Week 5 Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
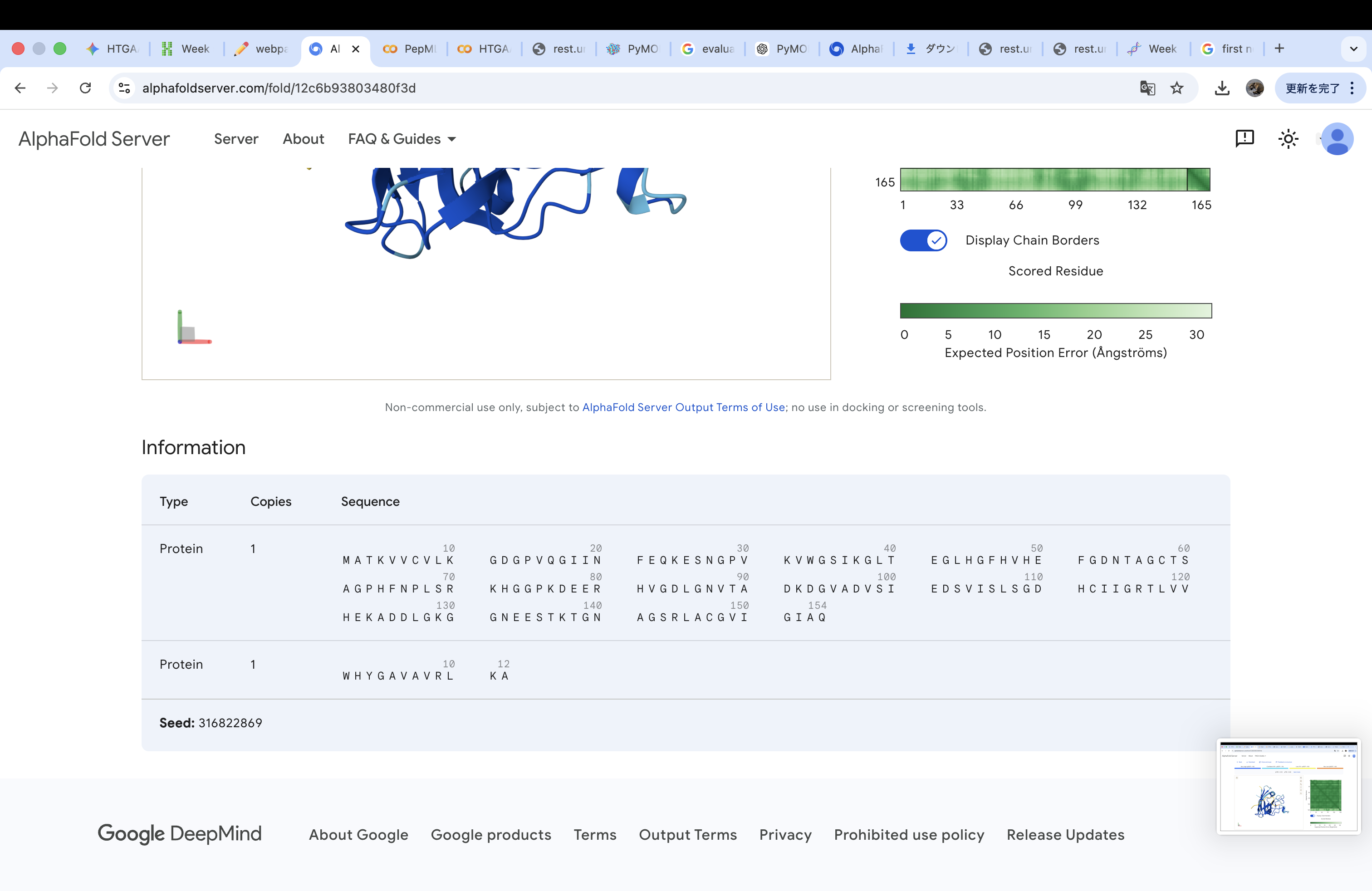
Amino acid sequence obtained from UniProt MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
A4V-mutated amino acid sequence MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
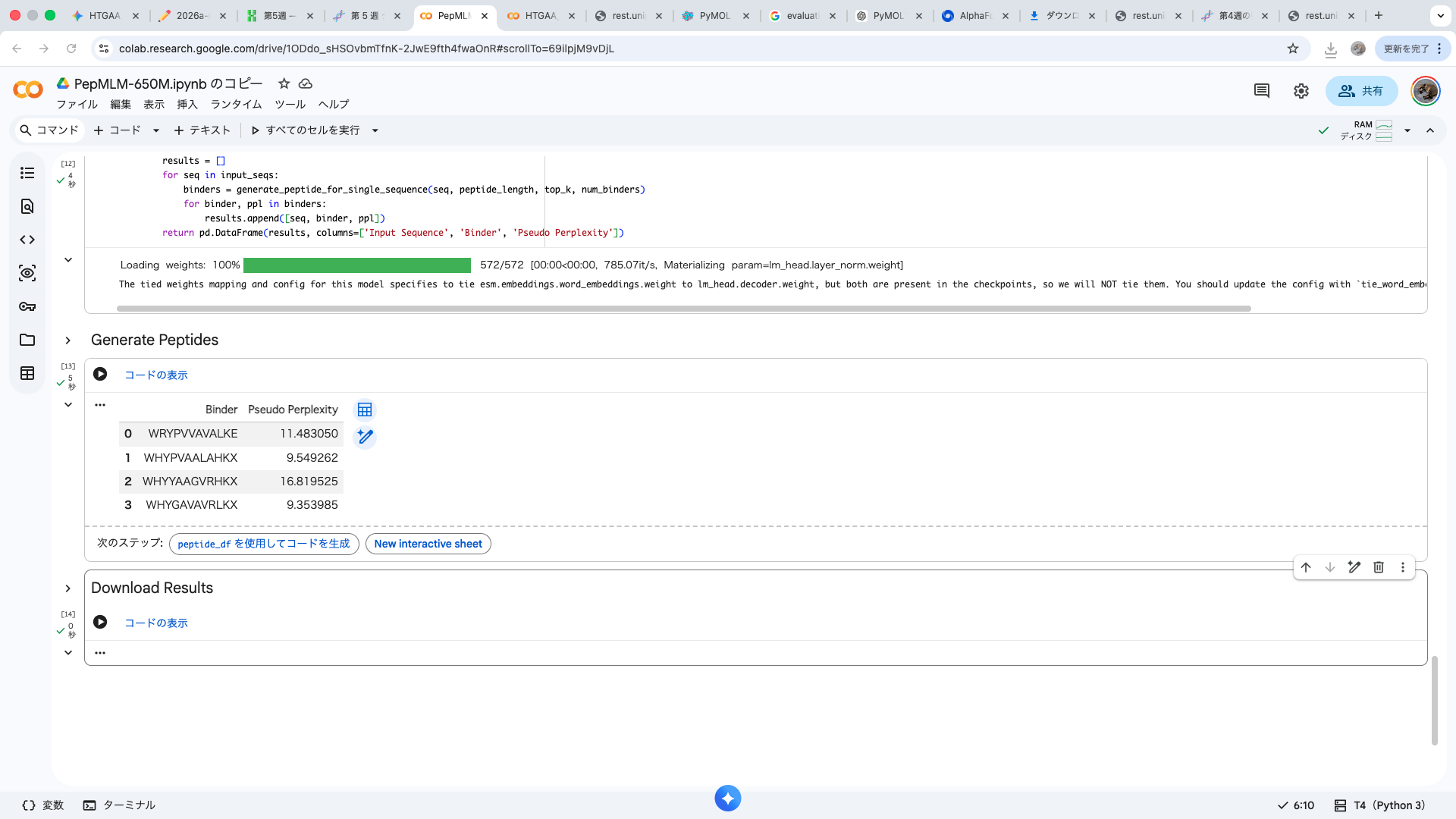
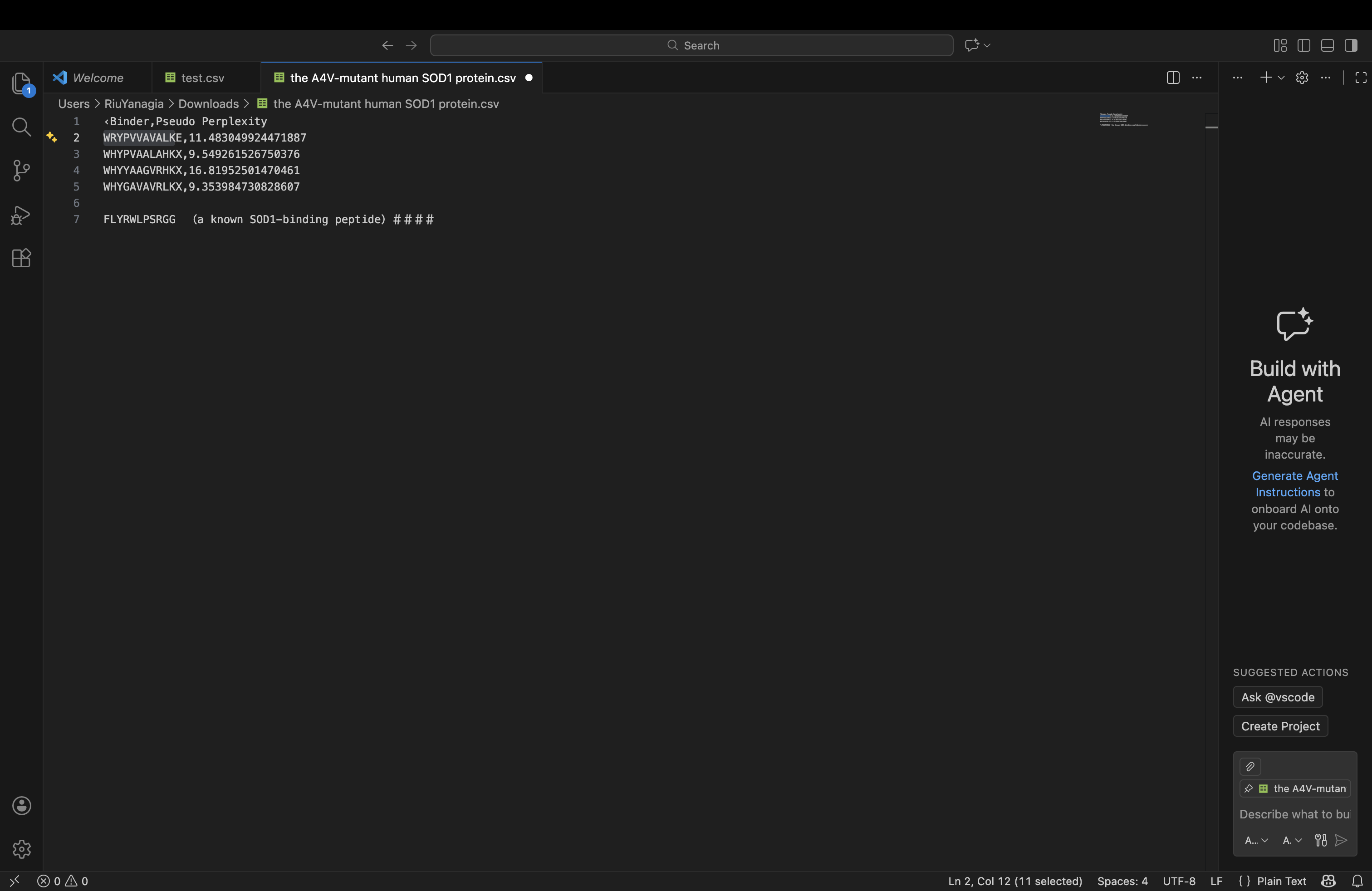
| index | Binder | Pseudo Perplexity |
|---|---|---|
| 0 | WRYPVVAVALKE | 11.483049924471887 |
| 1 | WHYPVAALAHKX | 9.549261526750376 |
| 2 | WHYYAAGVRHKX | 16.81952501470461 |
| 3 | WHYGAVAVRLKX | 9.353984730828607 |
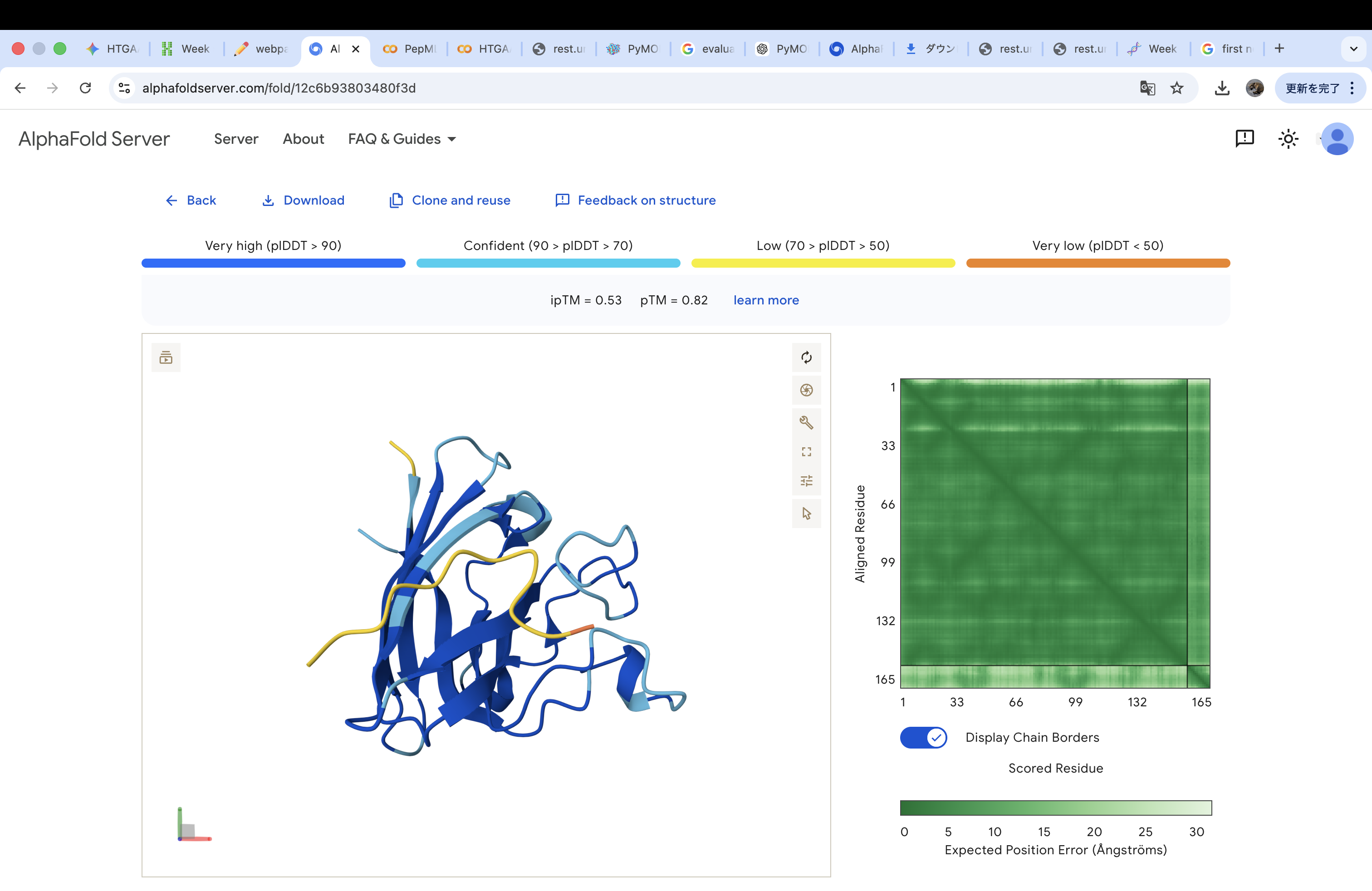
Part 2: Evaluate Binders with AlphaFold3
The A4V-mutant human SOD1 sequence and each peptide were submitted as separate chains to AlphaFold3 to model protein–peptide complexes. The predicted ipTM scores varied among the peptides, indicating differences in binding confidence. The peptides generally appeared to bind on the protein surface, with some localizing near the N-terminus where the A4V mutation is located, while others interacted with regions of the β-barrel structure. Most peptides were surface-bound rather than deeply buried
WRYPVVAVALKE
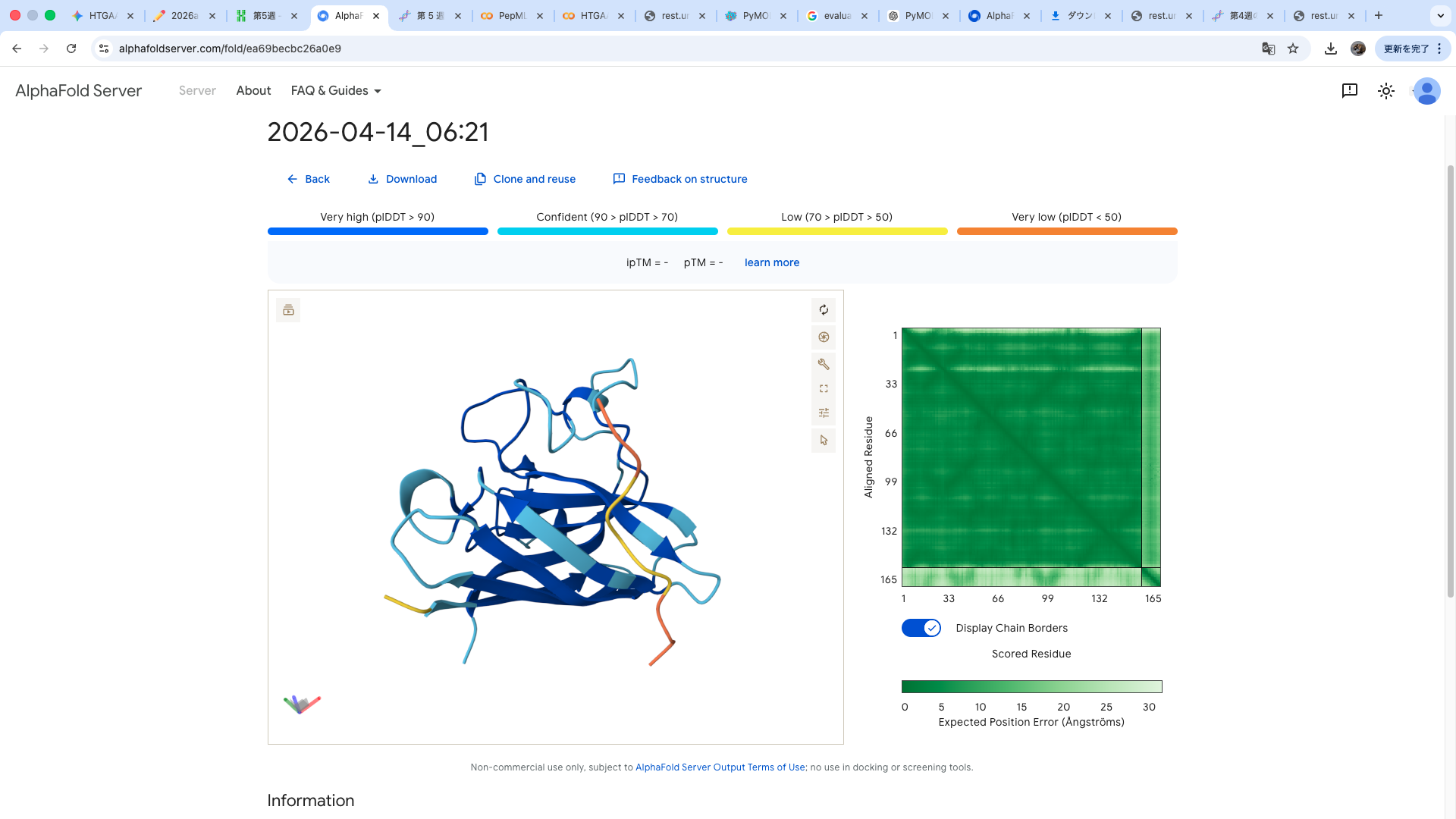
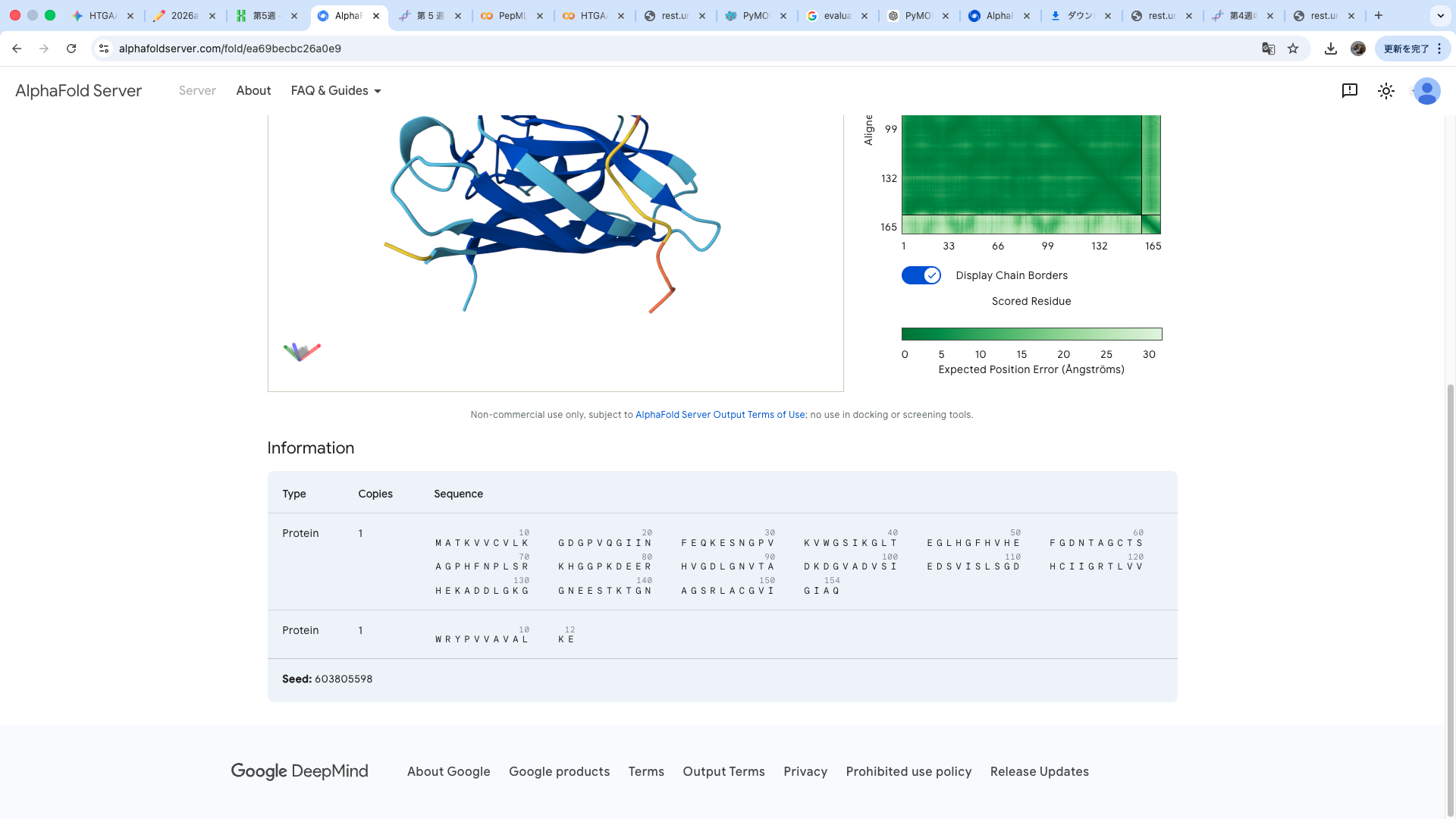
The predicted structure shows that the peptide binds weakly to the surface of the A4V-mutant SOD1 protein. The peptide appears to be loosely associated with the protein and does not form a well-defined binding interface. The interaction is mainly surface-bound rather than deeply buried, and it does not strongly localize near the N-terminus where the A4V mutation is located. The absence of a clear ipTM score suggests low confidence in stable binding.
HYPVAALAHKK
The invalid residue “X” was removed or replaced with a valid amino acid. In this case, it was substituted with “K” (lysine) to maintain the required peptide length of 12 amino acids.
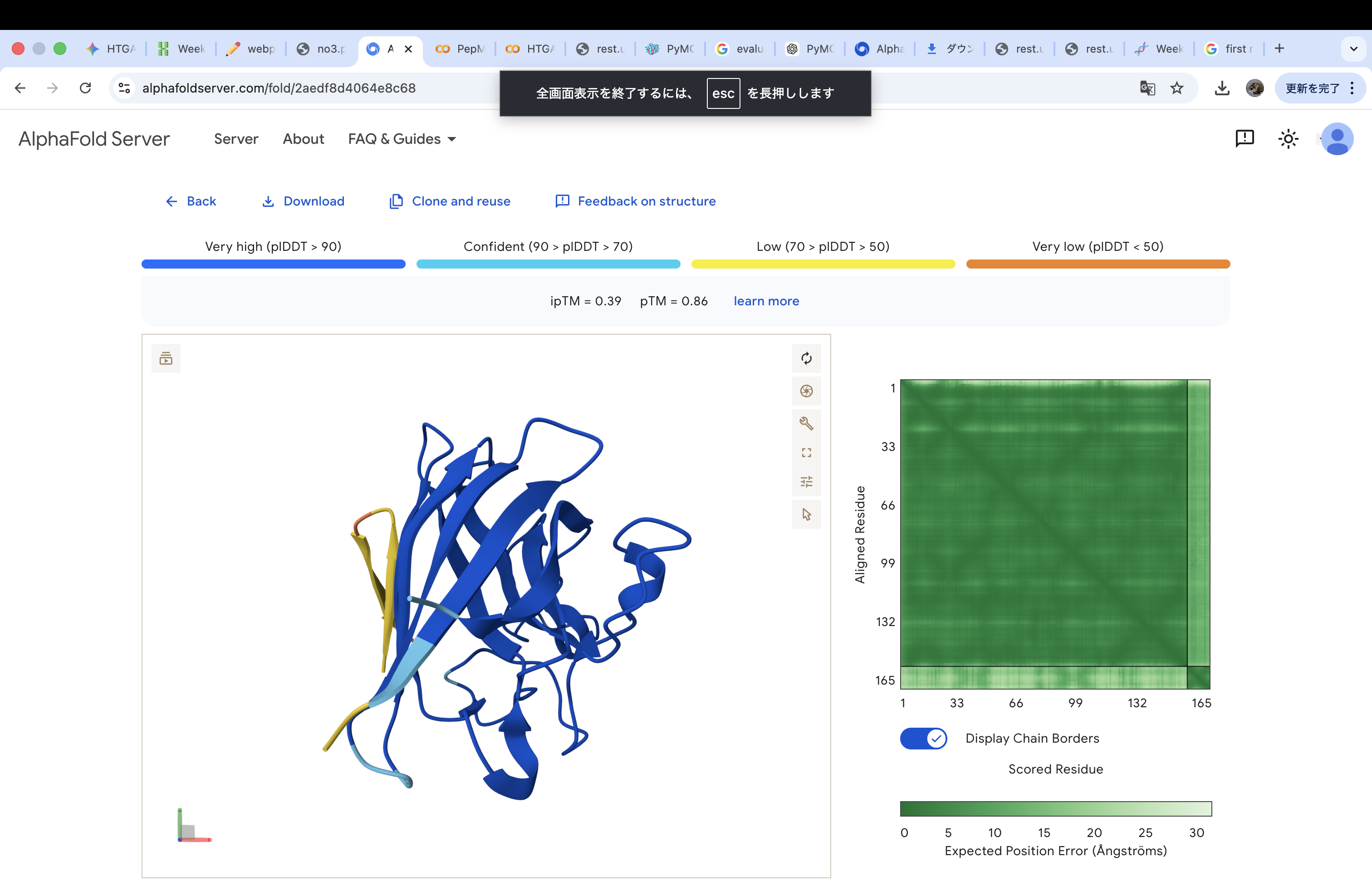
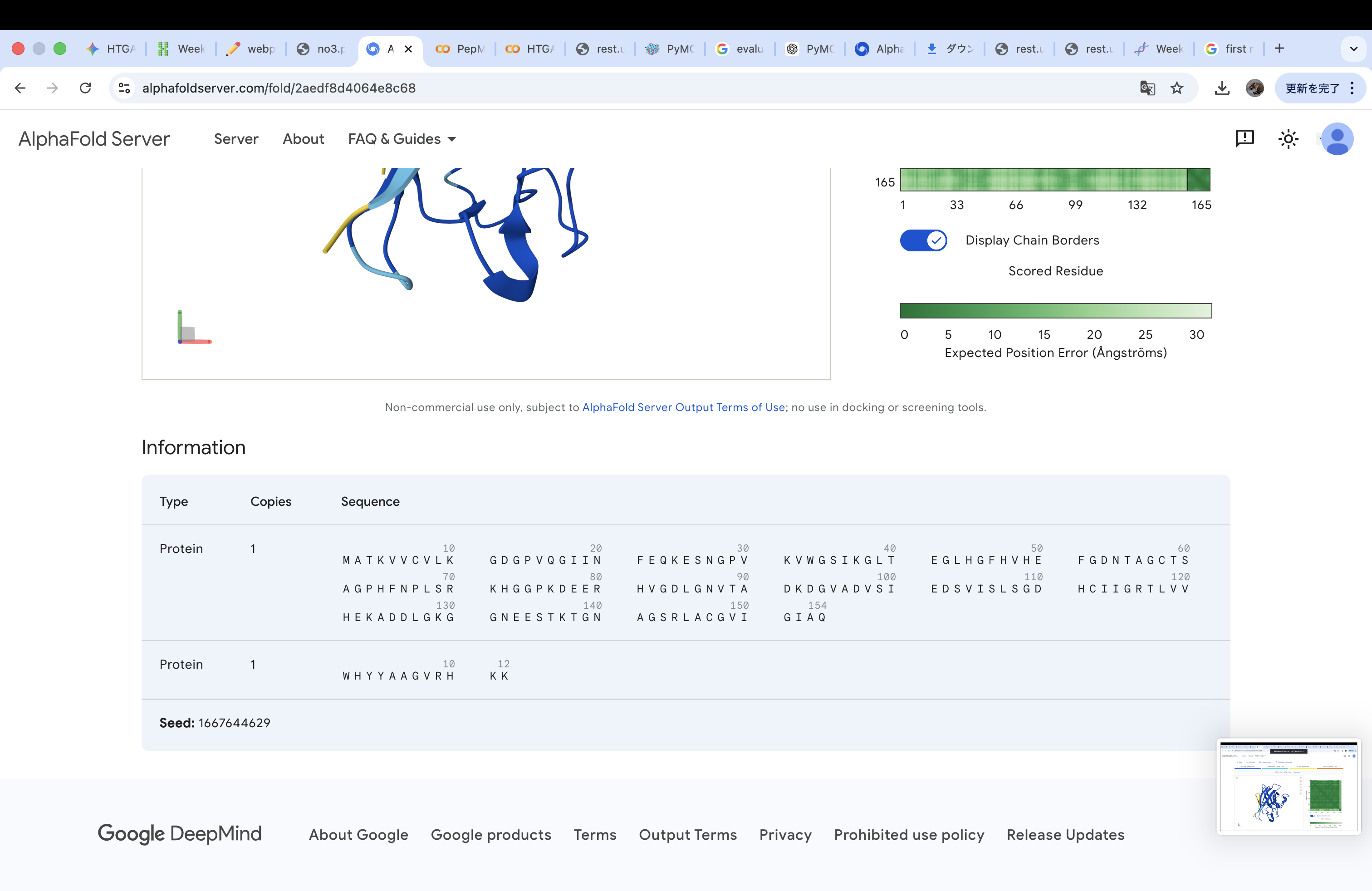
WHYYAAGVRHKK
The invalid residue “X” was removed or replaced with a valid amino acid. In this case, it was substituted with “K” (lysine) to maintain the required peptide length of 12 amino acids.
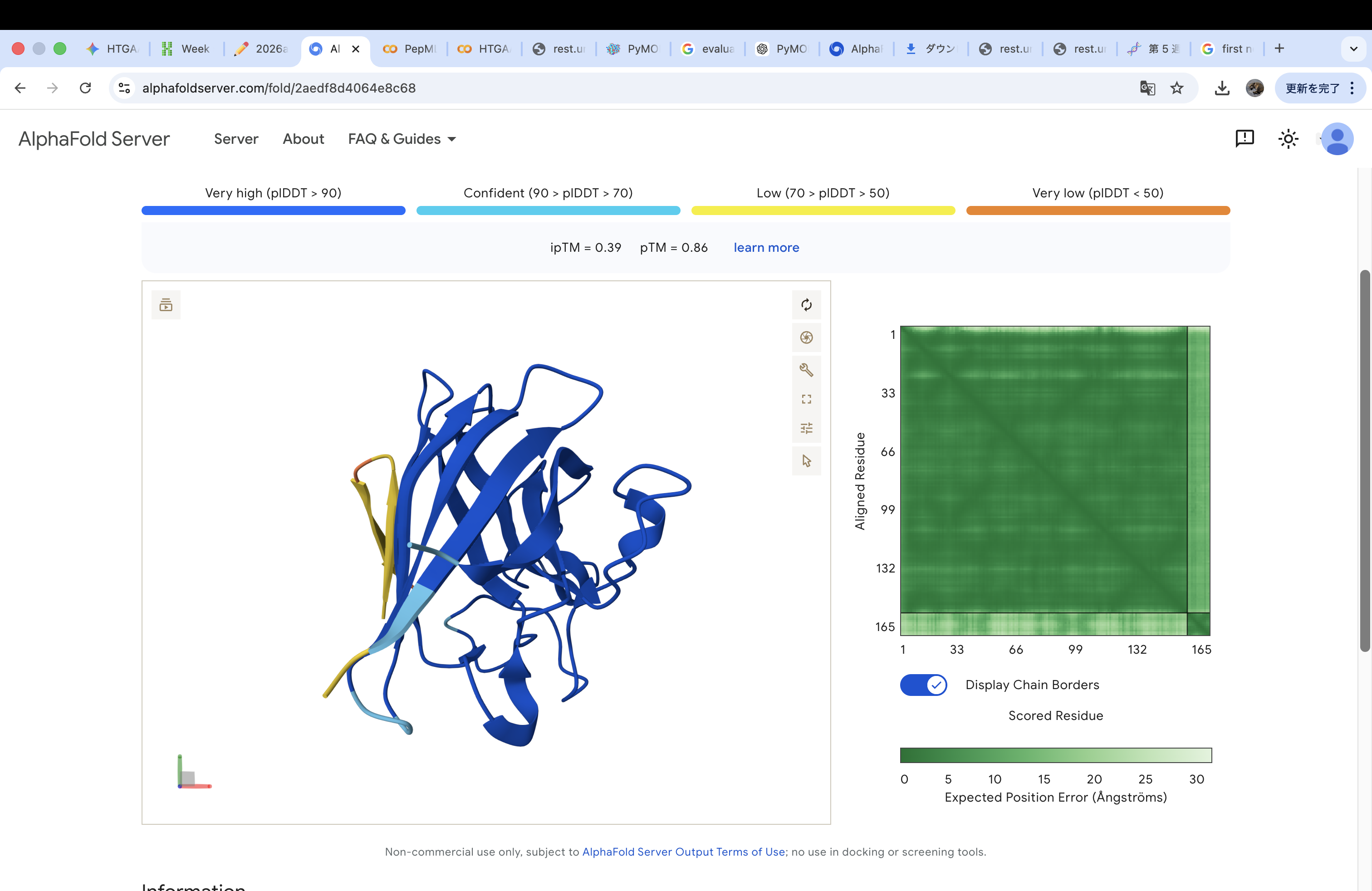
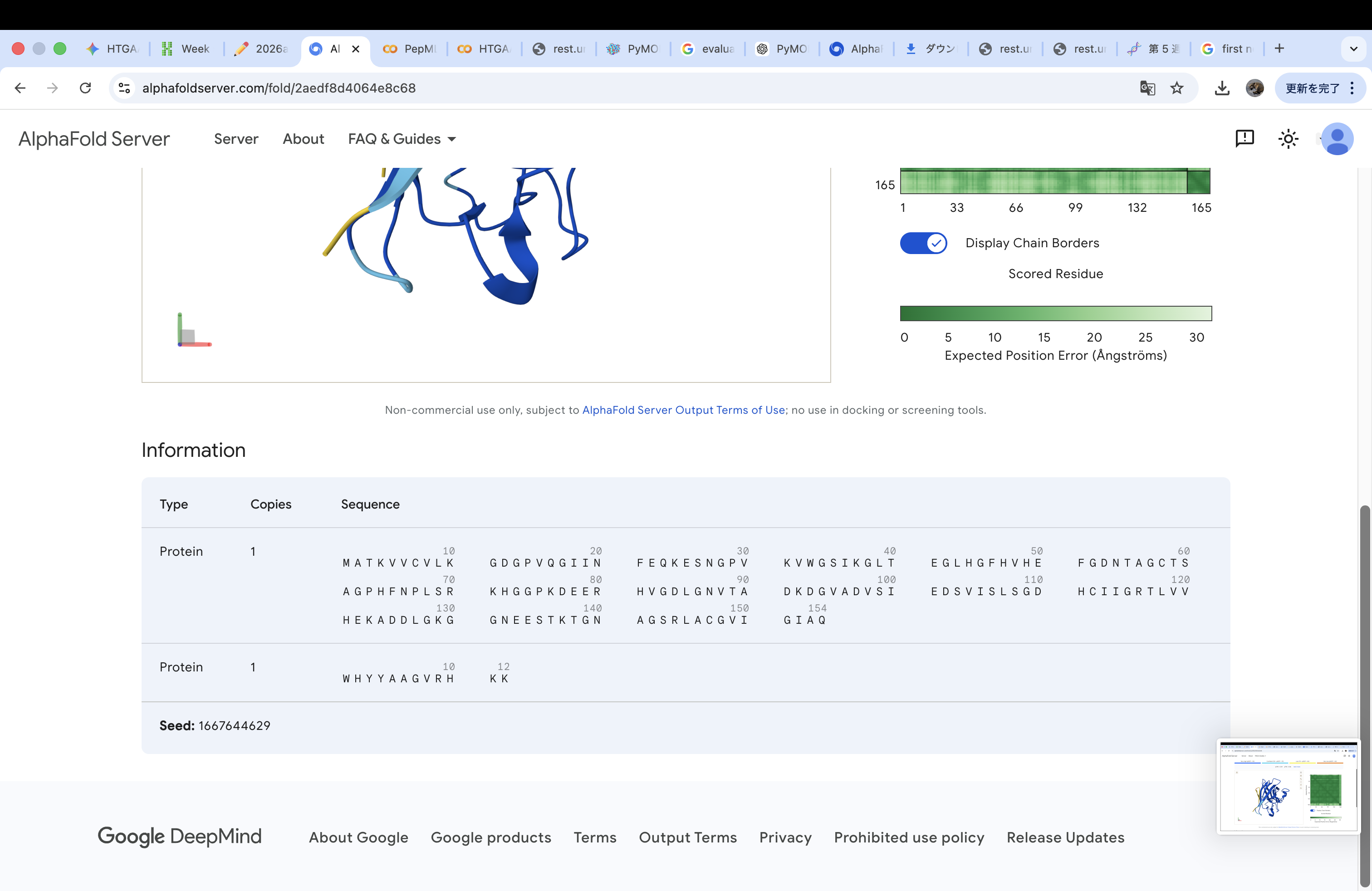
WHYGAVAVRLKX
The invalid residue “X” was removed or replaced with a valid amino acid. In this case, it was substituted with “K” (lysine) to maintain the required peptide length of 12 amino acids.