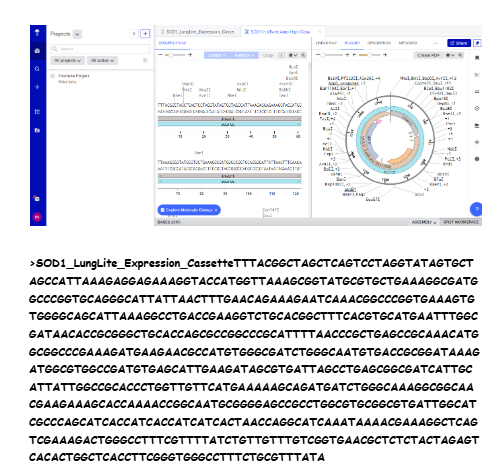
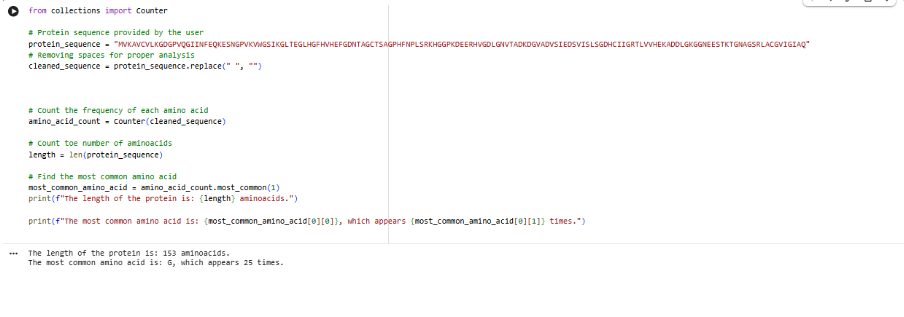
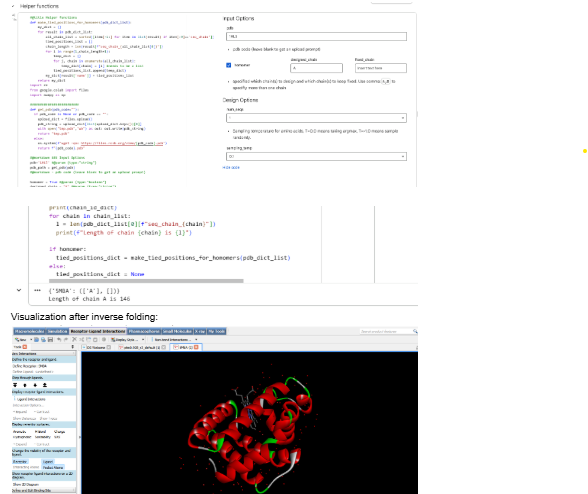
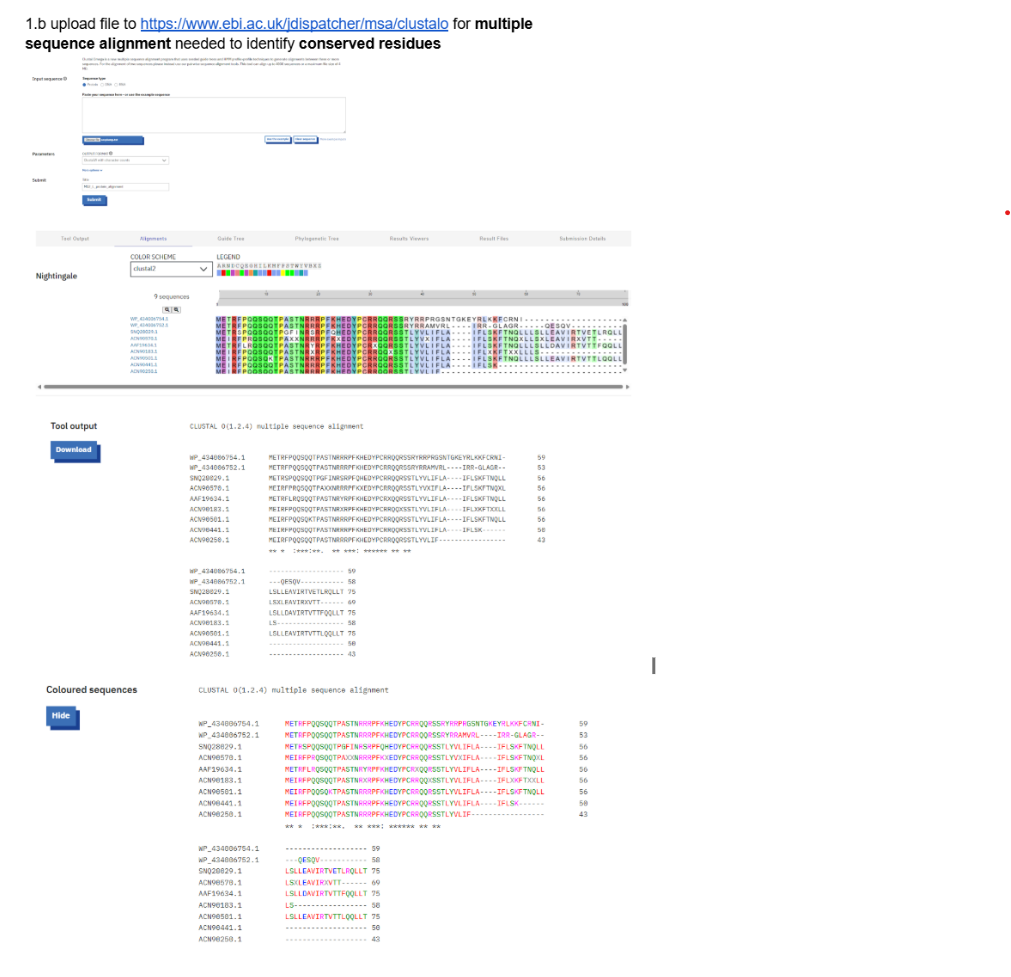
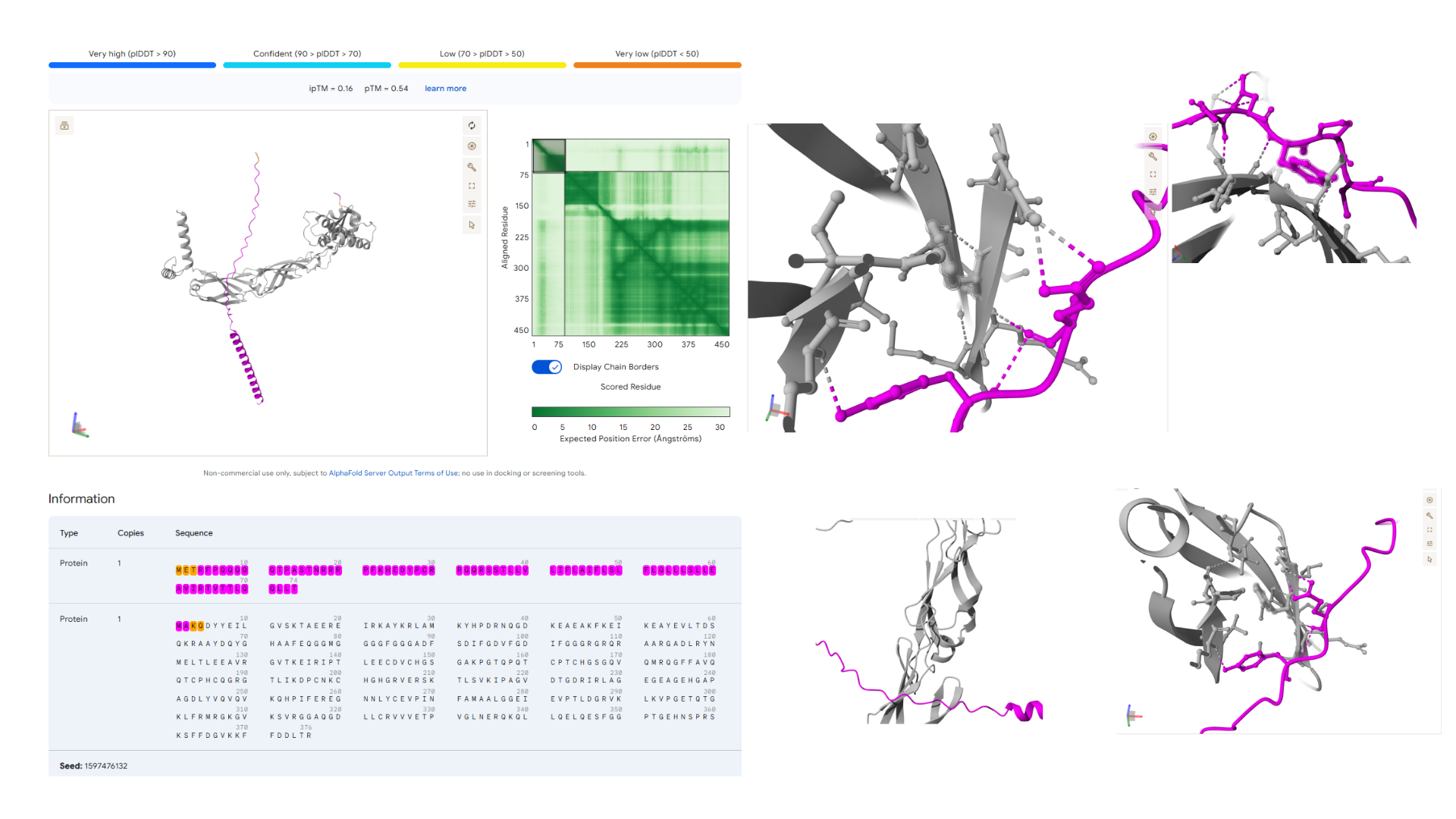
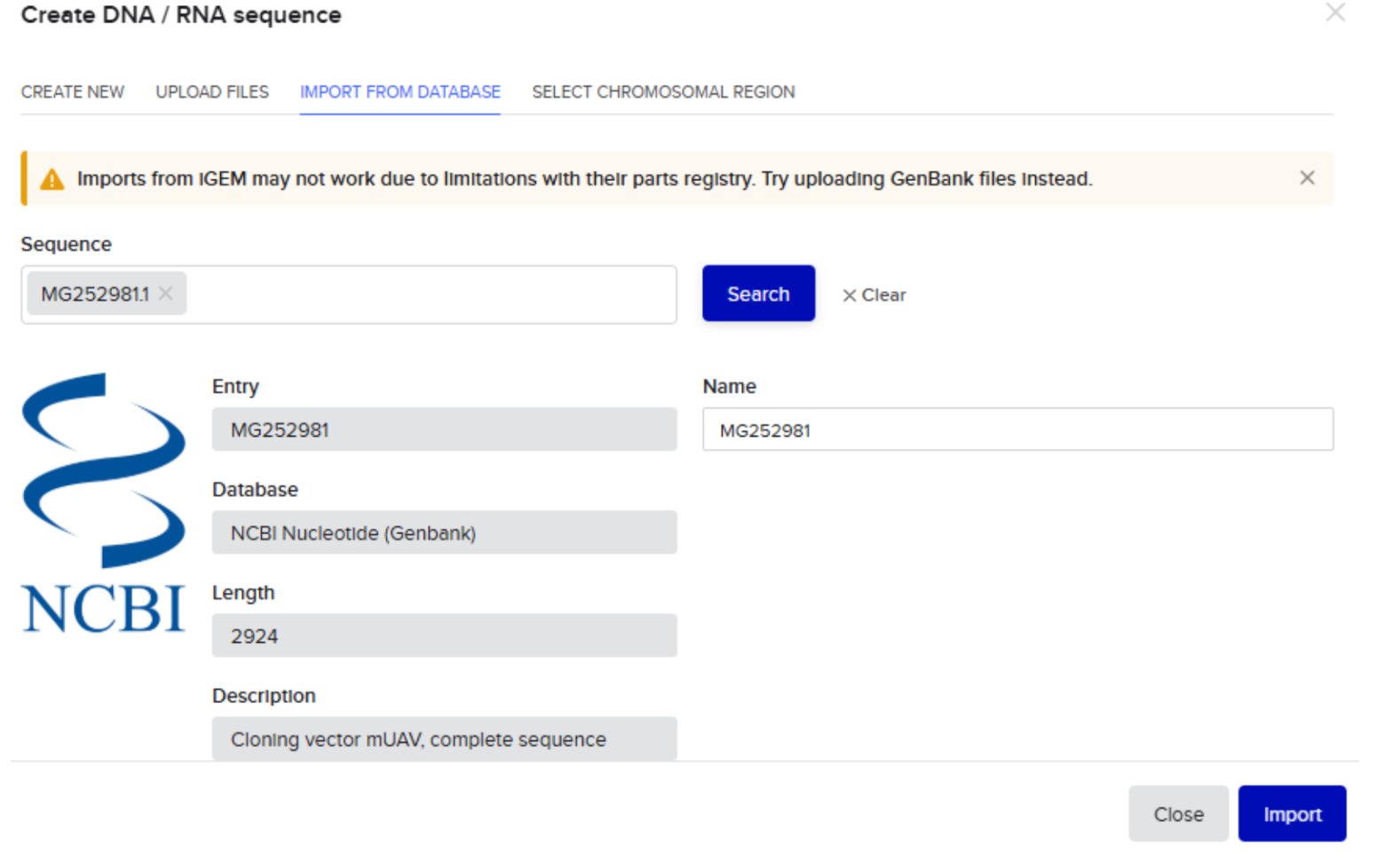
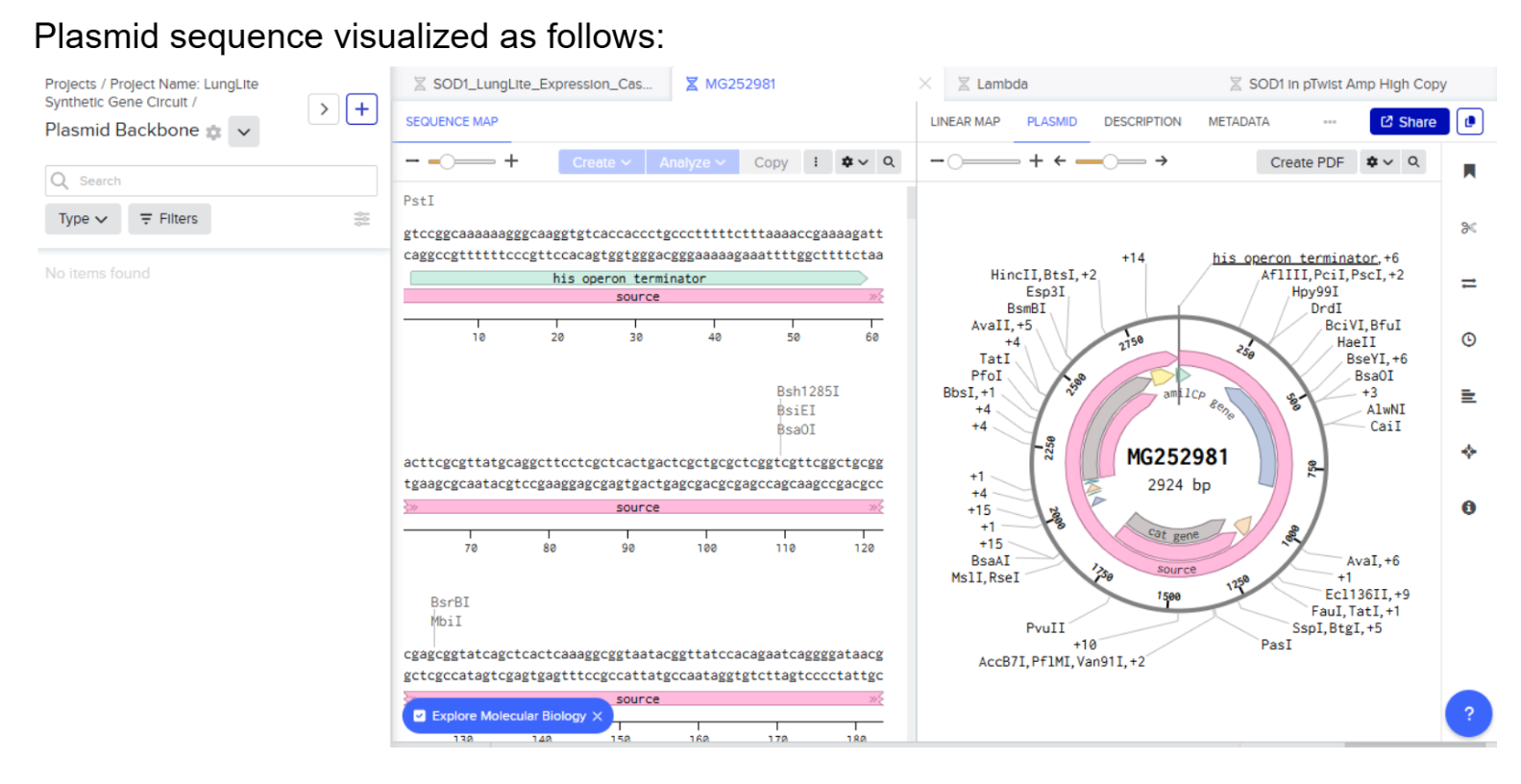
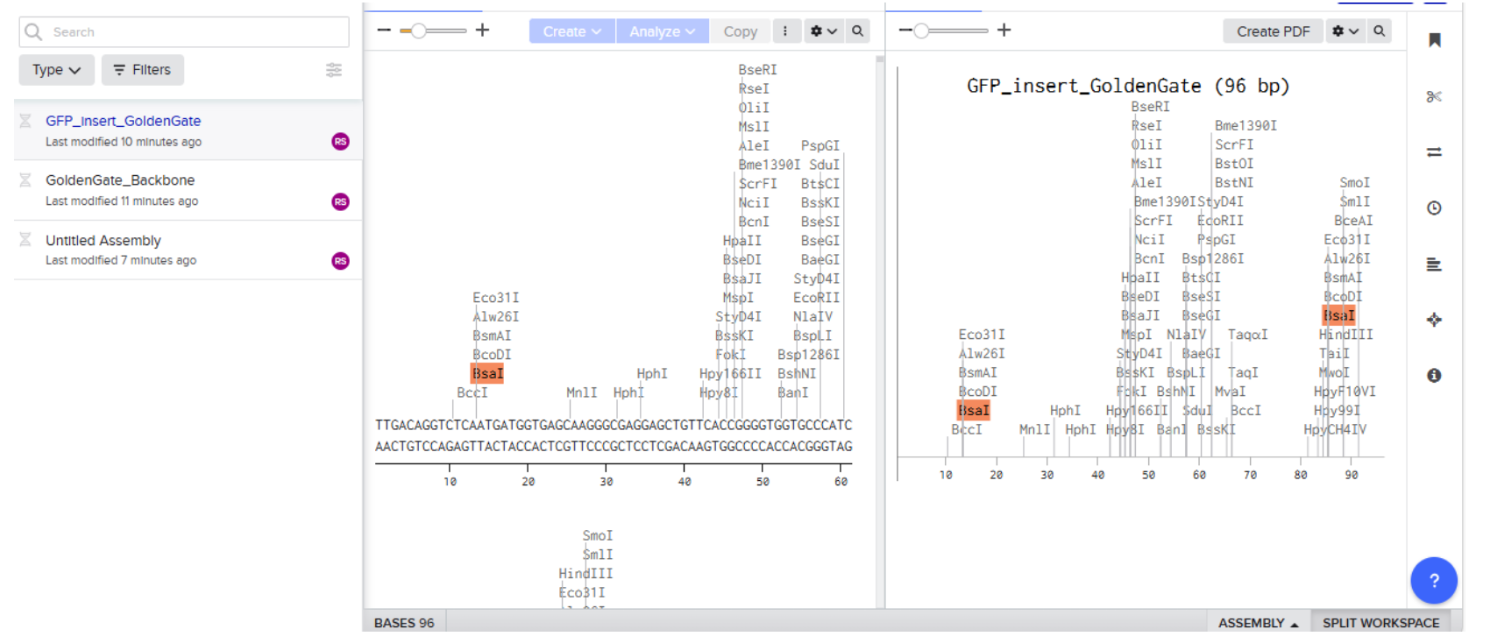
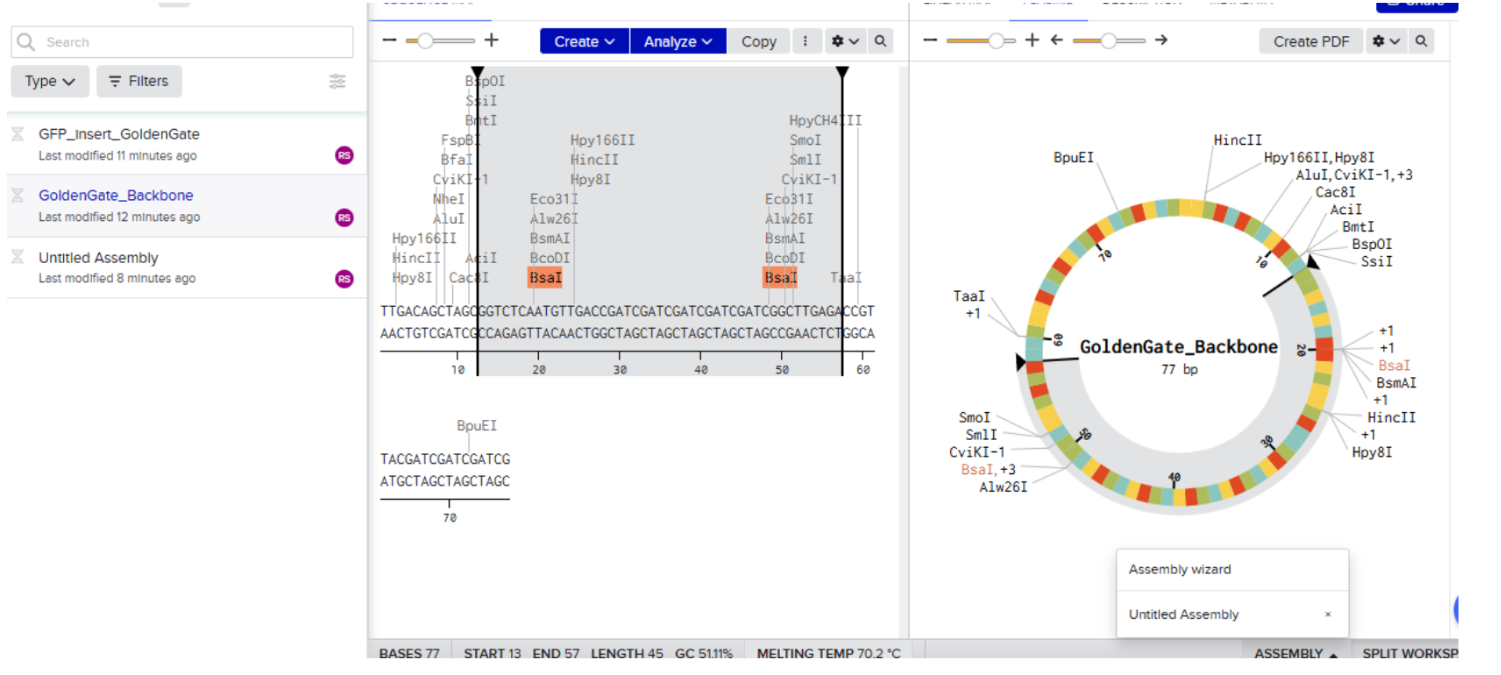
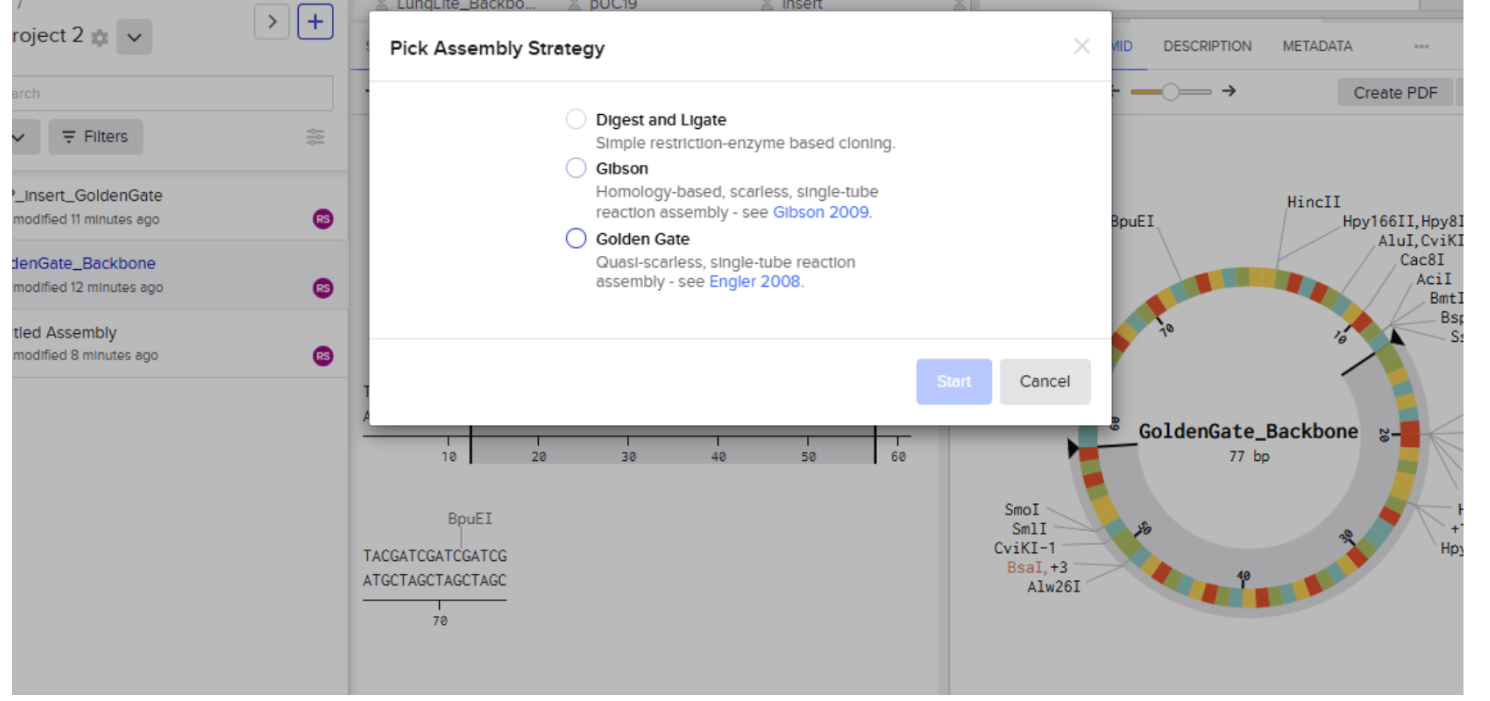
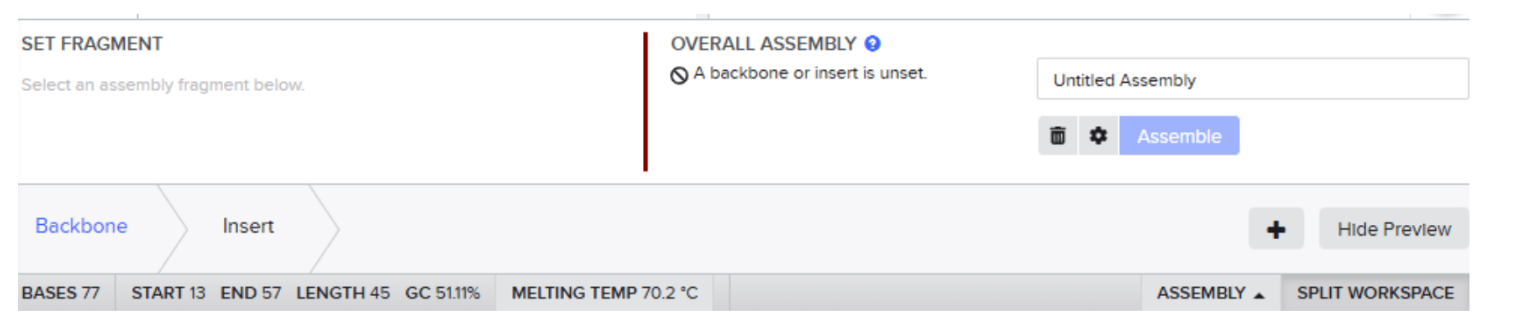
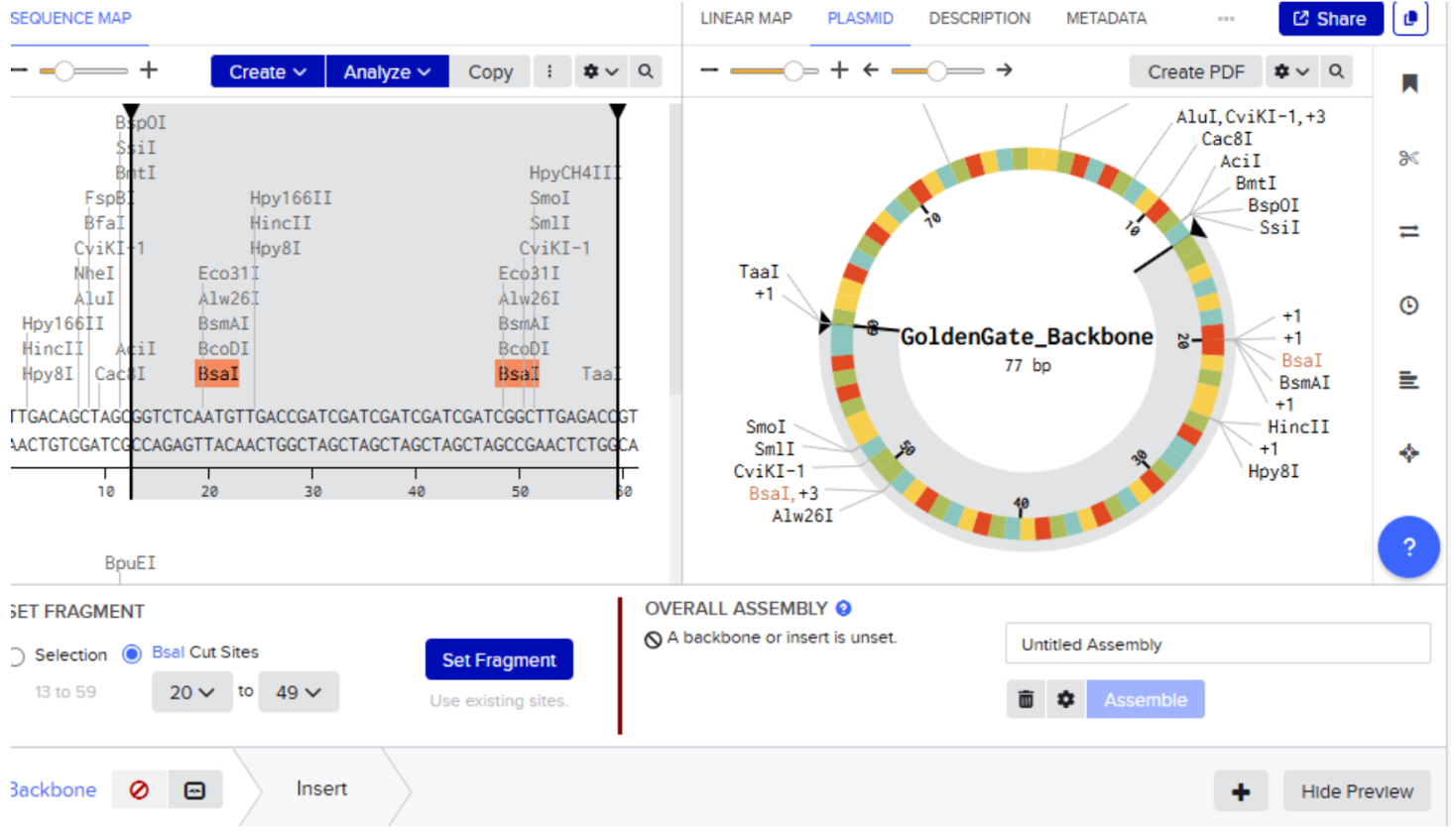
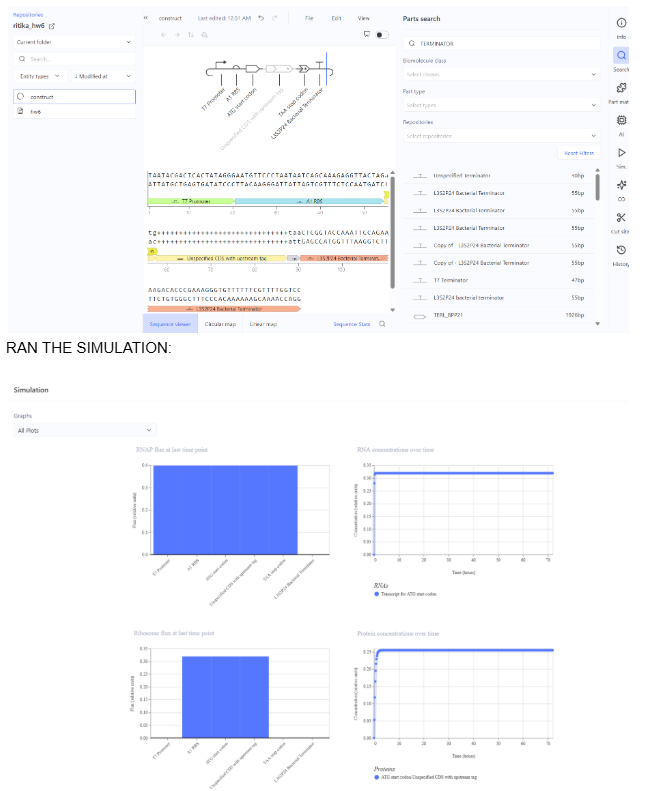
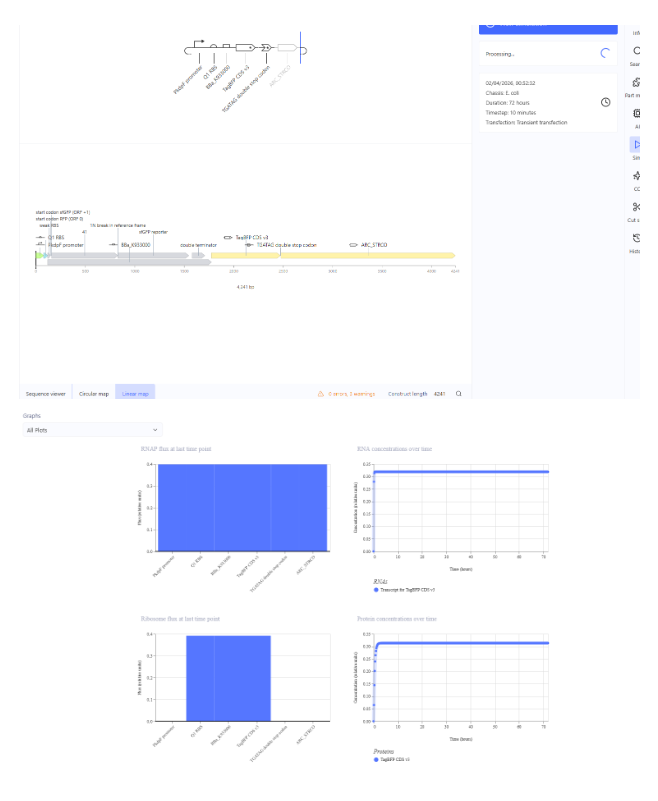
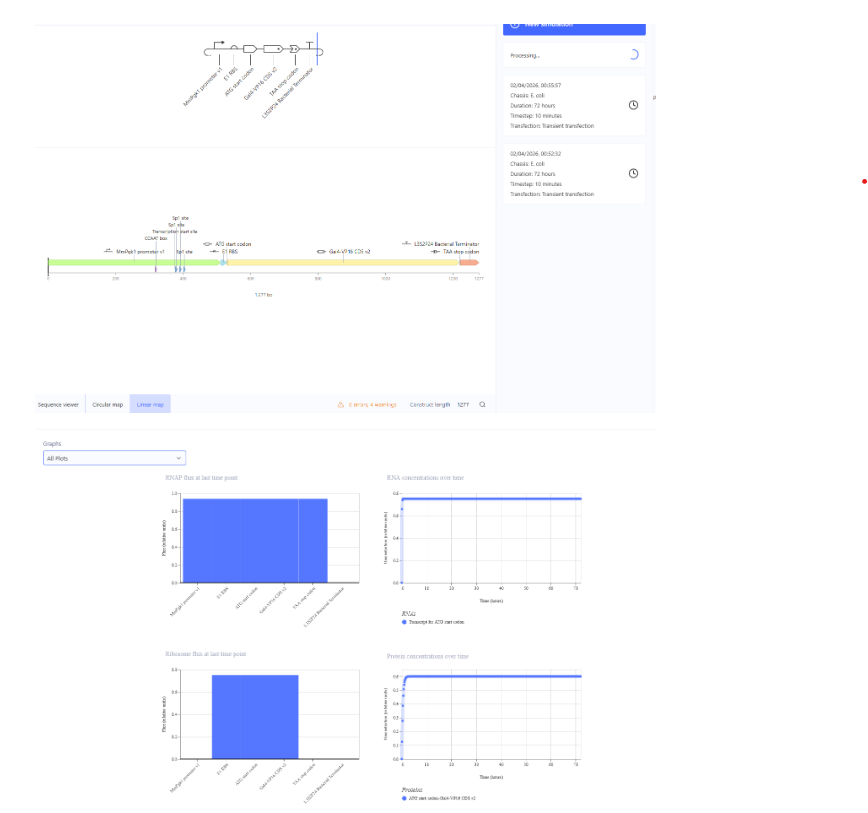
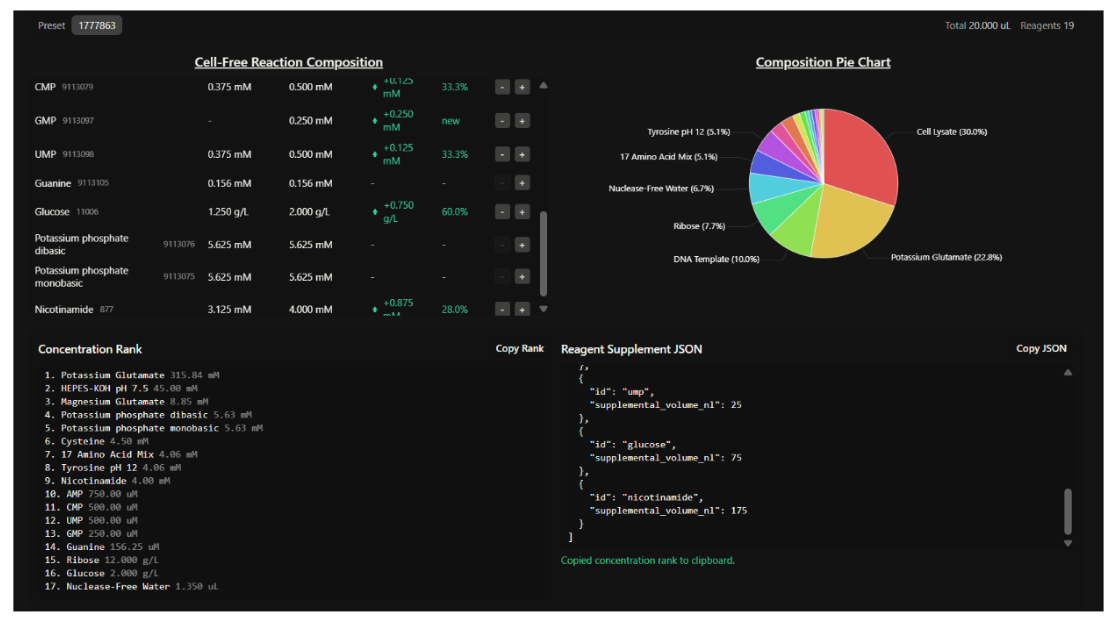
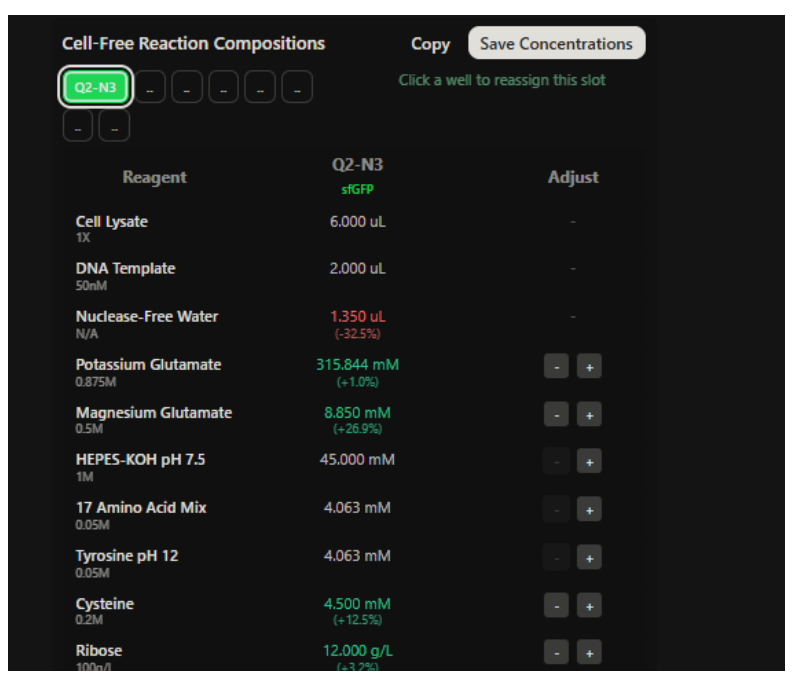
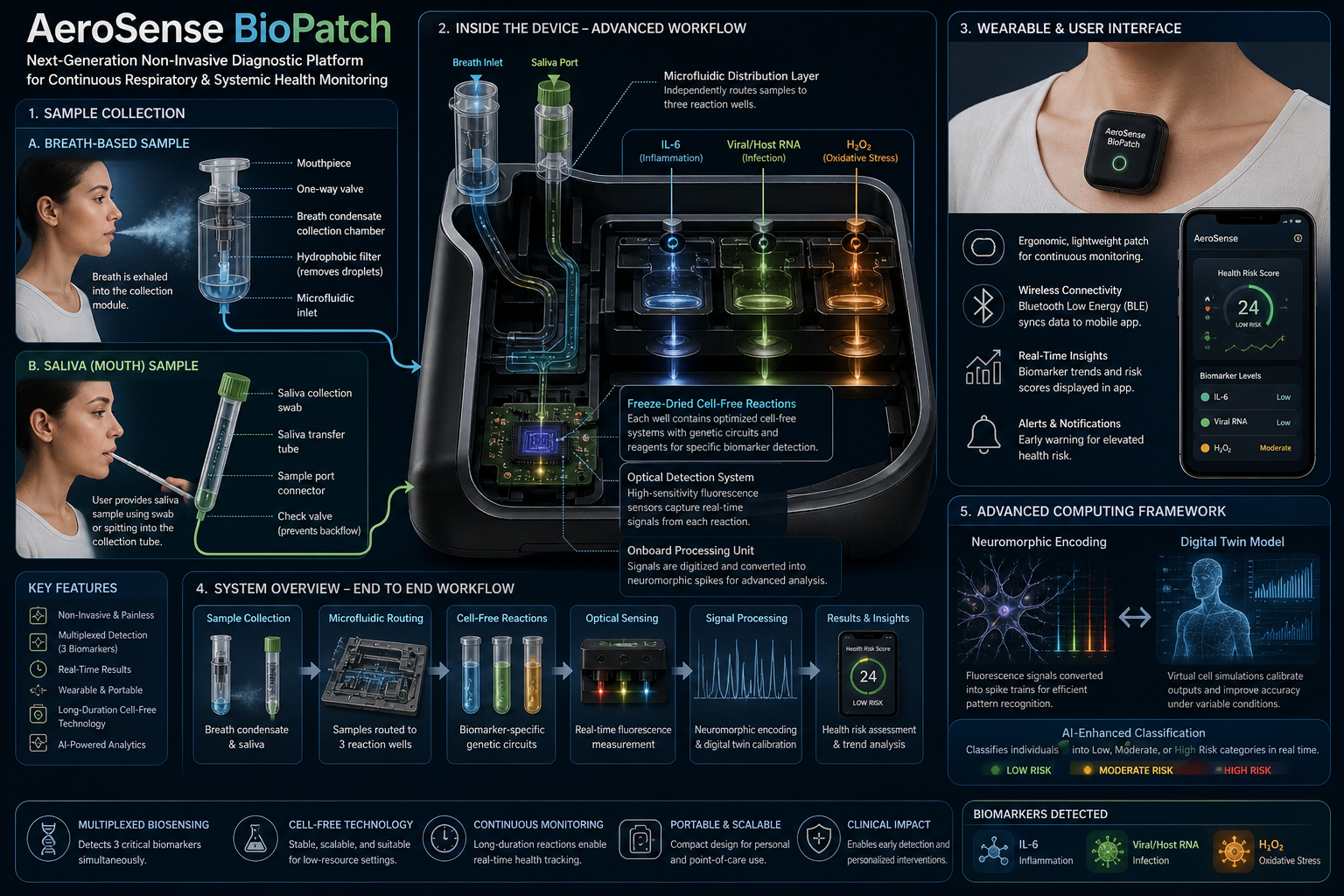
Our first task was to generate an artisitc design using the GUI at opentrons-art.rcdonovan.com.
My inspiration for this design was my dog shiro (although he is an Indian spitz), I ended up designing a dachshund-
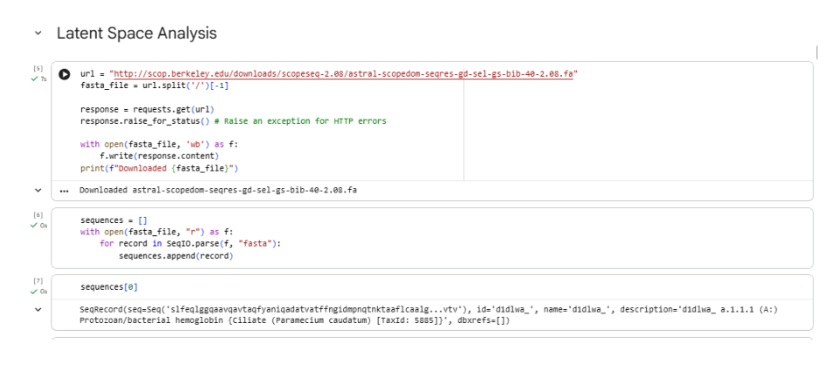
I, then exported the python script directly from the interface, as per the given instructions:
from opentrons import types
import string
metadata = {
'protocolName': '{YOUR NAME} - Opentrons Art - HTGAA',
'author': 'HTGAA',
'source': 'HTGAA 2026',
'apiLevel': '2.20'
}
Z_VALUE_AGAR = 2.0
POINT_SIZE = 1.25
mrfp1_points = [(23,31), (21,29), (23,29), (25,29), (19,27), (23,27), (21,23), (17,21), (19,21), (9,19), (11,19), (13,19), (15,19), (17,19), (1,11), (5,11), (1,9), (-1,7), (1,7), (-7,5), (-5,5), (-3,5), (-1,5), (-7,3), (-5,3), (-3,3), (-1,3), (-5,1), (-3,1), (-1,1), (-5,-1), (-3,-1), (9,-7), (-15,-9), (-11,-9), (15,-9), (23,-9), (25,-9), (27,-9), (25,-11), (27,-11), (-19,-13), (9,-13), (11,-13), (-5,-17), (-21,-19), (-7,-19), (-21,-21), (-9,-21), (-19,-23)]
mko2_points = [(19,29), (15,27), (17,27), (21,27), (13,25), (15,25), (17,25), (19,25), (21,25), (23,25), (11,23), (13,23), (15,23), (17,23), (19,23), (7,21), (9,21), (11,21), (13,21), (15,21), (5,19), (7,19), (5,17), (7,17), (9,17), (11,17), (13,17), (15,17), (17,17), (19,17), (7,15), (9,15), (11,15), (13,15), (15,15), (17,15), (7,13), (9,13), (11,13), (13,13), (15,13), (9,11), (11,11), (13,11), (15,11), (11,9), (13,9), (15,9), (13,7), (15,7), (7,3), (9,3), (7,1), (9,1), (11,1), (13,1), (15,1), (17,1), (7,-1), (9,-1), (11,-1), (13,-1), (15,-1), (17,-1), (7,-3), (9,-3), (11,-3), (13,-3), (15,-3), (17,-3), (9,-5), (11,-5), (13,-5), (15,-5), (17,-5), (17,-7), (21,-7), (23,-7), (25,-7), (27,-7), (-27,-9), (-25,-11), (19,-11), (-23,-13), (21,-13), (27,-13), (7,-15), (19,-15), (21,-15), (23,-15), (-7,-17), (-3,-17), (-11,-19), (-9,-19), (-5,-19), (-23,-21), (-13,-21), (-11,-21), (-7,-21), (-5,-21), (-23,-23), (-21,-23), (-15,-23), (-13,-23), (-11,-23), (-9,-23), (-7,-23), (-23,-25), (-21,-25), (-19,-25), (-17,-25), (-15,-25), (-13,-25), (-11,-25), (-9,-25), (-25,-27), (-23,-27), (-11,-27), (-9,-27), (-27,-29), (-25,-29), (-13,-29), (-11,-29)]
mscarlet_i_points = [(5,27), (7,27), (9,27), (11,27), (13,27), (5,25), (7,25), (9,25), (11,25), (3,23), (5,23), (7,23), (9,23), (-1,21), (1,21), (3,21), (5,21), (-3,19), (-1,19), (1,19), (3,19), (-13,17), (-11,17), (-9,17), (-7,17), (-5,17), (-3,17), (-1,17), (1,17), (3,17), (-15,15), (-13,15), (-11,15), (-9,15), (-7,15), (-5,15), (-3,15), (-1,15), (1,15), (3,15), (5,15), (-15,13), (-13,13), (-11,13), (-9,13), (-7,13), (-5,13), (-3,13), (-1,13), (1,13), (3,13), (5,13), (-15,11), (-13,11), (-11,11), (-9,11), (-7,11), (-5,11), (-3,11), (-1,11), (3,11), (7,11), (-15,9), (-13,9), (-11,9), (-9,9), (-7,9), (-5,9), (-3,9), (-1,9), (3,9), (5,9), (7,9), (9,9), (-15,7), (-13,7), (-11,7), (-9,7), (-7,7), (-5,7), (-3,7), (3,7), (5,7), (7,7), (9,7), (11,7), (-27,5), (1,5), (3,5), (5,5), (7,5), (9,5), (11,5), (13,5), (15,5), (-27,3), (1,3), (3,3), (5,3), (11,3), (13,3), (15,3), (-27,1), (1,1), (3,1), (5,1), (-1,-1), (1,-1), (3,-1), (5,-1), (-27,-3), (-3,-3), (-1,-3), (1,-3), (3,-3), (5,-3), (-27,-5), (-5,-5), (-3,-5), (-1,-5), (1,-5), (3,-5), (5,-5), (7,-5), (-27,-7), (-25,-7), (-13,-7), (-11,-7), (-9,-7), (-7,-7), (-5,-7), (-3,-7), (-1,-7), (1,-7), (3,-7), (5,-7), (7,-7), (11,-7), (13,-7), (15,-7), (19,-7), (-25,-9), (-23,-9), (-17,-9), (-13,-9), (-9,-9), (-7,-9), (-5,-9), (-3,-9), (-1,-9), (1,-9), (3,-9), (5,-9), (7,-9), (9,-9), (11,-9), (13,-9), (17,-9), (19,-9), (21,-9), (-23,-11), (-21,-11), (-17,-11), (-15,-11), (-13,-11), (-11,-11), (-9,-11), (-7,-11), (-5,-11), (-3,-11), (-1,-11), (1,-11), (3,-11), (5,-11), (7,-11), (9,-11), (15,-11), (17,-11), (21,-11), (-21,-13), (-17,-13), (-15,-13), (-13,-13), (-11,-13), (-9,-13), (-7,-13), (-5,-13), (-3,-13), (-1,-13), (1,-13), (3,-13), (5,-13), (7,-13), (23,-13), (-19,-15), (-17,-15), (-15,-15), (-13,-15), (-11,-15), (-9,-15), (-7,-15), (-5,-15), (-3,-15), (-1,-15), (1,-15), (3,-15), (5,-15), (-19,-17), (-17,-17), (-15,-17), (-13,-17), (-11,-17), (-9,-17), (-19,-19), (-17,-19), (-15,-19), (-13,-19), (-19,-21), (-17,-21), (-15,-21), (-17,-23)]
azurite_points = [(31,-9), (15,-13), (25,-13)]
mclover3_points = [(23,-11)]
point_name_pairing = [("mrfp1", mrfp1_points),("mko2", mko2_points),("mscarlet_i", mscarlet_i_points),("azurite", azurite_points),("mclover3", mclover3_points)]
# Robot deck setup constants
TIP_RACK_DECK_SLOT = 9
COLORS_DECK_SLOT = 6
AGAR_DECK_SLOT = 5
PIPETTE_STARTING_TIP_WELL = 'A1'
# Place the PCR tubes in this order
well_colors = {
'A1': 'sfGFP',
'A2': 'mRFP1',
'A3': 'mKO2',
'A4': 'Venus',
'A5': 'mKate2_TF',
'A6': 'Azurite',
'A7': 'mCerulean3',
'A8': 'mClover3',
'A9': 'mJuniper',
'A10': 'mTurquoise2',
'A11': 'mBanana',
'A12': 'mPlum',
'B1': 'Electra2',
'B2': 'mWasabi',
'B3': 'mScarlet_I',
'B4': 'mPapaya',
'B5': 'eqFP578',
'B6': 'tdTomato',
'B7': 'DsRed',
'B8': 'mKate2',
'B9': 'EGFP',
'B10': 'mRuby2',
'B11': 'TagBFP',
'B12': 'mChartreuse_TF',
'C1': 'mLychee_TF',
'C2': 'mTagBFP2',
'C3': 'mEGFP',
'C4': 'mNeonGreen',
'C5': 'mAzamiGreen',
'C6': 'mWatermelon',
'C7': 'avGFP',
'C8': 'mCitrine',
'C9': 'mVenus',
'C10': 'mCherry',
'C11': 'mHoneydew',
'C12': 'TagRFP',
'D1': 'mTFP1',
'D2': 'Ultramarine',
'D3': 'ZsGreen1',
'D4': 'mMiCy',
'D5': 'mStayGold2',
'D6': 'PA_GFP'
}
volume_used = {
'mrfp1': 0,
'mko2': 0,
'mscarlet_i': 0,
'azurite': 0,
'mclover3': 0
}
def update_volume_remaining(current_color, quantity_to_aspirate):
rows = string.ascii_uppercase
for well, color in list(well_colors.items()):
if color == current_color:
if (volume_used[current_color] + quantity_to_aspirate) > 250:
# Move to next well horizontally by advancing row letter, keeping column number
row = well[0]
col = well[1:]
# Find next row letter
next_row = rows[rows.index(row) + 1]
next_well = f"{next_row}{col}"
del well_colors[well]
well_colors[next_well] = current_color
volume_used[current_color] = quantity_to_aspirate
else:
volume_used[current_color] += quantity_to_aspirate
break
def run(protocol):
# Load labware, modules and pipettes
protocol.home()
# Tips
tips_20ul = protocol.load_labware('opentrons_96_tiprack_20ul', TIP_RACK_DECK_SLOT, 'Opentrons 20uL Tips')
# Pipettes
pipette_20ul = protocol.load_instrument("p20_single_gen2", "right", [tips_20ul])
# Deep Well Plate
temperature_plate = protocol.load_labware('nest_96_wellplate_2ml_deep', 6)
# Agar Plate
agar_plate = protocol.load_labware('htgaa_agar_plate', AGAR_DECK_SLOT, 'Agar Plate')
agar_plate.set_offset(x=0.00, y=0.00, z=Z_VALUE_AGAR)
# Get the top-center of the plate, make sure the plate was calibrated before running this
center_location = agar_plate['A1'].top()
pipette_20ul.starting_tip = tips_20ul.well(PIPETTE_STARTING_TIP_WELL)
# Helper function (dispensing)
def dispense_and_jog(pipette, volume, location):
assert(isinstance(volume, (int, float)))
# Go above the location
above_location = location.move(types.Point(z=location.point.z + 2))
pipette.move_to(above_location)
# Go downwards and dispense
pipette.dispense(volume, location)
# Go upwards to avoid smearing
pipette.move_to(above_location)
# Helper function (color location)
def location_of_color(color_string):
for well,color in well_colors.items():
if color.lower() == color_string.lower():
return temperature_plate[well]
raise ValueError(f"No well found with color {color_string}")
# Print pattern by iterating over lists
for i, (current_color, point_list) in enumerate(point_name_pairing):
# Skip the rest of the loop if the list is empty
if not point_list:
continue
# Get the tip for this run, set the bacteria color, and the aspirate bacteria of choice
pipette_20ul.pick_up_tip()
max_aspirate = int(18 // POINT_SIZE) * POINT_SIZE
quantity_to_aspirate = min(len(point_list)*POINT_SIZE, max_aspirate)
update_volume_remaining(current_color, quantity_to_aspirate)
pipette_20ul.aspirate(quantity_to_aspirate, location_of_color(current_color))
# Iterate over the current points list and dispense them, refilling along the way
for i in range(len(point_list)):
x, y = point_list[i]
adjusted_location = center_location.move(types.Point(x, y))
dispense_and_jog(pipette_20ul, POINT_SIZE, adjusted_location)
if pipette_20ul.current_volume == 0 and len(point_list[i+1:]) > 0:
quantity_to_aspirate = min(len(point_list[i:])*POINT_SIZE, max_aspirate)
update_volume_remaining(current_color, quantity_to_aspirate)
pipette_20ul.aspirate(quantity_to_aspirate, location_of_color(current_color))
# Drop tip between each color
pipette_20ul.drop_tip()
I also experimented with a Google Colab code file, where I worked on generating a design based on an image resembling the Earth.

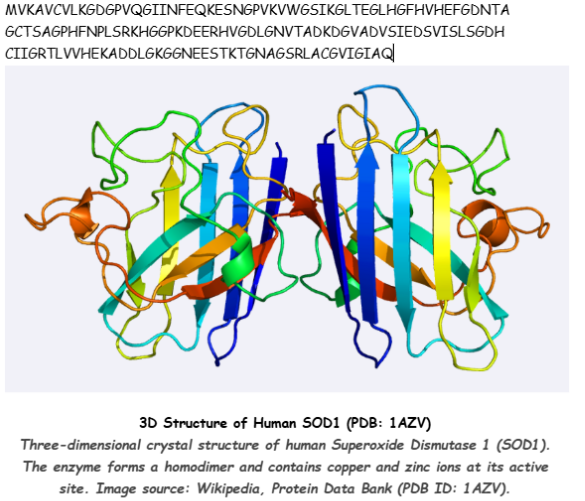
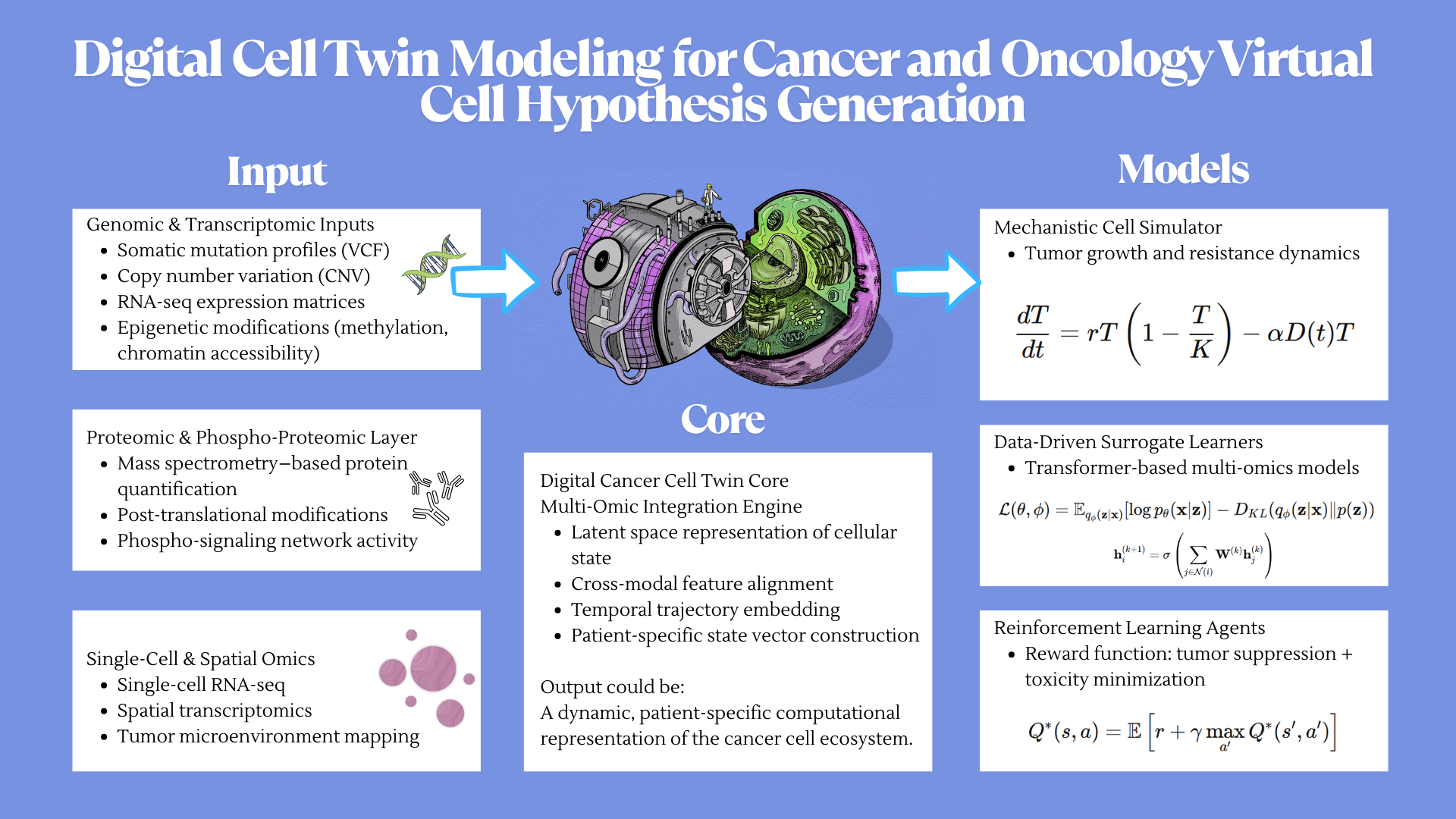
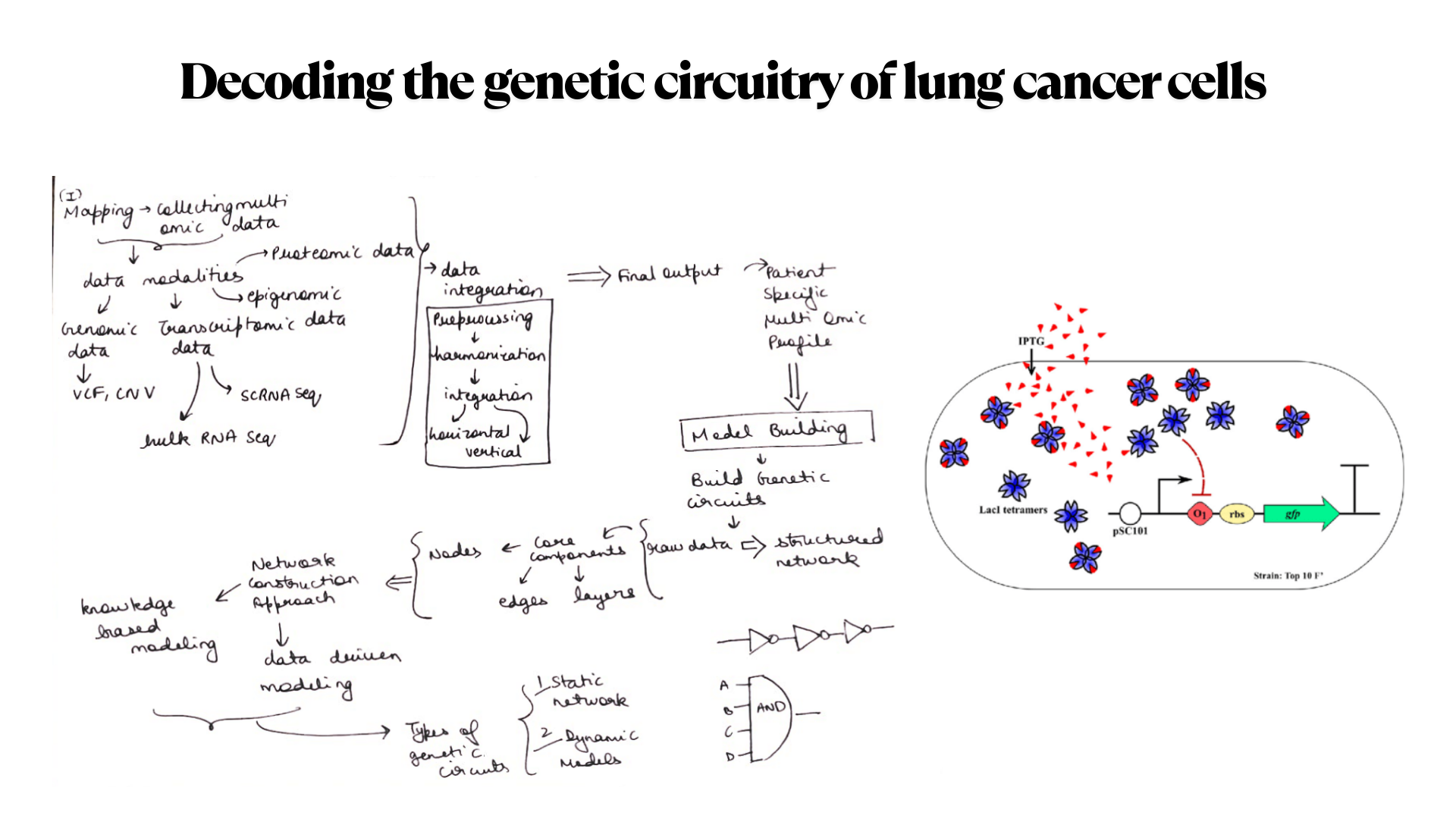
An Automated Versatile Diagnostic Workflow for Infectious Disease Detection in Low-Resource Settings
This paper presents an automated diagnostic workflow designed for detecting infectious diseases in low-resource settings. The system integrates microfluidics, biosensing, and automation to process biological samples efficiently. It focuses on creating a scalable and portable diagnostic pipeline that reduces manual intervention while maintaining accuracy.
The workflow incorporates automation tools to handle multiple steps of the diagnostic process, including sample preparation, reagent handling, and reaction execution. Automated systems ensure precise liquid handling, reduce human error, and enable reproducibility across multiple tests. The integration of microfluidic platforms further enhances throughput and minimizes reagent usage.
The key contribution of this work is the development of a versatile and low-cost automated diagnostic platform that can be deployed in resource-limited environments. It demonstrates how automation can bridge gaps in healthcare accessibility by enabling reliable and rapid disease detection.
This paper directly relates to this week’s focus on lab automation using Opentrons. It highlights how automated liquid handling and integrated workflows can transform biological experiments into scalable and reproducible systems, similar to how we programmed the Opentrons robot.
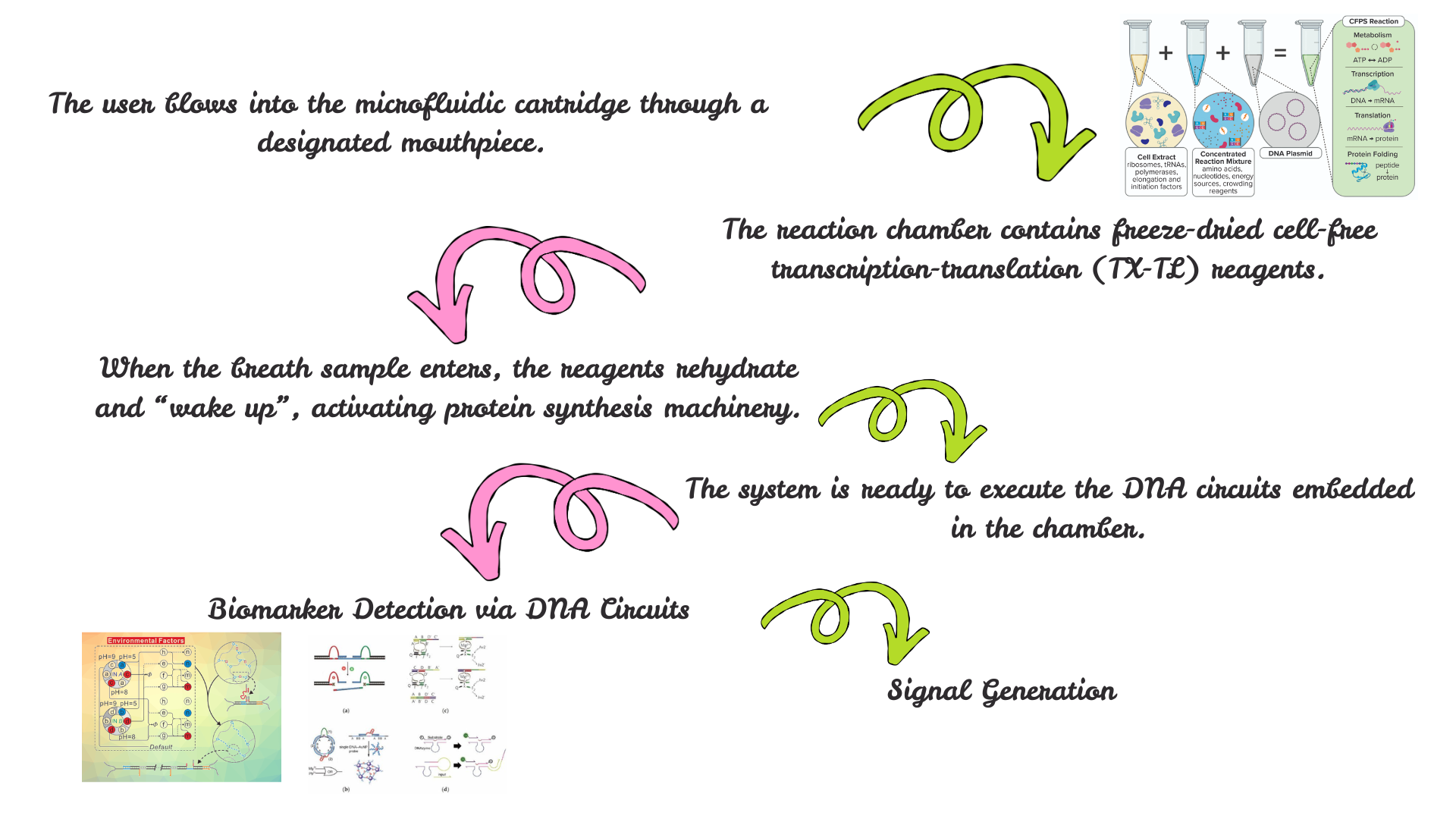
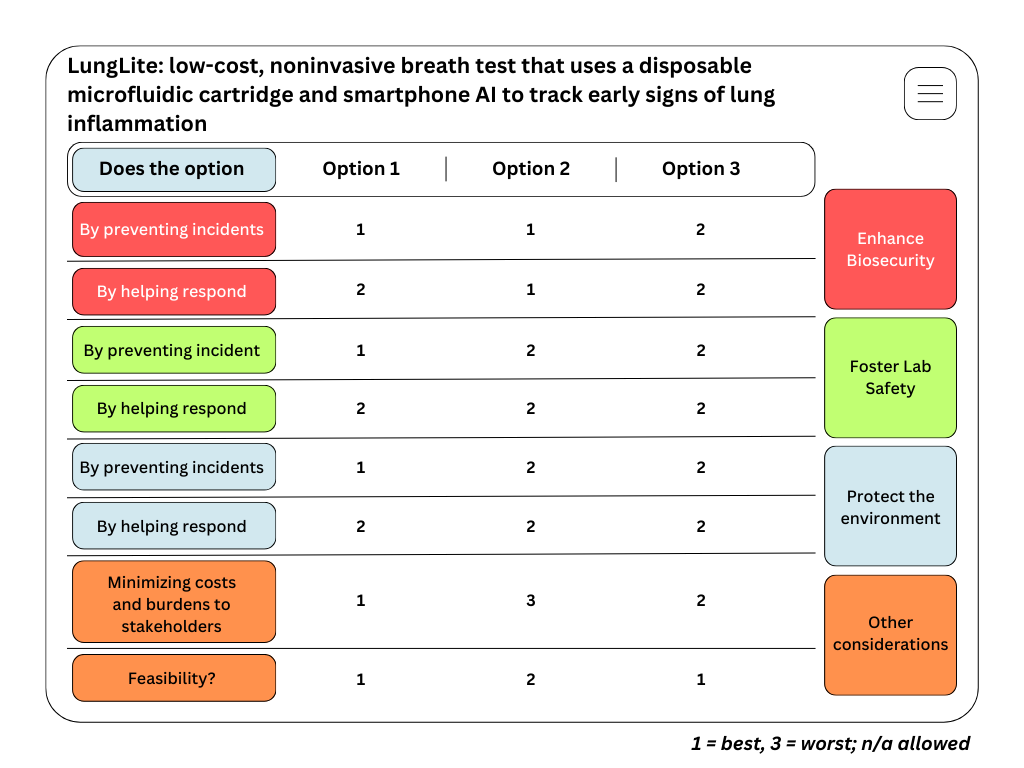
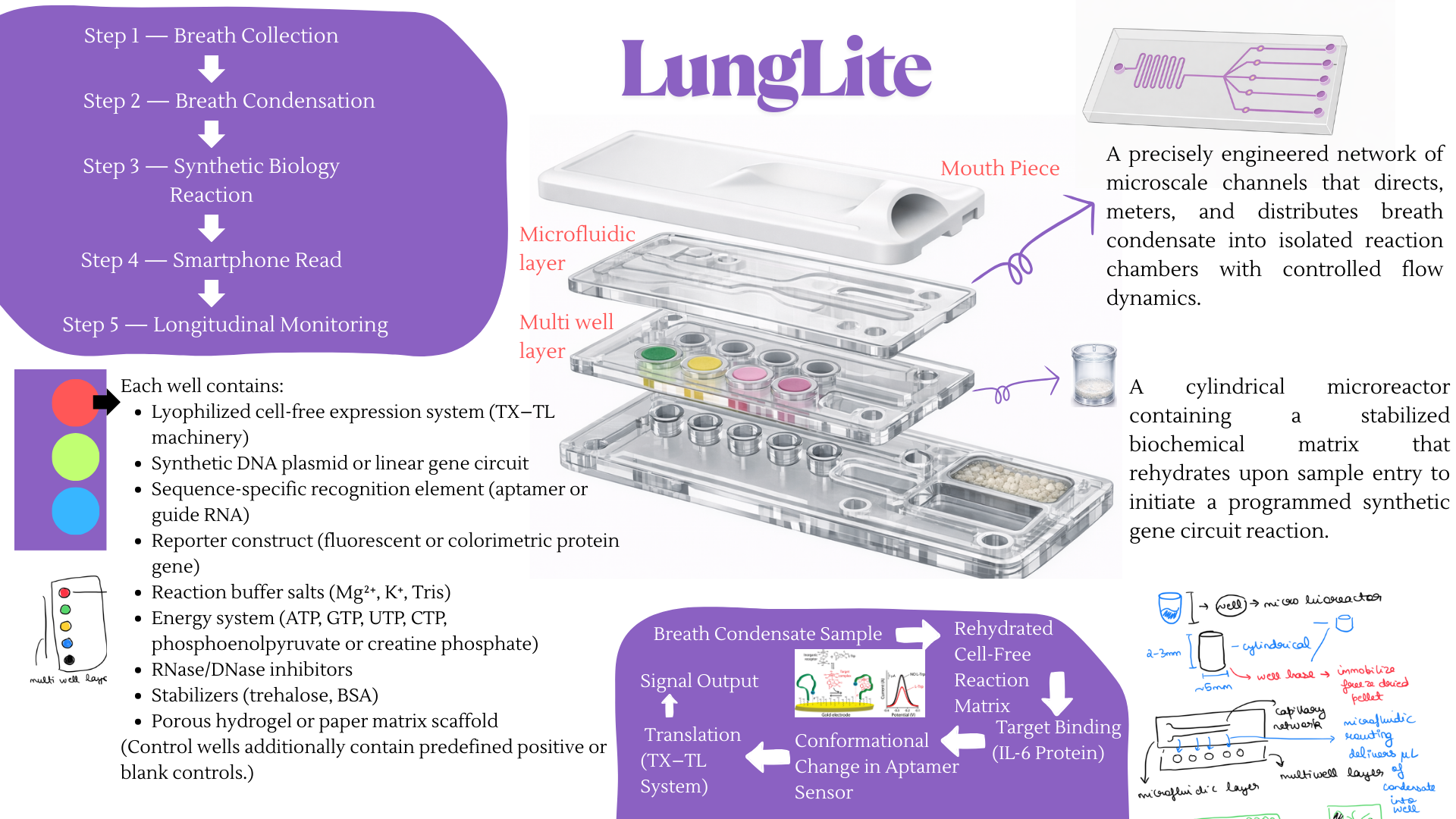
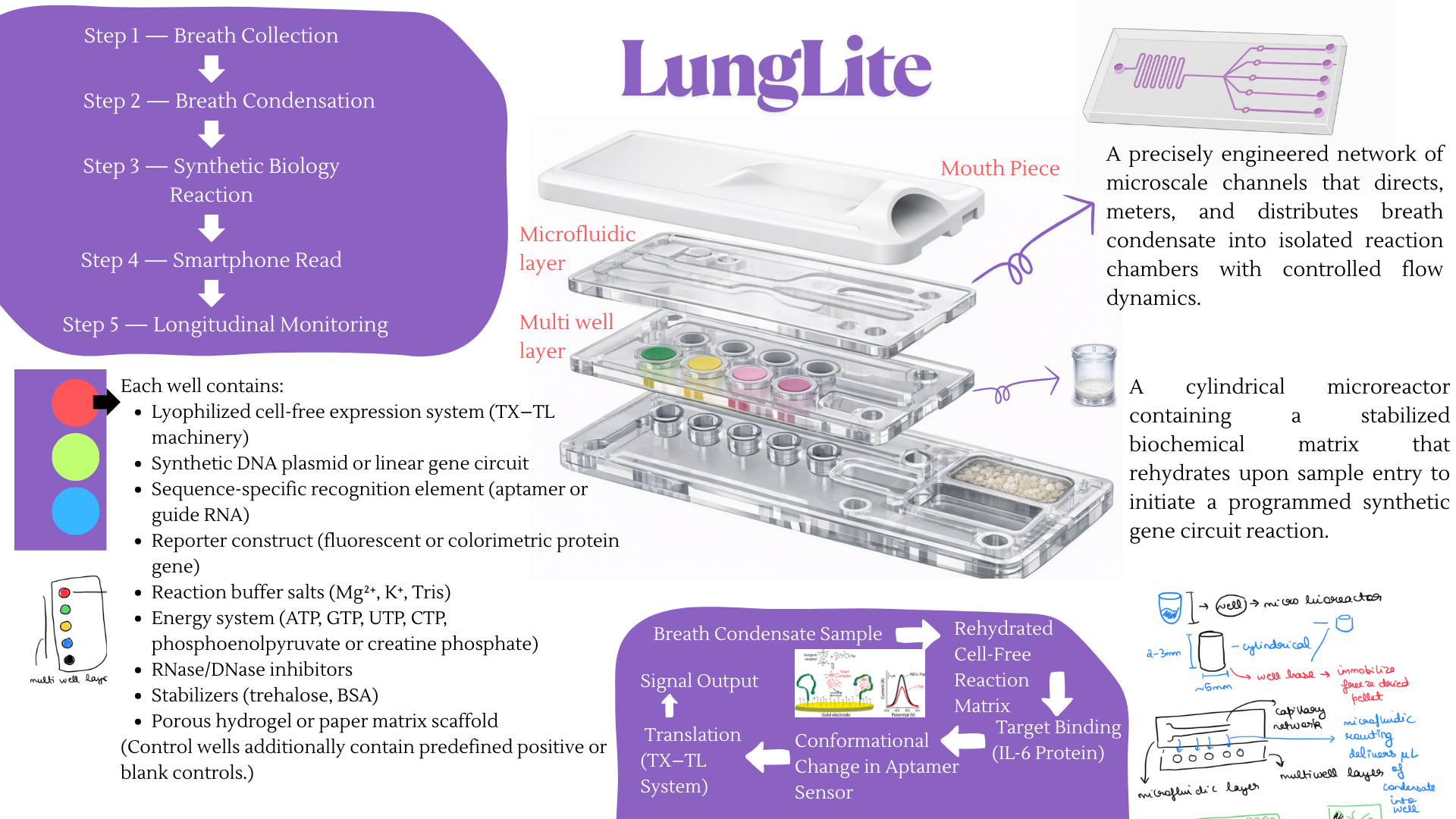
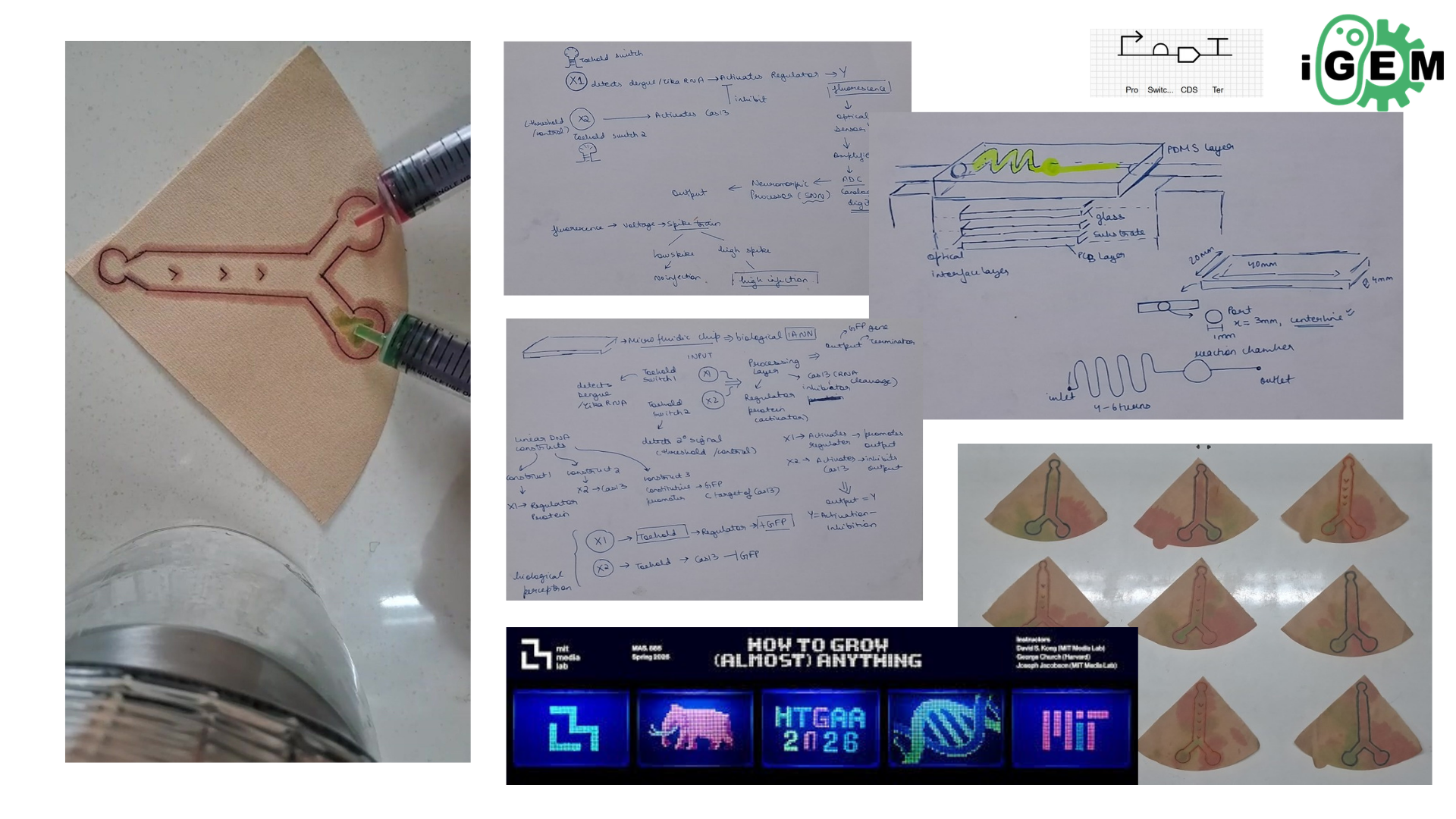
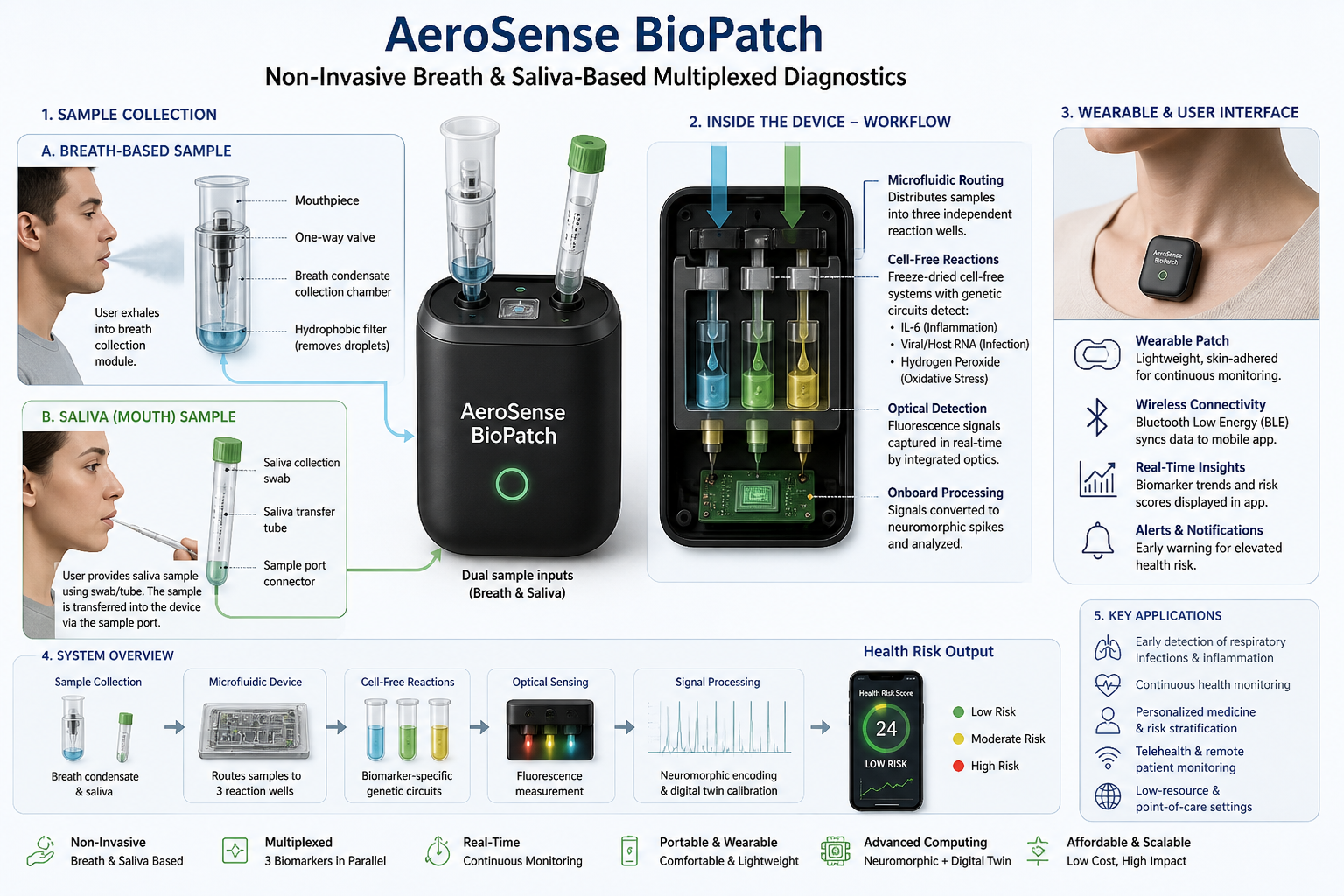
For the final project, I propose developing an automated diagnostic system that detects disease biomarkers from breath condensate samples using a microfluidic and cell-free synthetic biology platform.
Traditional diagnostic methods can be invasive, time-consuming, and require well-equipped laboratory settings. There is a need for a non-invasive, rapid, and scalable diagnostic solution that can work in low-resource environments.
The proposed system will combine breath-based sample collection with automated liquid handling and synthetic biology reactions. Using an Opentrons robot, the workflow will automate sample distribution, reagent addition, and reaction setup across multiple wells.
The system will enable rapid, automated, and non-invasive detection of biomarkers with high reproducibility. It will demonstrate how automation can be used to scale biological diagnostics.