Week 5 HW: Protein Design Part II
Time to do your homework- It’s always time to do homework in this course!
Part A: SOD1 Binder Peptide Design
Part 1: Generate Binders with PepMLM
Starting with the human SOD1 sequence from UniProt (P00441), and then introducing the A4V mutation, we get the following sequence where the fourth amino acid in the sequences is changed from K to V:
- MATVAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ.
- We then use PepMLM to produce the following binders:
Part 2: Evaluate Binders with AlphaFold3
- Loading the SOD1 A4V mutation into Alphafold with the generated peptide sequences produced the following results:
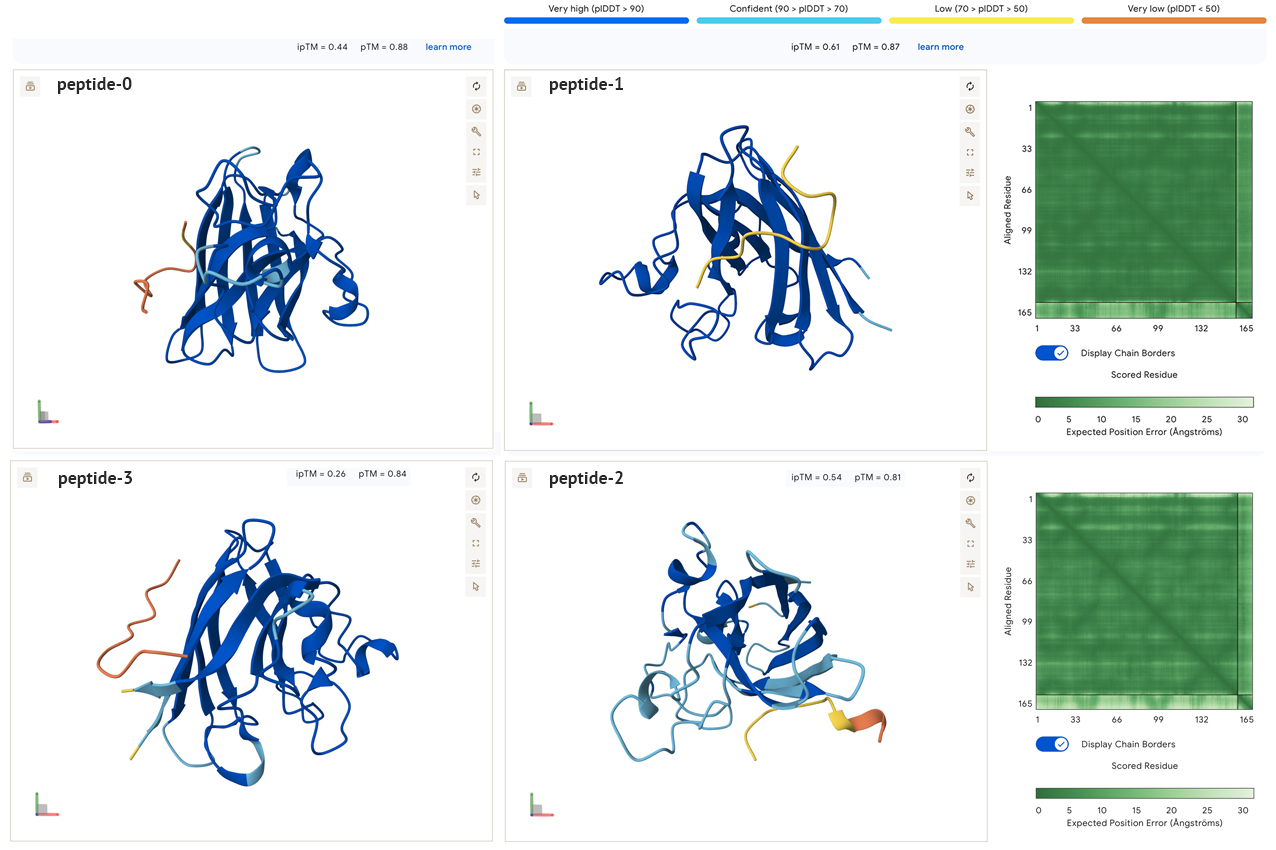
In the models above the ipTM and pTM values were:
- peptide 0: ipTM = 0.44, pTM = 0.88
- peptide 1: ipTM = 0.61, pTM = 0.87
- peptide 2: ipTM = 0.54, pTM = 0.81
- peptide 3: ipTM = 0.26, pTM = 0.84
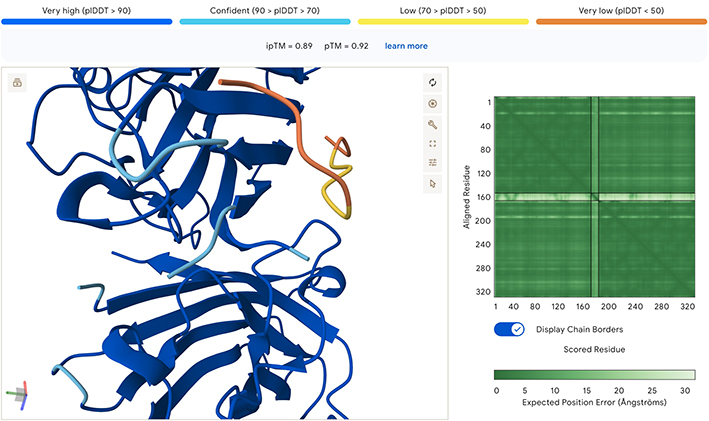
- peptide 4: ipTM = 0.89, pTM = 0.92 (known binder)
All the sidechain peptides generated had low to very low plDDT scores, and did not appear to be close to either ends of the SOD1. This seems to indicate to me that these peptides are not well suited for binding with SOD1. In addition, the low ipTM scores all seem to indicate low confidence of interaction between molecules. In the model below there are similar results when combining two SOD1 A4V molecules with the known binder. The pedicted local distance score for the known peptide is low, which is confusing, and makes me think it may be an incorrect reading by the AlphaFold model, especially if the peptide is a known binder.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
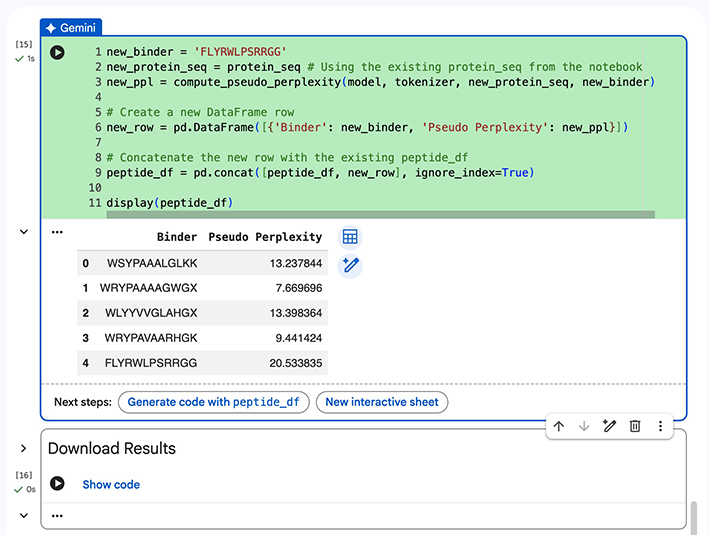
Peptiverse graphic results table for Peptide 0
-Peptiverse Results-
- Peptide 0 - Sequence: WSYPAAALGLKK. Realatively weak binding (5.1 pKd/pKi) with second to highest net charge, and the second lowest score for hydrophobicity. It’s non-hemolytic and soluble.
- Peptide 1 - Sequence: WRYPAAAAGWGX. Realatively weak binding (6.2 pKd/pKi) with the third highest net charge, and the third lowest score for hydrophobicity. It’s non-hemolytic and soluble.
- Peptide 2 - Sequence: WLYYVVGLAHX. Realatively weak binding (6.9 pKd/pKi) with the lowest net charge, and the highest score for hydrophobicity. It’s non-hemolytic and soluble.
- Peptide 3 - Sequence: WRYPAVAARHGK. Realatively weak binding (5.5 pKd/pKi) with the highest net charge, and the lowest score for hydrophobicity. It’s non-hemolytic and soluble.
Based on the results of this group, I don’t think I would advance any of them for further testing. The second peptide generated (peptide 1), seems closest to working with the highest iPTm score, third highest charge and the lowest hydrophobicity. Although, it is difficult for me to interpret these data sets at this point, and if I were actually looking for a binder to test I would continue looking for a set which had better predictions in the visualization models, which ideally would also match the Peptiverse scores.
Part 4: Generate Optimized Peptides with moPPIt
Functional parameters: Hemolysis, Non-Fouling, Solubility, Half-Life, Affinity, Motif, Specificity.
All set with parameters set with objective importance of 1.
Motif Positions were: 1-10, 144-154 (first ten positions, last ten positions)
Generated peptide sequences:
- KKKKKDKTTKWM
- EEVQKKQEWKTI
- ELIRWLQQRRTD
The following results are for the first peptide generated, Sequence - KKKKKDKTTKWM: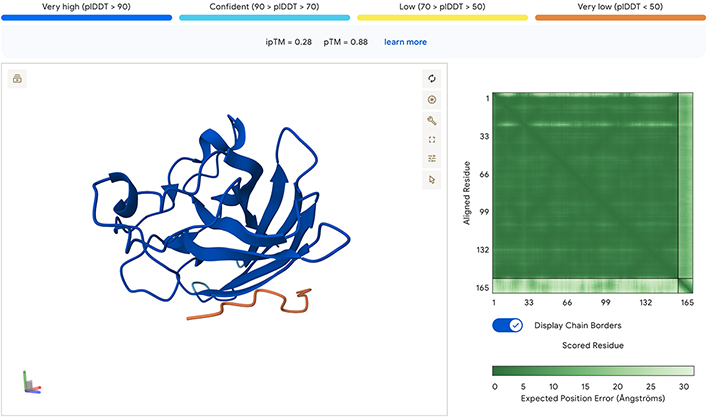
Based on these results, I am no more confident in this binder generated by moPPit than the first sets generated by PepMLM. Again, not sure if this is a result of the modeling, or perhaps I’m not doing this exercise correctly. It seems odd that none of the binders have very high ipTM or pTM scores.
Part C: Final Project: MS2L-Protein Mutants
Option 1: Unfortunately, I had technical issues with the colab notebook and was only able to run the first few sections. First, I generated the heat map, and the top ten mutations. As the goal of the assignment is to adapt the MS2L protein to fold on it’s own without the DnaJ chaperone, I used this data with Alphafold to improve it’s confidence in the model. I relize this is not how the assignment was intended to be done, but it was the best way I could proceed to generate five, hopefully better, mutations of the MS2L protein.
Working with AlphaFold to Generate New Sequences
Come up with 5 mutations along with how you came up with them and why you believe they would work. 2 of the variants you submit must have mutations in the transmembrane region (38-60, Sequence: LYVLIFLAIFLSKFTNQLLLSLL) and 2 of them must be in the soluble region.
AlphaFold of Original (non-mutated) MS2L Sequence
MS2L Heatmap of Likely Mutations
MS2L Top Ten Most Likely Mutations
| Position | Wild_Type_AA | Mutation_AA | LLR_Score |
|---|---|---|---|
| 50 | K | L | 2.561468 |
| 29 | C | R | 2.395427 |
| 39 | Y | L | 2.241780 |
| 29 | C | S | 2.043150 |
| 9 | S | Q | 2.014325 |
| 29 | C | Q | 1.997049 |
| 29 | C | P | 1.971029 |
| 29 | C | L | 1.960646 |
| 50 | K | I | 1.928801 |
| 53 | N | L | 1.864932 |
Mutational Tests
Test 1 and 2
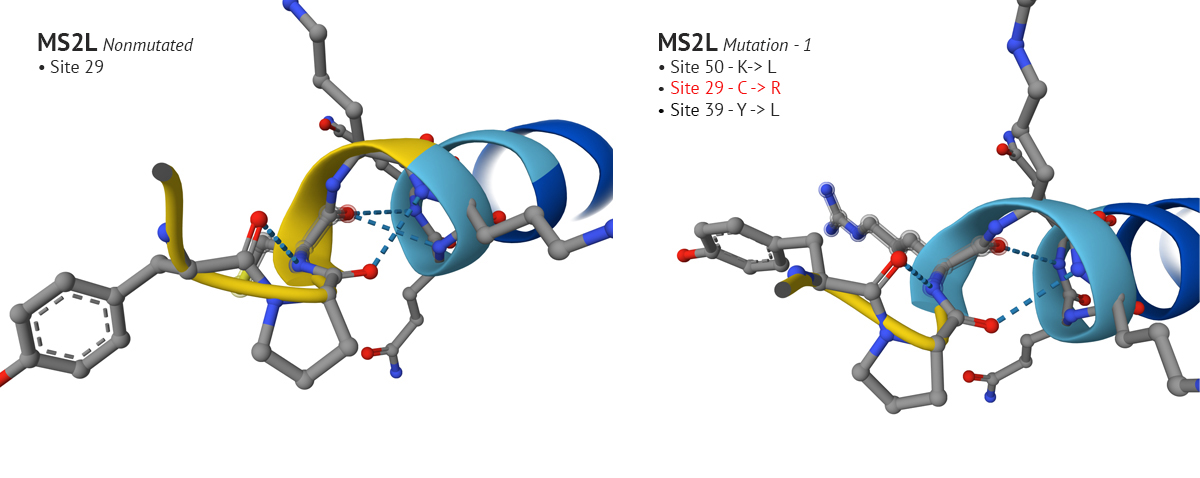
-Sites 50, 29, 39, and 9 were mutated using the top ten mutational LLR scores from ESM notebook.
-The first set of mutations were sites 50, 29, and 39 which produced a confident plDDT at site 29 compared to the original non-mutated MS2L sequence. Interestingly, when I continued with the mutation and switched site 9 from S to Q, the plDDT score dropped at sight 29.
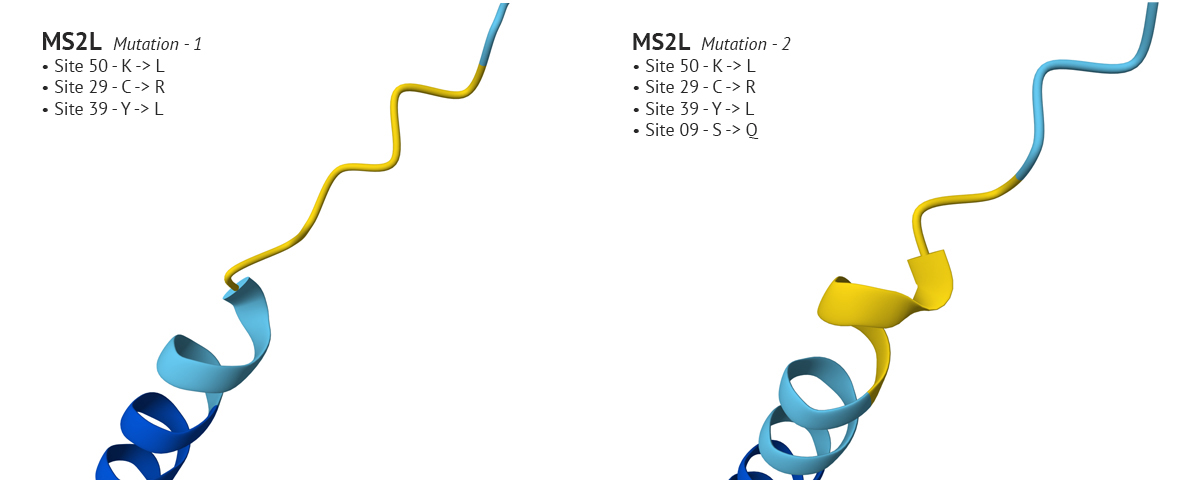 Above: Site 29 before S to Q mutation (lower-left), site 29 after S to Q (lower-right).
Above: Site 29 before S to Q mutation (lower-left), site 29 after S to Q (lower-right).
Test 3:
The third mutation keeps the changes made to sites 50 and 29 from mutation 1, which yielded a higher plDDT score, and modified the aa at site 8 from Glutamine (Q) to Threonine (T). I chose Threonine because it has a neutral charge like Glutamine but is freely soluble where Glutamine’s solubility score is 2.6. This affected the AlphaFold’s model with a higher plDDT score for that position, and seemed to positively affect other positions in the aa 1-29 region.
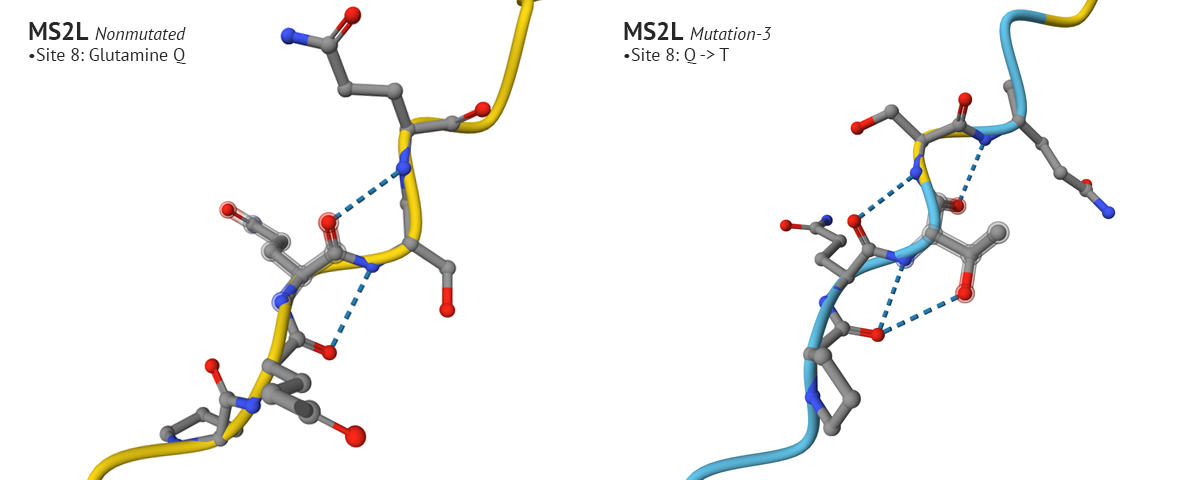
Test 4 and 5:
In mutation 4 site 8 changed from it’s original aa residue of Q to T, which had a positive affect on the plDDT score. In the fifth mutation I wanted to see if I could improve confidence around site 26. I went through several variations of changing the original residue, Aspartate (D), to G, then to E, and finally to Q which improved confidence. However, in my final analysis of the fifth variation, I realized that the improved confidence at site 8 in the fourth mutation changed back to a low confidence state. It’s an interesting problem which I don’t understand. It would make sense why changing residues which are in close proximity to one another would impact the region where they reside; but it’s odd to see how a change at site 26 would effect the confidence at site 8. Overall, by playing with MS2L in AlphaFold I was able to improve the model’s confidence in sites 1-29, but it still has a long way to go. And - I just now relized it would have been interesting to incrporate moPPIt into testing these mutational problems, instead of solely relying on AF. AHHH!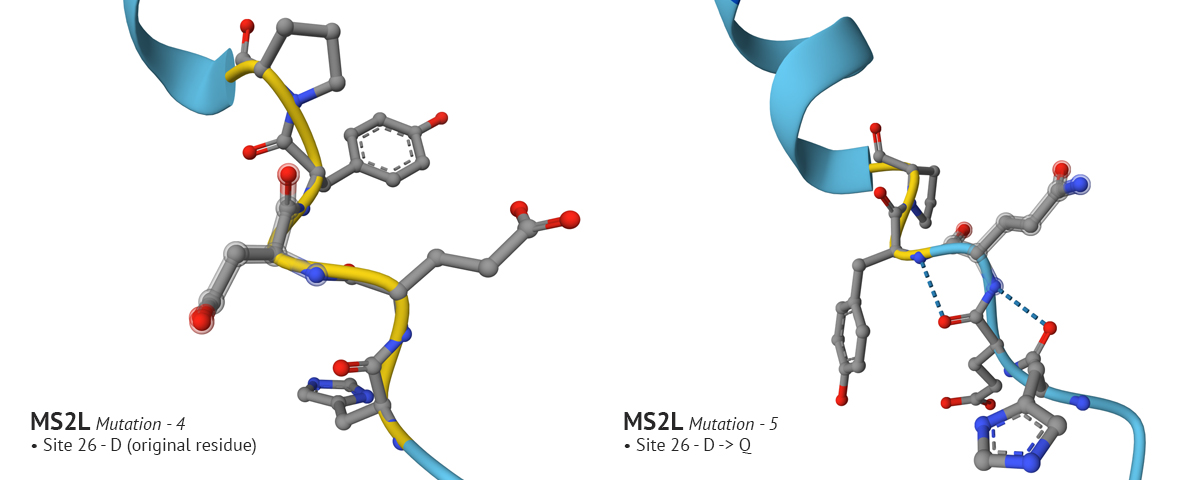
Conclusions
This assignemnt was particularily challenging because it required me to consider which of the protein design tools could I use to help approach the task of improving the folding characteristics of a protein, which would hopefully have a net positive affect on it’s functionality. Being a visual person, I found AlphaFold (AF) to be very helpful with it’s ability to model aa interaction. It’s certainly much faster than constructing a physical ball and stick model. However, I can see the value of those models too. In particular, it’s perplexing to see how AF interprets changes in design, where residues which are separated by several aa’s in sequence affect one another, and this is where (for me) a more traditional approach using physical models could help to work through compatability issues. I understand the importance of these insilico techniques, and I appreciate all the hard work behind their development!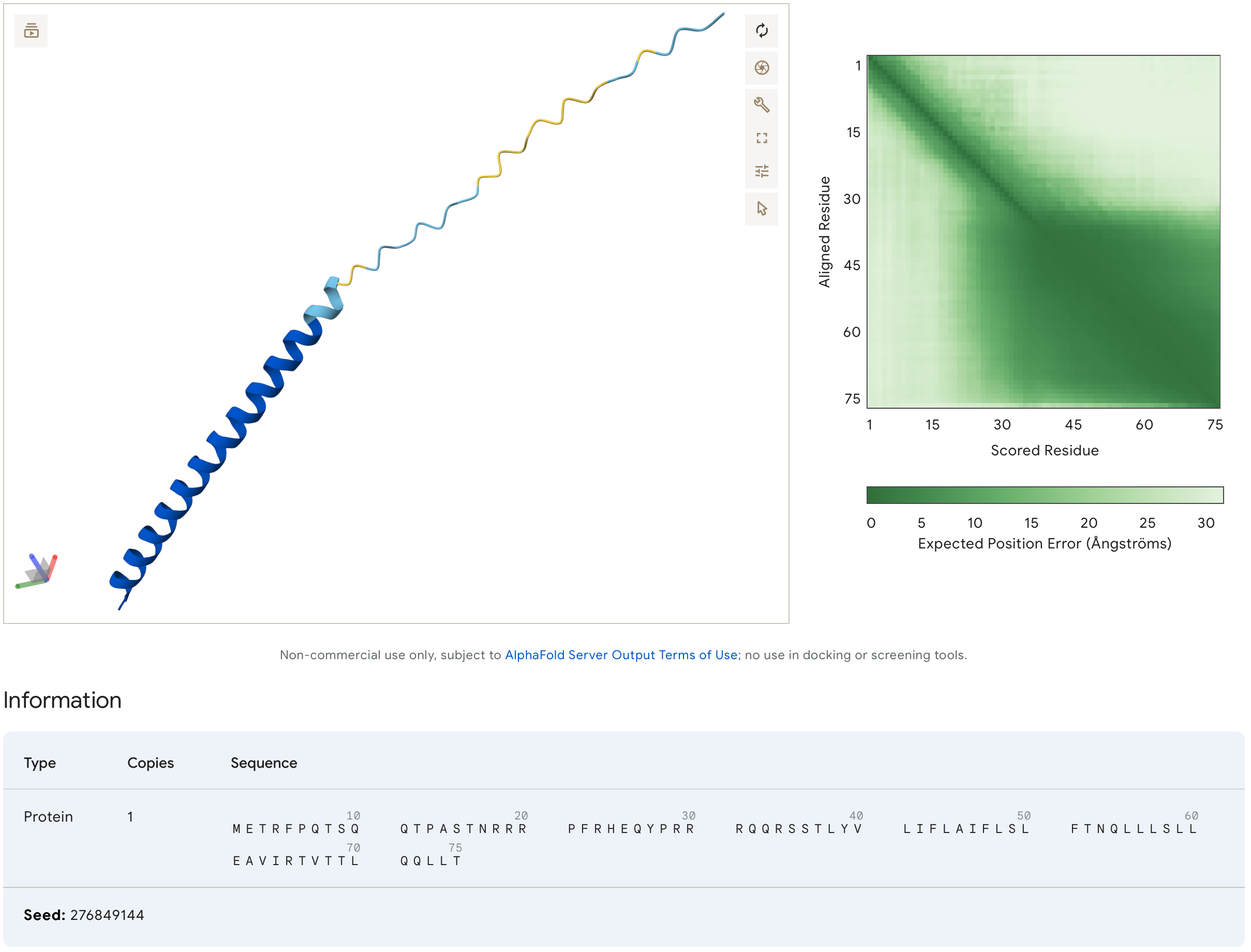
- Final mutational design. Changes to sequence- site 8: Q->T, site 23: K->R, site 26: D->Q, site 29: C->R, site 50: K->L.
Citation:
- ThermoFisher Scientific (2026). Amino Acid Physical Properties. Available at: https://www.thermofisher.com/uk/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/amino-acid-physical-properties.html (Accessed: 16 March 2026)
- Alphafold: Abramson, J et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature (2024).
- Peptiverse: @article {Zhang2025.12.31.697180, author = {Zhang, Yinuo and Tang, Sophia and Chen, Tong and Mahood, Elizabeth and Vincoff, Sophia and Chatterjee, Pranam}, title = {PeptiVerse: A Unified Platform for Therapeutic Peptide Property Prediction}, elocation-id = {2025.12.31.697180}, year = {2026}, doi = {10.64898/2025.12.31.697180}, publisher = {Cold Spring Harbor Laboratory}, URL = {https://www.biorxiv.org/content/early/2026/01/03/2025.12.31.697180}, eprint = {https://www.biorxiv.org/content/early/2026/01/03/2025.12.31.697180.full.pdf}, journal = {bioRxiv} }
- The UniProt Consortium UniProt: the Universal Protein Knowledgebase in 2025 Nucleic Acids Res. 53:D609–D617 (2025)