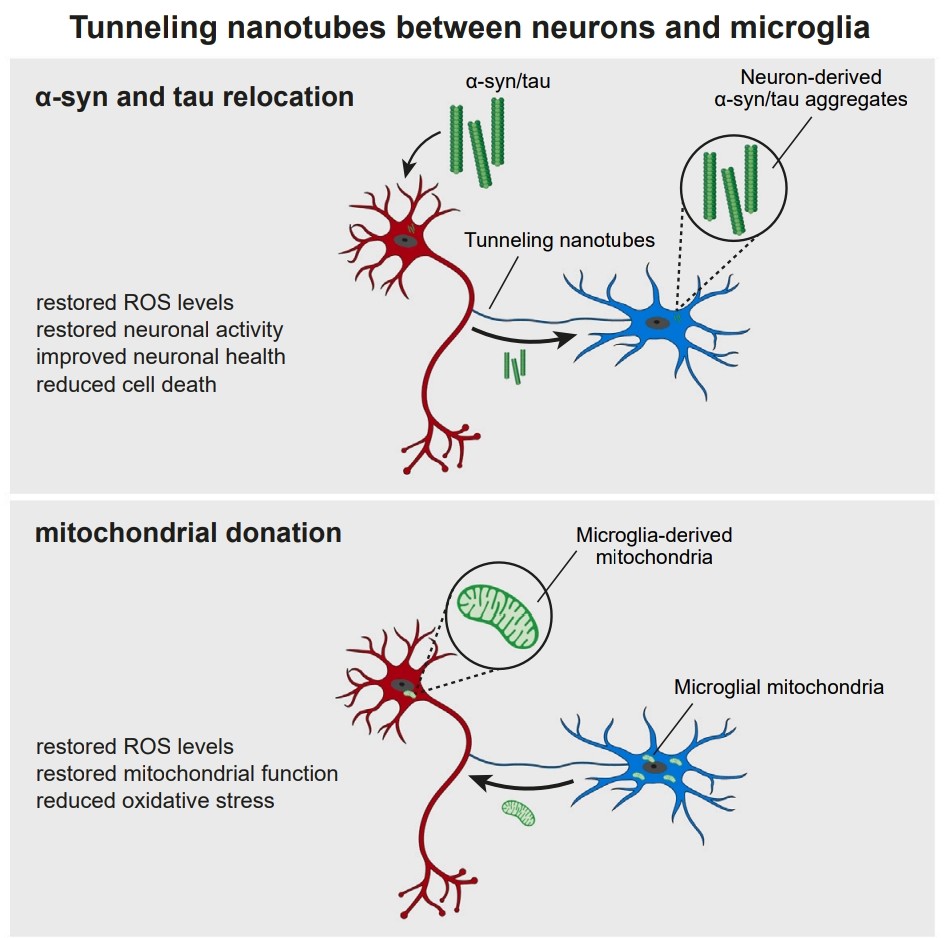
ARM-Net: Alzheimer’s Recovery Micro-TNT Network 1.Biological Engineering Application I am developing ARM-Net, a synthetic biology tool designed to overexpress CCT4 specifically in microglia. Unlike the full TRiC/CCT complex, CCT4 can function independently or as homo-oligomers to promote microtubule synthesis. By boosting these chaperonins, I aim to induce the formation of Tunneling Nanotubes (TNTs) between microglia, creating an “Intercellular Care Network” (ICN)
I have answered the following questions to prepare for the lecture on DNA design and synthesis. These answers are based on the slides provided by Professor Jacobson, Dr. LeProust, and Professor Church.
Question,1 Natures machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
This week, I focused on the design, simulation, and cloning workflow for CCT4, the core component of my project, ICN (Intercellular Care Network).
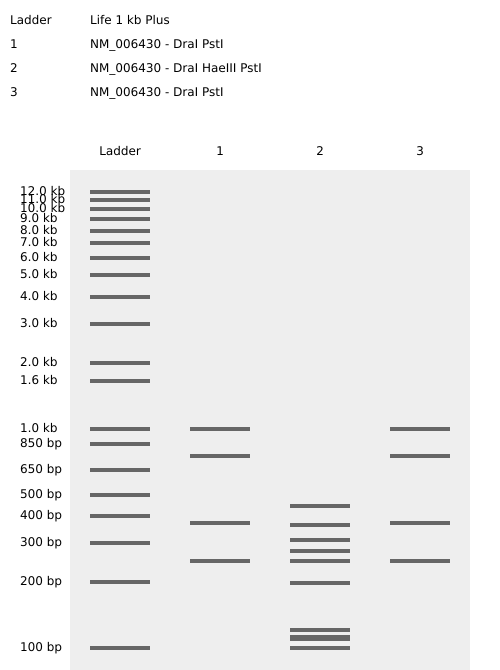
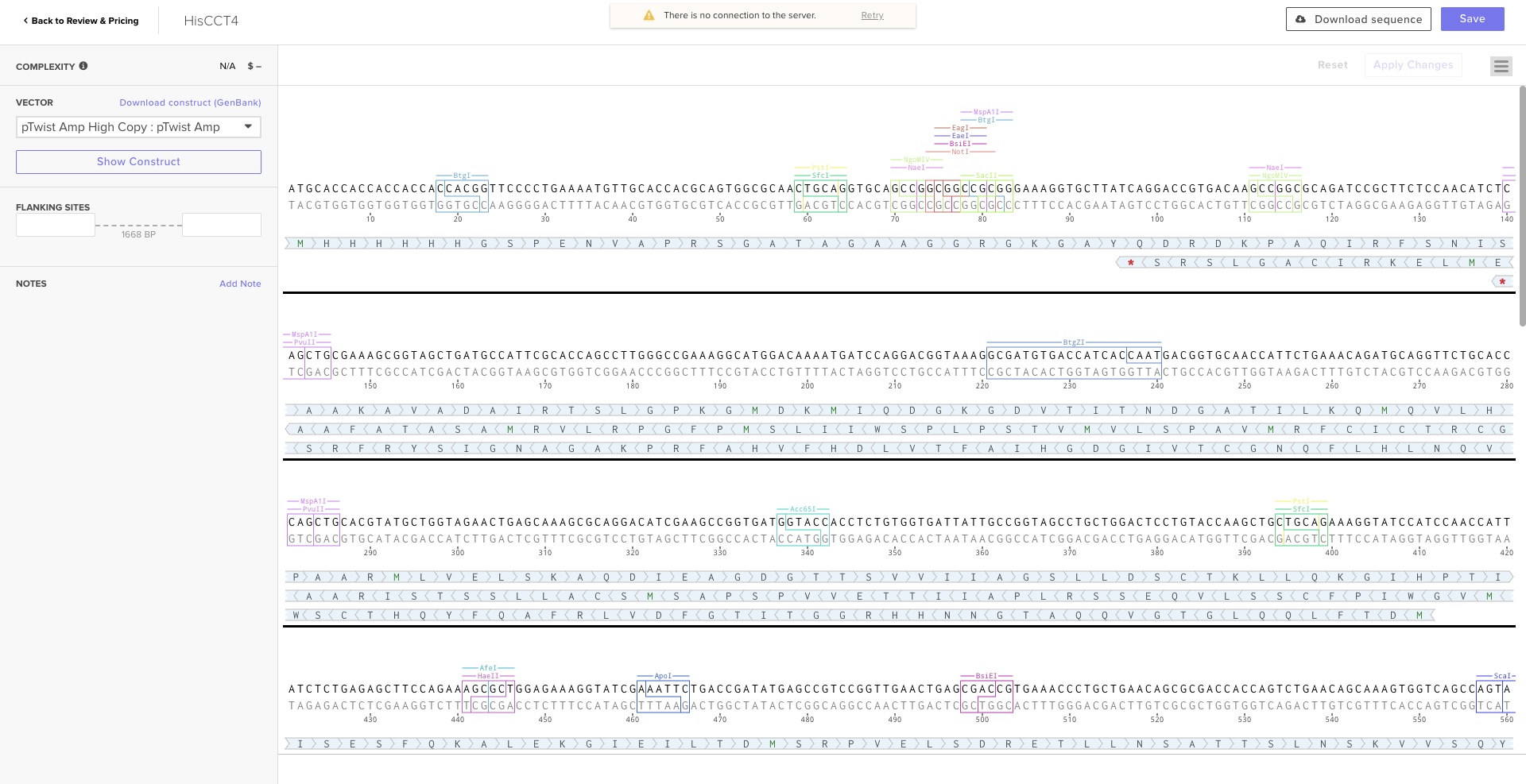
Part 1: Benchling and In Silico Gel Art Benchling Simulation Having long been interested in Benchling, I used this assignment to dive deep into the tool. Instead of the suggested Lambda DNA, I worked with the CCT4 protein sequence relevant to my project. I simulated restriction digests using the following enzymes:
Assignment: Telecaster Guitar Bio-Art This week’s task was to create a Python script for the Opentrons OT-2 robot to draw an artistic design on an agar plate using colored bacteria.
Design Concept: The Telecaster My design is based on the Fender Telecaster, an iconic electric guitar. I used different fluorescent proteins to represent the components:
Week 4: Protein Design I — Conceptual Questions Part A: Conceptual Questions The following nine questions were selected from the set provided by Professor Shuguang Zhang to explore the fundamental principles of protein structure and molecular evolution.
How many molecules of amino acids do you take with a piece of 500 grams of meat? (Assumed: 100 Daltons per amino acid, meat is ~20% protein) If 500g of meat contains approximately 100g of protein, and the average molecular weight of an amino acid is 100 g/mol:
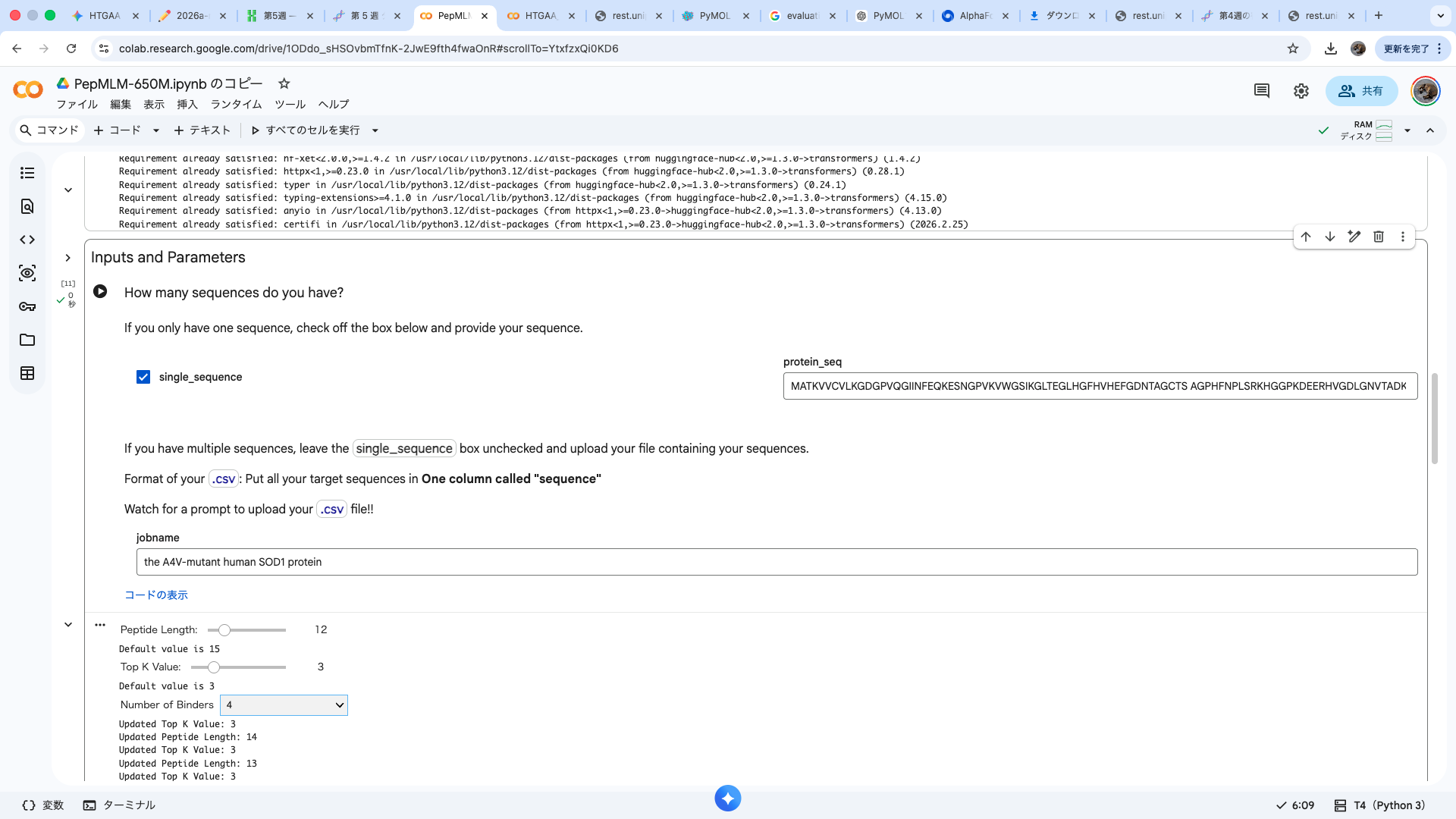
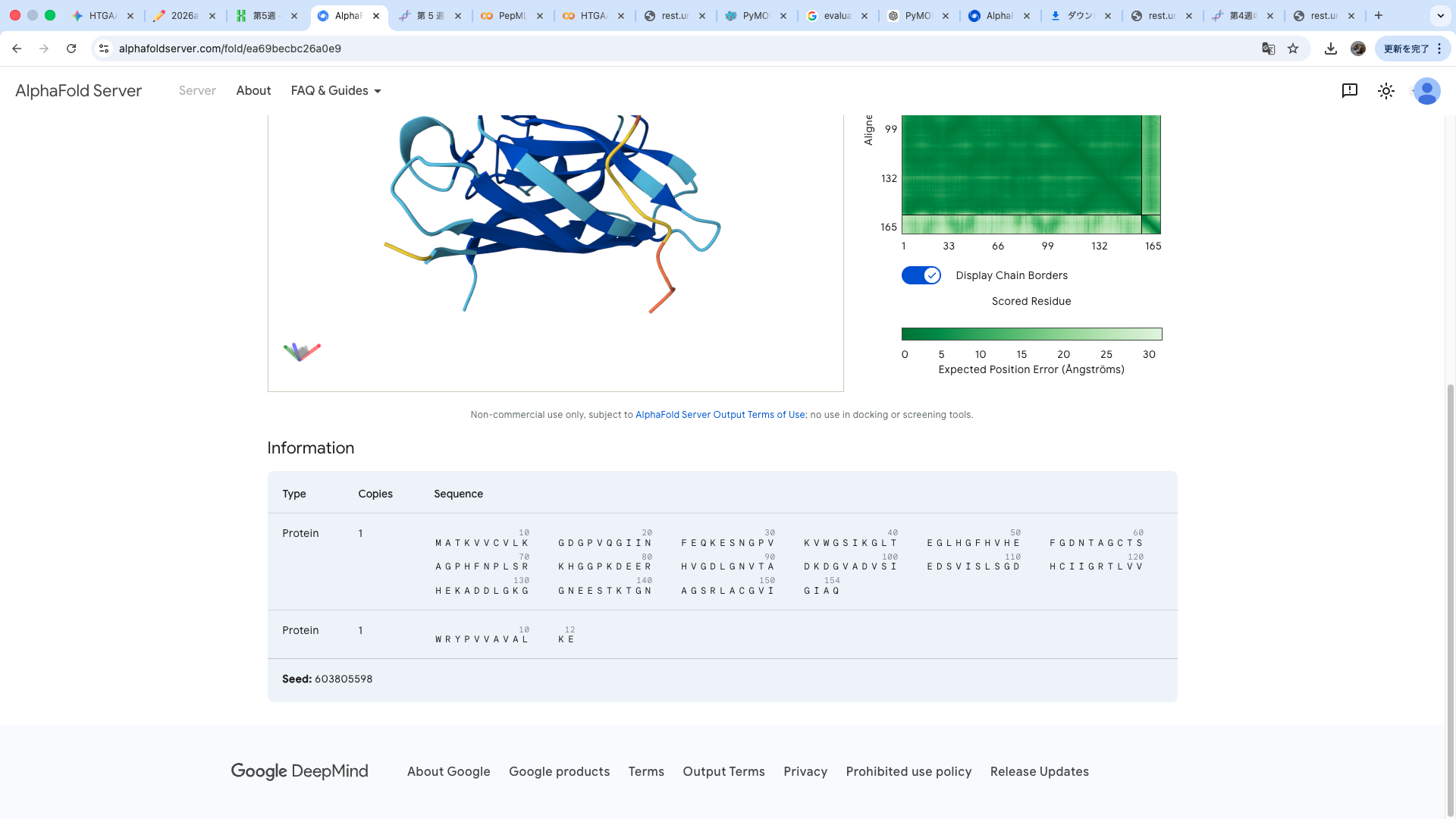
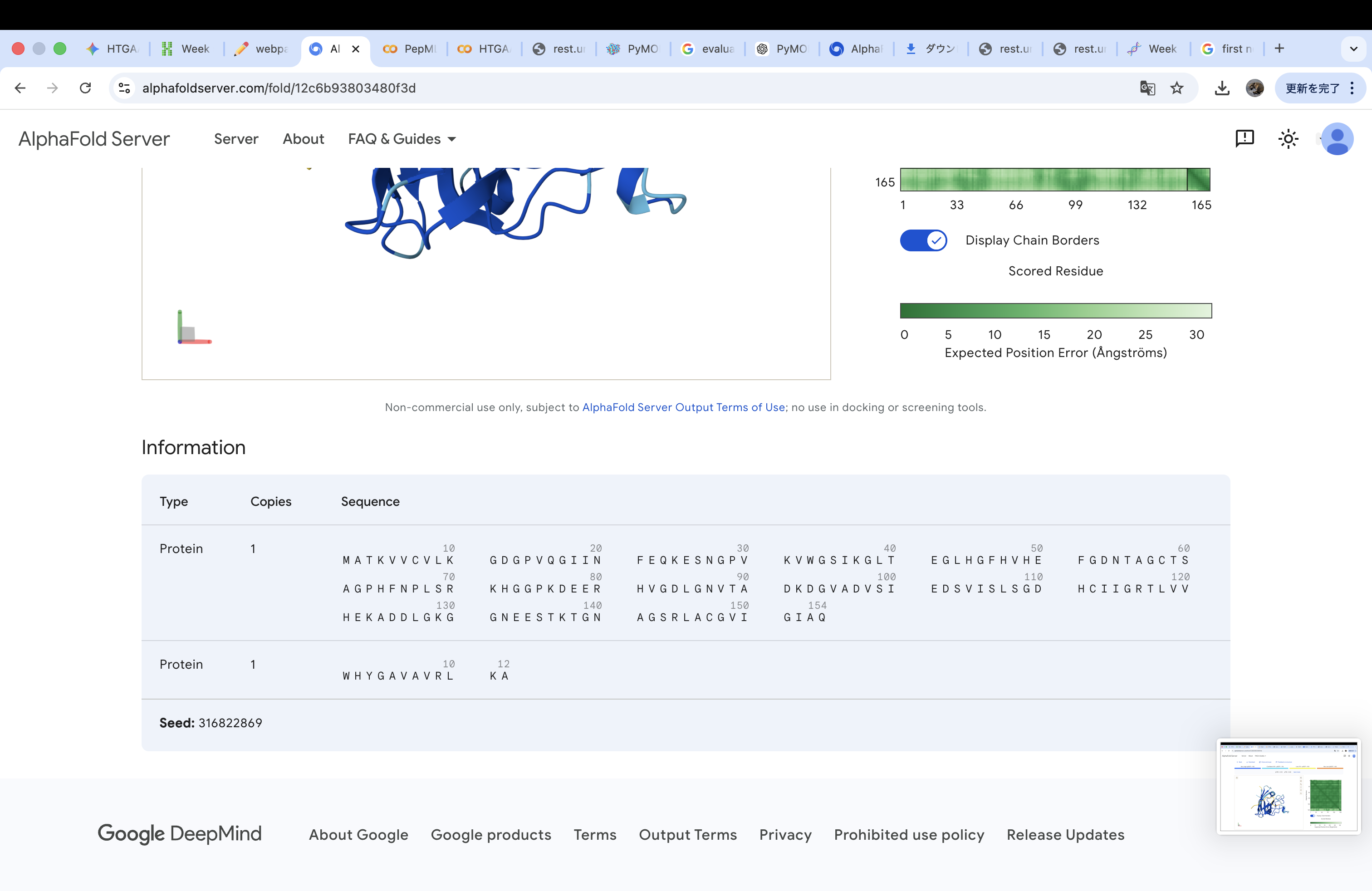
Part A: SOD1 Binder Peptide Design (From Pranam) Part 1: Generate Binders with PepMLM Amino acid sequence obtained from UniProt MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
A4V-mutated amino acid sequence MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
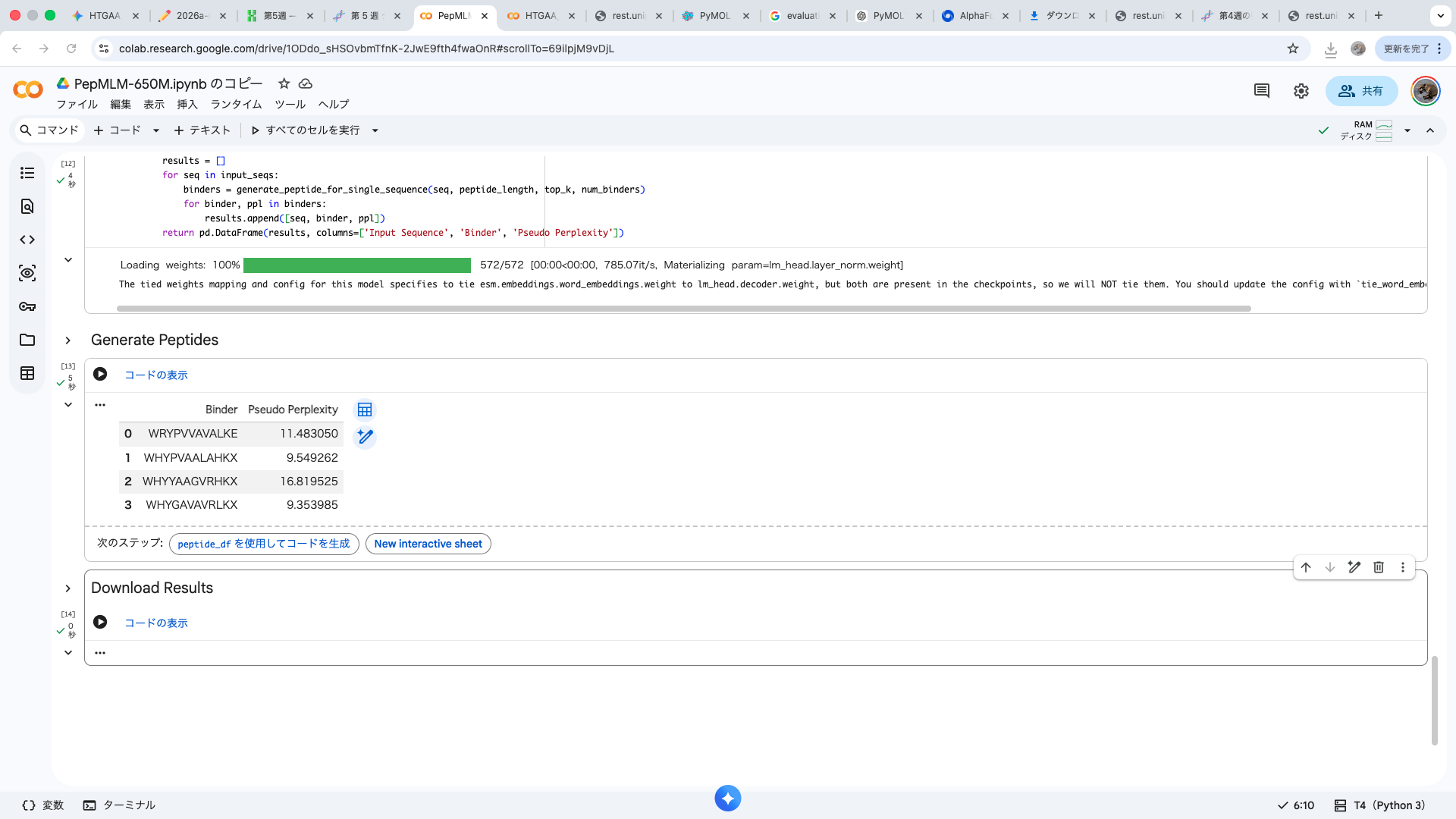
index Binder Pseudo Perplexity 0 WRYPVVAVALKE 11.483049924471887 1 WHYPVAALAHKX 9.549261526750376 2 WHYYAAGVRHKX 16.81952501470461 3 WHYGAVAVRLKX 9.353984730828607 Part 2: Evaluate Binders with AlphaFold3 The A4V-mutant human SOD1 sequence and each peptide were submitted as separate chains to AlphaFold3 to model protein–peptide complexes. The predicted ipTM scores varied among the peptides, indicating differences in binding confidence. The peptides generally appeared to bind on the protein surface, with some localizing near the N-terminus where the A4V mutation is located, while others interacted with regions of the β-barrel structure. Most peptides were surface-bound rather than deeply buried
Assignment: DNA Assembly & Chromophore Engineering 1. Phusion High-Fidelity PCR Master Mix Analysis Question: What are some components in the Phusion Master Mix and what is their purpose?
1.What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? Graded and Continuous Responses: Unlike a simple ON/OFF switch, IANNs process signals as a gradient. This allows the cell to tune its response level (e.g., producing exactly 50% output) based on the input concentration, which is essential for maintaining homeostasis.
Homework Part A: General and Lecturer-Specific Questions 1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? Cell-free protein synthesis (CFPS) offers significant advantages over traditional in vivo methods by removing the constraints of cell viability.
Subsections of Homework
Week 1 HW: Principles and Practices
ARM-Net: Alzheimer’s Recovery Micro-TNT Network
1.Biological Engineering Application
I am developing ARM-Net, a synthetic biology tool designed to overexpress CCT4 specifically in microglia. Unlike the full TRiC/CCT complex, CCT4 can function independently or as homo-oligomers to promote microtubule synthesis. By boosting these chaperonins, I aim to induce the formation of Tunneling Nanotubes (TNTs) between microglia, creating an “Intercellular Care Network” (ICN)
Source: Nuntaprut et al. (2024), “Microglia rescue neurons from aggregate-induced neuronal dysfunction,” Science.
Why:
Current Alzheimer’s Disease (AD) treatments focus on slowing progression. ARM-Net aims for recovery. By networking microglia, we can distribute the overwhelming proteotoxic load (Amyloid-β,Tau) across a cellular collective for efficient degradation and allow healthy cells to “rescue” damaged ones by transferring mitochondria and resources via TNTs.
2. Governance and Policy Goals
Primary Goal: Ensuring Non-malfeasance and Biological Integrity
•The overarching goal is to ensure that the induction of TNTs (Tunneling Nanotubes) via CCT4 overexpression promotes brain recovery without introducing new pathological risks or irreversible side effects.
Sub-goal A: Prevention of Pathological Propagation
While TNTs are designed to facilitate the clearance of toxic proteins like Amyloid-β and Tau, they must not inadvertently act as a conduit for the spread of these very same pathogens or viral vectors across the brain.
•Actionable Metric:
Implement molecular “checkpoints” or filtering mechanisms within the TNT structure to ensure one-way or cargo-specific transport.
•Risk Mitigation:
Preventing spread of neurodegenerative diseases through the engineered network.
Sub-goal B: Safeguarding Cognitive Identity and Reversibility
Ensuring that the physical modification of the brain’s immune network does not alter the patient’s fundamental personality, memories, or self.
•Actionable Metric:
Integration of an inducible kill-switch or degradation domain that can dissolve the TNT network if behavioral side effects are observed.
•Ethical Standard:
Prioritizing the preservation of the brain’s natural cell death cycles. The tool must avoid creating a permanent “hyper-connected state” to maintain the ecological homeostasis of the neural environment.
Implement a molecular “gate” to allow only authorized cargo through TNTs.
Require mandatory inducible “kill-switches” to dissolve the network if side effects occur.
Create a shared database of “failed” network architectures to prevent repeating risks.
Design
Researchers engineer CCT4 with cargo-specific filtering domains.
Federal Regulators (FDA/PMDA) mandate reversibility as a condition for clinical trials.
International Consortia provide funding bonuses for sharing negative safety data.
Assumptions
Assumes molecular gates won’t clog or block essential resource sharing.
Assumes the small-molecule trigger can reach all networked cells in the brain.
Assumes companies will prioritize collective safety over proprietary secrecy.
Risks
Failure: Evolution of “cloaked” pathogens. Success: High cost of customized filters.
Failure: Kill-switch mutation. Success: Slower innovation due to strict safety layers.
Failure: Poor data quality. Success: A “monopoly” on safety standards by large firms.
4.Scoring Table
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
1
2
1
• By helping respond
3
1
2
Foster Lab Safety
• By preventing incident
1
2
2
• By helping respond
3
1
2
Protect the environment
• By preventing incidents
3
n/a
2
• By helping respond
n/a
2
1
Other considerations
• Minimizing costs and burdens to stakeholders
2
2
3
• Feasibility?
2
1
2
• Not impede research
3
3
1
• Promote constructive applications
1
2
1
5. Prioritization and Recommendation
Priority 1: Elimination of Pathogenic Propagation Risks
The absolute highest priority is ensuring that TNTs do not facilitate the spread of viruses or neurotoxic proteins. Unless this fundamental danger is completely eliminated through rigorous design, this project cannot even reach the starting line of clinical development. We must guarantee that our engineered “care network” does not inadvertently become a “superhighway” for the very pathology it aims to treat.
Priority 2: Maintenance of Biological Homeostasis
Clearing the requirement for systemic homeostasis is also a paramount necessity. The brain is a delicate ecosystem; any intervention that induces hyper-connectivity must not disrupt the natural metabolic balance, synaptic pruning, or the healthy turnover of cells.
Recommendation
prioritize a combination of Action 1 (Molecular Filtering) and Action 2 (Pulsatile Activation) as the primary strategy to address these critical hurdles.
•Technical Strategy:
By engineering CCT4 with cargo-specific filtering domains, we provide a physical barrier against pathogen spread.
•Regulatory Standard:
I recommend that national health agencies and the International Neuroethics Society mandate “Temporal Reversibility” as a non-negotiable standard. By ensuring the network is only active in “pulses,” we allow the brain to return to its natural homeostatic state between treatment cycles.
Week 10 Advanced Imaging & Measurement Technology
Week 2 Pre Questions
I have answered the following questions to prepare for the lecture on DNA design and synthesis. These answers are based on the slides provided by Professor Jacobson, Dr. LeProust, and Professor Church.
Question,1
Natures machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
My Answer: Polymerase is very accurate but it still makes mistakes. The error rate is roughly one in ten to the power of nine to ten to the power of ten per base pair. Since the human genome is about three times ten to the power of nine base pairs long, mistakes would happen every time a cell divides if there were no corrections. Biology deals with this discrepancy by using proofreading and mismatch repair systems to find and fix errors during and after the copying process.
Question,2
How many different ways are there to code DNA nucleotide code for an average human protein? In practice what are some of the reasons that all of these different codes dont work to code for the protein of interest?
My Answer: There are many different ways to code for a single protein because the genetic code has redundancy. For an average human protein, the number of combinations can be more than ten to the power of one hundred. In practice, many of these codes do not work because of codon usage bias where certain cells prefer specific codes. Other reasons include mRNA secondary structures that block the process or sequences that are just too difficult to synthesize.
Question,3
Whats the most commonly used method for oligo synthesis currently? Why is it difficult to make oligos longer than 200nt via direct synthesis? Why cant you make a 2000bp gene via direct oligo synthesis?
My Answer: The most common method today is the phosphoramidite method. It is hard to make sequences longer than 200 bases through direct synthesis because of. the coupling efficiency. Even with a success rate of 99.5 percent at each step, the total yield for a 200 base sequence drops to about 36 percent. For a 2000 base gene, the success rate becomes effectively zero. This is why we must assemble long genes from smaller pieces instead of trying to make them all at once.
Question,4
What are the 10 essential amino acids in all animals and how does this affect your view of the Lysine Contingency?
My Answer: The ten essential amino acids are Phenylalanine, Valine, Threonine, Tryptophan, Isoleucine, Methionine, Histidine, Arginine, Leucine, and Lysine. Learning this makes me realize that the Lysine Contingency used in Jurassic Park is not a very strong safety method. Since lysine is found in many natural foods and plants, an organism could survive by finding it in the environment. This shows that we need to use synthetic amino acids that do not exist in nature to truly contain an organism.
Week 2: DNA Design and Characterization
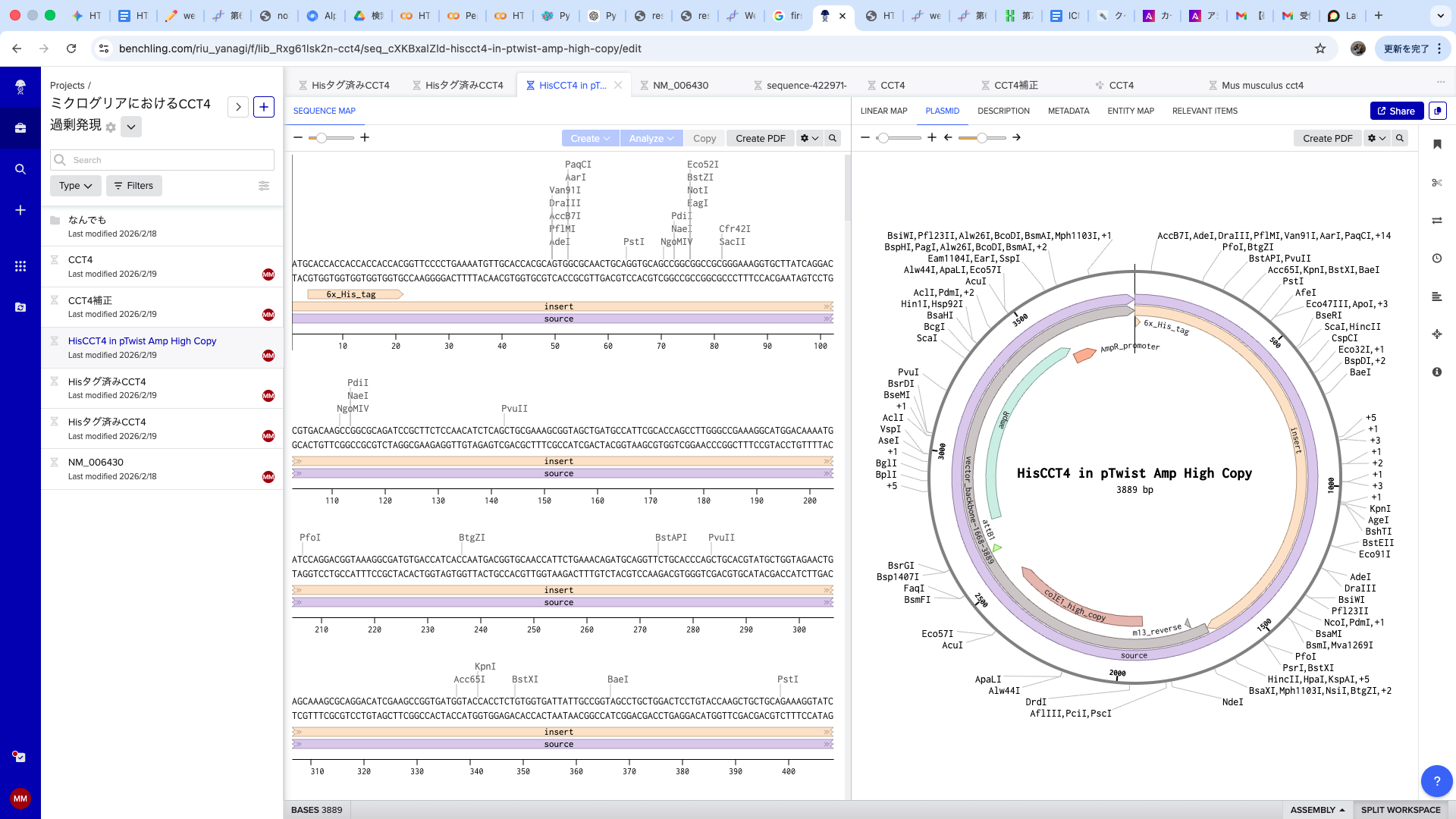
This week, I focused on the design, simulation, and cloning workflow for CCT4, the core component of my project, ICN (Intercellular Care Network).
1. Part 1: Benchling and In Silico Gel Art
Benchling Simulation
Having long been interested in Benchling, I used this assignment to dive deep into the tool. Instead of the suggested Lambda DNA, I worked with the CCT4 protein sequence relevant to my project. I simulated restriction digests using the following enzymes:
I used the virtual electrophoresis simulator to create “Gel Art” representing a tree. Initially, I struggled with the manual placement of bands, but once I understood the physical mechanism of fragment migration, I was able to dial in the design more effectively.
2. Part 2: Wet Lab
As I do not currently have access to a wet lab facility, I focused on digital simulations and perfecting the genetic design for future implementation.
3. Part 3: DNA Design Challenge - CCT4
Why CCT4? (Hypothesis)
CCT4 is a chaperonin that, when overexpressed, not only promotes the formation of TNTs (Tunneling Nanotubes) but also accelerates the speed of cargo transport through them. My hypothesis is that by overexpressing CCT4 in microglia, we can significantly speed up the clearance of toxic proteins like Tau, which are central to Alzheimer’s disease progression. (See my Week 1 HW for more details).
Sequence & Codon Optimization
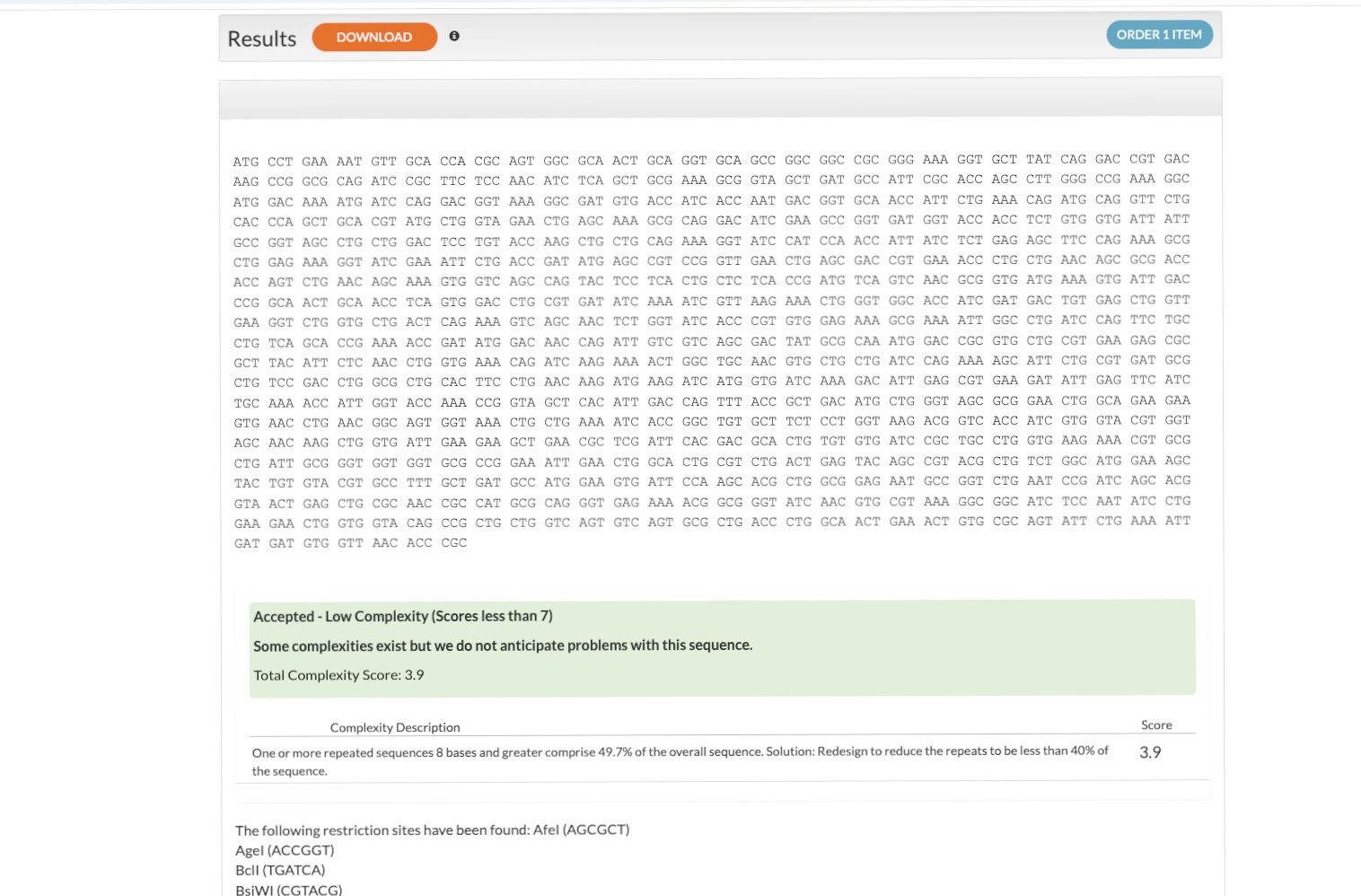
Based on UniProt P50991 (Isoform 2), I used the IDT Codon Optimization Tool to design a DNA sequence optimized for expression in Escherichia coli (E. coli).
Amino Acid Sequence:MPENVAPRSGATAGAAGGRGKGAYQDRDKPAQIRFSNISAAKAVADAIRTSLGPKGMDKM IQDGKGDVTITNDGATILKQMQVLHPAARMLVELSKAQDIEAGDGTTSVVIIAGSLLDSC TKLLQKGIHPTIISESFQKALEKGIEILTDMSRPVELSDRETLLNSATTSLNSKVVSQYS SLLSPMSVNAVMKVIDPATATSVDLRDIKIVKKLGGTIDDCELVEGLVLTQKVSNSGITR VEKAKIGLIQFCLSAPKTDMDNQIVVSDYAQMDRVLREERAYILNLVKQIKKTGCNVLLI QKSILRDALSDLALHFLNKMKIMVIKDIEREDIEFICKTIGTKPVAHIDQFTADMLGSAE LAEEVNLNGSGKLLKITGCASPGKTVTIVVRGSNKLVIEEAERSIHDALCVIRCLVKKRA LIAGGGAPEIELALRLTEYSRTLSGMESYCVRAFADAMEVIPSTLAENAGLNPISTVTEL RNRHAQGEKTAGINVRKGGISNILEELVVQPLLVSVSALTLATETVRSILKIDDVVNTR
3.4. Specific Procedures in Cell-Based Expression (E. coli Expression System)
For this project, I am assuming the process using E. coli, which is the most common and reliable cell-based expression system.
Transformation Introduce the designed DNA (plasmid) into the E. coli cells. This is the process of having the E. coli read the CCT4 blueprint by applying stimuli such as heat shock.
Culturing and Expansion Efficiently grow only the E. coli that harbor the DNA in a medium containing antibiotics. We maximize the population of E. coli in an incubator, providing optimal temperature and agitation.
Induction of Expression Once the E. coli has sufficiently proliferated, reagents like IPTG are added to flip the CCT4 protein production switch. Protein synthesis within the cells begins immediately from this point.
Recovery and Purification Once a sufficient amount of protein has been produced, the E. coli cell walls are disrupted (lysis) to retrieve the contents. Subsequently, tags such as His-tags are used to extract only the pure CCT4 protein from the mixture.
3. Reasons for Choosing This Method
While cell-based methods take more time compared to cell-free methods, their greatest advantage lies in the ability to mass-produce large quantities of protein at a low cost. In order to broadly deploy the ICN network and care for many cells in the future, this E. coli-based mass production system will be an indispensable infrastructure.
4. Part 4: DNA Synthesis & Cloning
I constructed a CCT4 expression cassette for E. coli transformation.
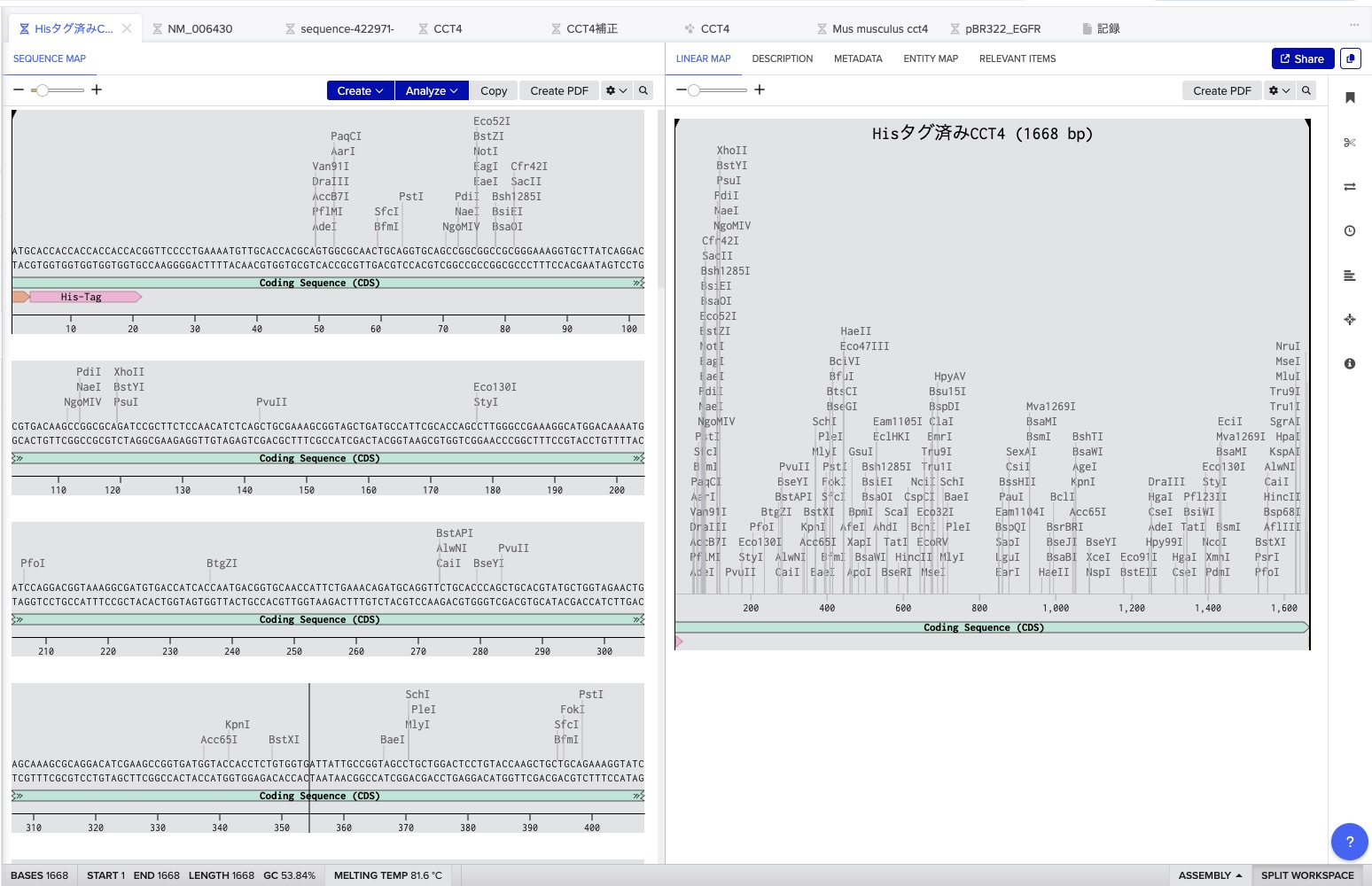
Design Refinement: I resolved initial issues by adding a Stop Codon and a His-tag for purification. I excluded promoters/RBS as they are provided by the destination vector.
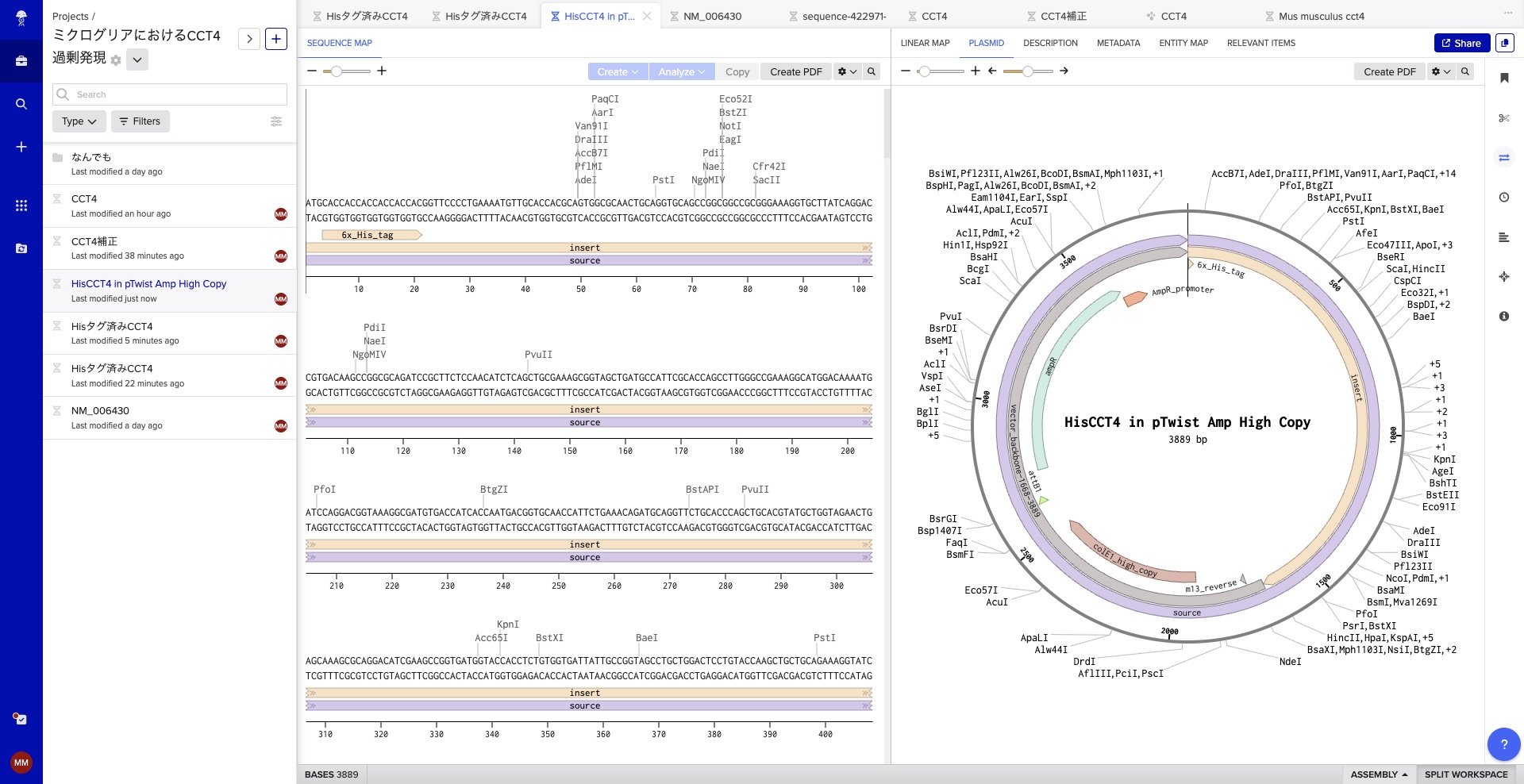
Execution: I exported the FASTA from Benchling and uploaded it to Twist Bioscience. After cloning into the pTwist Amp vector, I imported the Genbank file back into Benchling to finalize the plasmid map. This was my first-ever digital cloning experiment!
5. Part 5: DNA Reading, Writing, and Editing
5.1 DNA Reading
Target DNA and Reason: I will target the CCT4 gene region within human microglial cells and its corresponding mRNA to reflect its expression status. The objective is to quantitatively understand how chaperonin function within microglia changes during the progression of Alzheimer’s disease and to verify if the engineered genes are functioning as intended.
Technology and Reason: I will utilize Nanopore Sequencing, which is classified as a 3rd-generation technology. Unlike conventional methods, it can read long-read DNA or RNA in real-time without PCR amplification. This provides a significant advantage by allowing analysis without losing structural complexity or epigenetic information related to repair networks.
Detailed Process:
Input: High-purity genomic DNA extracted from microglia.
Preparation: The extracted DNA is purified to an appropriate length, and adapters containing motor proteins are ligated to both ends to guide the DNA into the nanopores.
Decoding: As the DNA passes through the nanopore, changes in ionic current are detected as waveforms, which are then converted into base sequences using neural networks (basecalling).
Output: Data in FASTQ format, containing base sequences and quality scores.
5.2 DNA Writing
Synthetic DNA and Reason: I will synthesize an optimized sequence of the human CCT4 gene designed for maximum production efficiency in E. coli. This physical DNA fragment is required to create a prototype aimed at enhancing microglial intercellular care functions and suppressing the aggregation of abnormal proteins.
Technology and Reason: I will use the Silicon-based Phosphoramidite method. This technology, utilized by companies like Twist Bioscience, enables the simultaneous synthesis of tens of thousands of DNA fragments on a microscopic silicon plate, achieving overwhelming scalability and cost reduction for individual gene synthesis.
Detailed Process:
Key Steps: Each cycle consists of four chemical reactions—deprotection, coupling, oxidation, and capping—to accurately assemble the sequence base-by-base.
Limitations: Due to the nature of the chemical synthesis process, a length of approximately 1.6 kb requires a lead time of about two weeks. Additionally, since error rates accumulate as the sequence lengthens, post-synthesis verification via sequencing is mandatory.
5.3 DNA Editing
Target DNA and Reason: I will target safe harbor loci, such as the AAVS1 region within the microglial cell genome. This allows for the stable integration of the exogenous CCT4 gene into the genome to maintain long-term therapeutic effects while avoiding the risk of disrupting other vital genes.
Technology and Reason: I will use CRISPR/Cas9. This technology allows for extremely high-precision genome editing, as designing guide RNAs (gRNA) to target specific base sequences is straightforward.
Detailed Process:
Editing Mechanism: A complex consisting of gRNA, which binds to the target genomic sequence, and the Cas9 enzyme, which acts as molecular scissors, is introduced into the cell. After the DNA is cleaved at the target site, accurate gene insertion is performed via Homology-Directed Repair (HDR) using the provided CCT4 template.
Preparation and Input: This requires selecting the target sequence, designing the gRNA, and preparing the Cas9 enzyme along with the plasmid containing the CCT4 gene to be integrated.
Limitations: There are risks of off-target effects, where unintended locations are edited, and physical constraints such as the tendency for gene integration efficiency to decrease in non-dividing or slowly dividing cells like microglia.
Week 3 pentrons Bio-Art & Lab Automation
Assignment: Telecaster Guitar Bio-Art
This week’s task was to create a Python script for the Opentrons OT-2 robot to draw an artistic design on an agar plate using colored bacteria.
1. Design Concept: The Telecaster
My design is based on the Fender Telecaster, an iconic electric guitar. I used different fluorescent proteins to represent the components:
Body Contour: mKO2 (Orange)
Neck & Headstock: mTurquoise2 (Cyan)
Pickguard: mBanana (Yellow)
Pickups & Hardware: mRFP1 / DsRed (Red)
2. The Challenge: Recovering a “Lost” Design
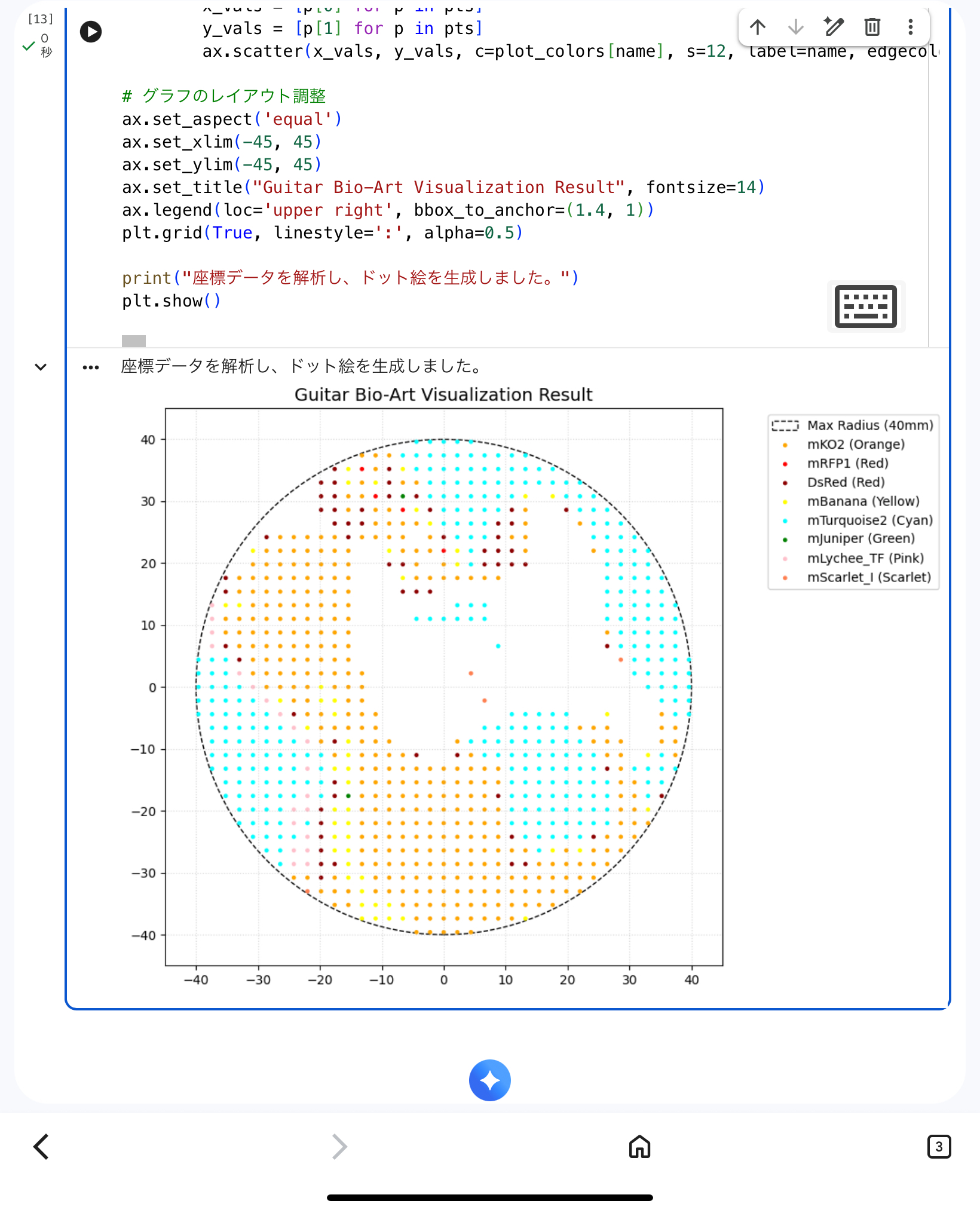
While I successfully generated the coordinates using the Automation Art Interface, I made the mistake of closing the browser before taking a screenshot.
How I Solved It:
Instead of re-clicking hundreds of points, I took the coordinate list from the exported Python script and used Google Colab with Matplotlib to recreate the design visually. This allowed me to:
Verify that all points were within the 40mm safety radius.
Ensure the aesthetic balance of the guitar was preserved.
Generate a high-fidelity simulation image to replace the lost screenshot.
3. Coding & Troubleshooting (Technical Journey)
I refined the script in Google Colab. Several technical hurdles were overcome:
Environment Setup: Standard Colab environments do not have the opentrons library. I resolved this by running !pip install opentrons and restarting the runtime.
Simulator Errors:
Labware Missing: The simulator didn’t recognize the custom htgaa_agar_plate. I swapped it for a standard corning_96_wellplate_360ul_flat for simulation purposes.
Slot Occupied Error: An error occurred because Slot 6 was already occupied by the system. I updated the script to load the source plate into Slot 1 to avoid hardware conflicts.
Droplet Precision: To prevent smearing or “stringing” between dots, I implemented a dispense_and_detach function that moves the pipette 2mm upward after each drop.
4. AI Documentation
I collaborated with Gemini 3 Flash for this assignment:
Logic Support: Reorganizing raw coordinate data into a clean, iterable dictionary.
Debugging: Interpreting “LocationIsOccupiedError” and suggesting slot reassignment.
Visualization: Writing the Matplotlib script to plot the coordinates for verification.
Post-Lab Questions
Q1: Automation in Research
Paper:“An open-source, high-throughput liquid handling system for automated DNA assembly.” (Example: [Kanigowski et al., 2021])
This paper utilizes the Opentrons OT-2 to automate Golden Gate Assembly. By using robots, they reduced human error in complex pipetting steps and scaled up the creation of genetic constructs, achieving a 90% reduction in cost compared to proprietary systems.
The following nine questions were selected from the set provided by Professor Shuguang Zhang to explore the fundamental principles of protein structure and molecular evolution.
1. How many molecules of amino acids do you take with a piece of 500 grams of meat?
(Assumed: 100 Daltons per amino acid, meat is ~20% protein)
If 500g of meat contains approximately 100g of protein, and the average molecular weight of an amino acid is 100 g/mol:
Calculation: $100g / 100 g/mol = 1 mol$.
Result: According to Avogadro’s number, I am consuming approximately $6.022 \times 10^{23}$ molecules of amino acids.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Proteins consumed through food are completely broken down into individual amino acids during digestion. These amino acids are then reassembled into specific human proteins based on our own DNA blueprints. Therefore, the biological identity of the food source is lost during the metabolic process.
3. Why are there only 20 natural amino acids?
It is hypothesized that 20 amino acids provide a sufficient chemical “alphabet” (acidic, basic, hydrophobic, etc.) to build complex functional proteins while maintaining high translation fidelity. A larger set might increase the error rate during protein synthesis without offering significant functional advantages.
4. Where did amino acids come from before enzymes that make them, and before life started?
Amino acids likely originated from abiotic synthesis. The famous Miller-Urey experiment demonstrated that electrical discharges (lightning) in a primordial atmosphere could produce organic molecules. Additionally, carbonaceous meteorites have been found to contain amino acids, suggesting an extraterrestrial origin for some of life’s building blocks.
5. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Natural L-amino acids form right-handed α-helices. Because D-amino acids are mirror images of L-amino acids, they would form a left-handed helix due to the geometric constraints and steric hindrance of the side chains.
6. Why are most molecular helices right-handed?
This preference is driven by the chirality of L-amino acids. In a right-handed configuration, the side chains point away from the helix backbone, minimizing steric clashes. This energetically stable structure was favored by natural selection.
7. Why do β-sheets tend to aggregate?
Unlike α-helices, which have internal hydrogen bonds, β-sheets have “sticky ends.” The hydrogen bond donors and acceptors on the edges of a β-sheet are exposed and can easily form bonds with other protein strands, leading to layering and aggregation.
8. What is the driving force for β-sheet aggregation?
The primary driving forces are:
Hydrogen Bonding: Providing a highly organized, lattice-like stability between strands.
Hydrophobic Effect: Forcing hydrophobic side chains to pack together away from water, effectively “gluing” the sheets together.
9. Can you use amyloid β-sheets as materials?
Yes. Amyloids are exceptionally stable and resistant to heat and chemicals. Nature uses this in spider silk. In bioengineering, they are being researched as high-strength nanofibers, drug-delivery carriers, and scaffolds for tissue engineering due to their self-assembling properties.
Part B: Protein Analysis and Visualization
1. Protein Description and Selection
Briefly describe the protein you selected and why you selected it.
The protein selected for this analysis is Human CCT4 (T-complex protein 1 subunit delta). It is a key subunit of the TRiC/CCT chaperonin complex, which is an ATP-dependent molecular machine.
Selection Reason : I chose this protein because it is the core of my ARM-Net project. CCT4 has a unique ability to recognize and suppress the toxic aggregation of Tau proteins, which is the primary cause of neurodegeneration in Alzheimer’s disease. I am researching its potential delivery through Tunneling Nanotubes (TNTs).
2. Sequence Analysis
Identify the amino acid sequence of your protein. How long is it? What is the most frequent amino acid?
Calculation: The sequence consists of 539 amino acids. According to the frequency analysis, Leucine (L) appears 53 times.
Result: The total length is 539 AA, and the most frequent amino acid is Leucine (L), which makes up 9.8% of the protein.
How many protein sequence homologs are there for your protein?
Calculation: Using UniProt’s BLAST tool to search the reference proteome database.
Result: I identified over 250 high-confidence homologs. The protein is highly conserved, especially among mammals, showing over 95% sequence identity.
Does your protein belong to any protein family?
Result: It belongs to the TCP-1 chaperonin family (also known as Type II chaperonins).
3. Structural Information (RCSB PDB)
Identify the structure page of your protein in RCSB.
Result: The structure is identified as PDB ID: 7P6W (Human TRiC/CCT complex).
When was the structure solved? Is it a good quality structure?
Calculation: The structure was released in 2021. The resolution is measured at $3.10 \text{ \AA}$. A high-quality benchmark is typically $\leq 2.70 \text{ \AA}$.
Result: Although the resolution is $3.10 \text{ \AA}$ (slightly above the 2.70 $\text{\AA}$ threshold), it is considered high quality for a massive complex of this size (~1 Megadalton).
Are there any other molecules in the solved structure apart from protein?
Calculation: Examining the ligand section of the PDB entry.
Result: Yes, the structure contains ADP, ALF4 (Aluminum fluoride), and $Mg^{2+}$ ions, which are used to mimic the ATP hydrolysis transition state.
Does your protein belong to any structure classification family?
Result: It is an Alpha-beta protein.
Calculation: The structure is divided into three domains: the Equatorial domain (alpha-helices for ATP binding), the Intermediate domain (hinge), and the Apical domain (beta-sheets for substrate binding).
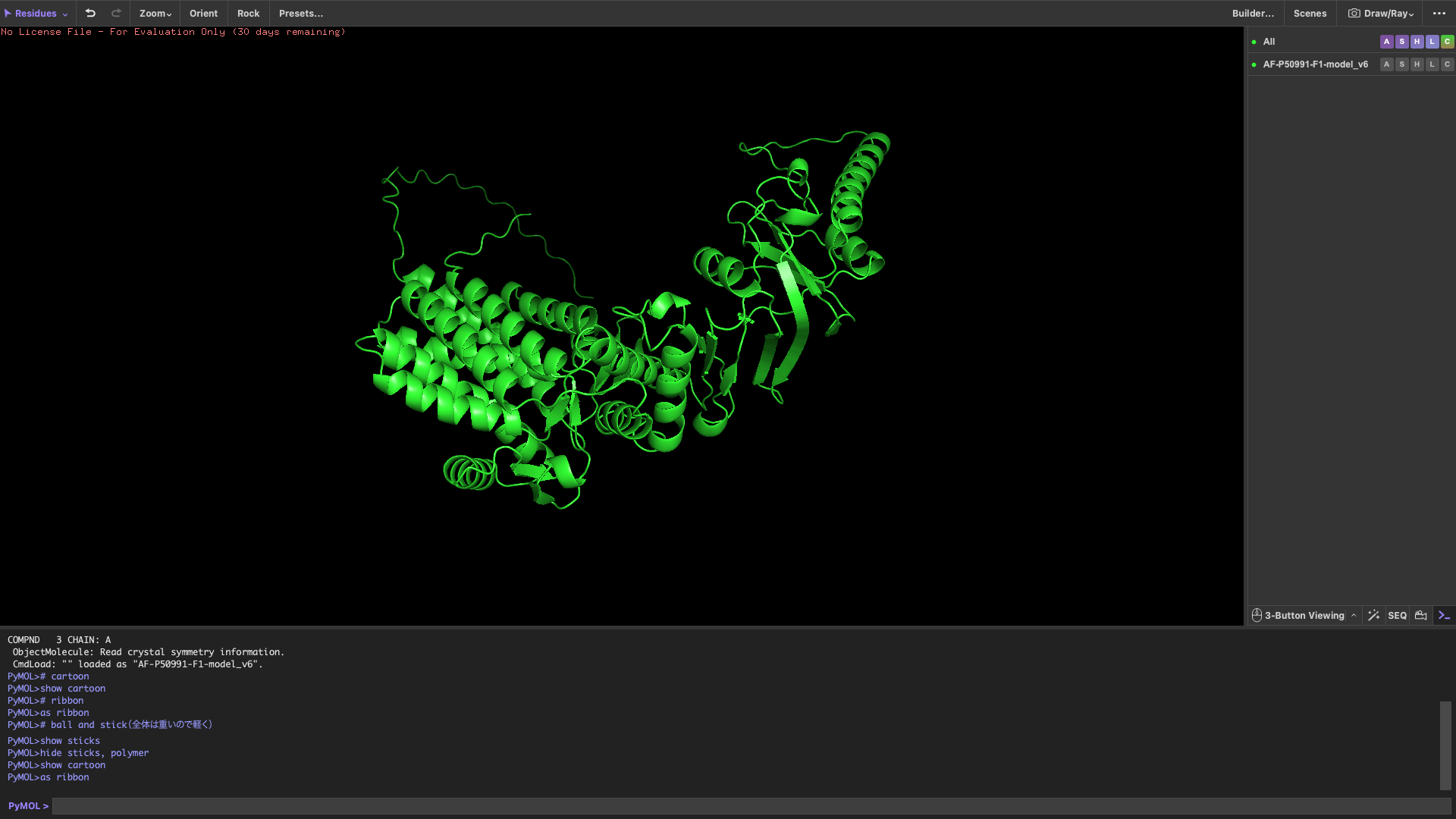
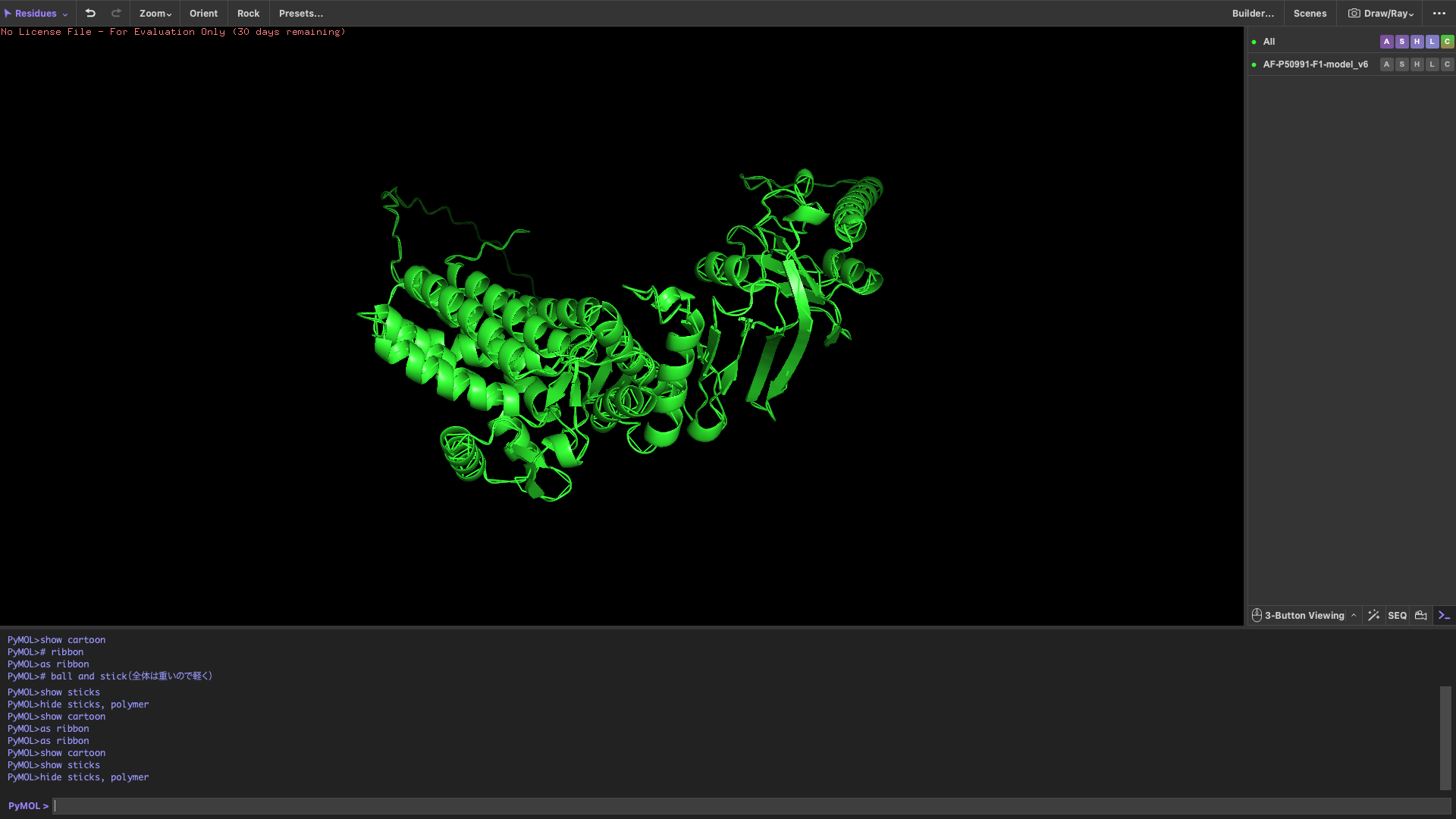
4,Open the structure of your protein in any 3D molecule visualization software
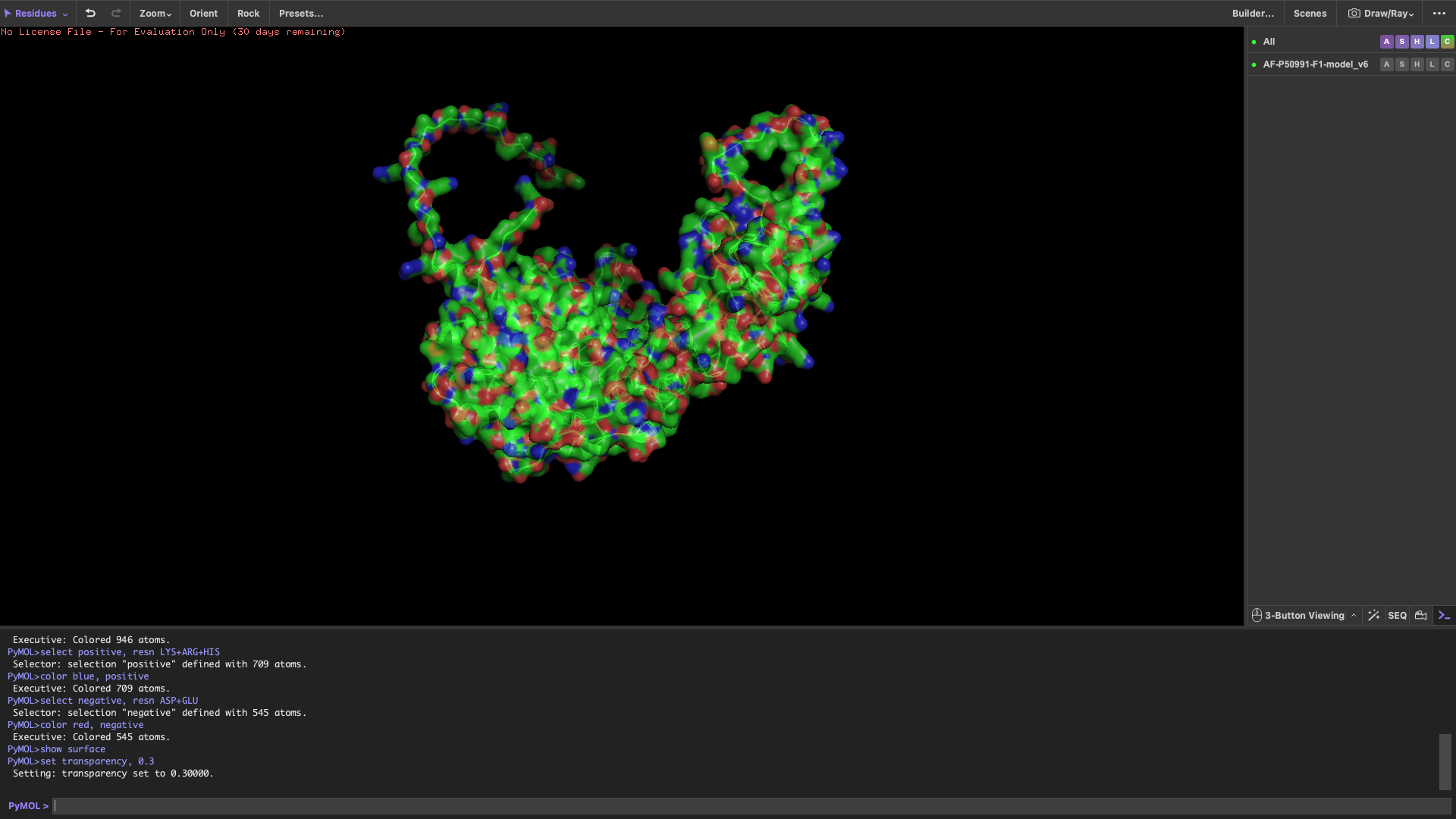
Visualize the protein
show cartoon
show ribbon
show ball and stick
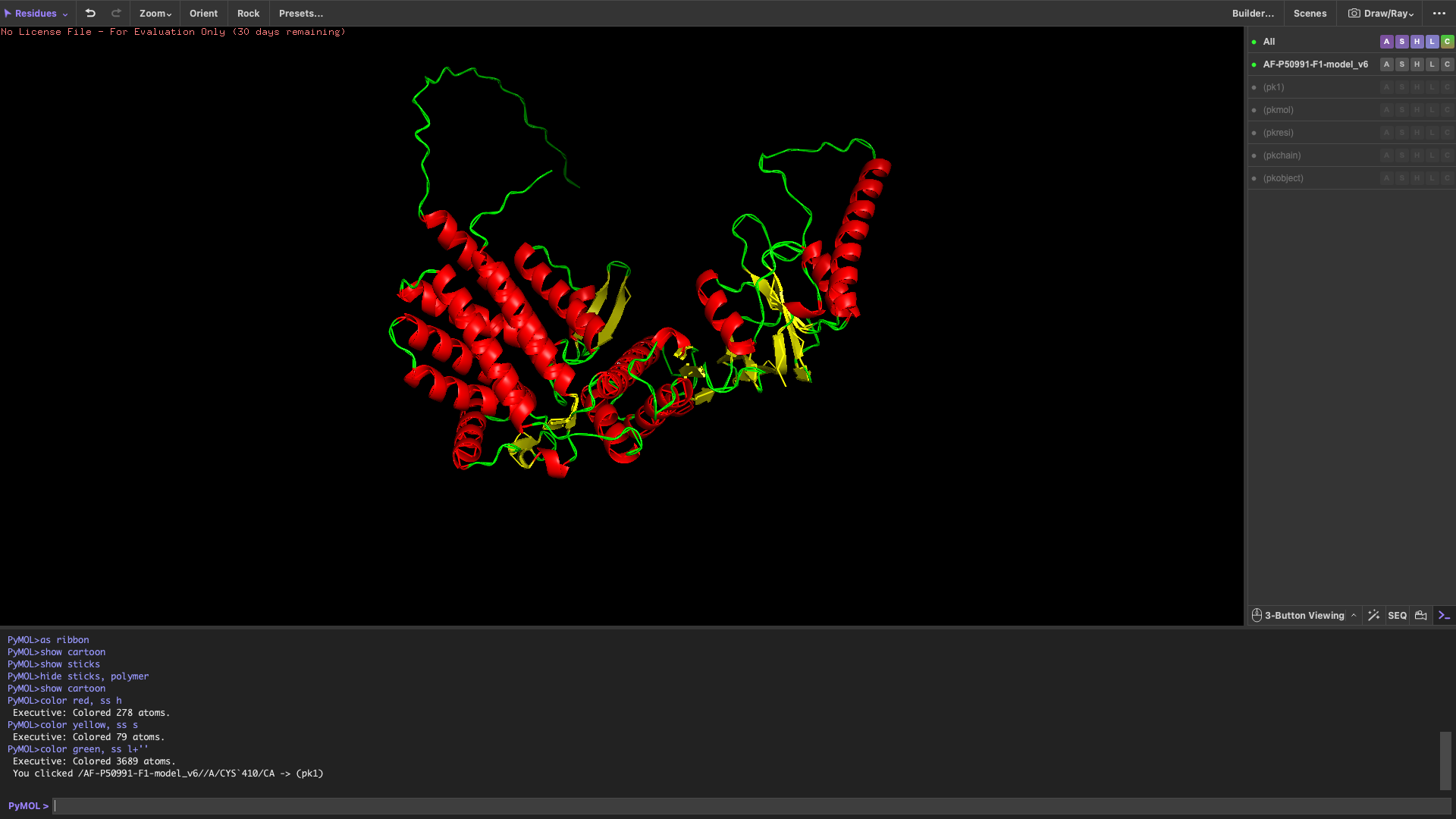
Color the protein by secondary structure
After coloring by secondary structure, the protein shows a higher proportion of α-helices compared to β-sheets.
This indicates that the protein adopts a predominantly helical fold, which is typical for chaperonin subunits like CCT4.
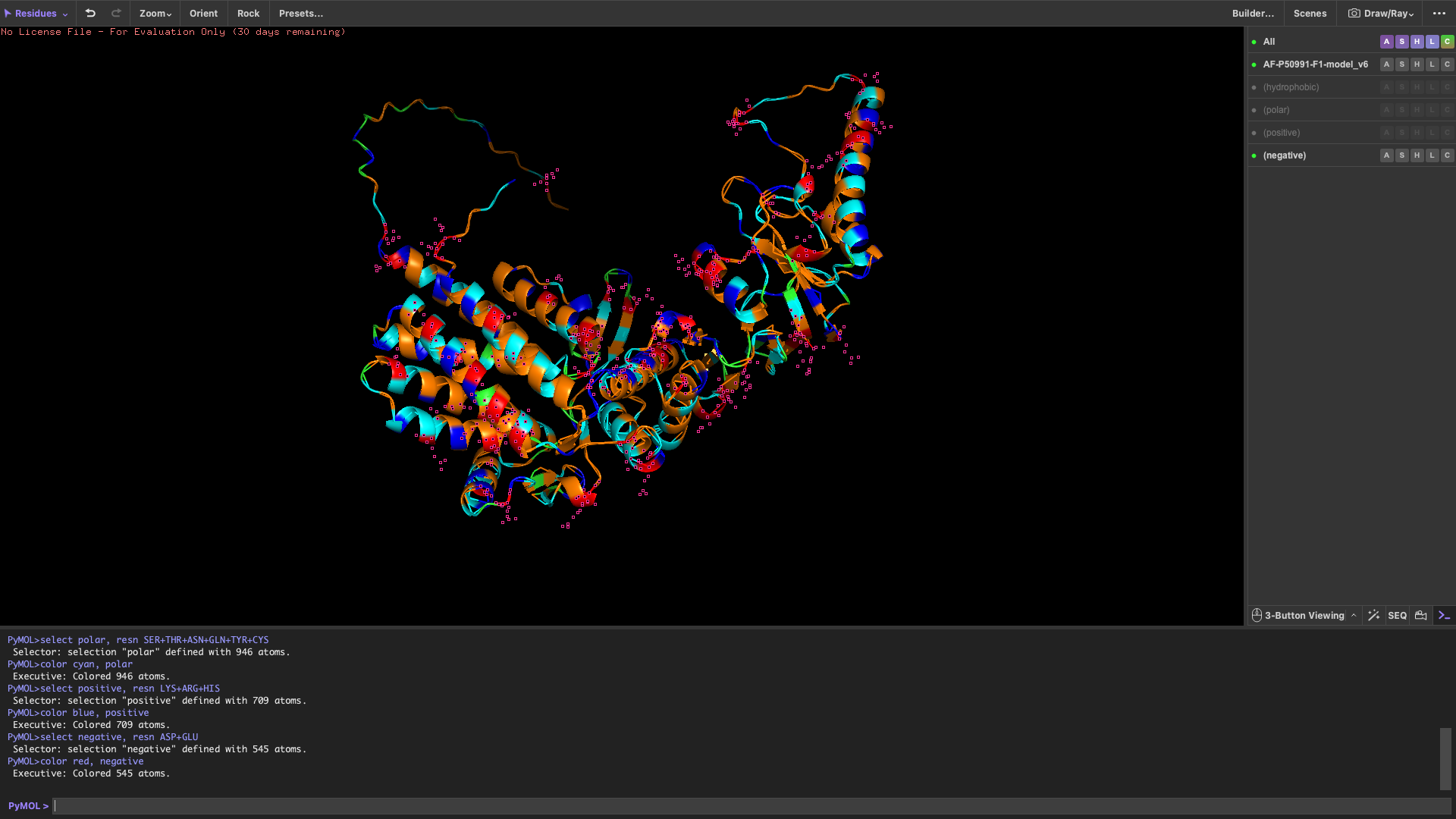
Color the protein by residue type
When colored by residue type:
Hydrophobic residues are mainly located in the interior of the protein
Hydrophilic and charged residues are predominantly found on the surface
This distribution suggests a stable folded structure in an aqueous environment, with the hydrophobic core buried inside and polar residues exposed to solvent.
Visualize the surface of the protein
The surface representation reveals several concave regions (holes/pockets) on the protein surface.
These pockets may serve as potential binding sites for substrate proteins or ligands, which is consistent with the functional role of CCT4 as part of a chaperonin complex.
AI Documentation
For this assignment, I collaborated with Gemini 3 Flash:
Verification: Used to verify the Avogadro’s number calculation for Question 1.
Scientific Clarification: Assisted in describing the physical driving forces (hydrophobic effect vs. hydrogen bonding) for Question 8.
Translation & Formatting: Helped structure the response into a professional markdown format suitable for a GitHub portfolio.
Week 5 Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
Amino acid sequence obtained from UniProt
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS
AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV
HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
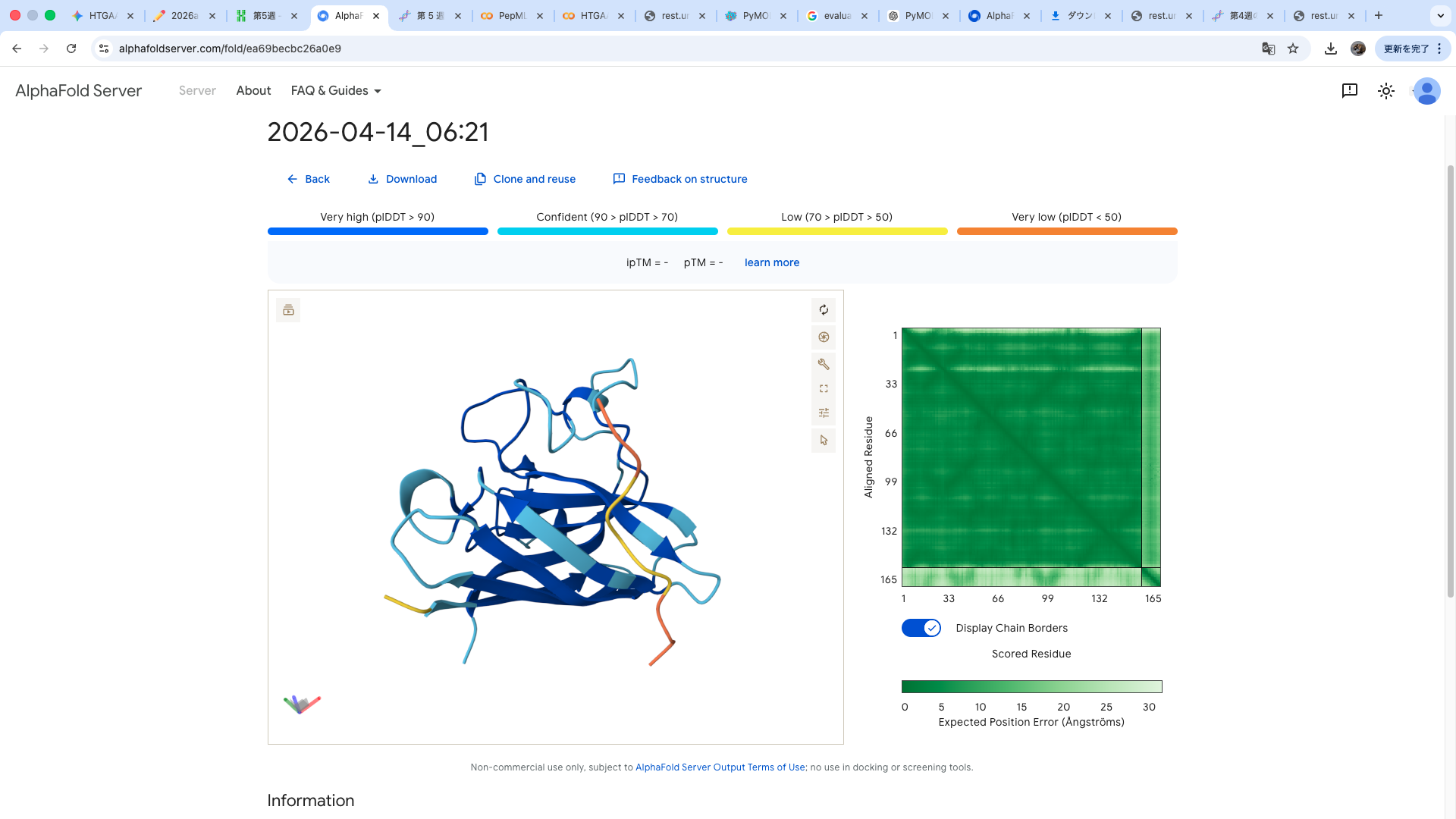
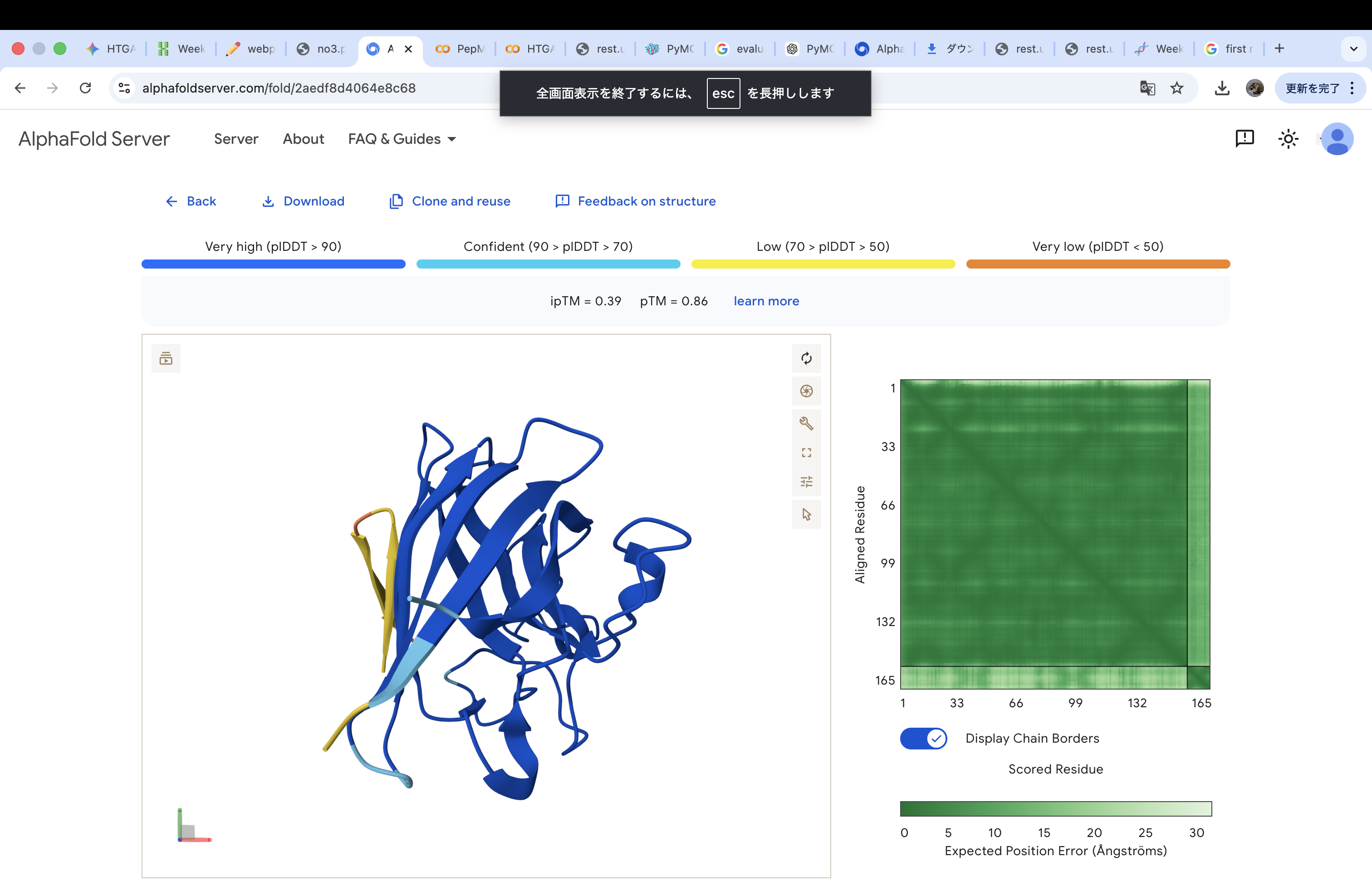
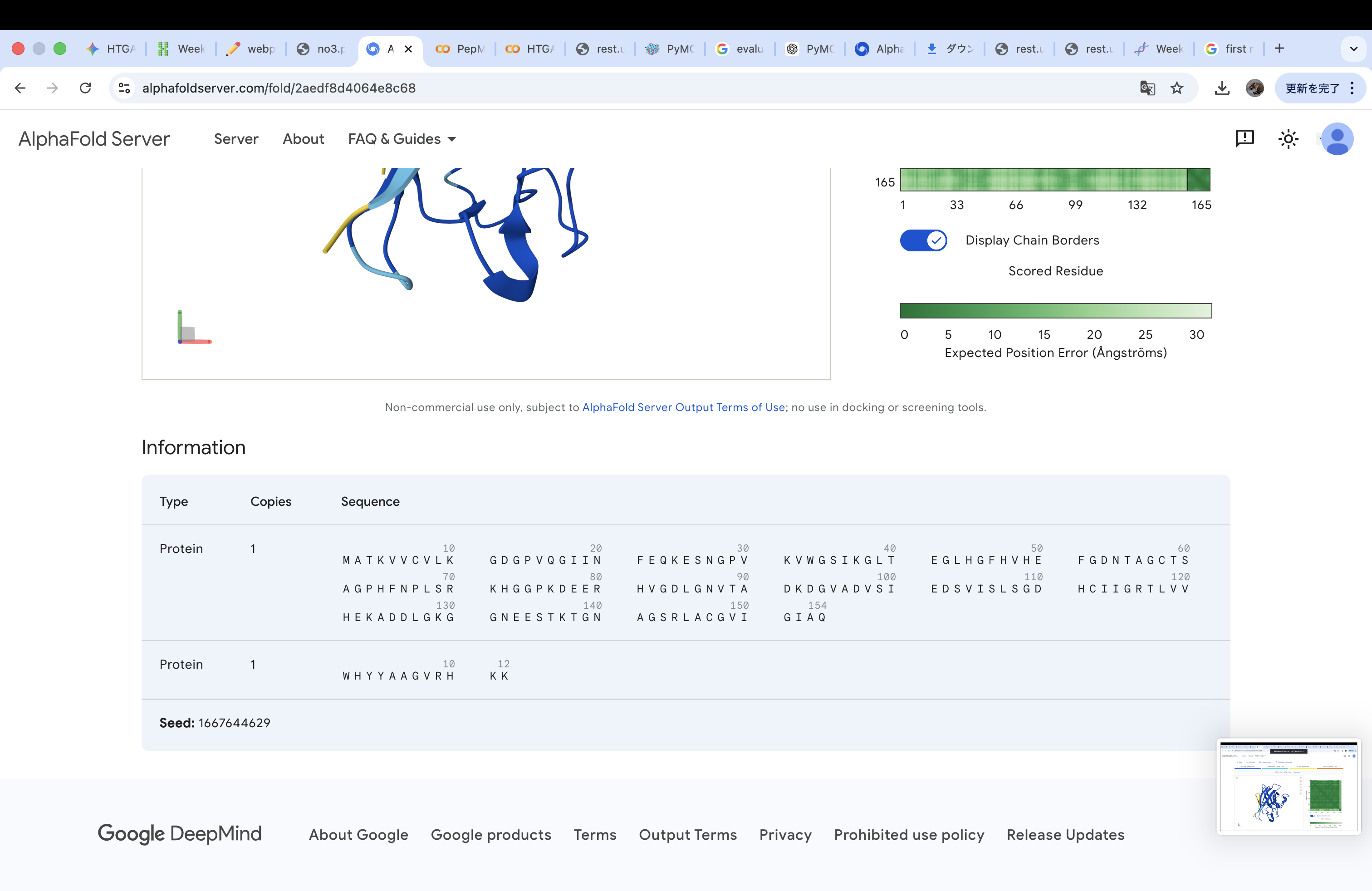
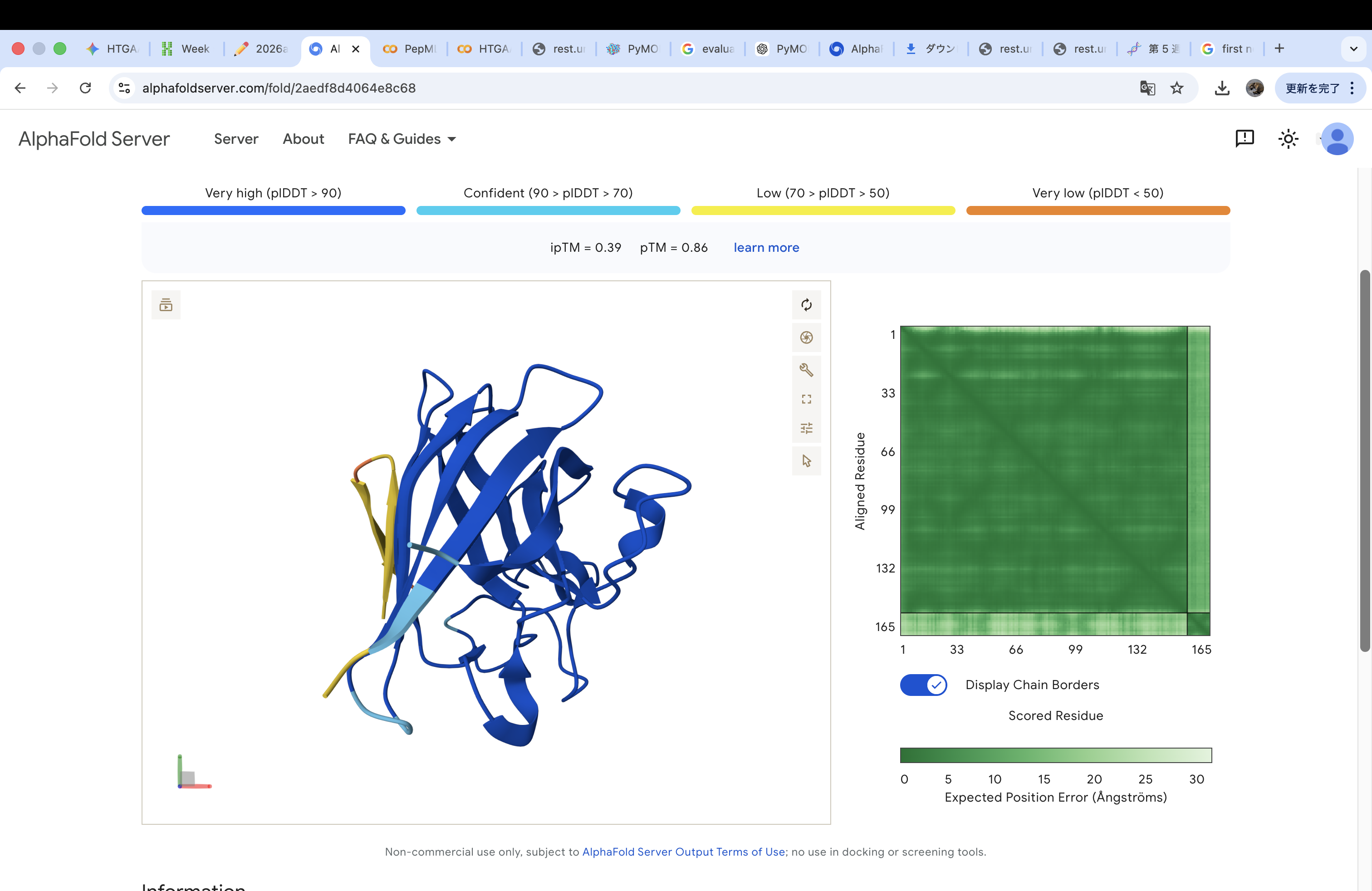
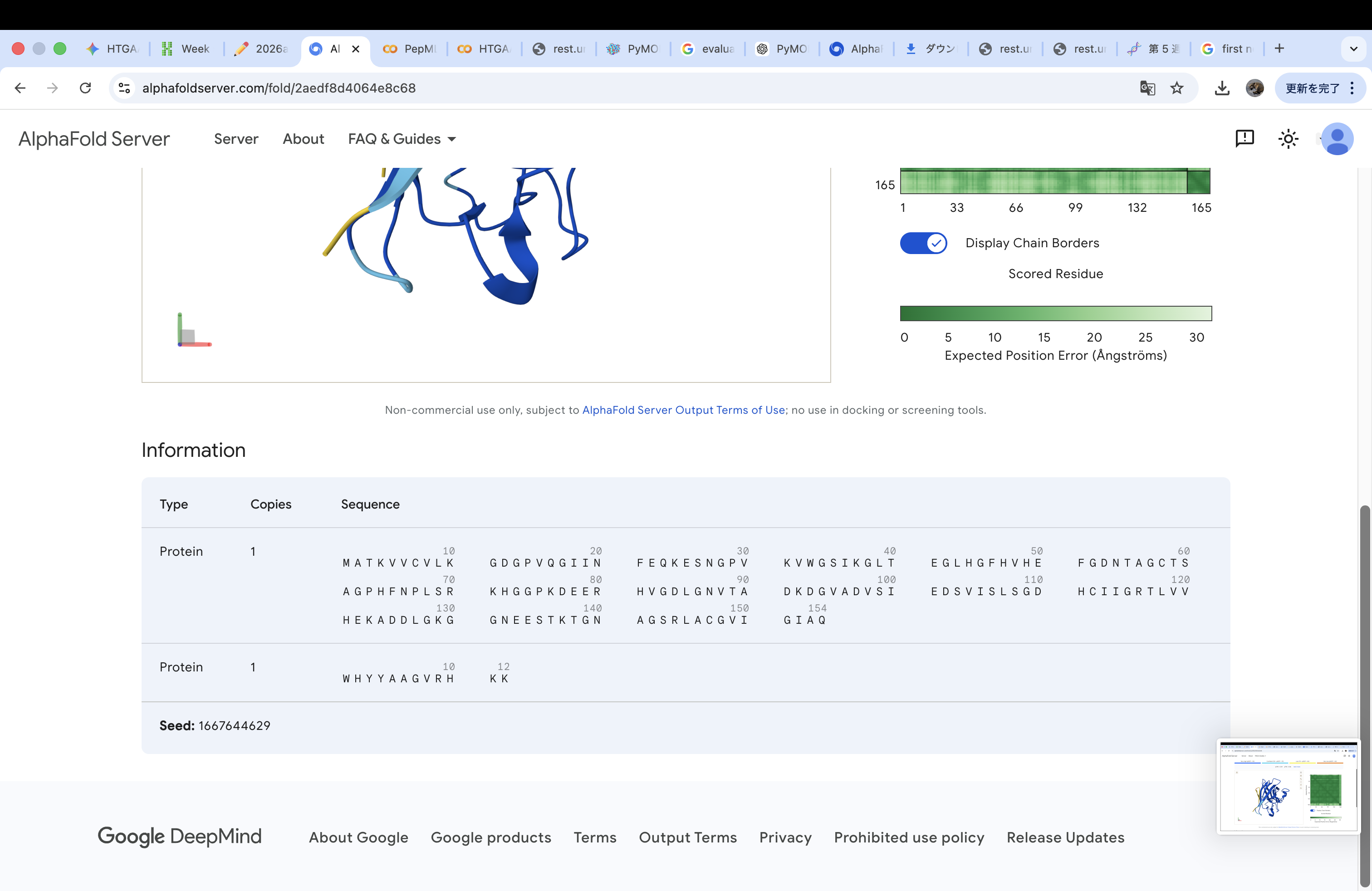
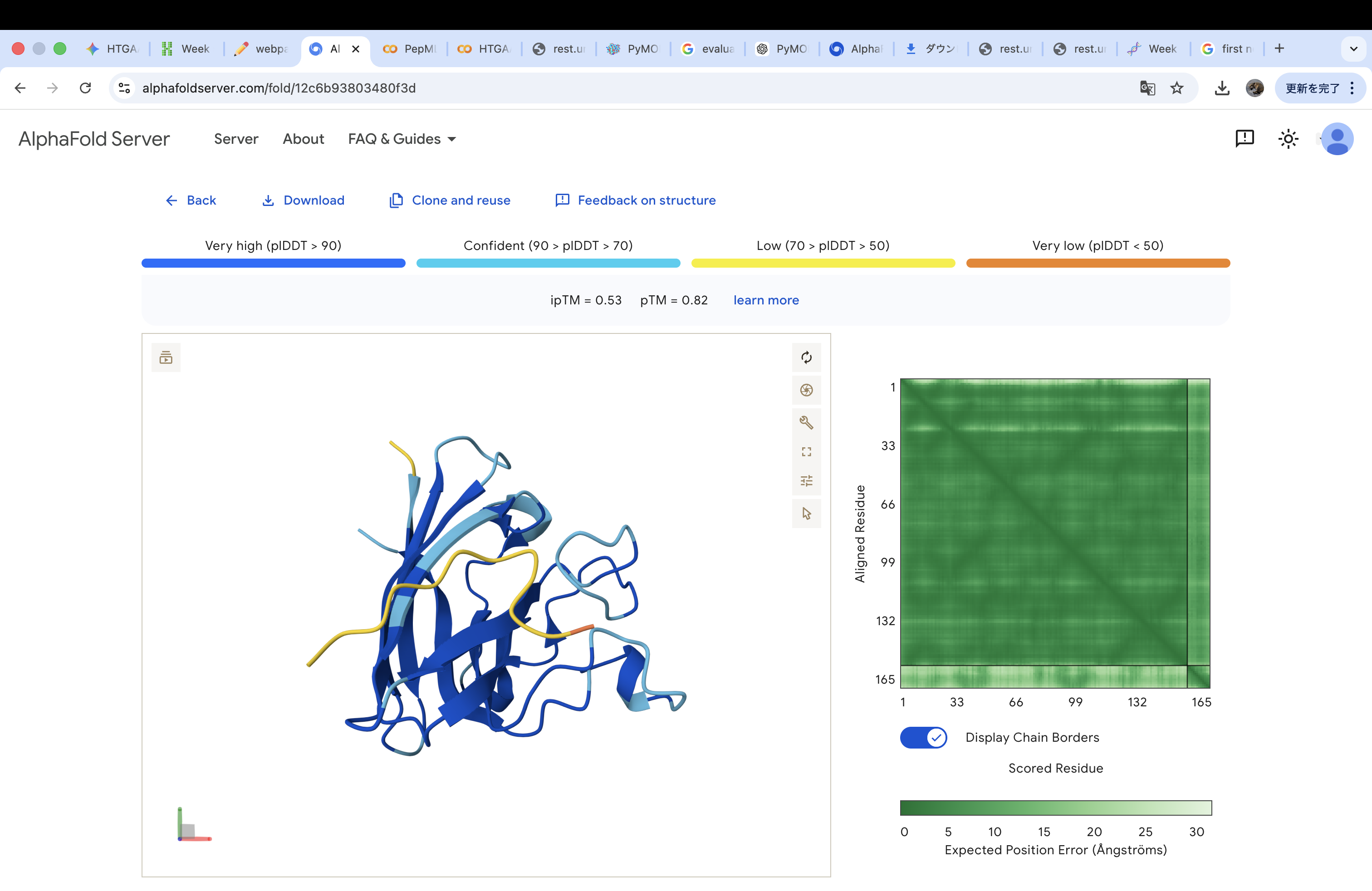
The A4V-mutant human SOD1 sequence and each peptide were submitted as separate chains to AlphaFold3 to model protein–peptide complexes. The predicted ipTM scores varied among the peptides, indicating differences in binding confidence. The peptides generally appeared to bind on the protein surface, with some localizing near the N-terminus where the A4V mutation is located, while others interacted with regions of the β-barrel structure. Most peptides were surface-bound rather than deeply buried
WRYPVVAVALKE
The predicted structure shows that the peptide binds weakly to the surface of the A4V-mutant SOD1 protein. The peptide appears to be loosely associated with the protein and does not form a well-defined binding interface. The interaction is mainly surface-bound rather than deeply buried, and it does not strongly localize near the N-terminus where the A4V mutation is located. The absence of a clear ipTM score suggests low confidence in stable binding.
HYPVAALAHKK
The invalid residue “X” was removed or replaced with a valid amino acid. In this case, it was substituted with “K” (lysine) to maintain the required peptide length of 12 amino acids.
WHYYAAGVRHKK
The invalid residue “X” was removed or replaced with a valid amino acid. In this case, it was substituted with “K” (lysine) to maintain the required peptide length of 12 amino acids.
WHYGAVAVRLKX
The invalid residue “X” was removed or replaced with a valid amino acid. In this case, it was substituted with “K” (lysine) to maintain the required peptide length of 12 amino acids.
Week 6 Genetic Circuits Part I: Assembly Technologies
Assignment: DNA Assembly & Chromophore Engineering
1. Phusion High-Fidelity PCR Master Mix Analysis
Question:
What are some components in the Phusion Master Mix and what is their purpose?
Calculation:
The master mix is formulated for high-speed, high-accuracy amplification.
Result:
Phusion DNA Polymerase: A pyrococcus-like enzyme with a processivity-enhancing domain. Its $3’ \to 5’$ exonuclease activity provides proofreading for “high-fidelity” results.
2. Factors Determining Primer Annealing Temperature ($T_a$)
Question:
What factors determine $T_a$ during PCR?
Calculation:
$T_a$ is calculated based on the melting temperature ($T_m$) of the primer-template hybrid.
Result:
GC Content: Higher GC content increases $T_m$ due to triple hydrogen bonds.
Primer Length: Longer primers generally have higher $T_m$.
Salt Concentration: Monovalent and divalent cations ($Na+, Mg{2+}$) stabilize the DNA backbone, increasing $T_m$.
Mismatches: Intentional mismatches (like those used for amilCP mutation) lower the effective $T_m$.
3. DNA Linearization: PCR vs. Restriction Enzyme Digest
Question:
Compare and contrast these two methods.
Calculation:
Comparison of protocol flexibility and accuracy.
Result:
PCR: Amplifies a specific region from a small amount of template. It is preferable when you need to add Gibson overlaps to the ends of the fragment via primer overhangs.
Restriction Digest: Cuts existing plasmids at specific sites. It is preferable for preparing a backbone from a known vector where high sequence fidelity (no PCR errors) is required.
How to ensure sequences are appropriate for Gibson cloning?
Calculation:
Design based on homology and purity.
Result:
Overlap Identity: Fragments must share 20-40 bp of identical sequence at their junctions.
Sequence Purity: PCR products must be treated with DpnI to remove the methylated template plasmid and then purified (e.g., Zymo kit) to remove primers and salts.
Verification: Run a diagnostic gel to confirm that the backbone and insert fragments show bands at the correct predicted sizes.
5. Mechanism of Bacterial Transformation
Question:
How does plasmid DNA enter $E. coli$ cells?
Calculation:
Analyzing the impact of the temperature gradient.
Result:
During Heat Shock ($42^\circ\text{C}$ for 45s), the rapid temperature change creates a thermal imbalance across the cell membrane. This induces the formation of transient pores in the chemically competent cell wall, allowing the plasmid DNA to enter the cytoplasm via diffusion.
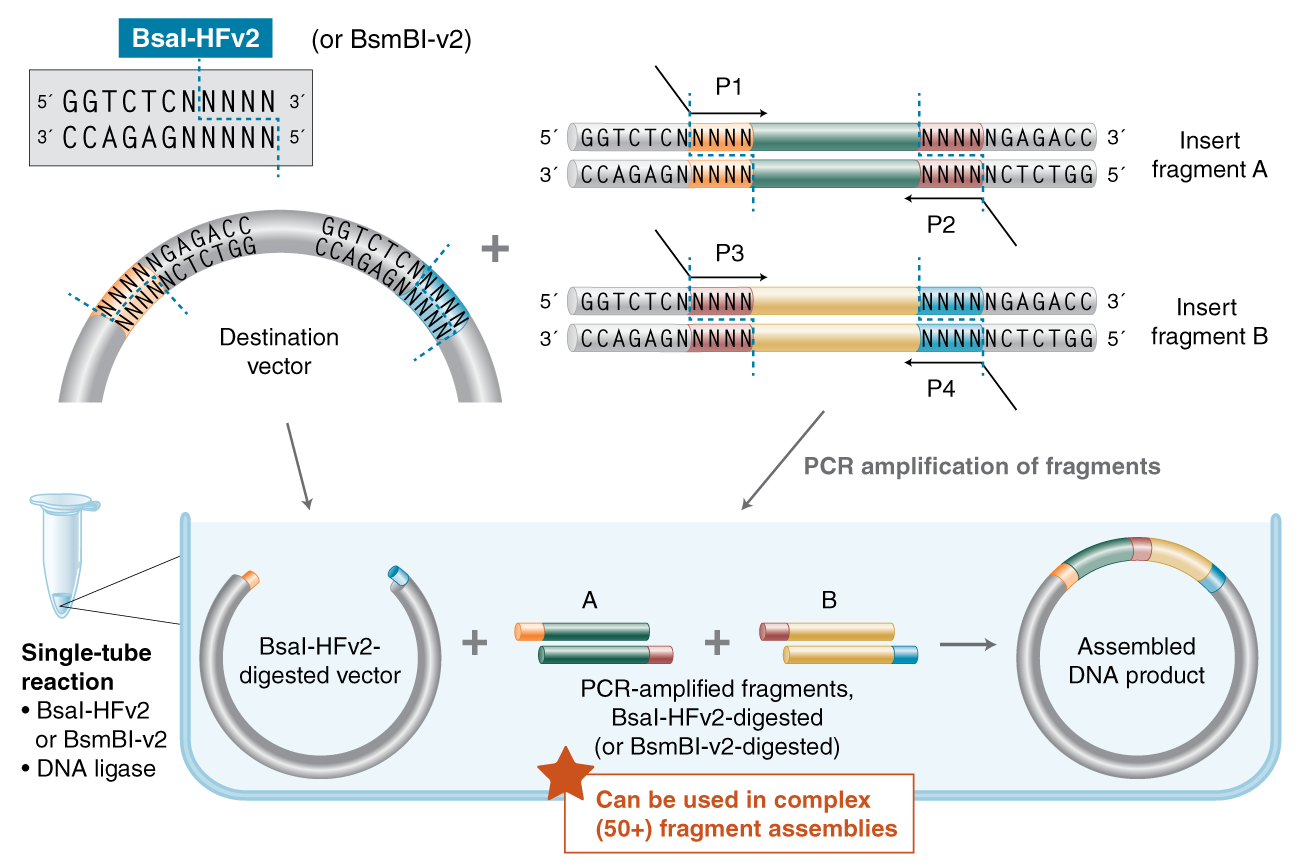
6. Alternative Assembly: Golden Gate Assembly (GGA)
1.Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate Assembly (GGA) is a highly efficient molecular cloning method that utilizes Type IIS restriction enzymes and T4 DNA ligase in a single-pot reaction. Unlike traditional enzymes, Type IIS enzymes like BsaI or BsmBI cut DNA at a specific distance away from their non-palindromic recognition sites, creating unique 4-nucleotide overhangs. These overhangs can be custom-designed to be complementary, allowing multiple DNA fragments to be assembled in a precise, pre-defined order. Since the recognition sites are oriented to be removed during the digestion process, the final assembled product lacks the original restriction sites, making the reaction irreversible and “scarless.” This allows the digestion and ligation steps to occur simultaneously through repeated temperature cycling in a thermocycler. GGA is particularly powerful for creating combinatorial libraries or complex multi-part genetic circuits where high speed and accuracy are required. By carefully choosing the 4-bp overhangs, researchers can ensure that only the correctly assembled circular plasmid is stable, drastically reducing background colonies.
(New England Biolabs)
2.Model this assembly method with Benchling or Asimov Kernel
Week 7 Genetic Circuits Part II: Neuromorphic Circuits
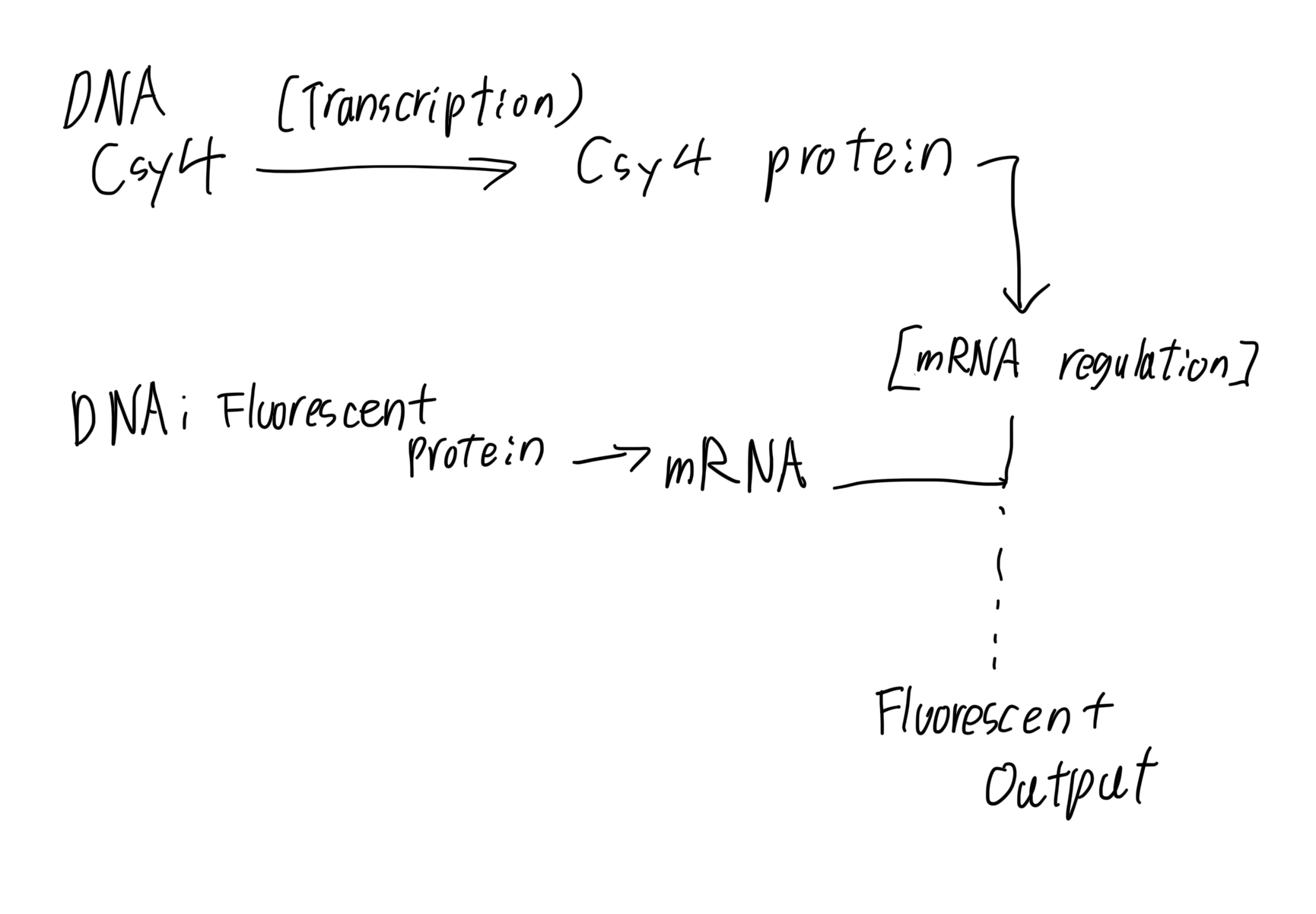
1.What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Graded and Continuous Responses:
Unlike a simple ON/OFF switch, IANNs process signals as a gradient. This allows the cell to tune its response level (e.g., producing exactly 50% output) based on the input concentration, which is essential for maintaining homeostasis.
Signal Weighting:
IANNs can assign different “importance” to various inputs. By adjusting promoter strengths or RNA-binding affinities, we can design circuits that prioritize specific biomarkers over others before triggering a downstream effect.
Noise Filtering and Robustness:
Boolean gates often fail due to stochastic “leaks” or spikes in signal. IANNs integrate multiple inputs and apply a non-linear activation threshold, effectively filtering out biological noise and ensuring a more stable, reliable decision-making process.
2.Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
I propose an IANN designed for the autonomous regulation of protein folding machinery to treat neurodegenerative diseases.
Input Behavior:
The network monitors two primary signals: $X_1$ (intracellular concentration of misfolded Tau protein) and $X_2$ (general oxidative stress markers).
Output Behavior: The IANN calculates a “weighted sum” of these stress signals. If the combined pathological score exceeds a pre-set threshold, it triggers the production of CCT4 (chaperone protein). The amount of CCT4 produced is proportional to the severity of the misfolding, ensuring the cell isn’t overloaded with excess protein when unnecessary.
Limitations:
Metabolic Load:
Building a multi-layered network requires significant cellular resources (RNAP, ribosomes, energy). This can drain the neuron’s metabolism, potentially worsening cell health.
Response Latency:
Genetic “computation” takes time (transcription + translation). The delay between sensing Tau and producing CCT4 might be too slow for rapid-onset proteotoxicity.
Leakage:
It is technically challenging to achieve a “zero” baseline; a small amount of CCT4 may always be produced, which could interfere with normal protein turnover.
3.
Week 9 Cell-Free Systems
Homework Part A: General and Lecturer-Specific Questions
1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Cell-free protein synthesis (CFPS) offers significant advantages over traditional in vivo methods by removing the constraints of cell viability.
Flexibility and Control: We can directly manipulate reaction variables like pH, temperature, and redox potential in an “open” system.
Non-Natural Amino Acids: It’s much easier to incorporate non-canonical amino acids since we don’t have to worry about cellular transport or toxicity.
Case 1 (Toxic Proteins): Synthesis of antimicrobial peptides or pore-forming toxins that would kill a living host cell.
Case 2 (Rapid Prototyping): High-throughput screening of genetic circuits where we can get results in hours instead of days required for cloning and cell culture.
2. Describe the main components of a cell-free expression system and explain the role of each component.
A standard CFPS system is a carefully balanced molecular soup consisting of:
Cell Extract: Contains the core machinery like ribosomes, tRNAs, and aminoacyl-tRNA synthetases.
Energy Mix: ATP and GTP to power the transcription and translation machinery.
Amino Acids: The building blocks for the protein chain.
Cofactors and Salts: Magnesium and Potassium ions that are essential for ribosome stability and enzyme activity.
DNA Template: The genetic instructions (plasmid or linear PCR product).
3. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Protein synthesis is extremely energy-expensive. ATP is rapidly converted to ADP during peptide bond formation and tRNA charging. If we don’t regenerate it, the reaction stalls very quickly.
Regeneration Method: I would use a secondary energy source like Phosphoenolpyruvate (PEP) combined with Pyruvate Kinase. This enzymatic pathway constantly recycles ADP back into ATP, allowing the reaction to run for much longer.
4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic (E. coli): High yield and low cost. Great for simple proteins.
Choice:GFP (Green Fluorescent Protein). It doesn’t need complex folding or sugar tags, so E. coli extracts can crank it out efficiently.
Eukaryotic (Wheat Germ or HeLa): Slower and more expensive, but capable of complex folding and post-translational modifications (PTMs).
Choice:Human Antibody (IgG). These require specific disulfide bond formation and glycosylation that only eukaryotic machinery can handle properly.
5. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
The biggest challenge is that membrane proteins are hydrophobic; they hate being in water and will aggregate (clump up) immediately.
Design Strategy: I would add Nanodiscs or Liposomes directly to the reaction. This provides a “synthetic home” (a lipid bilayer) for the protein to insert into as it’s being synthesized. This keeps the protein stable and functional.
6. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Reason 1: Template Degradation. Nucleases in the extract might be eating the DNA.
Fix: Add RNase inhibitors or use a more purified DNA template.
Reason 2: Energy Depletion. The ATP ran out too fast.
Fix: Switch to a dialysis-based system to continuously supply fresh energy and remove waste.
Reason 3: Misfolding/Aggregation. The protein is being made but it’s just clumping up.
Fix: Lower the incubation temperature or add exogenous chaperones (like CCT4) to help with the folding process.
Homework Question from Kate Adamala: Design a Synthetic Minimal Cell
1. Pick a function and describe it.
a. What would your synthetic cell do? What is the input and what is the output?
My synthetic cell functions as a “Neuro-Protective Sentinel.” It is designed to sense early-stage pathological markers of Alzheimer’s Disease and respond by releasing therapeutic chaperones.
Input: Extracellular Amyloid-beta (A$\beta$) oligomers or monomers.
b. Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
No. Without encapsulation, the transcription/translation (Tx/Tl) machinery would be diluted in the extracellular space and degraded by proteases. Encapsulation protects the PURE system and ensures the therapeutic protein is synthesized in a high local concentration before release.
c. Could this function be realized by genetically modified natural cell?
Yes, but using a minimal cell is safer for neuro-therapeutic applications. Since minimal cells are non-replicative and have a finite lifespan, they eliminate the risk of uncontrolled proliferation or potential tumorigenesis in the brain tissue.
d. Describe the desired outcome of your synthetic cell operation.
The local concentration of the CCT4 chaperone increases in the neuronal microenvironment, facilitating the refolding of misfolded proteins and preventing the formation of toxic tau aggregates or amyloid plaques.
2. Design all components that would need to be part of your synthetic cell.
a. What would the membrane made of?
A mixture of DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) and Cholesterol. Cholesterol is essential here to mimic the neuronal membrane environment and provide structural stability against mechanical stress.
b. What would you encapsulate inside? Enzymes, small molecules.
PURE system (cell-free Tx/Tl machinery).
DNA Template for the CCT4 gene under the control of an A$\beta$-sensitive riboswitch or aptamer.
Energy regeneration system (Creatine phosphate and Creatine kinase) to sustain protein synthesis.
c. Which organism your Tx/Tl system will come from?
A mammalian cell extract (e.g., HeLa or CHO) is preferred over a bacterial system. Since CCT4 is a large, complex eukaryotic protein, mammalian machinery is more likely to ensure proper initial folding and post-translational stability of the output.
d. How will your synthetic cell communicate with the environment?
The membrane will be embedded with alpha-hemolysin ($\alpha$HL) pores. These pores are large enough to allow the entry of A$\beta$ monomers (input) and the exit of the synthesized CCT4 protein (output) into the extracellular space.
3. Experimental details.
a. List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
Sensor: A customized A$\beta$-binding DNA aptamer linked to a T7 promoter.
b. How will you measure the function of your system?
I will use a Fluorescence Resonance Energy Transfer (FRET) assay. By encapsulating a FRET-labeled Tau protein reporter in nearby target vesicles, I can measure the reduction in Tau aggregation (indicated by a change in FRET signal) as a direct result of the CCT4 protein released by the minimal cells.
AI Disclosure
Conceptual development and technical writing for this assignment were supported by AI assistance.