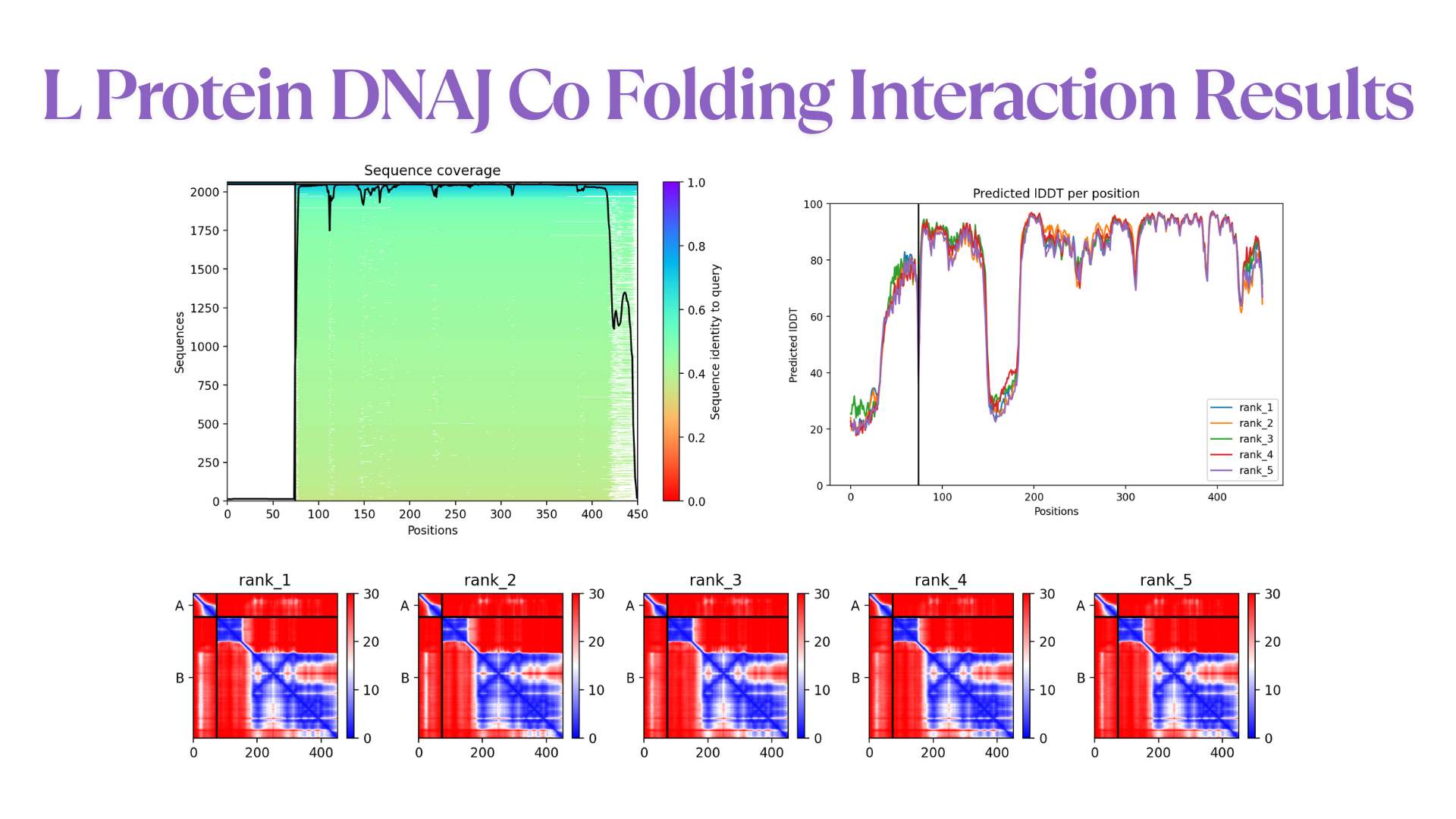
- Co-Folding Analysis
The mutated lysis protein sequence was further analyzed using co-folding simulations with additional protein sequences to investigate potential protein–protein interactions.
Structural visualization tools such as Discovery Studio were used to examine key structural and interaction features, including:
- Hydrogen bonding patterns
- Protein–protein interface interactions
- Membrane insertion orientation
Co-folding simulations were performed using both the AlphaFold Multimer v3 notebook and the AlphaFold Server to compare prediction consistency and interaction confidence across different platforms.
The results obtained from the AlphaFold Server are summarized as follows:
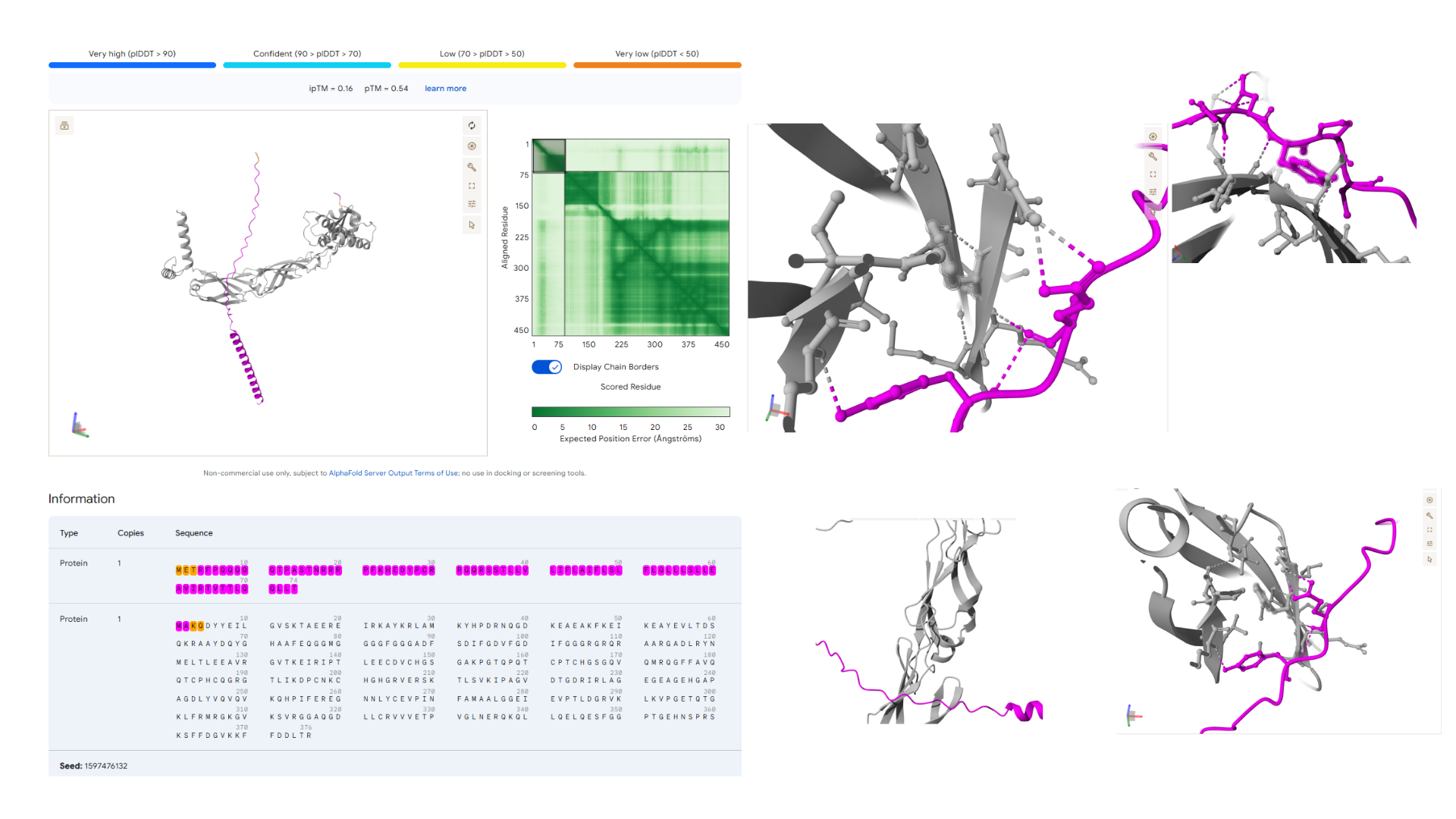
- Conclusion
This study applied evolutionary analysis, protein language models, and structural prediction to design mutations in the MS2 lysis protein.
Key findings:
The N-terminal region is highly conserved and involved in host interaction.
The C-terminal region forms a hydrophobic transmembrane helix.
Protein language model scoring identified favorable mutations.
Biological filtering ensured structural compatibility.
Final designed mutations
- S9Q
- Y39L
- K50L
- T52L
- N53L