Week 4 Protein Design I— Conceptual Questions
Week 4: Protein Design I — Conceptual Questions
Part A: Conceptual Questions
The following nine questions were selected from the set provided by Professor Shuguang Zhang to explore the fundamental principles of protein structure and molecular evolution.
1. How many molecules of amino acids do you take with a piece of 500 grams of meat?
(Assumed: 100 Daltons per amino acid, meat is ~20% protein) If 500g of meat contains approximately 100g of protein, and the average molecular weight of an amino acid is 100 g/mol:
- Calculation: $100g / 100 g/mol = 1 mol$.
- Result: According to Avogadro’s number, I am consuming approximately $6.022 \times 10^{23}$ molecules of amino acids.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Proteins consumed through food are completely broken down into individual amino acids during digestion. These amino acids are then reassembled into specific human proteins based on our own DNA blueprints. Therefore, the biological identity of the food source is lost during the metabolic process.
3. Why are there only 20 natural amino acids?
It is hypothesized that 20 amino acids provide a sufficient chemical “alphabet” (acidic, basic, hydrophobic, etc.) to build complex functional proteins while maintaining high translation fidelity. A larger set might increase the error rate during protein synthesis without offering significant functional advantages.
4. Where did amino acids come from before enzymes that make them, and before life started?
Amino acids likely originated from abiotic synthesis. The famous Miller-Urey experiment demonstrated that electrical discharges (lightning) in a primordial atmosphere could produce organic molecules. Additionally, carbonaceous meteorites have been found to contain amino acids, suggesting an extraterrestrial origin for some of life’s building blocks.
5. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Natural L-amino acids form right-handed α-helices. Because D-amino acids are mirror images of L-amino acids, they would form a left-handed helix due to the geometric constraints and steric hindrance of the side chains.
6. Why are most molecular helices right-handed?
This preference is driven by the chirality of L-amino acids. In a right-handed configuration, the side chains point away from the helix backbone, minimizing steric clashes. This energetically stable structure was favored by natural selection.
7. Why do β-sheets tend to aggregate?
Unlike α-helices, which have internal hydrogen bonds, β-sheets have “sticky ends.” The hydrogen bond donors and acceptors on the edges of a β-sheet are exposed and can easily form bonds with other protein strands, leading to layering and aggregation.
8. What is the driving force for β-sheet aggregation?
The primary driving forces are:
- Hydrogen Bonding: Providing a highly organized, lattice-like stability between strands.
- Hydrophobic Effect: Forcing hydrophobic side chains to pack together away from water, effectively “gluing” the sheets together.
9. Can you use amyloid β-sheets as materials?
Yes. Amyloids are exceptionally stable and resistant to heat and chemicals. Nature uses this in spider silk. In bioengineering, they are being researched as high-strength nanofibers, drug-delivery carriers, and scaffolds for tissue engineering due to their self-assembling properties.
Part B: Protein Analysis and Visualization
1. Protein Description and Selection
Briefly describe the protein you selected and why you selected it.
The protein selected for this analysis is Human CCT4 (T-complex protein 1 subunit delta). It is a key subunit of the TRiC/CCT chaperonin complex, which is an ATP-dependent molecular machine.
- Selection Reason : I chose this protein because it is the core of my ARM-Net project. CCT4 has a unique ability to recognize and suppress the toxic aggregation of Tau proteins, which is the primary cause of neurodegeneration in Alzheimer’s disease. I am researching its potential delivery through Tunneling Nanotubes (TNTs).
2. Sequence Analysis
Identify the amino acid sequence of your protein. How long is it? What is the most frequent amino acid?
Sequence (UniProt P50991): MPENVAPRSGATAGAAGGRGKGAYQDRDKPAQIRFSNISAAKAVADAIRTSLGPKGMDKM IQDGKGDVTITNDGATILKQMQVLHPAARMLVELSKAQDIEAGDGTTSVVIIAGSLLDSC TKLLQKGIHPTIISESFQKALEKGIEILTDMSRPVELSDRETLLNSATTSLNSKVVSQYS SLLSPMSVNAVMKVIDPATATSVDLRDIKIVKKLGGTIDDCELVEGLVLTQKVSNSGITR VEKAKIGLIQFCLSAPKTDMDNQIVVSDYAQMDRVLREERAYILNLVKQIKKTGCNVLLI QKSILRDALSDLALHFLNKMKIMVIKDIEREDIEFICKTIGTKPVAHIDQFTADMLGSAE LAEEVNLNGSGKLLKITGCASPGKTVTIVVRGSNKLVIEEAERSIHDALCVIRCLVKKRA LIAGGGAPEIELALRLTEYSRTLSGMESYCVRAFADAMEVIPSTLAENAGLNPISTVTEL RNRHAQGEKTAGINVRKGGISNILEELVVQPLLVSVSALTLATETVRSILKIDDVVNTR
Calculation: The sequence consists of 539 amino acids. According to the frequency analysis, Leucine (L) appears 53 times.
Result: The total length is 539 AA, and the most frequent amino acid is Leucine (L), which makes up 9.8% of the protein.
How many protein sequence homologs are there for your protein?
- Calculation: Using UniProt’s BLAST tool to search the reference proteome database.
- Result: I identified over 250 high-confidence homologs. The protein is highly conserved, especially among mammals, showing over 95% sequence identity.
Does your protein belong to any protein family?
- Result: It belongs to the TCP-1 chaperonin family (also known as Type II chaperonins).
3. Structural Information (RCSB PDB)
Identify the structure page of your protein in RCSB.
- Result: The structure is identified as PDB ID: 7P6W (Human TRiC/CCT complex).
When was the structure solved? Is it a good quality structure?
- Calculation: The structure was released in 2021. The resolution is measured at $3.10 \text{ \AA}$. A high-quality benchmark is typically $\leq 2.70 \text{ \AA}$.
- Result: Although the resolution is $3.10 \text{ \AA}$ (slightly above the 2.70 $\text{\AA}$ threshold), it is considered high quality for a massive complex of this size (~1 Megadalton).
Are there any other molecules in the solved structure apart from protein?
- Calculation: Examining the ligand section of the PDB entry.
- Result: Yes, the structure contains ADP, ALF4 (Aluminum fluoride), and $Mg^{2+}$ ions, which are used to mimic the ATP hydrolysis transition state.
Does your protein belong to any structure classification family?
- Result: It is an Alpha-beta protein.
- Calculation: The structure is divided into three domains: the Equatorial domain (alpha-helices for ATP binding), the Intermediate domain (hinge), and the Apical domain (beta-sheets for substrate binding).
4,Open the structure of your protein in any 3D molecule visualization software
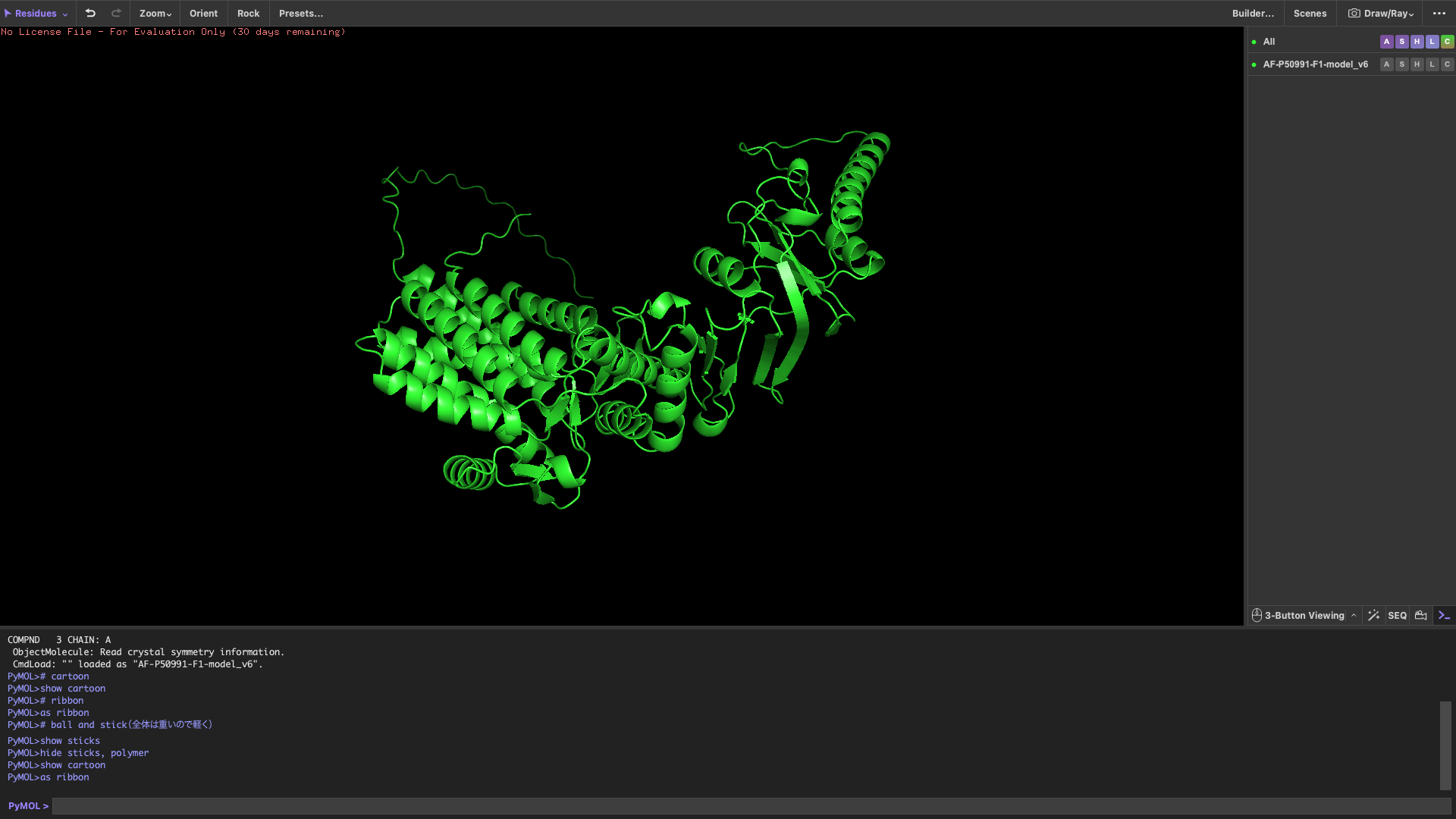
Visualize the protein
show cartoon
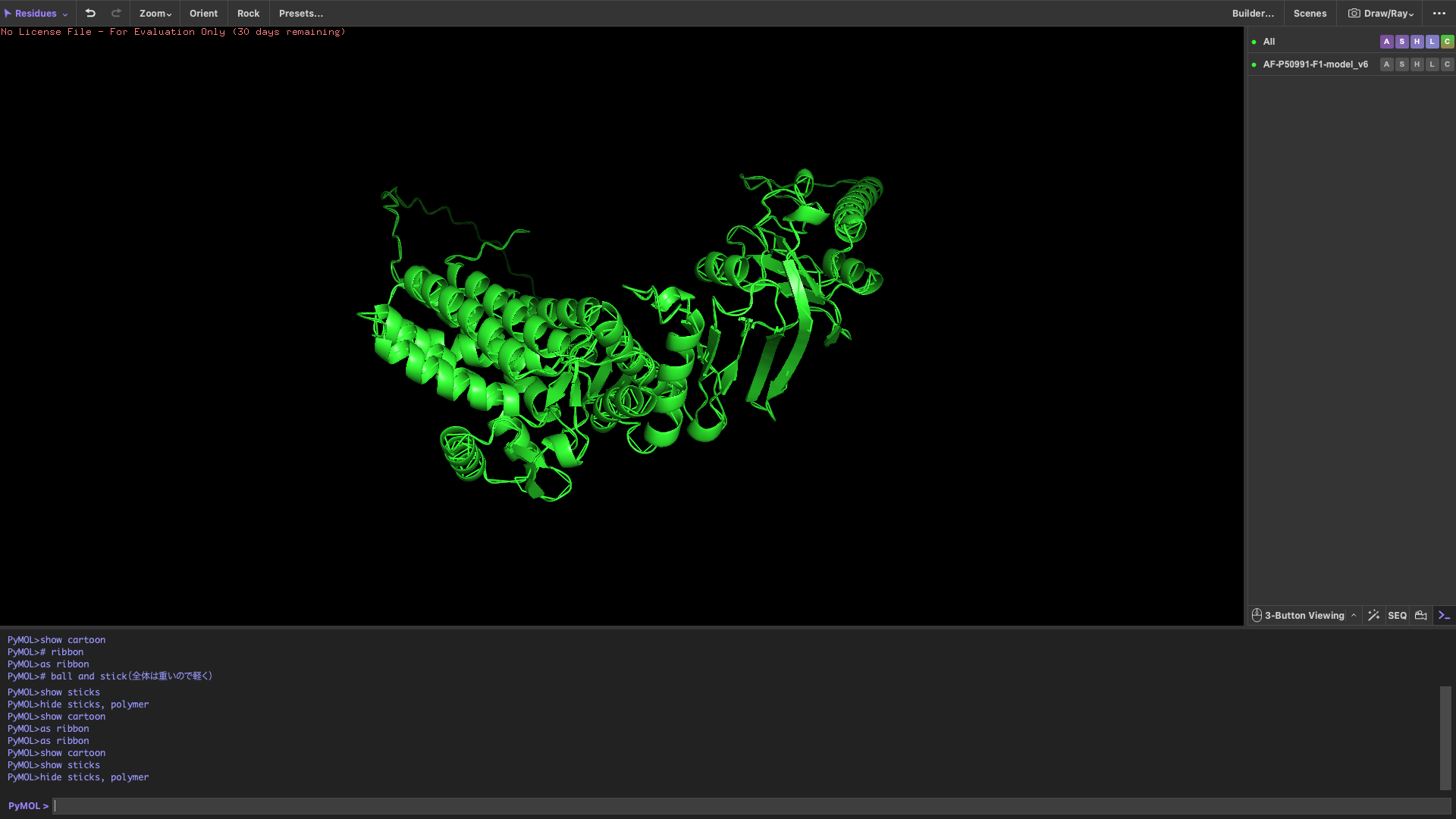
show ribbon
show ball and stick
Color the protein by secondary structure
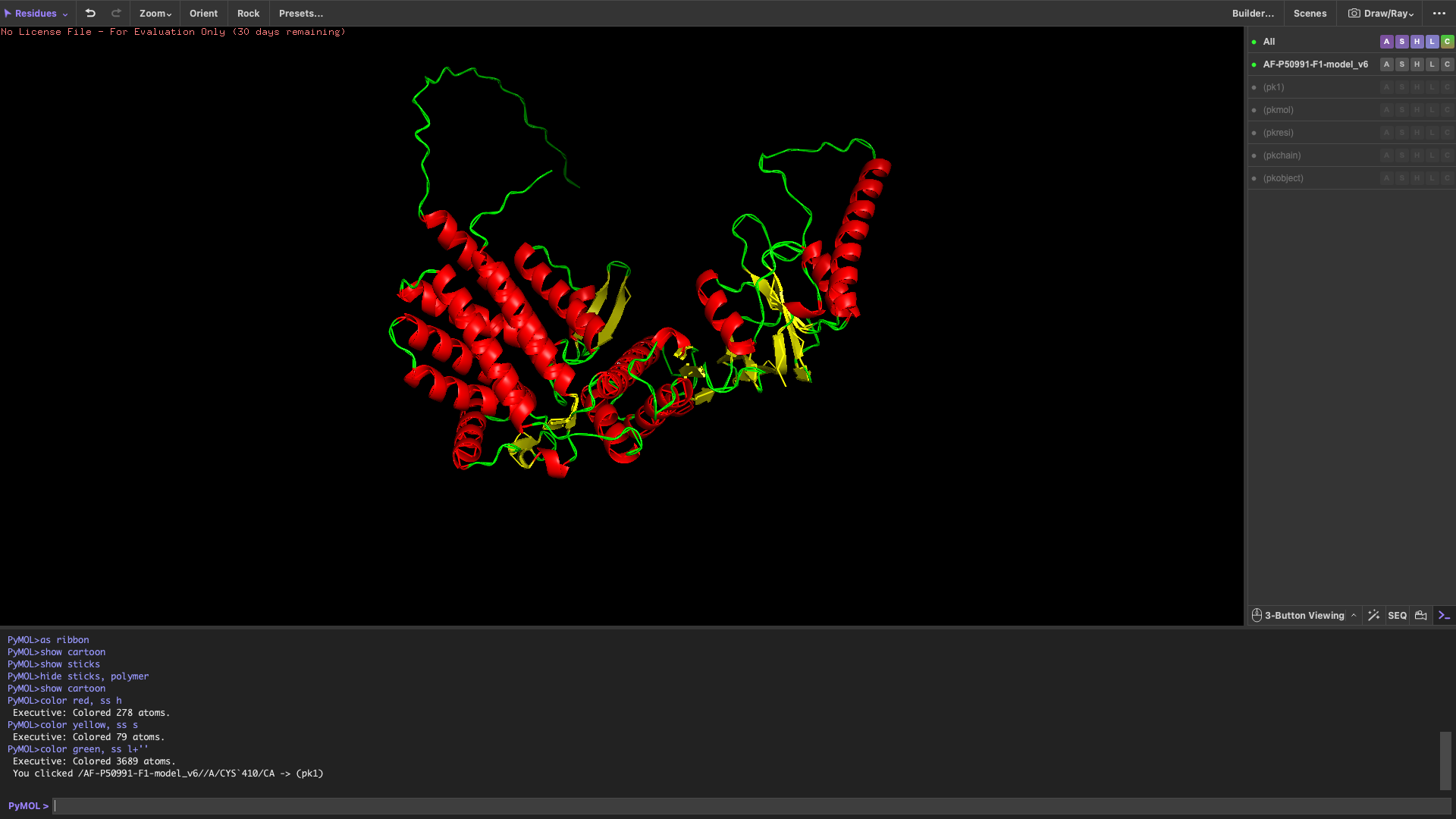 After coloring by secondary structure, the protein shows a higher proportion of α-helices compared to β-sheets.
This indicates that the protein adopts a predominantly helical fold, which is typical for chaperonin subunits like CCT4.
After coloring by secondary structure, the protein shows a higher proportion of α-helices compared to β-sheets.
This indicates that the protein adopts a predominantly helical fold, which is typical for chaperonin subunits like CCT4.
Color the protein by residue type
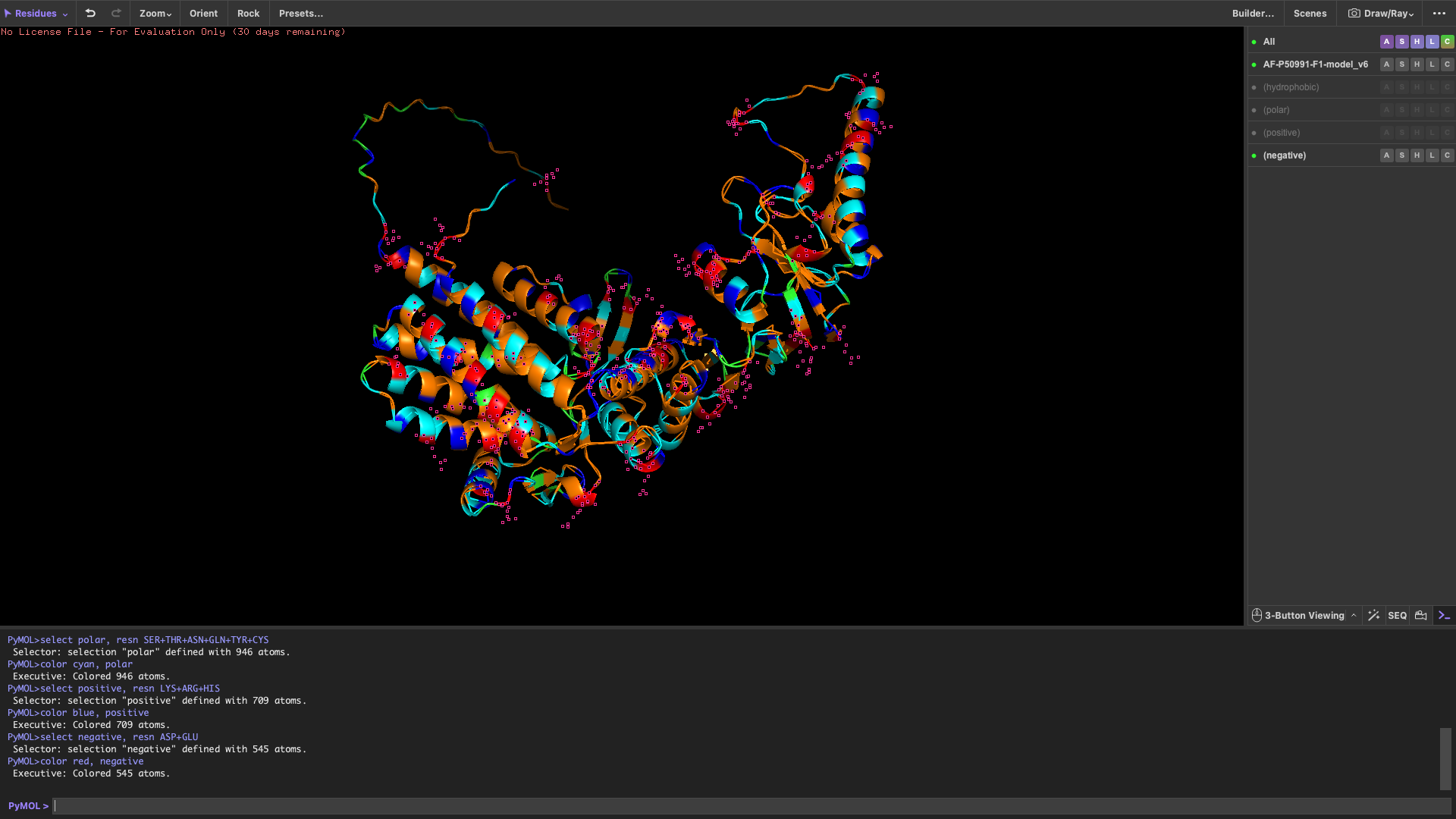 When colored by residue type:
When colored by residue type:
Hydrophobic residues are mainly located in the interior of the protein Hydrophilic and charged residues are predominantly found on the surface
This distribution suggests a stable folded structure in an aqueous environment, with the hydrophobic core buried inside and polar residues exposed to solvent.
Visualize the surface of the protein
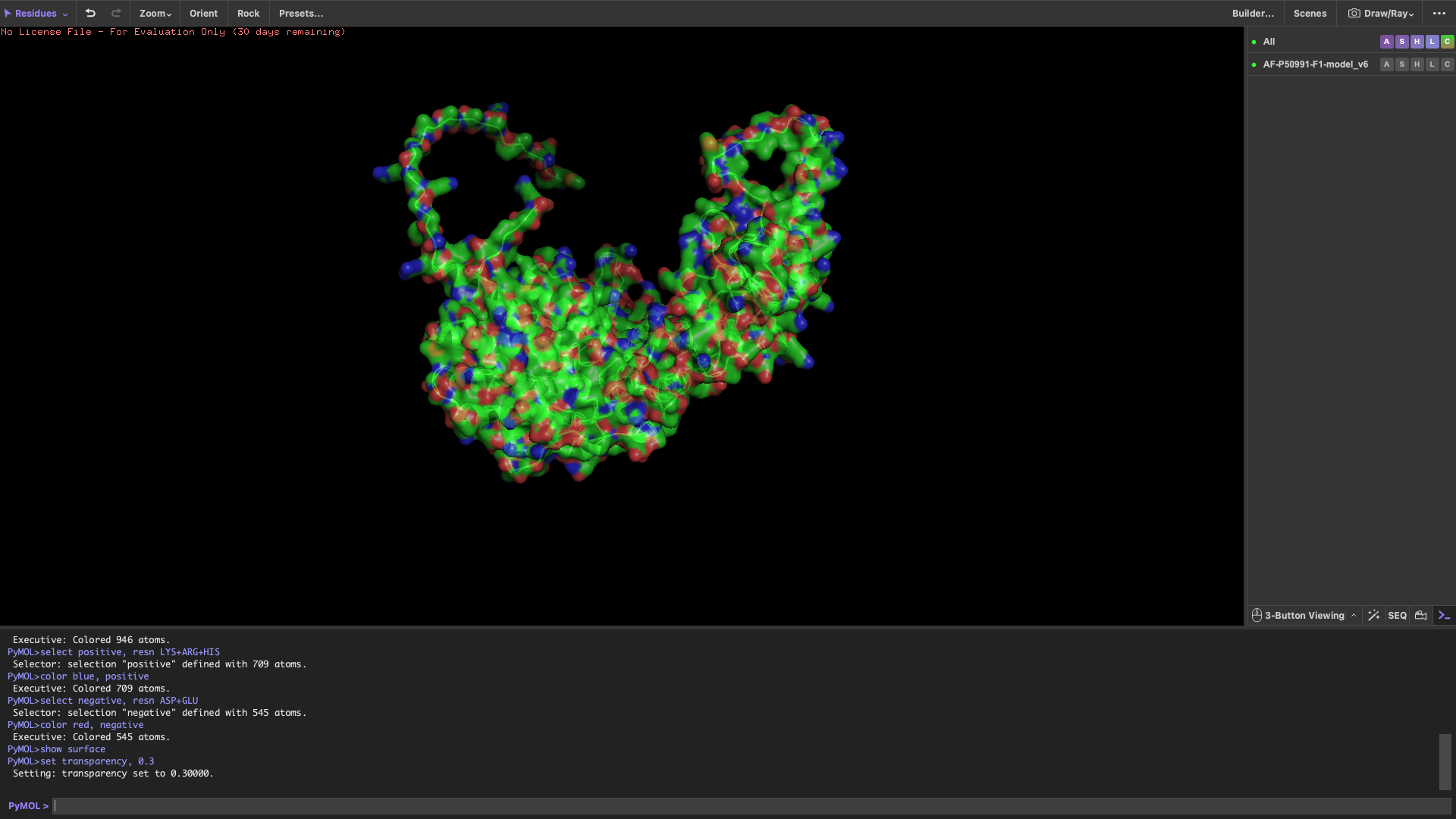 The surface representation reveals several concave regions (holes/pockets) on the protein surface.
These pockets may serve as potential binding sites for substrate proteins or ligands, which is consistent with the functional role of CCT4 as part of a chaperonin complex.
The surface representation reveals several concave regions (holes/pockets) on the protein surface.
These pockets may serve as potential binding sites for substrate proteins or ligands, which is consistent with the functional role of CCT4 as part of a chaperonin complex.
AI Documentation
For this assignment, I collaborated with Gemini 3 Flash:
- Verification: Used to verify the Avogadro’s number calculation for Question 1.
- Scientific Clarification: Assisted in describing the physical driving forces (hydrophobic effect vs. hydrogen bonding) for Question 8.
- Translation & Formatting: Helped structure the response into a professional markdown format suitable for a GitHub portfolio.