Hi- I’m Robert C Beck, a visual artist merging environmental science with experimental technology to challenge social perceptions of global health, ecology, and mutualism between human and non-human entities.
Photosynthetic Image Generation (PIG) “Art cannot be separated from life. It is the expression of the greatest need of which life is capable, and we value art not because of the skilled product, but because of its revelation of a life’s experience.”
-Robert Henri-
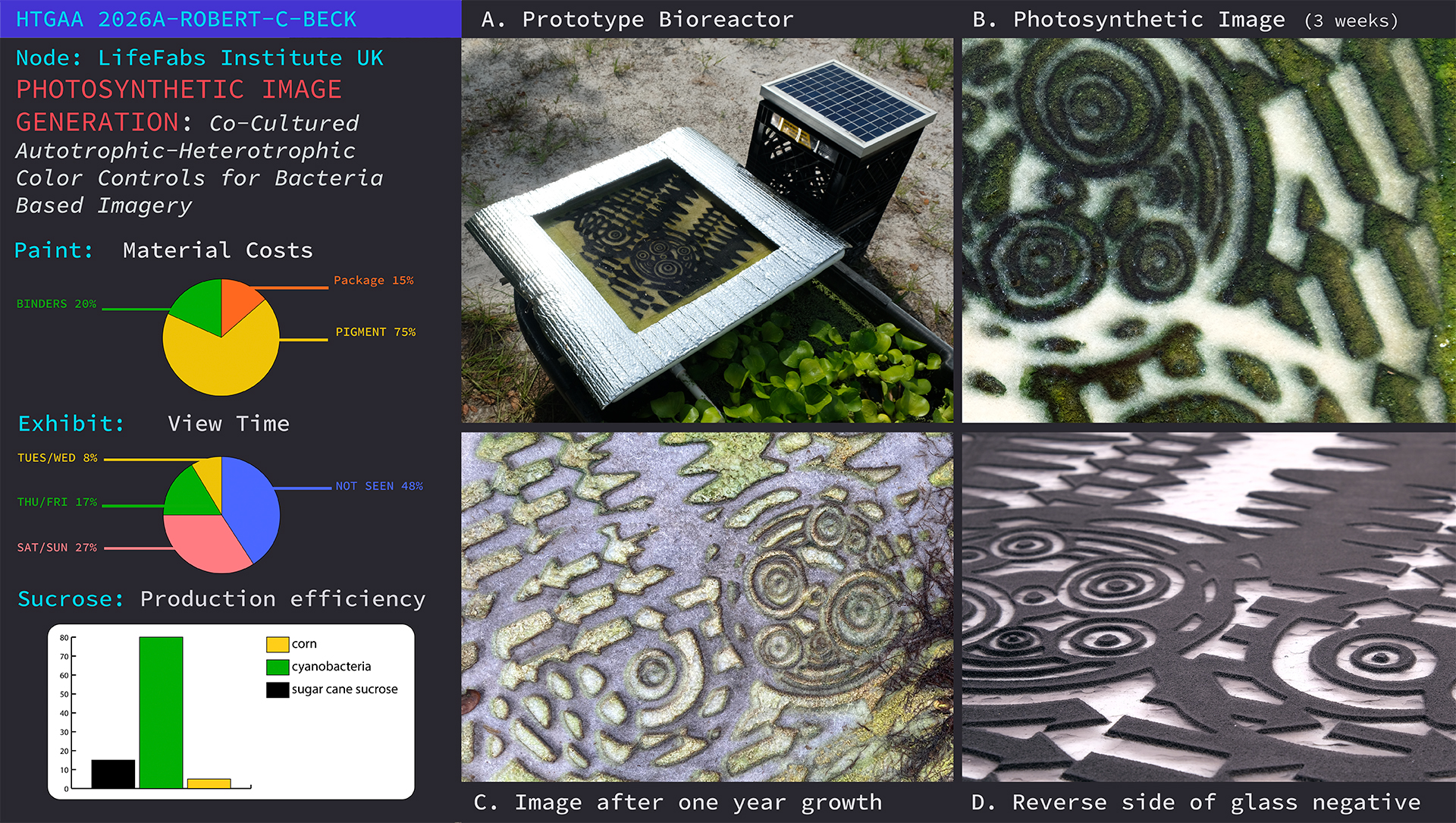
Overview PIG is an experimental combination of biology, glassmaking, printmaking, and photography which produces a living, two dimmensional surface of bacteria cultured as graphic imagery. The research behind the process to date has produced a small series of images grown on a synthetic felt substrate.
Start Your Homework! Part 1: Benchling & In-silico Gel Art Johnny Got His Gun -The sequence has been rearranged into the shape of a pistol. I wanted to keep the number of columns restricted to the original seven enzymes for this composition, but I’m sure the image could be improved with additional columns. Question? How many columns can be introduced to the gel matrix?
Do Your Homework!!! Links to Final Project Slides
Photosynthetic Image Generation Surface Design Bryoculture Automation Reading Review: -Nature- Advancing sustainable agricultural transformation through the synergy of automated experimental platforms and living labs
Forget the Opentron, it’s time for cranes, planes, and Agricutural Automation! Seriously though, not to hack on the Opentron, what an amazing tool for working in the lab. But let’s not forget about the outdoor living lab which is part of arguably one of the most important industries, Agriculture. The scale of an agricultural system can range from a backyard garden to industrial complexes spanning thousands of hectares. How can automation help manage systems across the scales of indusrty which ultimately translates into what does or doesn’t end up in our shopping carts?
Hi- It’s time to do your homework! Icosahedronic Molecular Modeling I -RCBeck
Part A Questions:
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) Solve- How many Daltons are in 500 grams? 1g = 6.022173643E+23 Daltons (6.022173643E+23 x 500)/100 = 4.29 x 10E+18
Time to do your homework- It’s always time to do homework in this course!
Part A: SOD1 Binder Peptide Design Part 1: Generate Binders with PepMLM
Starting with the human SOD1 sequence from UniProt (P00441), and then introducing the A4V mutation, we get the following sequence where the fourth amino acid in the sequences is changed from K to V:
Guess what. What? It’s time to do your homework. OMG!!! It’s never ending… I’m always doing my homework for HTGAA. Icosohedronic Molecular Model II -RCBeck
Assignment: DNA Assembly Answer these questions about the protocol in this week’s lab
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Answer: There are several components in the master mix. It contains - DNA polymerase, dNTP’s, reaction buffers, and MgCL2.
After this assignment, it will finally not be time to do your homework for one day… Happy Spring Break!
This is Your Brain on HTGAA- RCBeck
Assignment Part 1: Intracellular Artificial Neural Networks What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
They can be weighted with high and low input values which gives them unique capabilities. Unlike a Bool - which is if this then that - the specificity of an IANN means they can be designed to produce more specific outcomes in cellular function by responding to multiple control parameters. Boolean functions have one input. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
It’s that time again… RrrrrrraaAAHH!!! Glueless Icosahedron - RCBeck
General and Lecturer-Specific Questions General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
What happened to my brain?
The Only Question -RCBeck
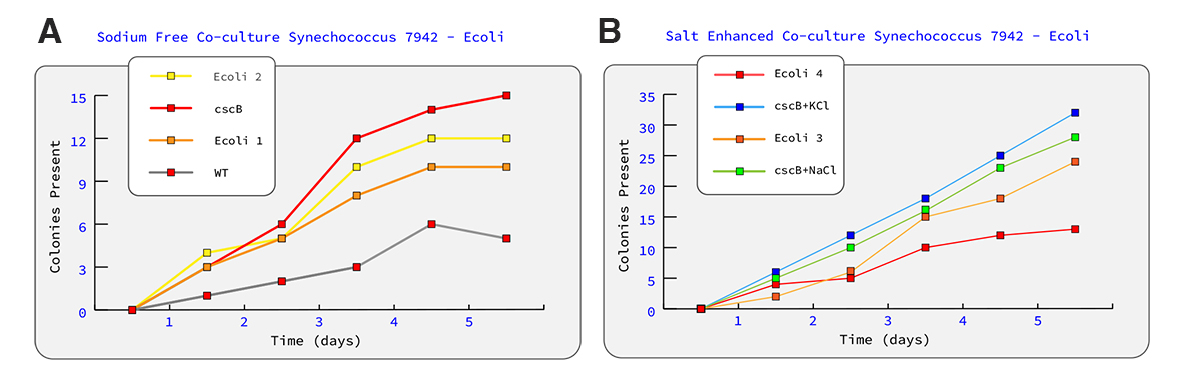
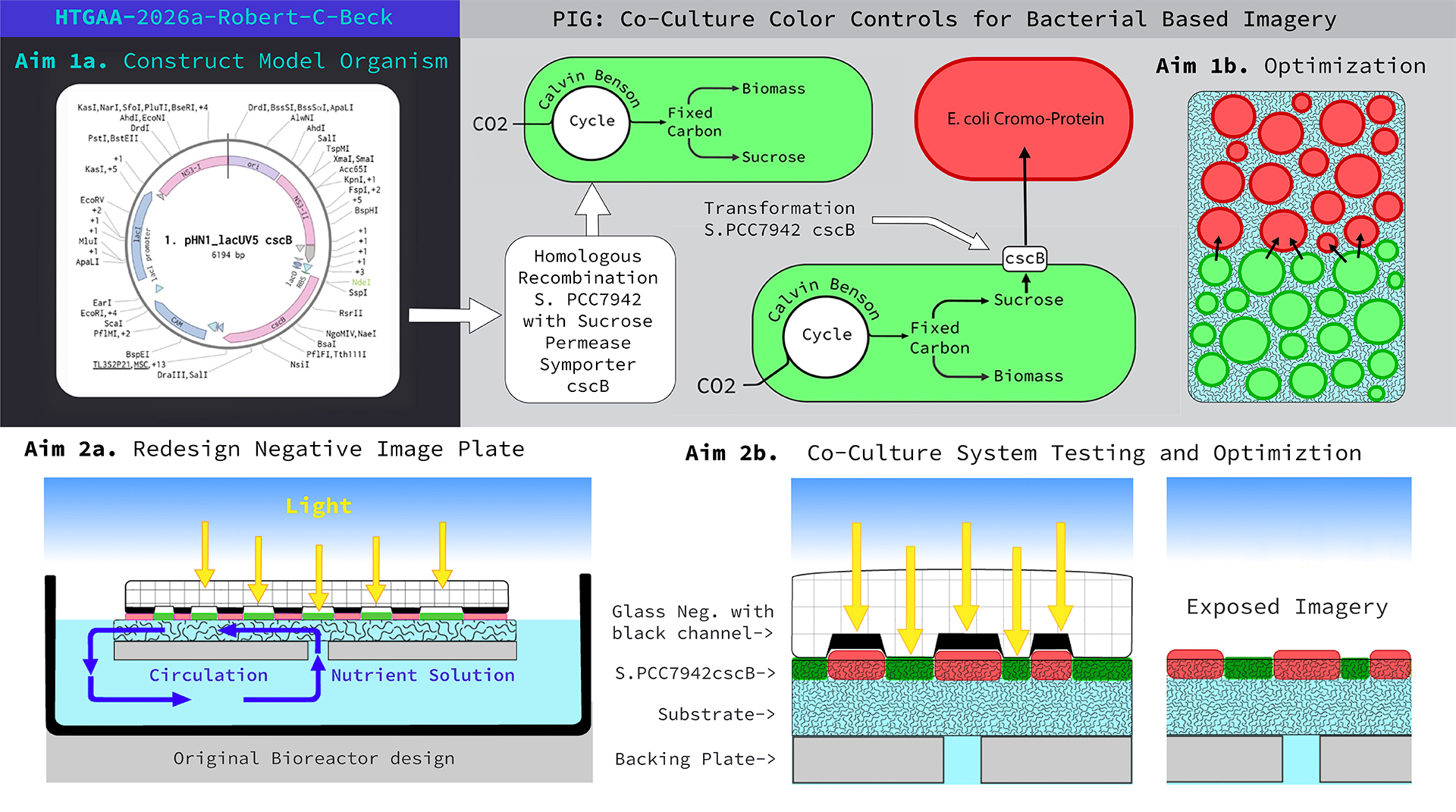
Final Project Homework For your final project: Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc. Aim One: Initial Transformation
OMG! Last One! Drawing Air (2026) Detail, Graphite on Paper -RCBeck
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork My contribution to the artwork was limited to one pixel, which was initially made to test interaction with the interface. I noticed how pixel contribution was rather competitive, where scripting was automatically writing over contributions. Unfortunately, this discouraged me from contributing any further, which lead me to focus on my final project alternatively.
Subsections of Homework
Week 1 HW: Principles and Practices
Photosynthetic Image Generation (PIG)
“Art cannot be separated from life. It is the expression of the greatest need of which life is capable, and we value art not because of the skilled product, but because of its revelation of a life’s experience.”
-Robert Henri-
Overview
PIG is an experimental combination of biology, glassmaking, printmaking, and photography which produces a living, two dimmensional surface of bacteria cultured as graphic imagery. The research behind the process to date has produced a small series of images grown on a synthetic felt substrate.
Background
The project emerged over the course of a two year period of experimental interactions with biological material on a Brooklyn rooftop, which began in 2016. It was the site of a collaborative project with my partner, Sarah Max Beck, called studioHydrostatic. It was located on top of a four story walkup in Bedford Stuyvesant, where we had set up a small greenhouse to carry out visual media experiments using an artificial wetland ecosystem. The system was a hybridized combination of aquaponic and terraponic technologies chemically driven by urine, seashells, and chelated iron as nutrient sources. The elevated levels of nitrogen in the water lead to cyanobacteria growing on wet surfaces exposed to sunlight. It was interesting to observe how the bacteria would form into very specific shapes and patterns depending on the qualities of whatever it was interacting with, and marked the beginning of this project. It reveals the collaborative nature of life, the ongoing exchange of resources between organisms -which is more than one life’s experience- it’s the sum of many in a vibrant display of living energy.
Process
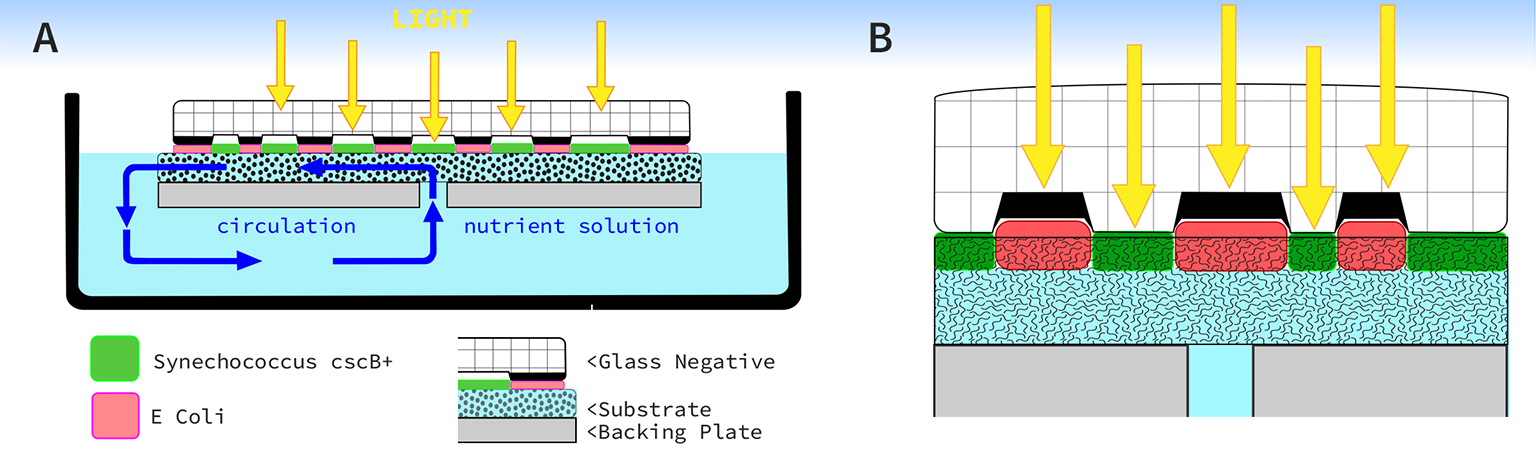
The live culture of cyanobacteria occurs in and around aquatic ecosystems, so I decided to set up a simple recirculating pond in a pasture to further experiment with the process. The main body of water was held by a fifty gallon, black plastic, water trough stocked with water hyacinth (Pontederia crassipes), which was harvested from a nearby body of freshwater. A small solar powered water pump moved water from the pond into a smaller processing tray which rested on top of the trough. Inside the tray was a piece of white polyester felt stretched over a foam board. I punched a small hole through the foam board and connected the water supply tube from the water pump. Resting on top of the felt was a plate of glass which had a negative image fused into its surface. The result was a water fed piece of felt sandwiched between foam and glass. As the sun shined, water moved from the pond up to the tray saturating the felt underneath the glass. The contraption exposed the felt to water, sunlight, and nutrients whereby, underneath the glass, cyanobacteria began to grow on the felt and produce an image pattern grown by photosynthetic bacteria.
Research with HTGAA
The beauty of this process is its ability to work with naturally occurring bio-cycles, organic matter, and the resources of its surroundings. These qualities do present a challenge to the idea of working with synthetic biology to augment a process which seems to work quite well without any manipulation. However, the current direction of the research is focused on the recyclability of the media, and since the current system utilizes a synthetic material to manifest the image on, I believe bacterial cellulose holds potential as an organic imaging substrate.
Perhaps differnt qualities of cellulose produced from certain strains of BC could be modified to obtain more desirable results, or perhaps change the process altogether. The possibility of creating something completely different also exist, and while my focus at this point is directed towards PIG, I am open to whatever I may discover through this course!
Strategies for Governance
The goal of my research with PIG is to produce a scalable, mobile technology for producing live surfaces as working media for visual artists in their studio practices as well as in exhibition environments. I believe it is important to research these potential pathways for humans to find new ways of interacting with ecology which are mutually beneficial, or symbiotic. However there are many potential hazards associated with the manipulation of any environment or material. Listed below are three real examples this project currently faces without synthetic alteration of organism genomes. Actions or considerations for GMO would be similar, but may be more procedurally specific.
Environmental protection from:
Release of foreign or exotic organisms from working displays into other ecosystems.
Equipment or device contamination from installations environments upon return to origin.
Release of foreign cultures from lab or experimental waste/water.
Self contamination from interaction with media:
Practioners could become infected by or transmit biological organisms from content/media to themselves or others.
Engaged public interaction poses similar risks.
Spread of Misinformation:
Public opposition to content insiting the spread of “bad” or incorrect information.
Misinterpreted information by gallerists, voluteers, or public.
Environmental Protection Policy
Environmental contamination could result from the improper handling or disposal of materials associated with work. While it is easier to safeguard in controlled laboratory settings, the potential still exists. There are additional risks once materials leave the lab for transportation between works sites, as well as for the work sites themselves.
Environmental Protection Goals
Protect localized environments from contamination by foreign organisms.
Protect people/personel interacting with experimental media.
Protect organizations hosting experimental media from harmful content.
Protection of Localized Environments
Actions to be taken
Follow Laboratory Protocals (this project is not currently active in any laboratories or public spaces.)
Follow BSL1 safety protocals and procedures.
Create list of ALL pertinent safety information specific to research for lab technicians/researcher working with media.
Awareness of experimental media’s location within the lab space: Use signs and markers to identify project space.
Be tidy and make sure surfaces are clean, organised, and clearly marked.
Make Visitors aware of active experiments. Remind them: please do not to touch anything w/o permission.
Post emergency contact information.
Design experiments and equipment for safe containment.
Equipment should be easy to clean and properly sealed.
Design traveling systems with primary and backup sealing/containment systems.
Improperly sealed systems could leak: always double check containment before transport.
Risk of containment breach is low with well maintained systems and adherence to protocals.
Clearly label shipping containers with contents and emergency contact information.
Maintain equipment to ensure systems and safety mechanisms are working properly.
Assess equipment before project initiation.
Replace any worn parts or seals and make any repairs.
Clean final assemblies.
Schedule maintenance with time for service before initiating project.
Does the Action:
Action 1
Action 2
Action 3
Enhance Biosecurity
• By preventing incidents
1
1
1
• By helping respond
1
2
3
Foster Lab Safety
• By preventing incident
1
1
1
• By helping respond
1
2
3
Protect the environment
• By preventing incidents
1
1
1
• By helping respond
2
1
3
Other considerations
• Minimizing costs and burdens to stakeholders
3
2
1
• Feasibility?
1
2
1
• Not impede research
1
3
2
• Promote constructive applications
2
1
2
—————————————————–
———-
———-
———-
Score
14
16
18
Conclusion
While I cannot imagine conducting any experimental research without following all the actions listed aboove, the exercise did make it clear that the research is still very much in a developmental phase, and there needs to be a little more care taken when considering the project’s deployment in non-laboratory environemnts. Overall, I believe the risk to benefit ratio to be very low, especially in its research and development phases. The application of the technology could be interesting as a bioprinting technique, but I’m more interested in exploring its application as an experiential/phenominological application within the visual arts. I’m not sure how synthetic biology will play into the research at this point, but I can see how this may change as I learn more about the organisms I’m working with, and the environmental challenges I may face when developing transport and display systems.
Week 2 HW: Reading and Writing DNA Sequences
Start Your Homework!
Part 1: Benchling & In-silico Gel Art
Johnny Got His Gun
-The sequence has been rearranged into the shape of a pistol. I wanted to keep the number of columns restricted to the original seven enzymes for this composition, but I’m sure the image could be improved with additional columns. Question? How many columns can be introduced to the gel matrix?
Part 3: DNA Design: Pick a Protein… Any Protein?
Drawing from HTGAA notebook
The organism - Chroococcidiopsis - contains the Orange Carotenoid Protein (OCP)
Microscopic detail of Chroococcidiopsis
As seen in the page from my notes above, I’m interested in Chroococcidiopsis, a type of cyanobacteria, which is the focal point of my research with PIG. A protein, OCP, is found in the Phycobilisome (PBS), which is a large light harvesting complex attached to the Thylacoid Membranes within the cyanobacteria cell body. It has 6483 genes, and I have no idea what any of this means, and I’m not sure if this protein - which was one of over 41,000 results to my search for “proteins commonly found in cyanobacteria” - will be of any importance to my project. Apparently, it acts as a sunscreen with its orange coloration for the thylacoid membrane, which is important for regulating photosynthesis within the cell. I chose this protein in particular for primarily two reasons, it’s a necessary part of a functional cyanobacterial cell, and it has been isolated as a plasmid, which seems to be important in context to the work we will be doing within HTGAA.
Reverse Translation of OCP Protein sequence to DNA sequence.
Reverse translation (most likely codons) of URD53675.1 orange carotenoid-binding protein (plasmid) [Chroococcidiopsis sp. CCNUC1] to a 957 base sequence: atgccgtataccattgaaagcgcgcgcagcatttttccggatacccaggtggcgagcgcg
gtgccgaccattgtggaaagctttgaacagctgagcgcggaagatcgcctggcgctgctg
tggtttgcgtataccgaaatgggcgtgaccattaccccggcggcgatgcaggtggcgaac
atgatgtttgcggaaaaaaccctggcgcagattgaacagattccggcggcggaacagacc
caggtgatgtgcgatctgattaaccataccgataccccgatttgccgcacctatagctat
tttggcatgaacgtgaaactgggcttttggtatcagctgggcgaatggatgaaacagggc
attgtggcgccgattccggaaggctataaactgagcgcgaaagcgagcaacgtgctgcag
accattcgccagctggaaggcggccagcagctgaccgtgctgcgcgatattgtggtgaac
atgggccatagcccgaccaccgcgacccagaaagtggaagaaccggtggtgccgccgaaa
gatctggcgccgcgcaccaaaattgtgattgaaggcattaacaacagcaccgtgctgagc
tatatggaaaacatgaacgcgtttgattttgaagcggcggtggcgctgtttgcggaagat
ggcgcgctgcagccgccgtttgaagaaccgattgtgggccaggaaagcattctggcgttt
atgcgcgaagaatgctatggcctgaaactgattccggaacgcggcattagcgaaccgggc
gaacgcggctttacccagattaaagtgatgggcaaagtgcagaccccgtgggcgggcgat
agcgtgggcattaacctggcgtggcgctttctgattaaccgccagggcaaaatttttttt
gtggcgattgatgtgctggcgagcccgcaggaactgctgaacctgggcctggtgaaa
Reverse translation (consensus codons) of URD53675.1 orange carotenoid-binding protein (plasmid) “"[Chroococcidiopsis sp. CCNUC1]” to a 957 base sequence: atgccntayacnathgarwsngcnmgnwsnathttyccngayacncargtngcnwsngcn
gtnccnacnathgtngarwsnttygarcarytnwsngcngargaymgnytngcnytnytn
tggttygcntayacngaratgggngtnacnathacnccngcngcnatgcargtngcnaay
atgatgttygcngaraaracnytngcncarathgarcarathccngcngcngarcaracn
cargtnatgtgygayytnathaaycayacngayacnccnathtgymgnacntaywsntay
ttyggnatgaaygtnaarytnggnttytggtaycarytnggngartggatgaarcarggn
athgtngcnccnathccngarggntayaarytnwsngcnaargcnwsnaaygtnytncar
acnathmgncarytngarggnggncarcarytnacngtnytnmgngayathgtngtnaay
atgggncaywsnccnacnacngcnacncaraargtngargarccngtngtnccnccnaar
gayytngcnccnmgnacnaarathgtnathgarggnathaayaaywsnacngtnytnwsn
tayatggaraayatgaaygcnttygayttygargcngcngtngcnytnttygcngargay
ggngcnytncarccnccnttygargarccnathgtnggncargarwsnathytngcntty
atgmgngargartgytayggnytnaarytnathccngarmgnggnathwsngarccnggn
garmgnggnttyacncarathaargtnatgggnaargtncaracnccntgggcnggngay
wsngtnggnathaayytngcntggmgnttyytnathaaymgncarggnaarathttytty
gtngcnathgaygtnytngcnwsnccncargarytnytnaayytnggnytngtnaar
Question 1. The translation results were produced using bioinformatics’ Sequence Analysis - Reverse Translate which produced the results above. In addtion it produced code for graphing the base probabilities. How is this code converted in a graph? Python? Question 2. What is the difference between most likely codons and consensus codons? Question 3. In addition to nitrogenous bases ACTG, there are also: n,y,h,r,w,s and m. These represent codon combos which form amino acids? - yes
Codon Optimization
Clearly there are advantages as well as disadvantages to Codon Optimization, and after reviewing the basic concept it seems there are two divergent camps, pro optimization vs. the purist. Let’s compare.
Some Advantages:
Optimization can increase protein production by making more efficient use of codons by using a simplified version of synonymous amino acid combinations. This could help improve the efficiency of producing therapeutic drugs, and potentially reduce production cost.
Certain codon combinations can potentially increase the expression of a gene thereby making more affective treatments.
Optimization helps to align a target gene’s codon usage with the preferred codons of the host organism.
Some Disadvantages:
Modification of protein structure can affect protein function or performance.
Potentially, there are deeper levels of code in specific codon combinations which could be related to rythms associated with protein folding patterns during elongation, or functional aspects which have not yet been discovered.
Optimization can also affect wobble of tRNAand potentially cause unwanted mutations or render proteins disfunctional.
Based off this surface research, I can see reasons for and against codon optimization - and as with any experimental research - we must carefully weigh the potential benefits of optimization relative to the application. For instance, if the optimization is for use in animal research, which could potentially impact the wellbeing of an organism with unintended “side effects”, then comprehensive multi level testing should be conducted to exhaust or reduce error within the application. If the optimization is purely lab based and not able to modify DNA replication, and will improve the speed of data and research assimilation then I can see this as a definite advantage to employ the technology.
Codon Optimization- novoprolabs.com, I used this because the Twist link was broken. The sequence type was: DNA/RNA with E.coli as expression host which produced the following result from most likely codon group:
Question 4. In the optimized sequence above the 25th character is T of TAC which would indicate RNA start codon AUG?
question 5. Then in the next line below I found the sequence ACTTAC, which would indicate a stop codon with another start directly after in the sequence. Everything in between the start and stop is a particular gene in the chain? Is that the promoter of the sequence?
Question 6. If AUG is start codon, which contains uracile, then start and stop codons only exist in RNA?
You have a sequence! Now what?
I don’t know, this is where it falls apart for me… WAH Wah wah… GAME OVER… INSERT ANOTHER TOKEN!!!
I’m failing to grasp the connection between the Orange Caratenoid Protein OCP, which I chose to explore, and how it relates to the host organism, Ecoli, in the codon optimization stage of homework.
Question 7. Am I trying to cut and paste the OCP into the ecoli genome using the benchling software? Or am I looking for the expression genes within the OCP sequence by identifying the starts and stops? Question 8. How do I determine where start and stop codons are within the sequence? I know Methionine (AUG) is start and there are three stop codons, UAA, UAG, UGA. Do I start reading in sets of three from the begining of optimized sequence looking for TAC, which would be on the DNA side of AUG?
Annotation of Sequence (I think I need a tutor!)
Prepare a Twist DNA Synthesis Order
I was able to create both Benchling and Twist accounts no problem. I imported the optimization sequence above as a new sequence, which marked all the restriction enzyme sites, but was not really able to distinguish promoter region etc. for annotation. Although, after following through with predicted regions and annotation, I arrived at the graphic above. When I imported the file into Twist Twist could not complile the sequence. I’m not sure where I went wrong.
Question 9. How do I determine promoter, RBS, etc. ? -RBS stands for Ribosomal Biding Site- It is a sequence of of nucleotides (codons) upstream of the start codon (AUG Methionine) in mRNA and has 5-7 nucleotides rich in A and G.
-Start codon in DNA is TAC, and it should be followed by a stop codon (ATT,ATC,ACT).
Question 10. Should I have fed the optimization results from the consensus codons into Benchling?
DNA Read
My Primary goal with HTGAA is to build my understanding cyanobacteria as the building block of not only my artwork, but also as a cellular organism which forms the foundational base of the ecological food chain. I cannot even begin to think or dream of how, what, or why I would want to alter the cellular systems of cyanobacteria at this point, but as my knowledge of these organisms develops I know my perspective will change.
DNA Write
What DNA would you want to synthesize (e.g., write) and why? When I started this homework assignement, over a month ago, I had no idea what I could or would want do with this opportunity, and to be honest, I’m not sure I feel much differently. Obviously, this technology has amazing potential to make changes which could, and have had a profound impact on people’s lives. As it relates to my research within visual art and its relationship with biological materials, I can see working with microorganisms to express complex concepts, which can enrich subject matter by creating dynamic relationships with other organisms. It’s really profound to think about it from this perspective. But, I’m at the begining of a new journey, and I would like to begin by writing an expression of color into a cyanobacterial cell. With this accomplishment I will have established enough understanding to begin exploring other methods to work with bacteria.
What technology or technologies would you use to perform this DNA synthesis and why?
“if I have seen further [than others], it is by standing on the shoulders of giants.” Isaac Newton
Refering to Newton, the research that’s been done in this field by others is really quite humbling. Especially when trying to work through some of these exercises. I follow the giant’s instructions, but I’m blinded by the light of perspective. Working with a known cynobacterial genome/sequence seems like the best place to start. Studying it in silico by working with Benchling will help me to understand how genes, and thier functional coding process produces protein for cellular operations. I hope this will lead to a basic comprehension of the systems by making simple adjustments one step at a time. I have to walk before I can run. Once I have taken my first step, and actually attempted to produce some form of DNA, probably by using restriction enzymes to cut and knock in a new sequence, I will need to test my work by sequencing it. If I am successful, then I would think using PCR to amplify the synthesized product to test in vivo would be the next step. As for scalability, I am not concerned with this yet. I think it would be amazing to have the ability to produce enough plasmid to modify a cyanobacterial image which is at least 30cm square. If I could do that, then I’m sure I will be able to work out larger scales.
DNA Edit
What DNA would you want to edit and why? I pretty much feel the same way about this as writing, and if I’m writing anything it will be something I’ve copied. In a certain sense I guess that’s what editing really is, little tweaks to something which already exists, and I would approach this one step at a time with the most appropriate technique. Since I would like to work with cyanobacteria, plasmids seem to be the most approachable form of modification. The goal is to shift the blue/green color spectrum of cyanobacteria from yellow to yellow-green, green to blue-green, and blue-green to blue. I would probably start by identifying a protein which is strong within the yellow spectrum. Then design a plasmid coded for the chosen protein. Order the plasmid, and test the design using ecoli, since it won’t produce any background color competition, to see if/how well it works. If I have positive results, I would then try it in cyanobacteria, by using either a heat shock or electroporation to introduce the plasmid. Culture cells on agar and compare results against an unmodified color control.
References
Image of Chroococcidiopsis -Villanueva, Chelsea & Hasler, Petr & Dvorak, Petr & Poulíčková, Aloisie & Casamatta, Dale. (2018). Brasilonema lichenoides sp. nov. and Chroococcidiopsis lichenoides sp. nov. (Cyanobacteria): Two novel cyanobacterial constituents isolated from a tripartite lichen of headstones. Journal of Phycology. 54. 10.1111/jpy.12621.
Sound JK, Bellamy-Carter J, Leney AC. The increasing role of structural proteomics in cyanobacteria. Essays Biochem. 2023 Mar 29;67(2):269-282. doi: 10.1042/EBC20220095. PMID: 36503929; PMCID: PMC10070481.
Shang,J. Chroococcidiopsis_sp._CCNUC1 genome. (2022). College of Life Sciences, Central China
Normal University, Central China Normal University, Wuhan, Hubei, 430079, China. Available at: https://www.ncbi.nlm.nih.gov/nuccore/CP097482.1 (Accessed: 23-3-2026)
Paremskaia AI, Kogan AA, Murashkina A, Naumova DA, Satish A, Abramov IS, Feoktistova SG, Mityaeva ON, Deviatkin AA, Volchkov PY. Codon-optimization in gene therapy: promises, prospects and challenges. Front Bioeng Biotechnol. 2024 Mar 28;12:1371596. doi: 10.3389/fbioe.2024.1371596. PMID: 38605988; PMCID: PMC11007035.
Forget the Opentron, it’s time for cranes, planes, and Agricutural Automation! Seriously though, not to hack on the Opentron, what an amazing tool for working in the lab. But let’s not forget about the outdoor living lab which is part of arguably one of the most important industries, Agriculture. The scale of an agricultural system can range from a backyard garden to industrial complexes spanning thousands of hectares. How can automation help manage systems across the scales of indusrty which ultimately translates into what does or doesn’t end up in our shopping carts?
Essentially, this article draws into focus many of the gaps which exist between actors working with biotech. As the pace of technology grows and areas of specialization become more specific - in terms of applications, economics, reqiured rescources etc. - a system of governance should be codeveloped between stakeholders, policymakers, and consumers alike to ensure that a reliable and equitable framework grows with industries as they develop. This is especially important to ensure that new research has a way to interact with development and implimentation within the overall framework of a rapidly transforming environment.
Citation: Hoffmann, M., Chen, C., Butterbach-Bahl, K. et al. Advancing sustainable agricultural transformation through the synergy of automated experimental platforms and living labs. Nat Commun 16, 8418 (2025). https://doi.org/10.1038/s41467-025-64450-7
Opentron Demo
Colab code for points genreated by the Ronan simulator:
First design for Opentron I’m not happy with this design, I quickly produced it as I was more concerned about the coding aspect of Colab. As a result, I never uploaded the design because I thought I would revisit it when I caught up with the homework. I never really caught up. This is interesting though because my experience with the class up to this point (week 7) has me reconsidering what my goals are for the final project. The opentron, and bacterial based imagery seems more approachable than my original goal of modifying a cynobacteria to exhibit color shifts by introducing a plasmid. I feel that plasmid design is not going to fall within the scope of what I can actually accomplish within this course. The learning curve is very steep given the amount of time relative to my level of experience.
Week 4 HW: Protein Design I
Hi- It’s time to do your homework!
Icosahedronic Molecular Modeling I -RCBeck
Part A
Questions:
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Solve- How many Daltons are in 500 grams? 1g = 6.022173643E+23 Daltons (6.022173643E+23 x 500)/100 = 4.29 x 10E+18
Answer- There are approximately 4.29 x 10E+18 amino acid molecules in 500 grams of meat.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Answer- The process of digestion uses acids and enzymes to break tissues down into simpler molecules for use in cellular mechanics. This process helps provide the rescources/energy to power our own cellular processes which reassemble the digested amino acids, sugars and such into the forms required by each cell to perform its function which is dictated by the organisms genetic code.
Why are there only 20 natural amino acids?
Answer- According to Andrew Doig, a chemical biologist at the University of Manchester in the UK, it goes all the way back to LUCA. The twenty natural amino acids found in all living organisms are the building blocks we share in common. It is speculated that these twenty in particular are the most stable do to their structure, and able to maintain their functional forms after being buried, or exposed to the environmental conditions of Earth 3.5–3.8 billion years ago when LUCA first emerged, probably from some archaic form of RNA. They are more hydrophobic than hydrophilic, and that may have something to do with their role in LUCA’s evolution because of the way they are able to fold and work together structuraly.
Can you make other non-natural amino acids? Design some new amino acids.
Answer- Yes, absolutely! The only problem though is when trying to incorporate synthetic amino acids into cell function/DNA-RNA the catalytic function of those new amino acids have to be incorporated into the cell’s chemical function of enzymatic activity.
Where did amino acids come from before enzymes that make them, and before life started?
Answer- Apparently, there are several theories which range from riding in on an astreroid to chemical reactions occuring through electrical activity-i.e., lightening striking in areas rich in oxygen, nitrogen, hydrogen, carbon and sulphur compounds. The astroid theory has been proven viable by the discovery of at least 86 amino acids on the Murichison meteorite, which landed in Australia in 1969. I beleive it was probably a combination of planetary chemical reactions and celestial seeding.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Answer- Left, because the a helix is usually right handed for the most common amino acids becuase the carbons have an L configuration, but in D-amino acids, the carbons have a D configuration, they mirror the L configuation.
x
Can you discover additional helices in proteins? In theory, yes. Although detecting structures outside the typical three is difficult, not to mention the limitation of the physical and chemical properties of helical structures. But with synbio, the possibility of discovering something new is certainly possible, maybe even likely.
Why are most molecular helices right-handed?
Answer- It’s a mystery… I think, but it has something to do with chirality. Perhaps polarity, too? Or maybe the ionic charge of particles, laws of attraction- opposites attract- who knows? Certainly, there must be an answer.
Why do β-sheets tend to aggregate?
What is the driving force for β-sheet aggregation?
Answer- It appears as though the proteing is programmed to fold in such a way that it wants to form aggregate structures. This behavior is probably caused by the alternating arrangement of charged and hydrophobic amino acids, which makes them inclined to gather through attraction of oppositely charged particles?
Part B
URD53675.1 orange carotenoid-binding protein (plasmid) Chroococcidiopsis sp. CCNUC1
For the sake of consistency I decided to stick with the first protein from the week two homework. It’s interesting to see how many different organisms this protein exists in, all with there own unique functions using this molecule.
It is 319 amnino acids long, most common is A with a count of 29. According to the UNIProt Homology, there are 239 homologs, and InterProscan identifies it as belonging to NTF2-like domain superfamily. Belongs to the orange carotenoid-binding protein family.
There were quite a few hits for OCP in RCSB PDB. The model above is a 3-D rendering of OCP2 Gloeocapsa sp. PCC 7428 from RCSB PDB.
PyMol Renderings: Stick and Ribbon (below)
Pymol - Orange Carotenoid-binding Protein
Clockwise from topleft: ribbon, secondary, stick, hyrdro
Number of helices: 10
Number of sheets: 6
Hyrophobic with 2,363 atoms in hydrophobic residues vs. Hydrophilic residues with a count of 782 atoms
Binding Pocket Yes, definately several pockets and even some holes leading to core.
Part C - Using ML-Based Protein Design Tools
C1. Protein Language Modeling
Deep Mutational Scans- pdb_00008qx5
Helical Carotenoid Protein 4 (HCP4) from Anabaena with bound Canthaxanthin• Heat map visuals indicate binding probabilities for:
Sites 43 and 112 exhibit high probability for following bases: A, D, E, G, K, N, Q, R, S, and T.
C has low probability across all sites except 43 and 112, where it is medium.
Y and X are fairly low across entire sequence.
Latent Space Analysis In the examples above the closest related matches were:
Cytochrome f subunit of the cytochrome b6f complex, transmembrane anchor (Green Alga, Chlamydomonas reinhardtii) TaxId: 3055
C2. Protein Folding
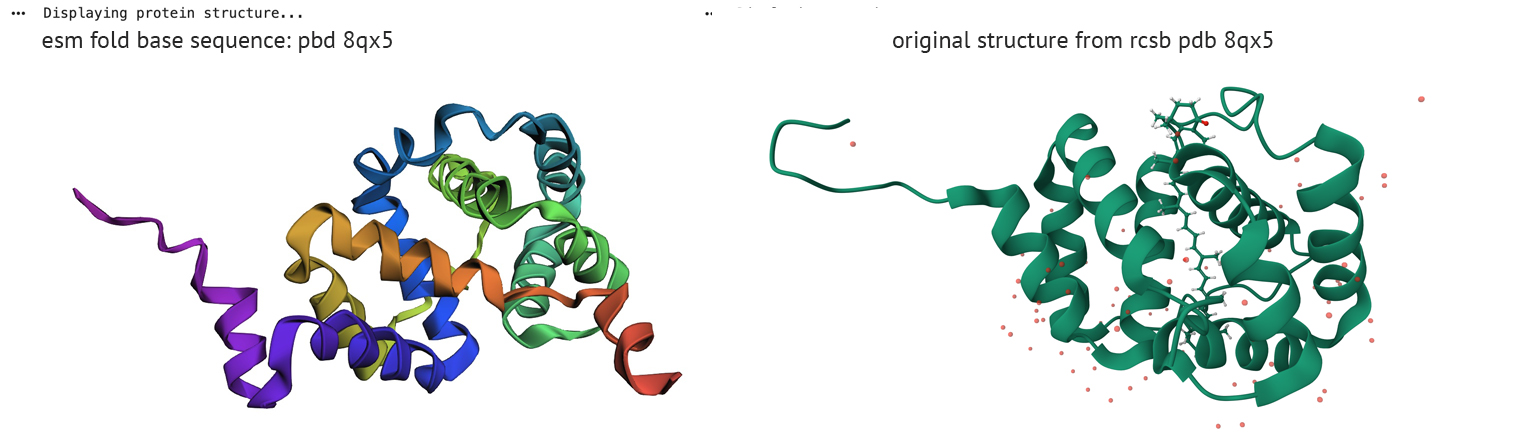
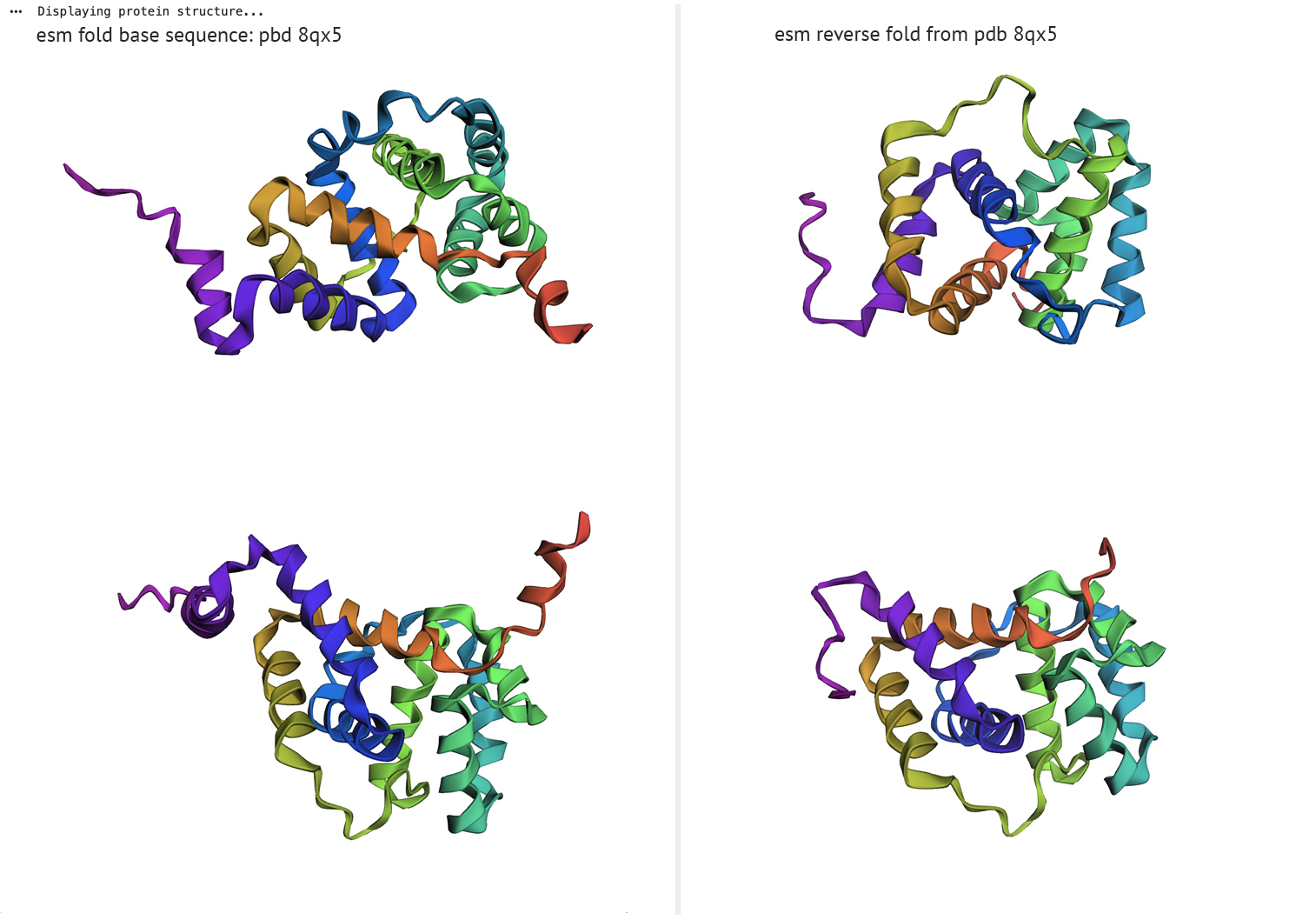
Amino Acid Probability - PDB 8qx5Folding a protein:
Fold your protein with ESMFold.
Do the predicted coordinates match your original structure?
ESM fold/reverse fold comparison
The left column represents ESM fold from PDB sequence-8qx5, right column displays reverse fold results
Part D: Group Brainstorm on Bacteriophage Engineering
Proposal by: Robert C Beck, Sameen Nasar
Concept
This is not an easy task - to approach Bacteriaphage theory with a brief introduction, and then cook up a conceptual modification of an extremely complex micro machine by altering its genetic code. In the spirit of HTGAA, here we go.
The first step is to consider what the phage does, what’s its purpose? It’s a virus which carries genetic information, and as a virus its job is to find a host which it can use to replicate itself and spread genetic information. As a machine it consists of different parts - a vessel which contains the genetic information, an attachment mechanism allowing it to connect itself to a host - and a system for transferring its genetic payload to the host. It’s pretty simple in theory, but how these different parts interact with one another to perform their function is very, very, very complex.
The problem with the phage mechanism is bacterial recognition of its presence, which leads to resistance. This raises a question about how a phage attaches to its host, and when it does, how the bacteria responds to the phage. This is what leads to resistance in the first place, bacterial recognition or memory of what the phage’s handshake is like. It reminds me of meeting people during and after covid, and how awkward it was. It was never really clear what to do. Do you bump fists or elbows, nod your head, or should we just wave at each other since you can’t see a polite smile from behind a mask? In short, it was a level of strangeness which comparatively could exist between bacterial cells and phages. Although this is rather tricky indeed, being that bacteria and viruses alike don’t have any feelings per say, but they do have parts which have to interact with one another in order to function.
So, what would it be like if a phage had hands to shake with its bacterial host? Obviously it doesn’t have hands, but it does have a protein which binds to a receptor site on the bacteria’s cell wall. Once it’s attached, it bridges the cell’s wall to inject its genetic payload. If the bacteria gets to know the phage it will no longer shake hands with it, or lean in for a hug and kiss.
What if a genetic circuit was designed and planted into the phage which would alter its handshake? What if that circuit was turned on and off through the action of attaching to bacteria or unloading its payload? The result then might be some form of an alternating handshake, a secret handshake, which would shift from generation to generation.
Group Project Goal:
Engineering a chaperone-independent efficient MS2 lysis protein
Project Rationale The efficacy of bacteriophage MS2 as an antibacterial agent is currently limited by the host’s ability to evolve resistance. Specifically, E. coli can mutate the molecular chaperone DnaJ (e.g., at position P330), disrupting the essential interaction required for the MS2 lysis (L) protein to fold and function [1.] This interaction is required for proper function of the lysis protein, as DnaJ binds to the N-terminal domain of MS2 lysis protein and alleviates its inhibitory effect on lytic activity.
We propose engineering a self-activating L protein by replacing its inhibitory, chaperone-dependent N-terminal region with a computationally designed, thermodynamically stable scaffold. As this original domain is dispensable for actual lysis but creates the DnaJ dependency [2], our redesign conceptually eliminates the need for the molecular “handshake” between host and phage, allowing MS2 to fold independently and bypass bacterial control mechanisms entirely.
Chamakura KR, Tran JS, Young R. MS2 lysis of Escherichia coli depends on host chaperone DnaJ. J Bacteriol. 2017;199(9):e00058-17. doi:10.1128/JB.00058-17.
Chamakura KR, Edwards GB, Young R. Mutational analysis of the MS2 lysis protein L. Microbiology (Reading). 2017;163(7):961–969. doi:10.1099/mic.0.000485.
Kirschning A. On the Evolutionary History of the Twenty Encoded Amino Acids. Chemistry. 2022 Oct 4;28(55):e202201419. doi: 10.1002/chem.202201419. Epub 2022 Jul 28. PMID: 35726786; PMCID: PMC9796705.
Huang, W., Wang, S., Wei, Y. et al. Design and evolution of artificial enzyme with in-situ biosynthesized non-canonical amino acid. Nat Commun 16, 8698 (2025). https://doi.org/10.1038/s41467-025-63733-3
Medeiros-Silva J, Dregni AJ, Hong M. Distinguishing Different Hydrogen-Bonded Helices in Proteins by Efficient 1H-Detected Three-Dimensional Solid-State NMR. Biochemistry. 2024 Jan 2;63(1):181-190. doi: 10.1021/acs.biochem.3c00589. Epub 2023 Dec 21. PMID: 38127783; PMCID: PMC10880114.
Greg Huber et al, Entropy and chirality in sphinx tilings, Physical Review Research (2024). DOI: 10.1103/PhysRevResearch.6.013227
The PROSITE database Sigrist CJA, Cuche BA, de Castro E, Coudert E, Redaschi N, Bridge A.
The PROSITE database for protein families, domains, and sites.
Nucleic Acids Res. 2026; doi: 10.1093/nar/gkaf1188 [In press]
PubMed:41263099 [Full text] [PDF version]
Week 5 HW: Protein Design Part II
Time to do your homework- It’s always time to do homework in this course!
Part A: SOD1 Binder Peptide Design
Part 1: Generate Binders with PepMLM Starting with the human SOD1 sequence from UniProt (P00441), and then introducing the A4V mutation, we get the following sequence where the fourth amino acid in the sequences is changed from K to V:
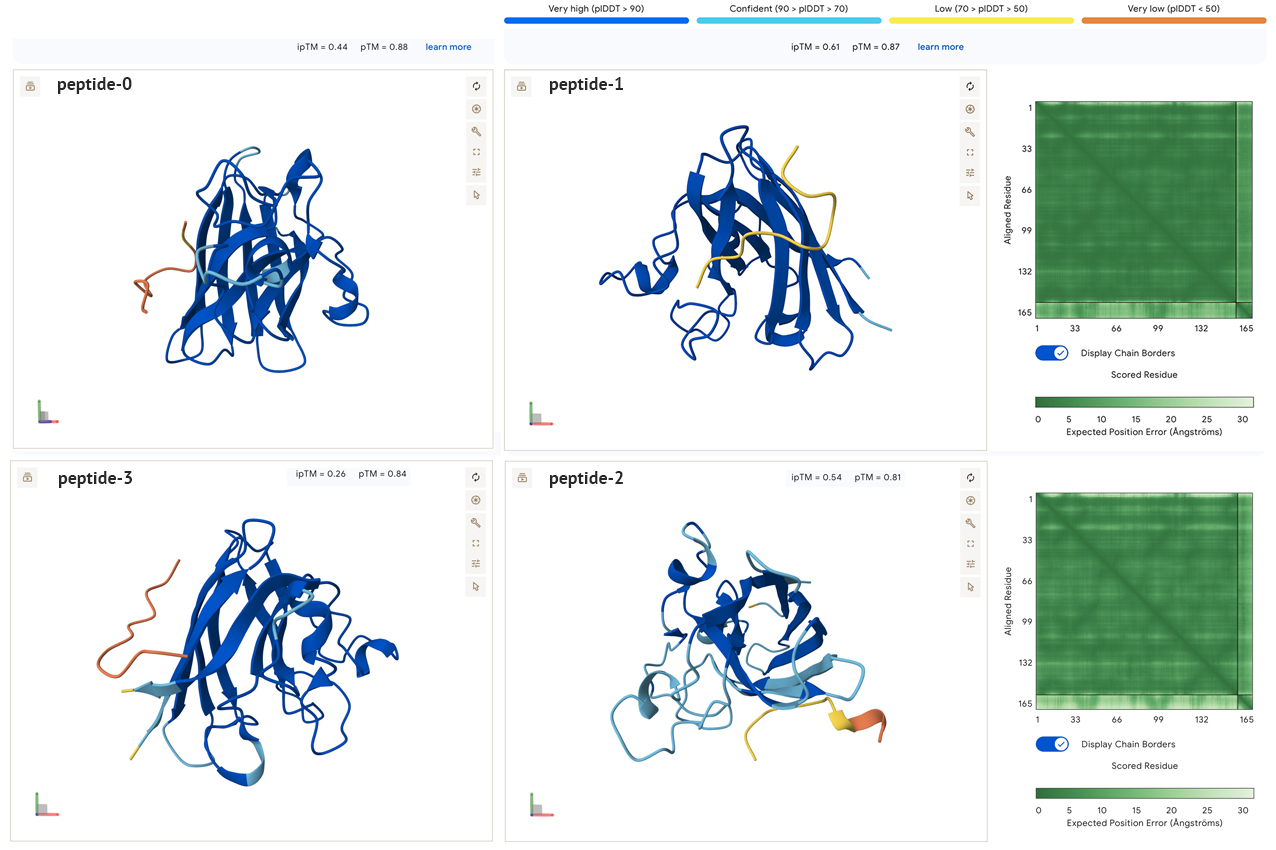
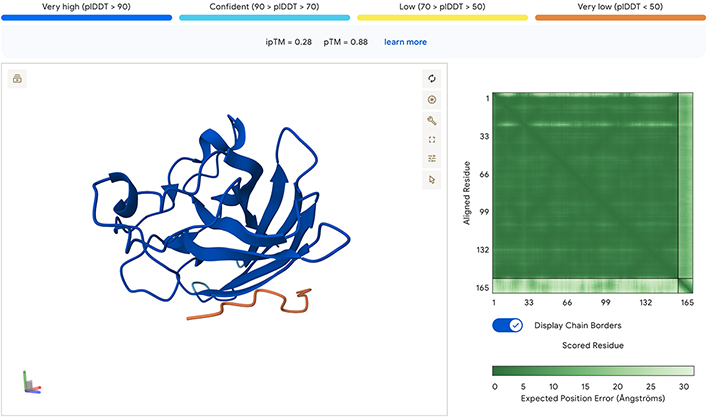
All the sidechain peptides generated had low to very low plDDT scores, and did not appear to be close to either ends of the SOD1. This seems to indicate to me that these peptides are not well suited for binding with SOD1. In addition, the low ipTM scores all seem to indicate low confidence of interaction between molecules. In the model below there are similar results when combining two SOD1 A4V molecules with the known binder. The pedicted local distance score for the known peptide is low, which is confusing, and makes me think it may be an incorrect reading by the AlphaFold model, especially if the peptide is a known binder.
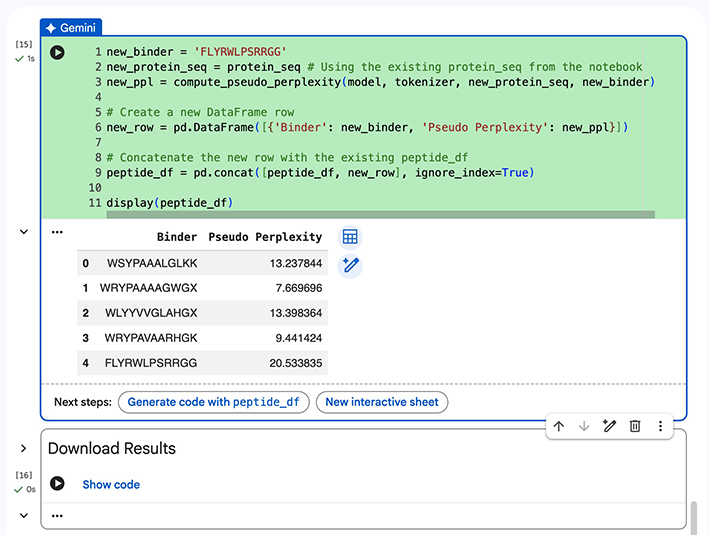
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Peptiverse graphic results table for Peptide 0
-Peptiverse Results-
Peptide 0 - Sequence: WSYPAAALGLKK. Realatively weak binding (5.1 pKd/pKi) with second to highest net charge, and the second lowest score for hydrophobicity. It’s non-hemolytic and soluble.
Peptide 1 - Sequence: WRYPAAAAGWGX. Realatively weak binding (6.2 pKd/pKi) with the third highest net charge, and the third lowest score for hydrophobicity. It’s non-hemolytic and soluble.
Peptide 2 - Sequence: WLYYVVGLAHX. Realatively weak binding (6.9 pKd/pKi) with the lowest net charge, and the highest score for hydrophobicity. It’s non-hemolytic and soluble.
Peptide 3 - Sequence: WRYPAVAARHGK. Realatively weak binding (5.5 pKd/pKi) with the highest net charge, and the lowest score for hydrophobicity. It’s non-hemolytic and soluble.
Based on the results of this group, I don’t think I would advance any of them for further testing. The second peptide generated (peptide 1), seems closest to working with the highest iPTm score, third highest charge and the lowest hydrophobicity. Although, it is difficult for me to interpret these data sets at this point, and if I were actually looking for a binder to test I would continue looking for a set which had better predictions in the visualization models, which ideally would also match the Peptiverse scores.
Part 4: Generate Optimized Peptides with moPPIt
Functional parameters: Hemolysis, Non-Fouling, Solubility, Half-Life, Affinity, Motif, Specificity. All set with parameters set with objective importance of 1. Motif Positions were: 1-10, 144-154 (first ten positions, last ten positions) Generated peptide sequences:
KKKKKDKTTKWM
EEVQKKQEWKTI
ELIRWLQQRRTD
The following results are for the first peptide generated, Sequence - KKKKKDKTTKWM:
Based on these results, I am no more confident in this binder generated by moPPit than the first sets generated by PepMLM. Again, not sure if this is a result of the modeling, or perhaps I’m not doing this exercise correctly. It seems odd that none of the binders have very high ipTM or pTM scores.
Part C: Final Project: MS2L-Protein Mutants
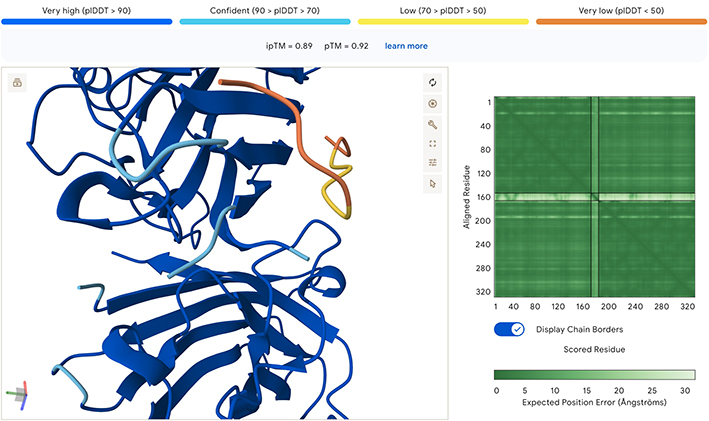
Option 1: Unfortunately, I had technical issues with the colab notebook and was only able to run the first few sections. First, I generated the heat map, and the top ten mutations. As the goal of the assignment is to adapt the MS2L protein to fold on it’s own without the DnaJ chaperone, I used this data with Alphafold to improve it’s confidence in the model. I relize this is not how the assignment was intended to be done, but it was the best way I could proceed to generate five, hopefully better, mutations of the MS2L protein.
Working with AlphaFold to Generate New Sequences
Come up with 5 mutations along with how you came up with them and why you believe they would work. 2 of the variants you submit must have mutations in the transmembrane region (38-60, Sequence: LYVLIFLAIFLSKFTNQLLLSLL) and 2 of them must be in the soluble region.
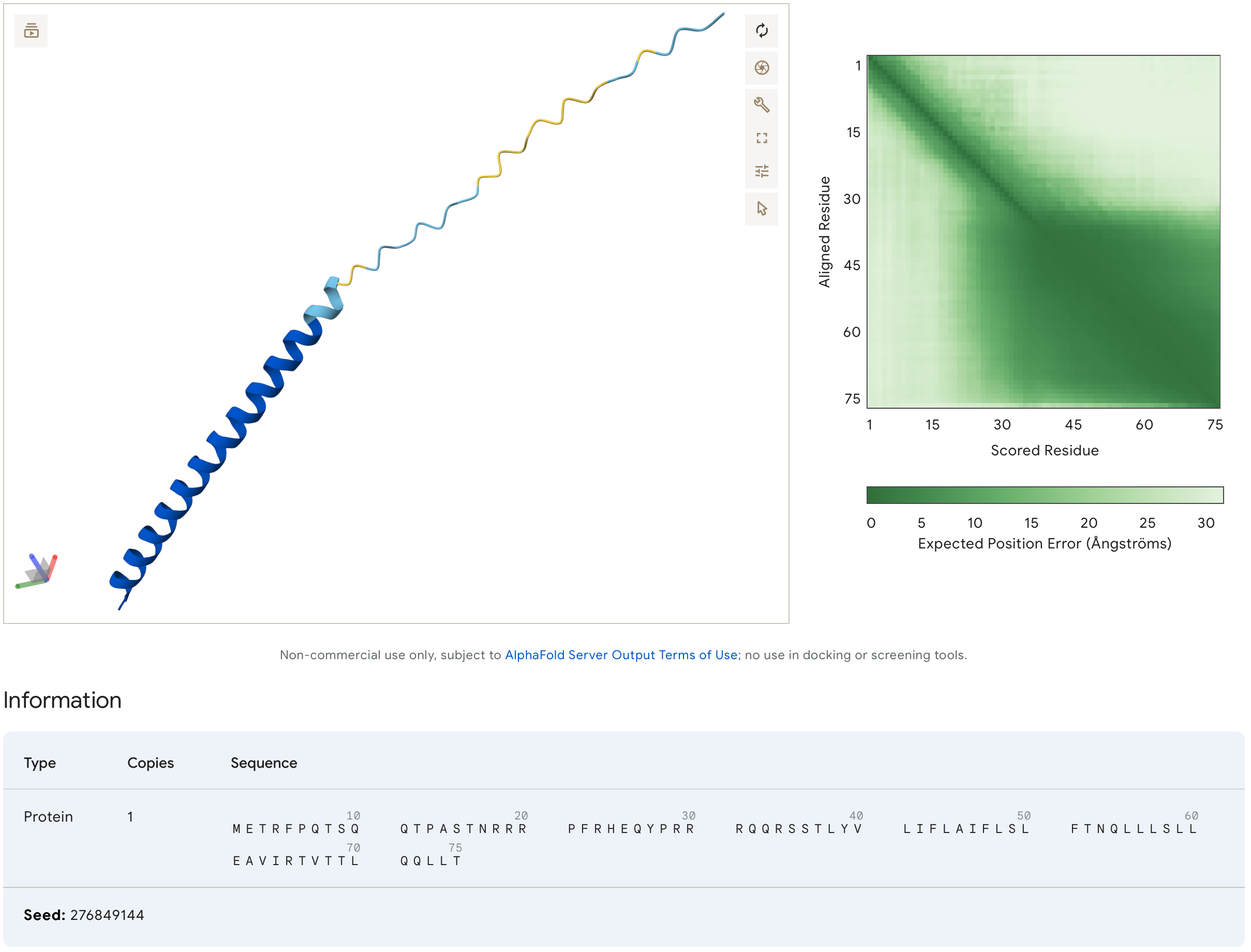
AlphaFold of Original (non-mutated) MS2L Sequence
MS2L Heatmap of Likely Mutations
MS2L Top Ten Most Likely Mutations
Position
Wild_Type_AA
Mutation_AA
LLR_Score
50
K
L
2.561468
29
C
R
2.395427
39
Y
L
2.241780
29
C
S
2.043150
9
S
Q
2.014325
29
C
Q
1.997049
29
C
P
1.971029
29
C
L
1.960646
50
K
I
1.928801
53
N
L
1.864932
Mutational Tests
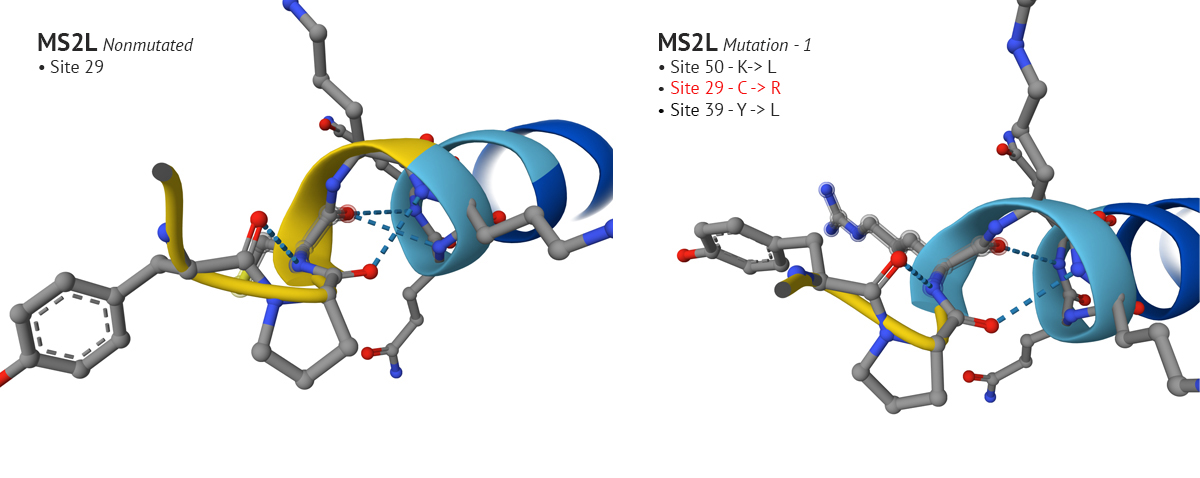
Test 1 and 2 -Sites 50, 29, 39, and 9 were mutated using the top ten mutational LLR scores from ESM notebook.
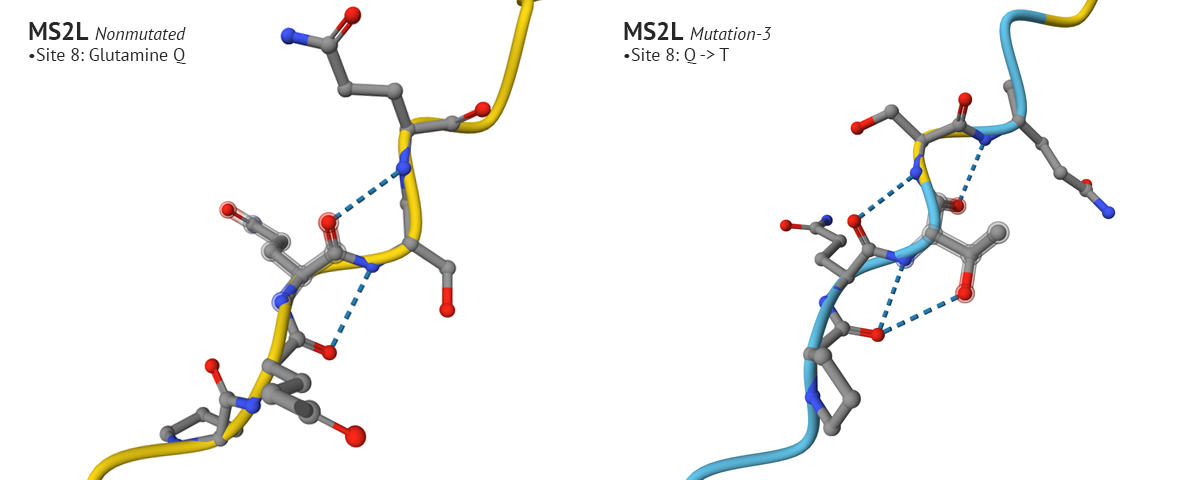
-The first set of mutations were sites 50, 29, and 39 which produced a confident plDDT at site 29 compared to the original non-mutated MS2L sequence. Interestingly, when I continued with the mutation and switched site 9 from S to Q, the plDDT score dropped at sight 29.
Above: Site 29 before S to Q mutation (lower-left), site 29 after S to Q (lower-right).
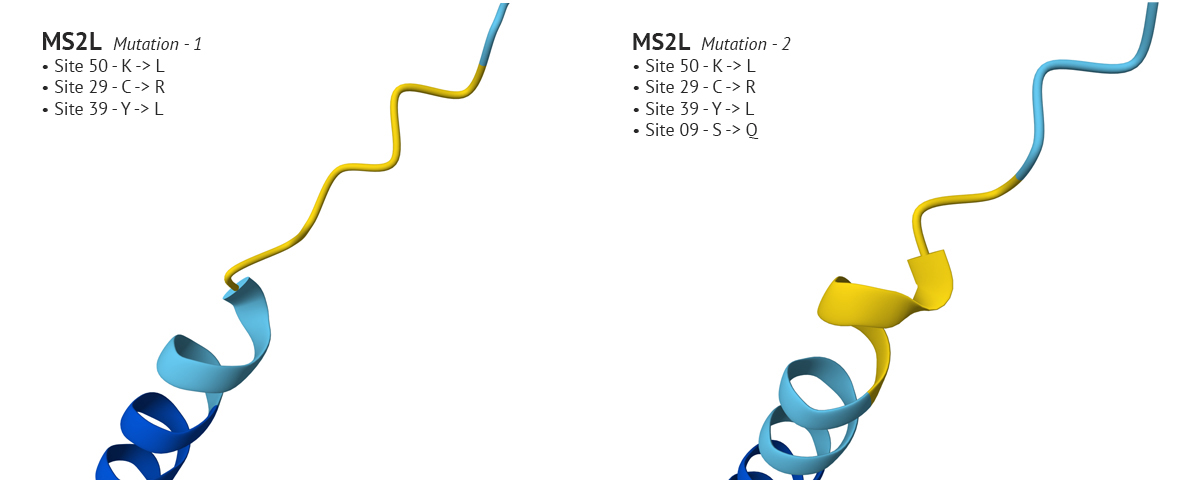
Test 3: The third mutation keeps the changes made to sites 50 and 29 from mutation 1, which yielded a higher plDDT score, and modified the aa at site 8 from Glutamine (Q) to Threonine (T). I chose Threonine because it has a neutral charge like Glutamine but is freely soluble where Glutamine’s solubility score is 2.6. This affected the AlphaFold’s model with a higher plDDT score for that position, and seemed to positively affect other positions in the aa 1-29 region.
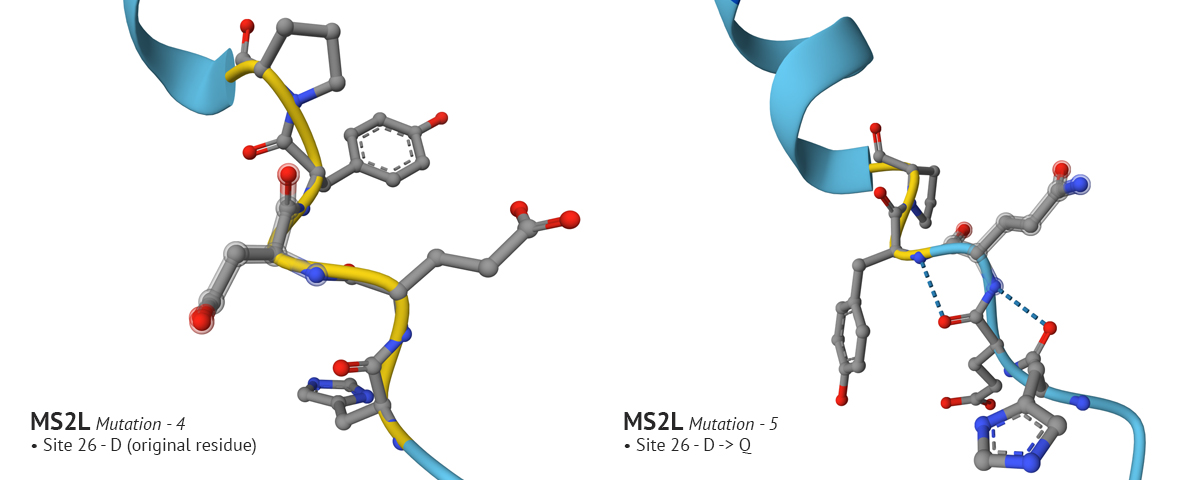
Test 4 and 5:
In mutation 4 site 8 changed from it’s original aa residue of Q to T, which had a positive affect on the plDDT score. In the fifth mutation I wanted to see if I could improve confidence around site 26. I went through several variations of changing the original residue, Aspartate (D), to G, then to E, and finally to Q which improved confidence. However, in my final analysis of the fifth variation, I realized that the improved confidence at site 8 in the fourth mutation changed back to a low confidence state. It’s an interesting problem which I don’t understand. It would make sense why changing residues which are in close proximity to one another would impact the region where they reside; but it’s odd to see how a change at site 26 would effect the confidence at site 8. Overall, by playing with MS2L in AlphaFold I was able to improve the model’s confidence in sites 1-29, but it still has a long way to go. And - I just now relized it would have been interesting to incrporate moPPIt into testing these mutational problems, instead of solely relying on AF. AHHH!
Conclusions
This assignemnt was particularily challenging because it required me to consider which of the protein design tools could I use to help approach the task of improving the folding characteristics of a protein, which would hopefully have a net positive affect on it’s functionality. Being a visual person, I found AlphaFold (AF) to be very helpful with it’s ability to model aa interaction. It’s certainly much faster than constructing a physical ball and stick model. However, I can see the value of those models too. In particular, it’s perplexing to see how AF interprets changes in design, where residues which are separated by several aa’s in sequence affect one another, and this is where (for me) a more traditional approach using physical models could help to work through compatability issues. I understand the importance of these insilico techniques, and I appreciate all the hard work behind their development!
Final mutational design. Changes to sequence- site 8: Q->T, site 23: K->R, site 26: D->Q, site 29: C->R, site 50: K->L.
Alphafold: Abramson, J et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature (2024).
Peptiverse: @article {Zhang2025.12.31.697180,
author = {Zhang, Yinuo and Tang, Sophia and Chen, Tong and Mahood, Elizabeth and Vincoff, Sophia and Chatterjee, Pranam},
title = {PeptiVerse: A Unified Platform for Therapeutic Peptide Property Prediction},
elocation-id = {2025.12.31.697180},
year = {2026},
doi = {10.64898/2025.12.31.697180},
publisher = {Cold Spring Harbor Laboratory},
URL = {https://www.biorxiv.org/content/early/2026/01/03/2025.12.31.697180},
eprint = {https://www.biorxiv.org/content/early/2026/01/03/2025.12.31.697180.full.pdf},
journal = {bioRxiv}
}
The UniProt Consortium
UniProt: the Universal Protein Knowledgebase in 2025
Nucleic Acids Res. 53:D609–D617 (2025)
Week 6 HW: Genetic Circuits Part I
Guess what. What? It’s time to do your homework. OMG!!! It’s never ending… I’m always doing my homework for HTGAA.
Icosohedronic Molecular Model II -RCBeck
Assignment: DNA Assembly
Answer these questions about the protocol in this week’s lab
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? Answer: There are several components in the master mix. It contains - DNA polymerase, dNTP’s, reaction buffers, and MgCL2.
DNA polymerase is an enzyme which catylizes the replication of DNA by reading the parent’s unzipped DNA aa sequence and pairing it
with its corresponding dNTP to synthesize a new strand of target DNA.
dNTP is short for Deoxyribonucleotide Triphosphate. They are the building blocks which allow DNA replication to take place. They consist of a deoxyribose sugar molecule, a nitogenous base (A,C,T,G), and a triphosphate group.
There are four dNTP’s - dATP, dCTP, dTTG, and dGTP.
The reaction buffers dissolve, or lyse the phospholipid cell membranes which hold the DNA inside the cell.
The MgCl2 is magnesium salt which neutralizes the charge on the sugar-phosphate backbone making the DNA less water soluble aiding in its precicipitation. It aslo helps remove proteins from DNA and keeps them dissolved in the lysed cell solution.
What are some factors that determine primer annealing temperature during PCR?
Answer: There are three tempererature cycles protocals important to PCR, denaturization, annealing, and extension.
The melting temperature (Tm) for primers is an important factor which initializes the PCR process, this is called denaturing. The increase in temperature is what separates the primers so they become single stranded.
The annealing temperature (Ta) is dependent upon the length, sequence and concentration of primers.
The extension process temperature is lower than first two and the last step where the 3’ ends are bound into finished PCR product.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR stands for Polymerase Chain Reaction and is widely used for its ability to amplify specific segments of DNA for study or testing. As stated above in question 2, it’s a thermocycling process using specific sets or a custom blend of primers, short single-stranded segments of DNA usually 18-25 nucleotides long, which bind to the target DNA strand in specific regions based on the primer sequence used. It is a relatively cheap and practical way to analyze DNA which requires a very basic “kitchen sink” lab to run protocal. It only requires small trace samples of DNA, which can be sourced from basically any tissue, and usually produces blunt end segments of DNA.
Restriction enzymes are proteins found in bacteria which can be used to cut DNA at specific target sites. They cut either blunt, or sticky ends - an advantageous quality for constructing recombinant DNA strands; there are three types.
Type I: recognize shorter sequences and do not cut at their recognition sites but rather at the unprotected ends.
Type II: Most widely used and available in many different forms. They cut at specific recognition sites to a predictable sequence.
Type III: Recognize short asymmetric DNA sequences and cut them nonspecfically into 25–28 nucleotide long sequences.
The main difference between these two methods is in the way they reform DNA segments. PCR seperates the strands and reforms them using DNA polymerase dNTP’s and designed primers, and is very good at generating millions of copies for testing. Restiction enzymes cut double stranded segments of DNA with either sticky or blunt ends. Sticky ends are ideal when assembling recombinant strands.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Answer: Gibson cloning works best when the PCR amplified fragments have overlapping ends which are 20-40 base pairs long with a high GC content. This can be achieved by optimizing the PCR conditions to ensure they are clean and specific, then checking the PCR product with gel electrophoresis before proceeding.
How does the plasmid DNA enter the E. coli cells during transformation? Answer: Heat shock and thermal cycling of transformed competent cells, or optimized reagents.
Describe another assembly method in detail (such as Golden Gate Assembly). Explain the method in 5 - 7 sentences plus diagrams. Model this assembly method with Benchling or Asimov Kernel!
Golden Gate Assembly is similar to Gibson in that it’s also a single step reaction however, it’s better for repetitive cloning tasks, whereas Gibson assembly is more appropriate for building longer sequences. Golden Gate is a restriction enzyme process using Type IIP enzymes which cut outside the recognition sites and create 3 or 4 nucleotide 5′-overhangs. The most common restriction enzymes are BsaI-HFv2, BsmBI-v2, and PaqCI, which recognize asymmetric non-palindromic sites which reduces the chances of creating oligimers. One of the current limitations of Golden Gate is the abvailabilty of Type IIS enzymes, there are only about a half dozen to choose from, most with base recognition sites.
Illustration by Tasha José (2024)
Model of Golden Gate in Benchling:
First I started by researching the restriction enzymes used. Then I downloaded a FASTA sequence from the Aanbaena genome and looked for these sites in Benchling. This lead me to conclude that approaching this manually was going to be very tricky.
During last homework session, Xavier suggested I try and use a backbone modeled for Golden Gate.
Below is the backbone sourced from Addgene: pLM433 (Empty Backbone) for creating CyanoTag vectors by Golden Gate cloning (BspQI version). I was able to locate the BsaX1 restriction cut site, but I’m not sure how to relocate the promoter to the cut site. I’m not sure if I should use the reverse DNA sequence as the promoter appears to going in 3’ to 5’ direction, and when I tried to paste it in front of cut site the sequnce became unselected. Very frustrating to say the least.
I chose this particular backbone because it’s modeled for cyanobacteria, which falls within the focus of my final project.
Benchling is not an intuitive application. I had trouble with it in the week two homework, and I’m not sure I will be able to successfully use it without some specific instruction- YouTube doesn’t really cut it, pardon the pun.
Tag / Fusion Protein
mNeonGreen - 3x FLAG (C terminal on insert)
Growth in Bacteria
Bacterial Resistance(s) Ampicillin, 100 μg/mL
Growth Temperature 37°C
Growth Strain(s) ccdB Survival
Citation:
Lorenz TC. Polymerase chain reaction: basic protocol plus troubleshooting and optimization strategies. J Vis Exp. 2012 May 22;(63):e3998. doi: 10.3791/3998. PMID: 22664923; PMCID: PMC4846334.
pLM433 was a gift from Luke Mackinder (Addgene plasmid # 217293 ; http://n2t.net/addgene:217293 ; RRID:Addgene_217293)
Week 7 HW: Gentetic Circuits Part II: Neuromorphic Circuits
After this assignment, it will finally not be time to do your homework for one day… Happy Spring Break! This is Your Brain on HTGAA- RCBeck
Assignment Part 1: Intracellular Artificial Neural Networks
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
They can be weighted with high and low input values which gives them unique capabilities.
Unlike a Bool - which is if this then that - the specificity of an IANN means they can be designed to produce more specific outcomes in cellular function by responding to multiple control parameters. Boolean functions have one input.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
In an application working with cyanobacteria, I think it would be interesting to use an IANN to sense the enviromnetal conditions with designed responses to stimuli.
For instance, in a freshwater ecosystem, the bicarbonate cycle of the water body can produce radical swings in pH depending on the water’s carbon and oxygen content. During photosynthesis plants absorb carbon from water to produce glucose and oxygen, which raises water pH from an increase of hydroxide. Without sunlight for photosynthesis, plants burn glucose producing CO2 which lowers pH. In a healthy ecosystem this realtionship is in balance.
In systems where there is excessive cyanobacteria growth from high nutrient levels (pollution), water pH can swing from >8 during peak photosynthesis to <6 during the night. Oxygen levels are affected in a similar way where there is high dissolved O2 during day and very low at night, which can be lethal to fish populations.
I’m realizing that releasing a synthetic cyanobacteria with IANN has some significant ethical issues, but perhaps it could be useful in a biofuel application where the goal is to produce dense populations of bacteria in closed loop systems. In this case, bacteria which could stop photosynthesizing in response to increasing pH and oxygen levels could be very useful in production systems, which in theory would have no need for artificial buffering.
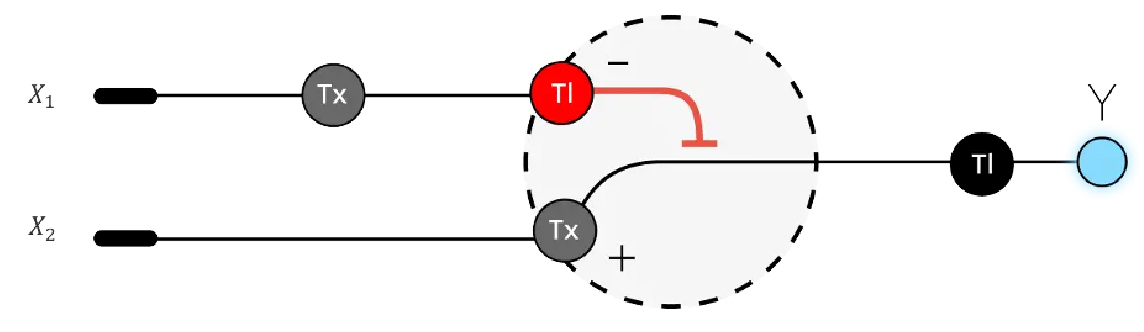
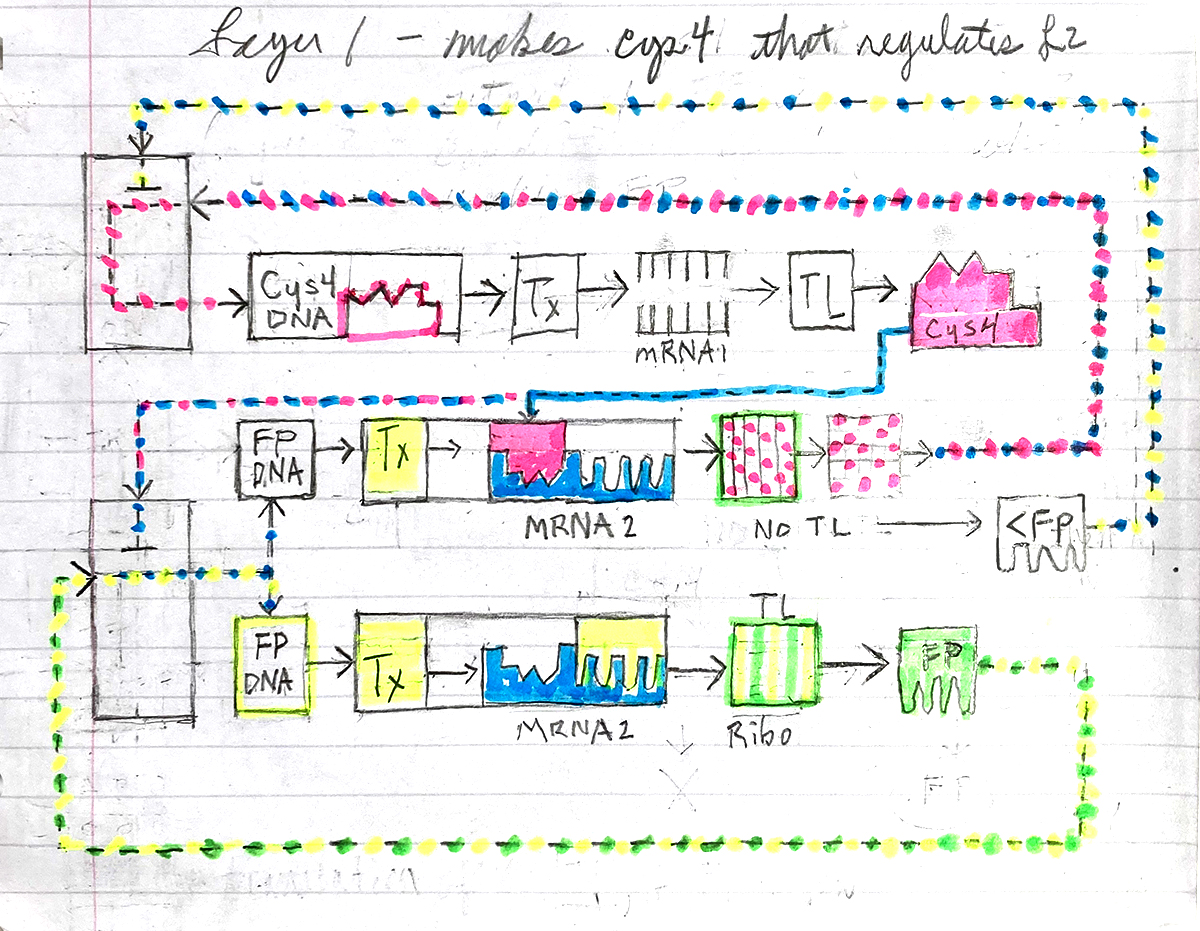
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Attempted Design
Logic failed me on this part of the assignment. The visual compartmentalization of it helped to describe the relationship between enzymes, which block the production of others, but I failed to design a proper theoretical logic circuit.
The idea of what constitutes an input in terms of X1 and X2 and creating a functional loop with them is not clear. An electrical circuit is a complete loop of electron flow. There are no loose ends, and trying to conceptualize the inside of a cell as a circuit loop is very confusing.
An anology which seems fitting is how an electrical appliance works. It doesn’t work unless its plugged in, turned on, and receives input from an external variable/user.
In the drawing above, FP stands for Fluorescent Protein which is regulated by the Cys4 enzyme.
The second or middle line represents the instance where Cys4 binds to a post transcription (Tx) RNA site preventing translation (TL) of mRNA into FP. The Cys4 is released from the site, breaks up, and returns to the cell unorganised for reassembly by DNA.
The bottom line represents the normal processing of FP.
I attempted to produce a weighted function where low FP (<FP) would interupt Tx of Cys4 mRNA thereby allowing normal processing of FP. If there is too much Cys4, limiting FP production, normal FP production continues.
In retrospect, this represents Cys4 regulation by FP, not FP regulated by Cys4, which was assigned task. And it’s not a proper logic circuit design in my opinion.
Assignment Part 2: Fungal Materials
Golden Top (Psilocybe cubensis) -RCBeck
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Aside of making tasty treats and good times, they can be used in many different material applications. They can make bricks from organic material substrates by using it as a casting medium, and probably mold making as well.
Apparently they can even function as a refractory material under thermal extremes, since it can be used as insulation.
In the biological sense, they break down and process minerals, act as nutrient delivery systems, and convey information between plants, soil and bacteria.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
I can imagine using mycelium to mine and transport specific minerals, or to collect and extract pollution.
Fungi has been prolifically studied since Alexander Fleming’s discovery of Penicillin 1928. This has lead to the development of a wide range of synthetically produced “natural products” or NPs. From this perspective, the groundwork for research with fungi is in place. These compounds are generally produced using biosythetic pathways, which are closely related to one another; sometimes refered to as biosynthetic gene clusters (BGCs).
Research within the fungal domain of BGCs leads researchers to believe the number of unknown compounds which have yet to be synthesised is staggering in number. This is promising for the development of many discoveries in research which could produce treatments, medications, material technologies and industrial applications.
Citation:
Valiante V. Advances in Synthetic Biology of Fungi and Contributions to the Discovery of New Molecules. Chembiochem. 2023 Jun 1;24(11):e202300008. doi: 10.1002/cbic.202300008. Epub 2023 May 4. PMID: 36862368. Available at: https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/cbic.202300008 (Accessed: 3-29-26).
Huang M, Hull CM. Sporulation: how to survive on planet Earth (and beyond). Curr Genet. 2017 Oct;63(5):831-838. doi: 10.1007/s00294-017-0694-7. Epub 2017 Apr 18. PMID: 28421279; PMCID: PMC5647196.
Week 9: Cell Free Systems
It’s that time again… RrrrrrraaAAHH!!!
Glueless Icosahedron - RCBeck
General and Lecturer-Specific Questions
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
CFPS have many advantages over traditional in vivo processes. Since there’s not a cell to keep alive, it doesn’t have the constraints of live culture, such as time, cell stabiltiy, cytotoxicity etc.
It’s also an on demand process, which makes accessing the materials for reactions easier, and it’s scalable from low output to industrial manufacturing.
Describe the main components of a cell-free expression system and explain the role of each component.
DNA Polymerase: Enzymes which catylize nucleoside molecules in DNA replication.
RNA Polymerase: Enzyme for replicating RNA.
Ribosome: Assembles protein from mRNA coding.
Plasmid DNA: Contains instructions for specific/desired protein construction.
Amino Acids, buffers, cofactors, salts: Provide building blocks for assembly of desired product. [1]
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy is requred to drive chemical reactions both in and outside of cells. ATP is the main source of energy for cellular reactions and it’s synthesis is critical to functionality.
In cell free systems, there are pathways which function similar to cellular ATP production, but are more affordable to produce.
Based on quick research, d-fructose to d-erythrose and AcP in a PKT-catalyzed reaction [2]seems to be a promising solution for producing ATP in a stable, cost affective system.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
The simplicity of prokaryotic cells streamlines the process of CFPS for more efficient output versus complex eukaryotic cells, which are more complex and require more attention to process design.
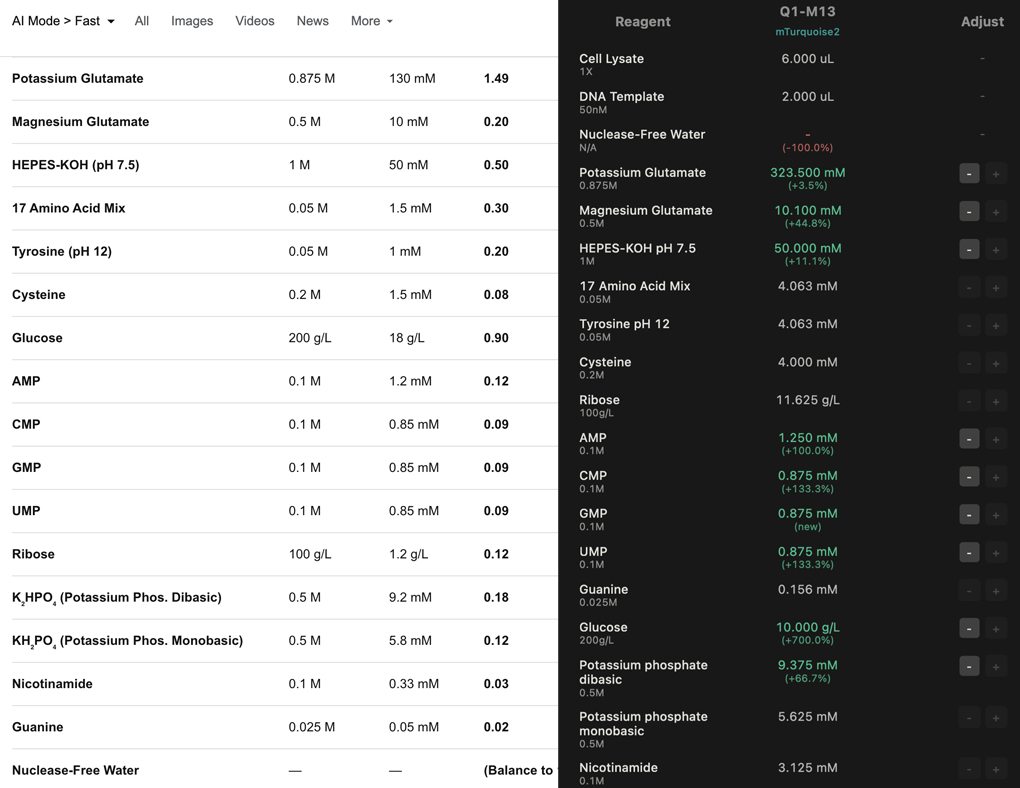
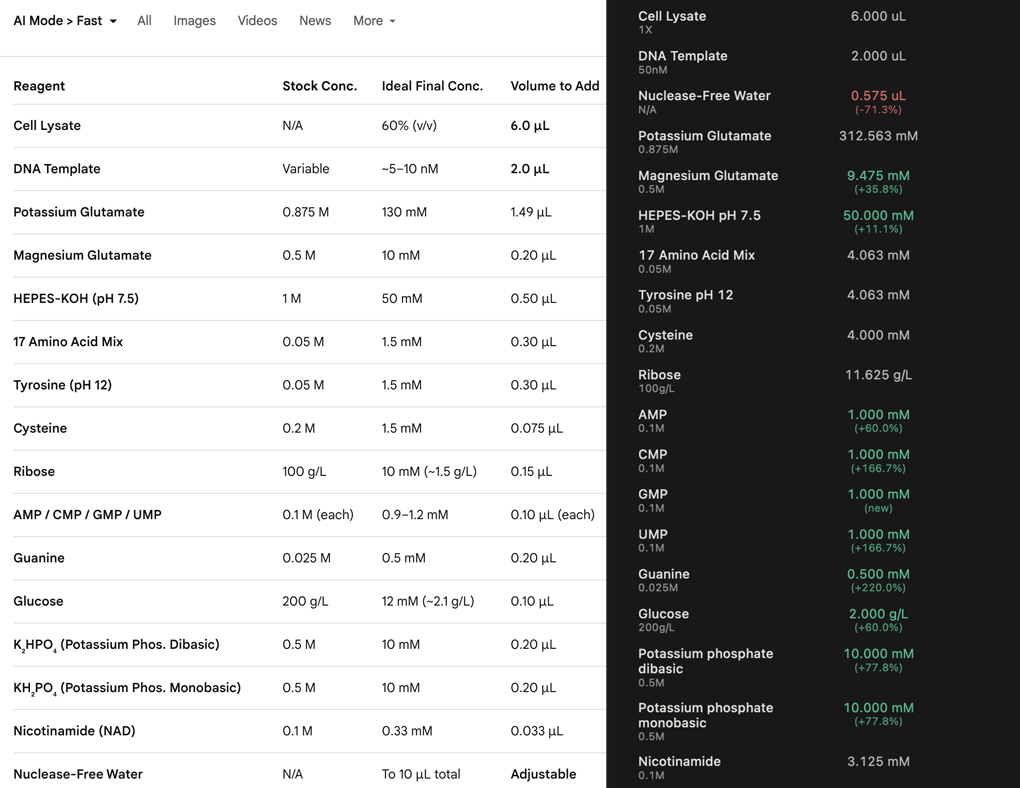
Prokaryotic could be any number proteins, something I would want to produce a lot of. My final project proposal is based on GFP, which would benefit from CFPS for its on demand production and streamlined process.
Eukaryotic proteins are certainly just as important to the future development of the research, and if I were to move away from bacteia towards plants the Bryophytes would be an interesting area of research. I would like to focus on the resurrection plants [3], and perhaps draw a correlation between protein producion and the characteristics which give this particular group their ability to survive desiccation.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
This is an interesting question being that cell free systems are inherently membrane free. The first thing to consider are the building blocks of a membrane system which will determine the type/indgredients of the CFPS. What are the chemical characteristics of the membrane and what type of environment would be most hospitable for production? pH, temp, charge?
Challenges would be producing functional membranes which incorporate pore systems and all the dynamics of a working membrane. This is dependent upon the function/type of cell the membrane is encapsulating and the what the cell needs to exchange for function.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Problems could include insufficient material for desired output, system contamination, or lack of control over process variables.
The first thing to check for would be any signs of contamination, check the state of reagents used, and trace back executed steps with protocal. Was anything missed, out of date, or compromised?
If so, then how or what needs to be changed in the setup?
Triple check new set up before proceeding and streamline the process to avoid confusion which could lead to error.
If available, perhaps building an Opentron protocal would help streamline.
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
Pick a function and describe it.
A cell which would group/bind together with other similar/same cell types to form a collective would be an interesting place to start as it could potentially be used to build larger more complex structures based on a known architecture.
What would your synthetic cell do? What is the input and what is the output?
This is a color shifting cell, which can assume a high or low level of color intensity depending on environmental feedback, such as temperature change.
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
If one of it’s characteristics is to join together and create more complex structures then it seems like encapsulation would be necessary.
It’s color shifting response could be achieved without encapsulation but organised structure not.
Could this function be realized by genetically modified natural cell?
Possibly, I did read a paper recently which described how Orange Carotenoid Proteins can shift their color from orange to red which helps with heat dissipation. But the structural aspects of design may be limited by cell type and their functional relationship with OCP.
Also, OCP is only one example of a color shifting protein, so if the goal was to produce a more diverse range of color shifting protein then maybe not.
Describe the desired outcome of your synthetic cell operation.
With exposure to environmental stress such as heat or intense radiation, the cell would change color as desiccation progressed giving a visual indicator of drought and its intensity.
Design all components that would need to be part of your synthetic cell.
What would be the membrane made of?
Most cell membranes seem to be made of a glycerophospholipids which are hydrphylic on both the inside and outside of cell with a hydrophbic interior. If the cell is supposed to be capable of rehydrating after desication then the membrane would need to be very elastic to allow for expansion and contraction during this process. I would imagine that’s a function of what type of proteins are embedded into the phospholipid bilayer.
What would you encapsulate inside? Enzymes, small molecules.
First it needs to have genetic information for replication, and since it’s elastic there should be some kind of osmotic pressure sensor/regulator which will help it manage water and other molecules pass through its membrane.
A nucleous containing genetic information which is protected by the outer membrane which expands and contracts with changing water conditions.
It will also need some ribisomes for protein assembly, which will help with its color changing ability also linked to an osmotic pressure switch.
Enzymes for activating color regulation relative to photosynthesis, and osmotic pressure.
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
The elasticity of the outer membrane would probably benefit from an integral membrane protein system where channels are fixed into the flexible membrane which holds them, allowing transport of water, minerals, and materials for cellular function.
Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
How will you measure the function of your system?
In silico models would be the best place to start.
Once a working theoretical model is established, then individual systems could be tested for isolated functionality.
Building on functional test data, more complex system to system interactions wwould be next.
Combine systems in a stacking order of functionality when building actual cell starting with most basic functions first.
Complete cell tested for multifunctionality with controlled input and measured output.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Healthy Air- it’s in your control with the AQ+ test strip, real time indoor air quiltiy data.
Proposal: Indoor air quility conditions are a moving target for both new structures as well as older ones. The increased dependance on HVAC to maintain conditions is growing in demand, and while these mechanical systems do help, they have functional limitations. Air filtration systems deal with the mechanical filtration of particles, but they are often ineffective due to lack of maintenance, which can lead to poor indoor air quality and affect resparatory health. This could easily be improved with the addition of an affordable paper based, or other biodegradable substrate, sensor, which would indicate air quality parameters. These sensors could offer a variety of conditions to monitor, as well as a built in exporation indicator for the test. Based on specific needs/environment, the sensor could be incorporated into an existing AC filter or placed on the exterior of air supply intakes. They could aslo help HVAC technicians test ducting and harware for contaminants in systems. Access to this technolgy would give people knowledge of their environment, and the ability to take appropriate action before their health or business is impacted.
Homework question from Ally Huang
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
The ability to grow food in space is essential for human health on long term missions. Further, the recyclability of material is paramount, which includes the food/digestion relationship between plants and humans. There needs to be a clear and functional system developed for food production and recycling in closed loop environments such as space.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
Veggie depends on fertilizers for plants. Nitrifying bacteria, Nitrosospira and Nitrosomonas are key to recycling nutrients in space. This project explores the complete ammonia oxidizing bacteria (Comammox), Nitrospira inopinataammonia, for producing fertilizer in space.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
Designing a closed loop biological system is the key to growing food in space. A critical part of this is the nitrogen cycle. This experiment will test Comammox enzymes which should convert ammonia into a bioavailable form of nitrogen for plant growth.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
How will food be grown in space for long term missions? By working with cell free systems, enzymes produced by nitrifying bacteria can convert toxic ammonia into food for plants. The focus of this project isolates bacterial genes associated with enzyme production to produce fertilizer needed for plant cultivation. Comammox bacteria has been found to efficiently convert ammonia into nitrite/nitrate through its amoA gene sequence. Using restriction enzymes, gel electrophoresis, and PCR, this project will attempt to produce the amoA enzyme responsible for nitrification. Successful synthesis of this enzyme in space could play a critical role in establishing closed loop biological systems for space travel.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
Part 1: Hydrate Complete ammonia oxidizing bacteria (Comammox), Nitrospira inopinata. Using preprepared restriction enzymes, isolate target gene sequences amoA, Ca. N. Inopinata. Select sequence using gel electrophoresis.
Part 2: PCR amplify amoA, combine with reagents, produce protein. Microdose samples with three different dilutions of ammonia - H2O, test for nitrite/nitrate ppm.
Part 3: Based on results from part 2, optimize cell free reaction protocol, produce nitrate/nutrient mix for Veggie plant pillow.
Part 4: Inoculate plant pillow with mix, compare plant growth against veggie system.
Citation:
Gregorio NE, Levine MZ, Oza JP. A User’s Guide to Cell-Free Protein Synthesis. Methods Protoc. 2019 Mar 12;2(1):24. doi: 10.3390/mps2010024. PMID: 31164605; PMCID: PMC6481089.
Cell-Free Reaction System for ATP Regeneration from d-Fructose
Franziska Kraußer, Kenny Rabe, Christopher M. Topham, Julian Voland, Laura Lilienthal, Jan-Ole Kundoch, Daniel Ohde, Andreas Liese, and Thomas Walther
ACS Synthetic Biology 2025 14 (4), 1250-1263
DOI: 10.1021/acssynbio.4c00877
Commisso M, Guarino F, Marchi L, Muto A, Piro A, Degola F. Bryo-Activities: A Review on How Bryophytes Are Contributing to the Arsenal of Natural Bioactive Compounds against Fungi. Plants (Basel). 2021 Jan 21;10(2):203. doi: 10.3390/plants10020203. PMID: 33494524; PMCID: PMC7911284.
Gechev T, Lyall R, Petrov V, Bartels D. Systems biology of resurrection plants. Cell Mol Life Sci. 2021 Oct;78(19-20):6365-6394. doi: 10.1007/s00018-021-03913-8. Epub 2021 Aug 14. PMID: 34390381; PMCID: PMC8558194.
Clark, I.M., Hughes, D.J., Fu, Q. et al. Metagenomic approaches reveal differences in genetic diversity and relative abundance of nitrifying bacteria and archaea in contrasting soils. Sci Rep 11, 15905 (2021). https://doi.org/10.1038/s41598-021-95100-9
Hayatsu, M., Katsuyama, C., & Tago, K. (2021). Overview of recent researches on nitrifying microorganisms in soil. Soil Science and Plant Nutrition, 67(6), 619–632. https://doi.org/10.1080/00380768.2021.1981119
Week 10: Advanced Imaging and Measurment Technology
What happened to my brain? The Only Question -RCBeck
Final Project Homework
For your final project:
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
Aim One: Initial Transformation
The first aim of this project transforms a strain of Synechococcus elongatus 7942 (PCC7942) into the sucrose secreting, model organism S. elongatus cscB+. The original PCC7942 colony will need to be grown to an optical density (OD) 750 of ≥1 before transformation may take place. A spectrophotometer will be used to obtain this measurement. - First growth media needs to be prepared, which will require measuring agar, BG11 medium, AS Trace Elements, water, pH, and temperature. Plates are poured, streaked and incubated. Colonies are then selected and transfereed to new grow media. - Colonies are incubated until an OD of 750 is reached, then natural transformation may take place.
- Following procedure, we will use GeneArt® Synechococcus Engineering Kit 14259 to preform natural transformation of S. 7942 into s7942 cscB+. - Transformed samples are then recultured with spectinomycin treated plates, selecting for transformed colonies. - Gel electrophoresis will confirm transformation.
Sub-Aim: Optimize growth conditions: Growth of transformed S.7942 cscB+ to be tested with NaCL, KCl, and salt free conditions. Essay will allow selection of optimal lighting for S.7942 cscB+ and influence of salts (Fig 1.). Figure 1. Comparitive analysis of s. 7942 growth under NaCl, KCl, fluorescent and LED light respectively.
Aim Two: Co-culture Optimization
Setting up the co-culture between S. 7942 cscB+ with chromo-protein E. coli. This phase will test model organism S. 7942 cscB+ with diffent starting concentrations of E. coli chromo-protein on LB agar plates prepared with BG-11 media as control. - Positive control plate A will measure colony growth under control IPTG with NaCl induced stress. - Positive control plate B will measure colony growth under control IPTG with KCl induced stress. Figure 2. Comparitive analysis of S. 7942 under co-culture with E. coli chromo-protein.
Aim Three: Reactor Optimization
Testing the co-culture system with photo-negative plates. 14 day trial culture. - Adjusting/modifying bio-reactor setup to optimize reactions. We will be measuring DO, pH, light intensity (EV), temperature, salt, nitrate, and chelated iron concentrations. - Nutrient essays will confirm optimal conditions relative to visual inspections of co-culture under salt stress, and IPTG.
Successful co-cultures will then undergo a stasis study where pH, temperature, DO, light, and nutrient concentrations are tested without control of negative imaging plate. - Goal is to optimize life support conditions for long term display of cultures with minimal image distortion.
Waters Part I — Molecular Weight
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight?
Results based on calculator for average and monoisotopic:
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1):
To calcultate MW using master charge states (left)= 848.9758, and (right)= 875.4421 we calculate for n:
n = (848.9758-1)/(875.4421-848.9758)
n = 847.9758/26.4663
n = 32.0398
MW = (32.0398 x 875.4421 - 32.0398)
MW = 28,016.9499
Comparatively to the calculator’s weight, there’s 1,092.46 difference between my calculation and the average MW value of 26924.48.
Accuracy = (MWexp - MWtheory)/MWtheory
Accuracy = (28,016.9499 - 26,924.48)/26,924.48
Accuracy = .0405
Accuracy = .0405 x 1000000
Accuracy = 40500 Definately something wrong here.
Calculated again using different peaks and ended up with 36,972… obviously, there’s a problem with the way I’m calculating this.
I adjsuted theoretical weight to include His tag and linker which changed score to 344, which is better, but still way outside target range.
Waters Part III — Peptide Mapping - primary structure
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above.
How many peptides will be generated from tryptic digestion of eGFP?
There are nineteen masses generated from the tryptic digestion.
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes?
In the graph, the base peak has is equal to 24 units on the y scale. 10% of 24 is 2.4.
There are nineteen peaks over 10%, or 2.4 on the y scale in between .5 and 6 minutes.
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above?
Yes! There are 19.
Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ([M+H]+) based on its m/z and z.
We use values of the peakes in fig 5b. which are: 526.25918 (right peak) and 525.76712 (left peak) to calcultate charge state:
526.25918 - 525.76712 = .49206
1/.49206 = 2.0322
Charge state = 2
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement?
Based on the charge state comparison, the single charge state is 1050.
Therefore this should be the peptide: FEGDTLVNR, with mass of 1050.5214, position 115-123
Mass accuracy = [(1050.53438 - 1050.5214)/1050.5214] x 1,000,000
Mass accuracy = 2.83 ppm
What is the percentage of the sequence that is confirmed by peptide mapping?
According to Expasy Mass Peptide results, we covered 90.7%.
Waters Part IV — Oligomers
Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
Waters Part V — Did I make GFP?
Theoretical molecular weight: 27,875.41 (without start codon, average MW)
Observed/measured on the intact LC-MS: 27,919.9307
PPM mass error:.00159713 x 1000000 = 1597.1316 ppm
It does not appear to be eGFP
Week 11: Bioproduction and Cloud Labs
OMG! Last One!
Drawing Air (2026) Detail, Graphite on Paper -RCBeck
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
My contribution to the artwork was limited to one pixel, which was initially made to test interaction with the interface. I noticed how pixel contribution was rather competitive, where scripting was automatically writing over contributions. Unfortunately, this discouraged me from contributing any further, which lead me to focus on my final project alternatively.
For the optimization part of the assignment I decided to use AI since my overall understanding of CFPS is still fairly limited. This decision lead me to reserve my interaction with the optimization protocal. Additionaly, I switched gears and chose to work with AI for the sake of time as well as the complexity of the 16 or so variables. Through my interaction with the AI I noticed how certain variables within the protocol were more or less fixed, as certain starting concentrations were more or less set targets.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
E. coli - BL21 (DE3) Star Lysate: The cell lysate approach is advantageous as it releases cellular contents directly into bioreactor where product of interest may be synthesized. The reactions can be optimized with addition of protease to eliminate specific pathways by breaking proteins down into peptides or even smaller amino acids.
Salts:
Potassium Glutamate- Key component in E. coli cytoplasm important to protein folding. In CFPS it’s important as a stablizing agent which promotes efficient protein production.
HEPES-KOH pH 7.5- The primary buffering agent used to keep pH stable in CFPS, which is important to maintaining optimal conditions for protein production.
Magnesium Glutamate- Improtant cofactor in enzyme activity and energy systems, such as ATP, required for reactions to take place.
Potassium phosphate monobasic- Important buffering agent neede to maintain pH as well as phosphate nutrient supply supporting biochemical reactions in protein production.
Potassium phosphate dibasic- Another important buffering agent, which in contrast to potassium phosphate monobasic, provides potassium and phosphate ions required for enzymatic activity and metabolic functions.
Energy / Nucleotide System:
Ribose- Essential component of RNA backbone which translates DNA to protein. It’s also a component of ATP, a neccessary reagent which provides energy in protein synthesis.
Glucose- Required for the synthesis of ATP, which as previously stated, a neccessary energy source to drive chemical reactions in protein construction. Glucose also helps sustain reactions in CFPS.
AMP, CMP, GMP, UMP- are nucleotides important to CFPS during RNA transcription.
Guanine- One of the main nucleotide bases required for production of GTP, which provides energy in the translation process.
Translation Mix (Amino Acids)
17 Amino Acid Mix- Amino acids are the building blocks of protein; without them the translation process will not have product to build proteins.
Tyrosine- Amino acid with important post transcriptional properties which are an integral part of protein desgin.
Cysteine- Key amino acid involved in disulfied bonds required for proper protein formation/folding. It’s also important for proper cell function as many cellular proteins require Cysteine for their construction.
Additive:
Nicotinamide- Also known as NAD, or adenine dinucelotide, is a coenzyme involved in redox reactions making it an important factor in metobolic processes. It’s also important for DNA repair, cell senescence and imune function required for healthy cell aging.
Backfill:
Nuclease free water- nuetral additive important for acheiving desired concentrations of reagents, which helps prevent nucletide degredation and improves efficiency of CFPS.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems.
sfGFP- SF stands for super folder which is one of this green fluorescent protein’s main strengths, and why it’s extensively used in research. Because of its molecular stability, it is one of the most reliable systems used for successful gene delivery verification.
mRFP1- Monomeric Red Fluorescent Protein, optimal tempertature for maturation within one hour is 37°C. Overnight incubation improves can improve its brightness.
-mKO2- mKusabira-Orange2, recombinant monomer derived from Fungia concinna, it is oxygen-dependent for maturation, and has a higher phototoxicity compared to other FPs.
mTurquoise2- Has a fast muturation rate < one hour under optimal conditions with long fluorescence lifetime and resistance to photobleaching.
mScarlet_I- Red fluorescent protein characterised by rapid maturatation (~35m) and superior brightness. Optimal temperature 32-37C.
Electra2- Monomer, optimal tempreature range is 30°C or 37°C, and can reach maturation within several hours.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation.
mTurquoise2- Given it’s fast muturation rate (< one hour under optimal conditions), lowering incubation temperature to ~30 will effect maturation rate and slow down the reaction. The long incubation period should be carried out with a sealed plate to avoid evaporation. Adjusting magnesium glutamate for ideal concentration is important in the transcription/translation processes in CFPS. Having too much can cause DNA/RNA build up, whereas too little can slow down cellular machinery thereby inhibiting protein production. Correct nucleotide and amino acid concentrations are also important as they drive both transcription and translation processes.
The second phase of this lab will be to define the precise reagent concentrations for cell-free experiment:
Considering complexity of designing a cell free master mix containing over 16 reagents with conditional variables I experimented with the following AI prompts to generate optmized mixtures.
Well M14
AI prompt: Design cell free protein synthesis protocol for fluorescent protein mTurquoise2 starting with: 6.0 microLiter cell lysate, 2.0 microLiter DNA template, Potassium Glutamate- 0.875M stock with 312.5 millimolar concentration. Adjust amount of Nuclease-Free water in mixture to keep total volume of mixture equal to 10 microliters. Calculate ideal millimolar concentrations for following reagents in mixture:
AI prompt: Design cell free protein synthesis protocol for fluorescent protein mTurquoise2 starting with: 6.0 microLiter cell lysate, 2.0 microLiter DNA template. Adjust amount of Nuclease-Free water in mixture to keep total volume of mixture equal to 10 microliters. Calculate ideal millimolar concentrations for following reagents in mixture:
There are several differnt size pipettes and tips to use depending on the amount of fluid you want to dispense or extract.
Pipettes sizes: P20: 1-20uL of liquid P200: 20-200uL P1000: 100-1000uL
Pipette tips: 10uL, 200uL, 1000uL
They are adjustable within their specified range using the adustment wheel located at top of pipette.
If you have a 1-20uL pipette and the set read out is: 015, then you have it set to aspirate/dispense 1.5uL.
The smallest tip will only fit on the small pipette.
There are are two stops on the plunger, the first stop measures the amount the pipette is set to, the second stop (plunger depressed all the way) will blow out the pipette.
There is a button on the top of the pipette which will detach/eject the tip from the pipette.
Tips are single use only!
Do not invert the pipette! This can damage the mechanism, especially if there is fluid in the tip. If there is no tip attached you can set it down.
Pipette Practice
Always pipette with a steady hand, and avoid touching the walls of Eppendorf or PCR tubes.
You can mix a solution by aspirating and dispensing samples in the tube. Do this slowly to avid introducing bubbles in the mixture.
Week 2 Lab: Preparing Gels for Art
Agar solutions cooling down under the hood
In this LifeFabs lab we focused on preparing gels for:
Electropheresis- Mixing reagents to produce gel, and pouring gel into tank.
Agar plates- to streak with E. Coli.
Agrose Gel for Electropheresis
Prepared by mixing a 250mL batch of agarose gel solution. Solution contained TAE buffer and Red safe DNA stain, heated in the microwave to dissolve agarose. Once solution was completely disolved the gel was poured into tank with comb in place and set aside to cool.
Agar Plates
Getting ready to add antibiotic to solution
First step was to mix the agarose solution. We prepared two 250mL batches, one with black activated charcoal, and a clear one without antibiotic.
Both were heated in the microwave (45 sec pulses) until the agar was completley dissolved. I believe we added the activated carbon after heating.
Solutions were placed under the hood with thermometer waiting for the solution to cool down to 55C.
Antibiotic was added to solution, then plates were poured.
Each plate was poured with approximately 25mL, about halfway full. You have to move through this process quickly as the agarose starts to congeal as it cools.
The lids are placed on top of the dishes once they’re poured, but with space for condensation to escape. Once they’re cooled off they’re ready to be streaked.
Using a culture collector, we each sampled a different E. Coli colony/color from its culure dish and streaked a black plate with the sample.
There are two basic patterns used for streaking; the commonality between methods is they’re both linear, and connected by crossing over the end point of the former with the starting point of the ladder.
Best practice is to gently swab the surface of the agrose gel without piercing it’s surface.
Once the plates are swabed, they’re labeled, sealed with parafilm, and placed in the incubator.
PHOTOSYNTHETIC IMAGE GENERATION: Co-Cultured Autotrophic-Heterotrophic Color Controls for Bacteria Based Imagery
HTGAA TA’s: Danny Chen, Inger Le Gué, Juan Diego Unda, Digby Usher
SECTION 1: ABSTRACT:
Currently, biological art and engineered living material systems are often based on heterotrophic microorganisms such as E. coli, which require continuous external nutrient supplementation to function. In contrast this project looks at cyanobacteria as a potential solution to nutrient supply since they are capable of using light and CO₂ to naturally generate organic carbon through photosynthesis. However, cyanobacteria is less suitable for engineering visual outputs. The goal of this project is to combine cyanobacteria and E. coli into a synthetic co-culture, where cyanobacteria generates sucrose to support engineered E. coli for the production of chromo-proteins in a continuously recirculating visual media system.
SECTION 2: PROJECT AIMS
Aim I: Design and construct plasmids for homologous recombination of cyanobacterial strain, Synechococcus elongatus PCC 7942 with the sucrose permease cscB gene for secretion of sucrose.
Design functional cscB plasmids and submit order to Twist.
With arrival of plasmids, complete first transformation of S. 7942, and transfer microorganism to cold storage.
Redesign negative image plate for co-culture bioreactor.
Aim II: The co-culture system will require optimization of environment and media before attempting the co-culture.
First subaim- will test culture of S. 7942 cscB+ under control of salt stress and IPTG for sucrose production. Control variables will include, but are not limited to, the following:
- Nutrient concentrations of BG11 media.
- NaCl induced salt stress vs. KCl.
- Growth under LED and fluorescent lighting.
Second subaim- builds on results from the optimized media tests to construct the co-culture system with E. coli chromo-protein.
Third subaim- fabricates the bio-reactor negative image plates from Aim 1.
Aim III: Multi color expressions in the co-culture system, alternative salt free cyanobacteria systems, and mobile lab construction.
First subaim- tests multiple E. coli chromo-proteins within the co-culture system to generate multi color expressions.Design and fabricate mobile lab system for transportation of work to exhibition venues.
Second subaim- Design and fabricate mobile lab system for transportation of work to exhibition venues.
Third subaim- exhibits the project outside of the lab in alternative setting.
SECTION 3: BACKGROUND
Based on research of co-culturing Cyanobacteria Synechococcus elongateus 7942 cscB (S. cscB 7942) with Escherichia coli for bio-chemical synthesis [1,2], this project will explore the adaptation of the co-culturing process to produce visual color modifications within an image by generating muti-colored bacteria based pixels in a continuous reaction. The origin of this theoretical approach to image cultivation began through a series of visual experiments in collaboration with my partner, Sarah Max Beck, entitled studioHydrostatic. The project was set on top of a Brooklyn rooftop where we were experimenting with aqauponic technology to create a recirculating, human based, organic food production system. Two large flood tables grew a variety of edible plants and herbs in nitrogen rich water. This is where I first discovered a blue-green microorganism growing in a polka-dot like pattern on a sheet of bubble-foil insulation within the system. Recognition of the pattern lead to the developent of a glass plate negative (figure 2) for growing wild caught cyanobacteria in a contolled visual experiment.
Field tests were conducted in 2020 -2021, which proved the viability of the concept. It was during this initial trial when the first full size plate, WTF, was tested in a prototype bioreactor producing a graphic image (figure 1). As the cyanobacteria growth continued to progress in the WTF image, the density of bacteria progressively darkened the image. It then started to build on itself creating a slightly elevated surface of growth; it appeared to be different from the cyanobacteria. As it progressed the biomass reached a thickness of aproximately 2-3 mm after one year of growth. Eventually, it took over the image, and significantly suppressed the cyanobacteria’s chlorophyll pigmentation (figure 3). While I can only speculate, I believe the image was overtaken by some form of E. coli judging from the color and texture of the biomass.
My speculation is based on lab grown E. coli cultures observed during HTGAA labs with Lifefabs Institute, which indicated there’s a natural relationship between these organisms. Searching for evidence to support the connection, the paper entitled, Cross-feeding between cyanobacterium Synechococcus and Escherichia coli in an artificial autotrophic-heterotrphic cocuture system revealed by integrated omic analysis, demonstrated that cyanobacteria could be engineered to export sucrose and provide energy for E. coli [1].
The possibility of creating a symbiotic relationship between two different organisms to enhance the graphic functionality of PIG is the driving motivation behind this project. With the discovery of the co-culture technique came the task of understanding the mechanisms behind creating a similar relationship, where cross-feeding would result in a multi-color image of chromo-proteins supported by cyanobacteria’s ability to supply carbon to E. coli for the production of chromo-protein. In doing so, this project will reveal a different relationship between these organisms, and advance the field of biological art with a new system for growing visual media, which exceeds the time limitations of gel art, by co-culturing organisms under the control of a photonegative plate engineered specifically for this process.
PAPER #1:Cross-feeding between cyanobacterium Synechococcus and Escherichia coli in an artificial autotrophic-heterotrophic coculture system revealed by integrated omic analysis
In aquatic environments, the ecological interaction between photoautotrophic and heterotrophic species is based on cross-feeding and metabolite exchange. To date, multiprotein complexes that cross cell membrane(s) and extracellular vesicles have been evaluated in photoautotrophs for transporting materials from the interior to the exterior of the cell, which facilitate the secretion of various chemicals ranging from targeted photosynthetic intermediates, such as glycolate, osmolytes and fatty acids, and extracellular polymeric substances, to the products of cell lysis, including sugars, proteins, lipids and nucleic acids. These organic compounds could support the cell growth of the heterotrophic species [1].
The research driving this paper defines the metabolic efficiency which results when cyanobacterium Synechococcus 7942 cscB+ is co-cultured with E. coli engineered to produce 3-HP acid. The S. 7942 cscB+ strain was produced in a prior study proving that the wild type S.7942 could be engineered to export sucrose across its cellular membrane and support growth of other heterotrophic organanisms. This study confirmed the enhanced performance of both S. 7942 cscB+ and E. Coli 3-HP in a co-culture system through comparative analysis. Further, it demonstrated the consortium relieved oxidative stress and increased CO₂ availability improving the cell growth of cyanobacteria [1]. After reviewing this research we concluded it may be possible to reproduce similar results by engineering E. coli to produce chromoproteins instead of 3-HP acid.
PAPER #2:Engineering Cynaobacterium To Synthesize and Export Hydrophylic Products
Metabolic engineering of cyanobacteria has the advantage that sunlight and CO₂ are the sole source of energy and carbon for these organisms. However, as photoautotrophs, cyanobacteria generally lack transporters to move hydrophilic primary metabolites across membranes. To address whether cyanobacteria could be engineered to produce and secrete organic primary metabolites, Synechococcus elongatus PCC7942 was engineered to express genes encoding an invertase and a glucose facilitator, which mediated secretion of glucose and fructose [2].
This paper revealed some of the initial research behind the co-culture process, as well as the original patent (which has expired) confirming the results of the study. It introduced us to the S. 7942 genome, and its neutral sites 1, 2 and 3, which allow for the introduction of heterologous genes without impacting the genomic functionality of the cell [2]. The paper included the plasmid backbone, DS21, which was used to introduce the invA and glf genes from Z. mobilis as well as lacI with an IPTG regulated promoter [2]. This modification allows S. 7942 to convert CO2 into glucose and fructose from sucrose, and support their diffusion across the cell membrane. An important part of the process is an external stress which is placed on cyanobateria cells by introducing salt. This causes the cell to increase sucrose production, which helps balance osmotic pressure and maintain hydration. By engineering cyanobacteria to export glucose and fructose through a symporter protein under salt stress, the system will provide E. coli with a source of carbon for growth.
PAPER #3:Rerouting Carbon Flux To Enhance Photosynthetic Productivity
We demonstrate the efficient production of sucrose from a cyanobacterial species, Synechococcus elongatus, heterologously expressing a symporter of protons and sucrose (cscB).
In the second paper S.7942 was modified to export sucrose to E. coli using invA, which cleaves sucrose into glucose and frustose, and glf which tranports those products across the cell membrane. However, E. coli can directly uptake sucrose as a carbon source which renders invA superfluous as part of the cellular machinery. In effort to streamline and improve the efficiecy of sucrose production in S.7942, this research study introduces a sucrose permease symporter, cscB, into the S. 7942 genome through homologous recombination . The study compared the efficiency of the two variants by knocking out invA and glf, and measuring exportation of sugar molecules between them. S.7942 cscB+ exported 28% more sugar than its predecessor.
Further more, the study points out that “this productivity is the highest photosynthetic rate of microbial target product formation reported in academic literature for cyanobacteria or algae, and it exceeds (by ∼30-fold) that of a previously patented strategy for extracting sucrose from cyanobacteria” [3]. This is significant when considering how cynabacteria could revolutionize the biochemical production of chemicals for industry, especially since its carbon source is atmospheric CO₂. Given this advancement, cyanobacteria could compete with other fuel sources, and do so in an environmentally beneficial way.
SECTION 4: EXPERIMENTAL DESIGN, TECHNIQUES, TOOLS, AND TECHNOLOGY
Subaim 1.1. Plasma Design The concept of plasma design is challenging, especially when considering how the collective sum of its many components determine the functionality of the plasmid relative to its host. The following image (Fig. 4) comes from my notes on the second paper, which is referenced by Cross-feeding between cyanobacterium Synechococcus and Escherichia coli. It illustrates the progression of learning plasmid theory, design, and function.
Figure 4. Plasma from article 2 (supplementary material). On the right, grasping concepts of a plasma backbone/vector.
The next steps were to replicate the design in Benchling, which further challenged my understanding of synthetic biology as a practice. In the image (Fig.5) on the left hand side is my first Benchling construct for the project. Originally, when I inserted the cscB into the plasmid it pushed the number of base pairs to over 6000, which presents a problem as it relates to Twist. I thought “nuetral” sites could be substituted for functional genes since they have no effect on the target organism. My TA, Juan Diego Unda, made the functionality of the nuetral sites clear by explaining they are a neccessary component of the plasmid for it to be recognized by the target DNA and its positioning within the sequence. Figure 5. From the left, first plasma design attempt. Then with TA, Juan Diego Unda, we corrected the design (right).
Subaim 1.2. Construction of Model Organism S.7942 cscB+ (Protocol from: Specialized techniques for site-directed mutagenesis in cyanobacteria [3].)
Plasmid Construction for Mutagenesis by Homologous Recombination
Using standard molecular biology techniques (8), clone the cyanobacterial sequence
to be mutated into an E. coli cloning vector, such as pUC18 or pBR322 (we will be using pHN1-LacUV5).
Clone an antibiotic-resistance cassette within the sequence homologous to the
cyanobacterial chromosome, making sure to leave at least 300 bp of homologous
DNA flanking either side of the resistance gene for efficient recombination.
Synechococcus Transformation
Grow 100 mL of the S. elongatus strain to be mutated in liquid BG-11M to an
OD750 of 0.7.
Harvest 15 mL of cyanobacterial cells by centrifugation for 10 min at 6000g (see
Note 6).
Resuspend the cell pellet in 10 mL of 10 mM NaCl and harvest by centrifugation
for 10 min at 6000g.
Resuspend the cell pellet in 0.3 mL of BG-11M and transfer to a microcentrifuge
tube (see Note 6).
To each 0.3 mL of cells, add between 50 ng and 2 μg (typically, we use 1–2 μL
from a preparation of 100–200 ng/μL) of the recombinant plasmid that carries
the mutagenized cyanobacterial gene.
Wrap the tubes in aluminum foil to shield the cells from light and incubate them
overnight at 30°C with gentle agitation.
Plate the entire 0.3-mL cell suspension on a BG-11M plate containing the appro-
priate selective medium (see Note 2).
Incubate the plates at 30°C in constant light for approx 5 d until single colonies
appear.
Restreak isolated colonies that have the appropriate phenotypes, maintaining the
selection to favor complete segregation of mutant cyanobacterial chromosomes.
Grow mutant clones in 100 mL of BG-11M with the appropriate antibiotic to an
OD750 of 0.7. Extract the chromosomal DNA (see Subheading 3.1.3.) and verify
the presence of the mutation and its segregation on the cyanobacterial chromosome
by PCR, restriction enzyme analysis, or other technique (see Notes 4 and 7).
Extraction of Chromosomal DNA From S. elongatus
Pellet approx 10 mL of a liquid culture of cyanobacteria or scrape cells from a
plate.
Resuspend the pellet in 500 μL of 120 mM NaCl and 10 mM EDTA, pH 8.0, and
transfer the suspension to a microcentrifuge tube.
Re-pellet the cells and resuspend them in 340 μL of 25% sucrose, 50 mM Tris-
HCl, pH 8.0, 10 mM EDTA, pH 8.0. Add lysozyme to a final concentration of
2 mg/mL. Incubate the cell suspension for 45 min at 37°C.
Add 2 μL of proteinase K (from a 10 mg/mL stock solution) and 20 μL of 20%
sarkosyl, and vortex for 20 s. Incubate the mix at 55°C for 30 min.
Add 57 μL of 5 M NaCl and 45 μL of 10% cetyltrimethylammonium bromide in
0.7 M NaCl. Mix well and incubate for 10 min at 65°C.
Extract the suspension with 500 μL 24:1 chloroform:isoamyl alcohol.
Carefully transfer the upper aqueous phase to another tube and extract with 500 μL
of equilibrated phenol. Vortex for 20 s and spin for 10 min at 16,000g. The high
NaCl concentration may cause the phases to flip, placing the aqueous phase on
the bottom after the centrifugation step; the aqueous solution can be identified by
its pink hue.
Transfer the aqueous phase to another tube and extract with 500 μL of 24:1
chloroform:isoamyl alcohol mix. Vortex quickly and spin for 10 min at 16,000g.
Take the upper aqueous phase and precipitate the DNA by adding 2 v of 100%
ethanol. Mix by inverting the tube several times.
Spin down the DNA for 15 min at 16,000g in a microcentrifuge. Carefully remove
all the liquid (which contains significant salt) and wash the pellet with 1 mL of
70% ethanol. Spin again for 5 min and remove the ethanol from over the pellet.
Dry the pellet by leaving the tube open or by applying vacuum.
Resuspend the DNA in 50 μL of water and add 20 ng/μL of RNAse A (optional).
From this 50-μL final solution, 0.5 to 1 μL is typically used for a standard polymerase chain reaction (PCR).
Subaim 1.3. Negative image plate design for co-culture system The original imaging system (Fig. 2) used in the 2020-2021 field experiments (Figs. 1, 3) consists of a clear glass plate with an opaque, black, raised graphic surface. The graphic is a pourous layer of low fired powdered glass which was designed to form shallow channels for cyanobacteria to grow in. After the field experiments, I noticed how the cyanobacteria grew into the substrate, and over time was over taken by another form of bacteria which grew on the surface of the substrate media (Fig 3).
My observational hypothesis is the bacteria, which took over the image, was a form of E. coli growing in the space it was afforded by the channel. If the cyanobacteria can grow into the substrate, then a new plate (Fig. 6B) could be designed to support a co-culture where engineered organisms would not compete with one another. In the illustration below (Fig. 6A), the original bio-reactor and plate design is not optimized for a co-culture system since the transparency of the channel was designed to supply light for cyanobacteria. I believe compression of the opaque areas by the raised negative graphic will inhibit E. coli formation on the substrate’s surface. The new design, reverses the spatial relationship, where opaque-negative channels provide space for chromo-protein E. coli to grow with protection from light. Adjacent to the opaque channels, clear glass will allow light to reach S. 7942 cscB+ facilitating its growth into the substrate.
Figure 6. From the left, profile of original reactor system, then redesigned image plate on right.
Aim 2. Optimization of the co-culture environment
Subaim 2.1. Test culture of S. 7942 cscB+
With a successful transformtion of S. 7942 into the model organism S.7942 cscB+, a co-culture medium will need to be designed along side a fluid delivery system for a recirculating bio-reactor. We will begin by optimizing the environment for culturing the model organism by following established protocols from previous research. In addition to previous research, we will test KCl as an alternate to NaCl salt (Fig. 7).
Figure 7. (Hypothetical Data). Graph A represents growth curve of sodium free cultures of PCC 7942 and E. coli expressing chromoprotein. Graph B represents two cultures which test the difference between NaCl salt induced stress in comparison with KCl as a potential alternative.
Subaim 2.3. Negative image plate fabrication Glass fabrication is expensive and energy intensive, therefore we will begin with a series of small plates designed for experiments in petri dishes (Fig. 8). If we are able to confirm the functionality of the new design, the next step (aim 3) would scale the project to accomodate larger and more complex imagery. Part of this initial research will adapt data from previous research to produce a liquid grow media formula for establishing the co-culture system in a simple petri dish bioreactor.
Figure 8. Cut glass for negative image plate tests.
Aim 3. Multicolor Systems and Mobile Technology
Subaim 3.1. Test multiple E. coli chromo-proteins within the co-culture system The possibility of generating multi color expressions with PIG is very exciting, and the ability to sustain them for extended periods of time would have a significant impact on the field of bio-art imaging. The first problem I see, past establishing a successful co-culture, is controlling the growth rate of multiple organisms within the same culture. An effective plate design may help with maintenance, but there will be other factors which will influence the system. Temperature, pH, light, CO₂, salts, and fluid dynamics all play roles in the system, and we will have to deal with them through further research and development.
Subaim 3.2. Alternative salt free systems In a seperate study exploring a different strain of Synechococcus elongatus, Utex 2973, revealed alternative engineering strategies for producing sucrose without salt stress by overexpressing sps and spp genes in addition to cscB. Without salt stress, this organism’s sucrose production was over 2-fold higher than that from the S. 7942 strain [5]. While elevated sucrose production may not be required for a chomo-protein co-culture, reducing biological stress from salt may have beneficial effects on the overall health of the system.
Subaim 3.3. Mobile lab systems for transportation of work to alternative locations While there is a considerable amount of material which needs to be researched, analysed and compiled into results, it is important to consider how the original goal of PIG, which is to produce living visual media, will function as viable form of bio-art. A portable lab for culturing imagery under a wide range of variable conditions will need to be developed. The original PIG system is in a mobile form (Fig. 9), however it will require modifications, such as the addition of a co-culture module for multi-color imagery.
Expectations Obviously, there is much research and development ahead of PIG and its future as a potential biologically controlled multi-color imagery system. Previous research with co-culturing autotrophic-heterotrophic organisms suggests the color control hypothesis is promising for bacteria based imagery. With a model organism to work with, we will be able to better define the relationships between E. coli chromo-proteins and metabolically engineered cyanobacteria.
Ethics The biotechnology driving the potential development of the project is synthetic, and must be handled in a manner applicable with the environmental laws and standards associated with handling synthetically designed organisms in any particular city, state or country. Both cyanobacteria and E. coli are water born organisms which can have significant impacts on the environment. The organsims designed for this study are lab organisms, and to our knowledge, have no modifications which could be harmful to the environment. However, as with any lab organism, they are not intended for release into the environment under any circumstances.
Further, previous research in the arena of metabolically engineered cyanobacteria reveals some very interesting characteristics these organisms can possess when they are introduced to heterologous genes. While the natural world of biology has been evolving for over four billion years, the gentic relationships we share as descendants of LUCA may hold promissing solutions to the environmental challenges many organisms face on Earth today. As humans have modified the environment, and in many cases exploited it, the potential synthetic biology holds may not only help us develop a better understanding of the environment, it could also offer many opportunities to reciprocate in a beneficial way with careful consideration.
SECTION 6: ADDITIONAL INFORMATION:
Materials and Supplies
Twist clonal gene order Q-648659
Plasmid #1: $802.88
Plasimd #2: $154.70
GeneArt® Synechococcus Engineering Kit For expression of recombinant proteins in Synechococcus elongatus
Catalog Numbers A14259
Kit A14259: 310.00bp
DH5α Competent Cells
EC0112: 128.65bp
Alternative Materials for recombinant transformation:
E. coli cloning vector, such as pUC18 or pBR322.
Modified BG-11 medium (BG-11M). Liquid medium (7): 1.5 g/L NaNO3, 0.039
g/L K2HPO4, 0.075 g/L MgSO4·7H2O, 0.02 g/L Na2CO3, 0.027 g/L CaCl2, 0.001
g/L EDTA, 0.012 g/L FeNH4 citrate, and 1 mL of the following microelement
solution: 2.86 g/L H3BO3, 1.81 g/L MnCl2·4H2O, 0.222 g/L ZnSO4·7H2O, 0.391 g/L
Na2MoO4, 0.079 g/L CuSO4·5H2O, and 0.0494 g/L Co(NO3)2·6H2O. Solid medium
Site-Directed Mutagenesis in Cyanobacteria 157
(1): equal volumes of twice-concentrated BG-11M liquid medium and Difco agar
solution (3% in sterile water) autoclaved separately and mixed together; add fil-
ter-sterilized Na2SO3 to 1 mM final concentration.
Antibiotics: kanamycin (Km), spectinomycin (Sp), cloramphenicol (Cm), strep-
tomycin (Sm), and gentamicin (Gm) (see Note 2).
10 mM NaCl (sterile).
120 mM NaCl.
10 mM EDTA, pH 8.0.
25% Sucrose (w/v in water; sterile).
50 mM Tris-HCl, pH 8.0.
10 mg/mL Lysozyme in water.
20% Sarkosyl (w/v in water).
10 mg/mL Proteinase K in water.
5 M NaCl.
10% Cetyltrimethylammonium bromide in 0.7 M NaCl.
24:1 Chloroform:isoamylic alcohol.
Equilibrated phenol (8).
100% Ethanol.
70% Ethanol.
10 mg/mL RNAse A in water.
Hit-and-run vector (see Subheading 3.3.).
E. coli strain carrying conjugal vector for conjugation (see Subheading 3.1.4.).
E. coli strain carrying helper vector for conjugation (see Subheading 3.1.4.).
Ma J, Guo T, Ren M, Chen L, Song X, Zhang W. Cross-feeding between cyanobacterium Synechococcus and Escherichia coli in an artificial autotrophic-heterotrophic coculture system revealed by integrated omics analysis. Biotechnol Biofuels Bioprod. 2022 Jun 22;15(1):69. doi: 10.1186/s13068-022-02163-5. PMID: 35733176; PMCID: PMC9219151.
Niederholtmeyer H, Wolfstädter BT, Savage DF, Silver PA, Way JC. Engineering cyanobacteria to synthesize and export hydrophilic products. Appl Environ Microbiol. 2010 Jun;76(11):3462-6. doi: 10.1128/AEM.00202-10. Epub 2010 Apr 2. Erratum in: Appl Environ Microbiol. 2010 Sep;76(17):6023. PMID: 20363793; PMCID: PMC2876443.
Ducat DC, Avelar-Rivas JA, Way JC, Silver PA. Rerouting carbon flux to enhance photosynthetic productivity. Appl Environ Microbiol. 2012 Apr;78(8):2660-8. doi: 10.1128/AEM.07901-11. Epub 2012 Feb 3. PMID: 22307292; PMCID: PMC3318813.
Clerico EM, Ditty JL, Golden SS. Specialized techniques for site-directed mutagenesis in cyanobacteria. Methods Mol Biol. 2007;362:155-71. doi: 10.1007/978-1-59745-257-1_11. PMID: 17417008.
Lin, PC., Zhang, F. & Pakrasi, H.B. Enhanced production of sucrose in the fast-growing cyanobacterium Synechococcus elongatus UTEX 2973. Sci Rep 10, 390 (2020). https://doi.org/10.1038/s41598-019-57319-5


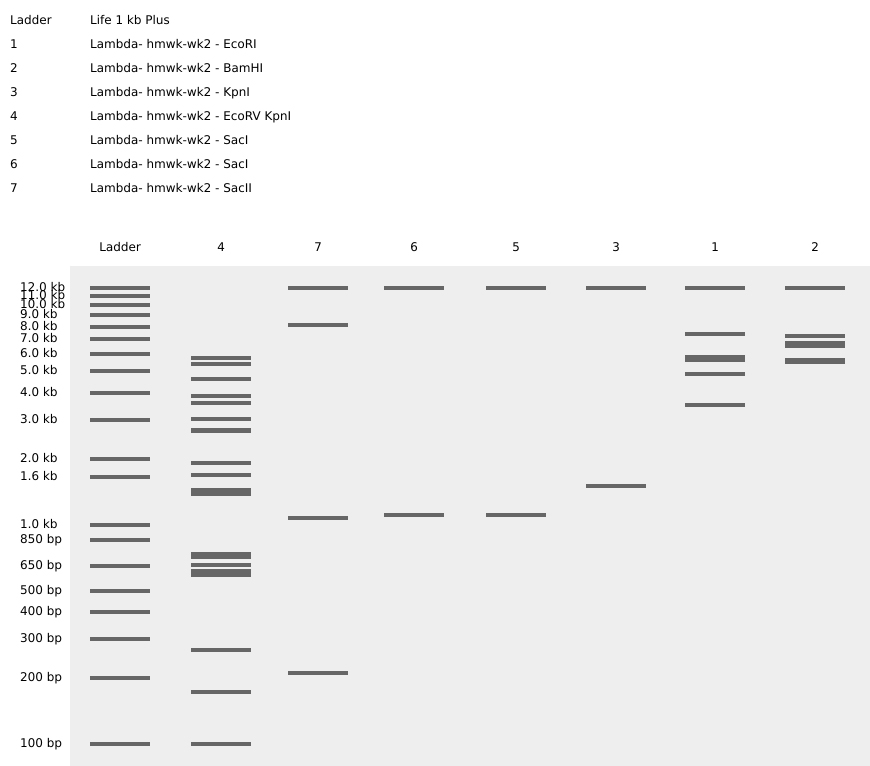
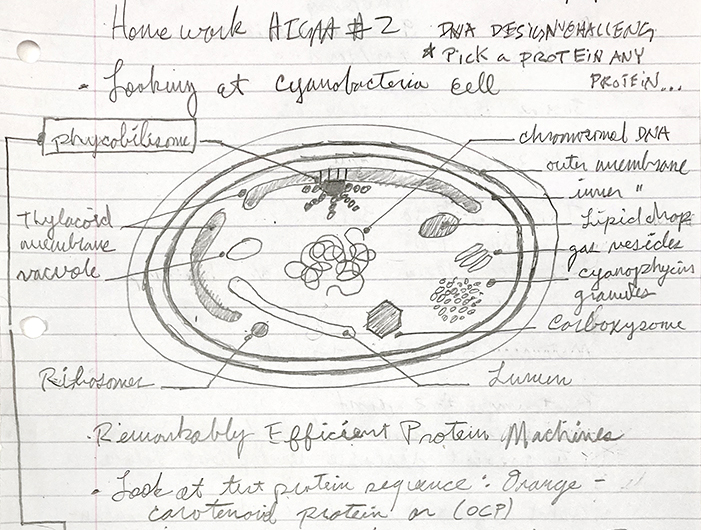


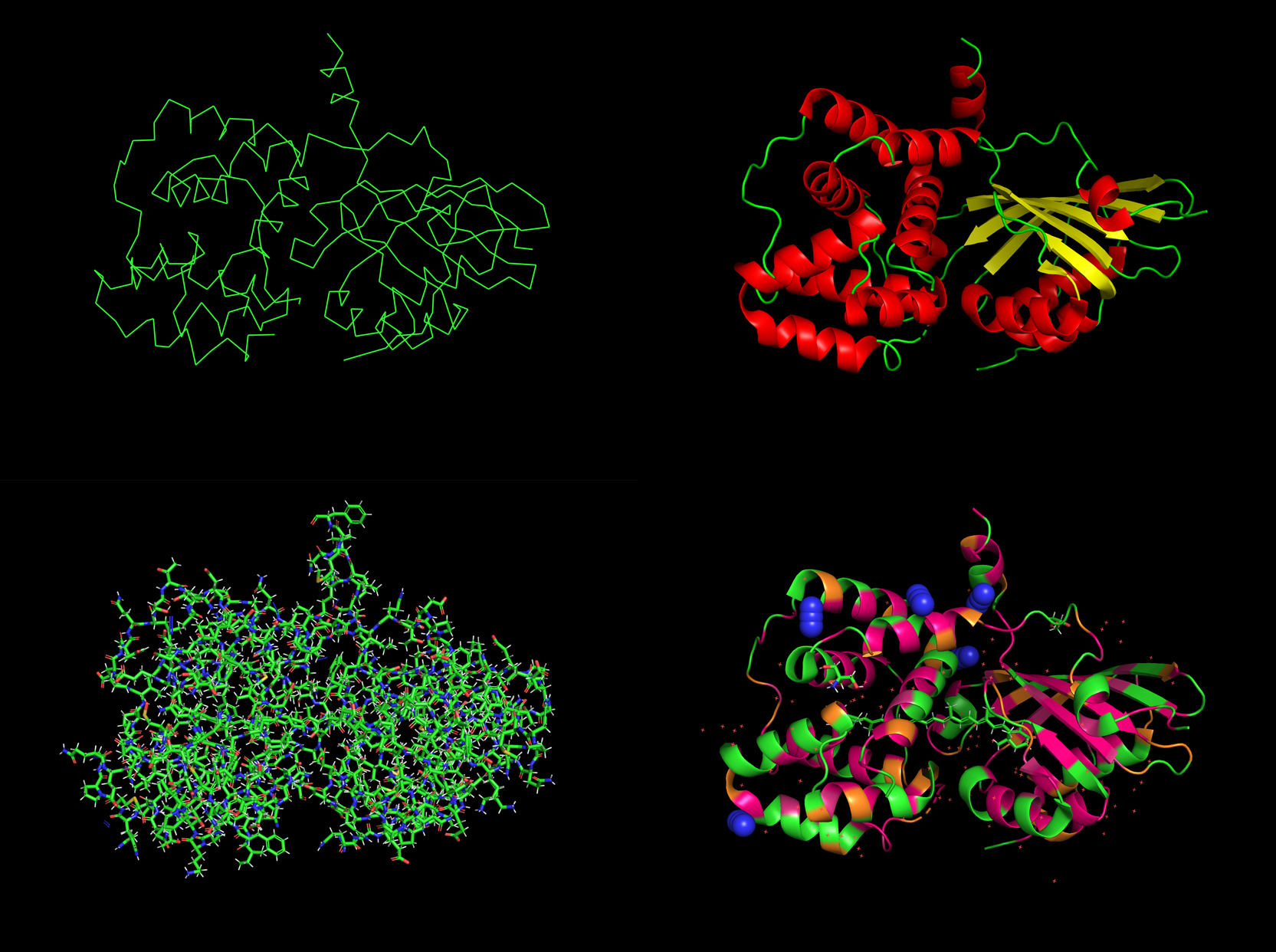
.png)
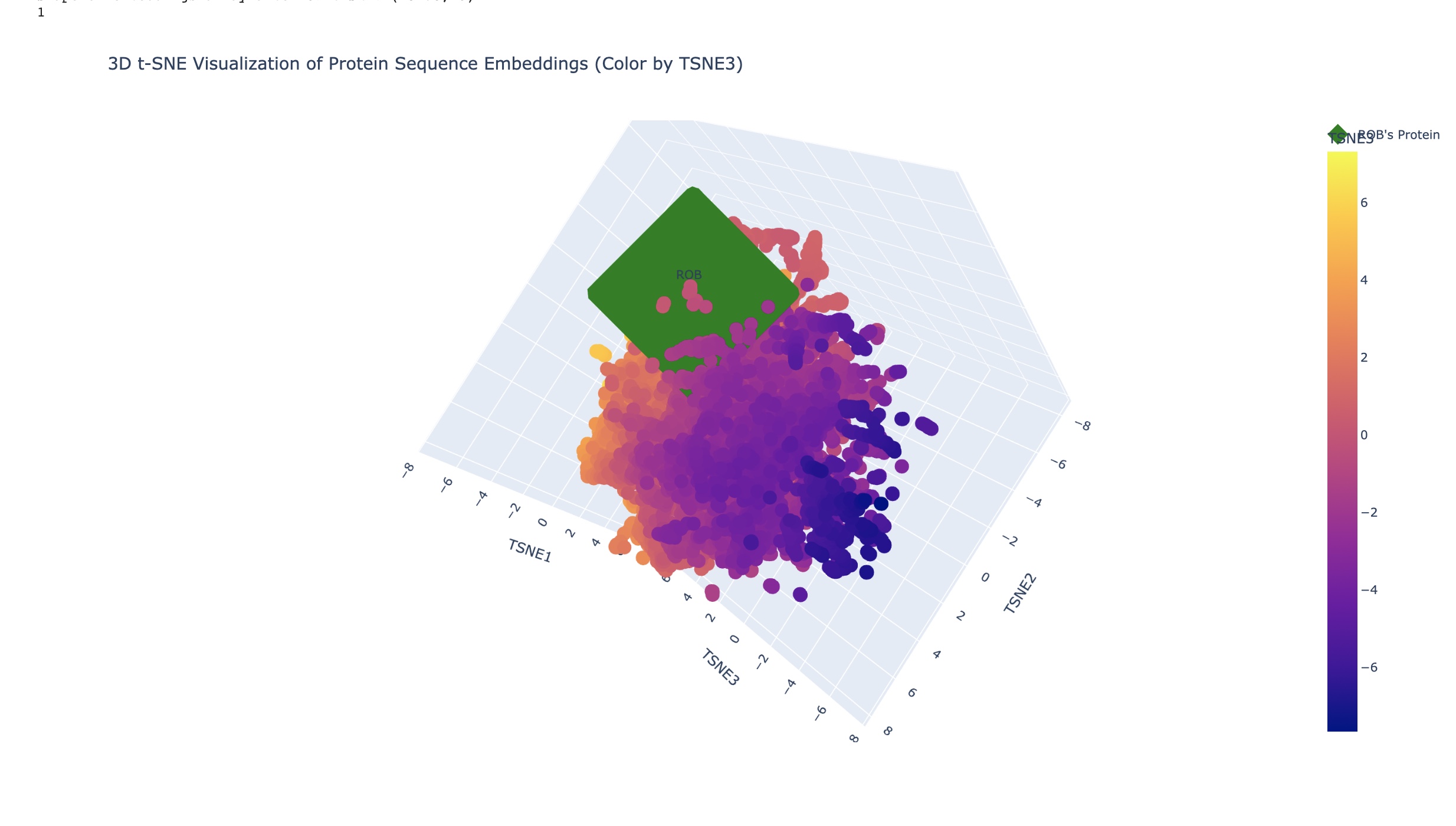
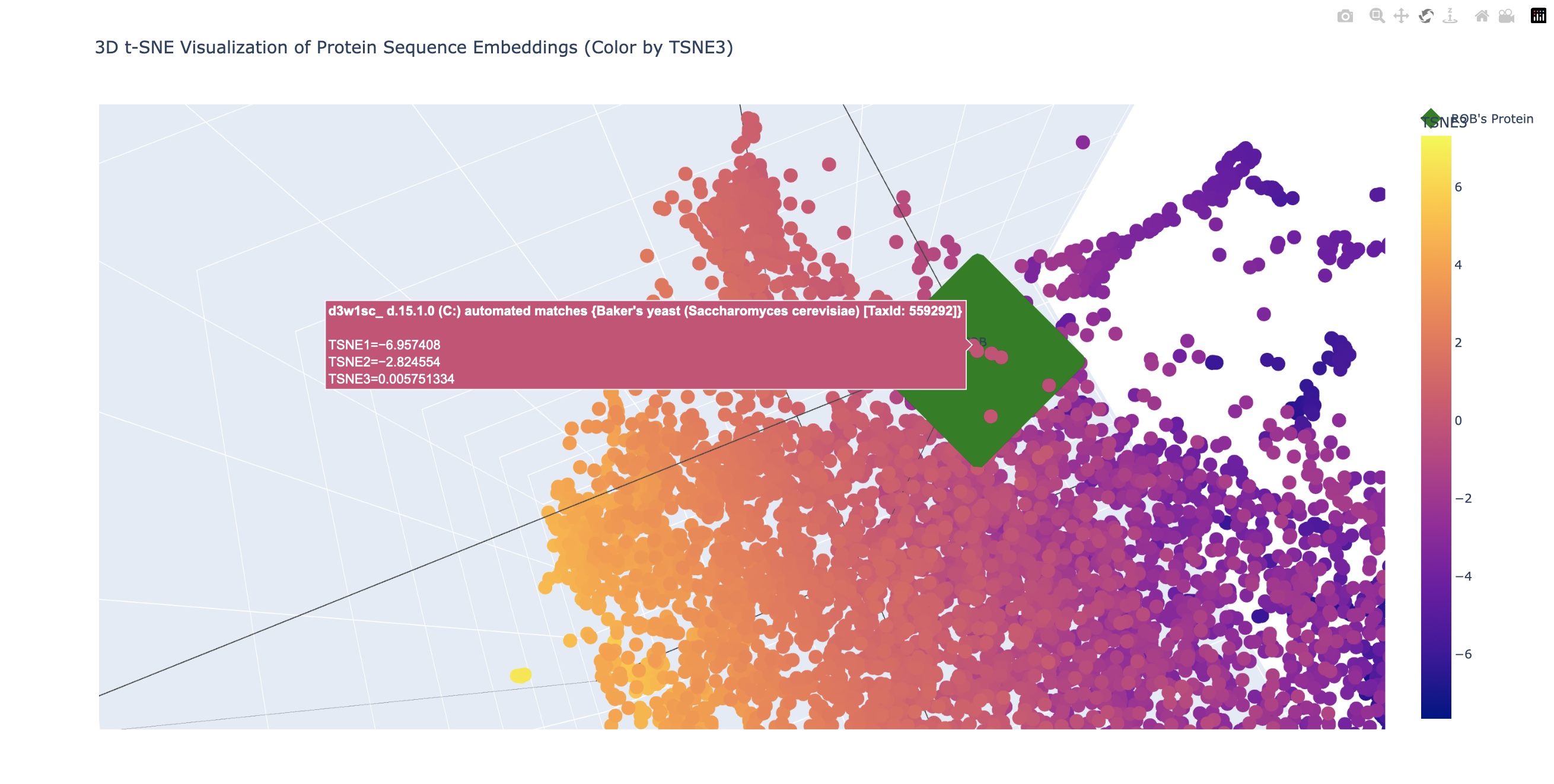
.png)