Week 5 HW: Protein Design II
Part A: SOD1 Binder Peptide Design
1. Background and Design Goal
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, SOD1 forms a stable homodimer and binds copper and zinc cofactors. Mutations in SOD1 are associated with familial ALS; among them, the A4V variant is one of the aggressive disease-associated variants and has been linked to SOD1 destabilization, altered folding behavior, and toxic aggregation.
In this section, I designed 12-amino-acid peptide binders against A4V mutant SOD1 and evaluated which candidates showed enough computational evidence to justify experimental validation. I treated this as an early-stage computational screening exercise rather than proof of therapeutic activity.
2. A4V Mutant SOD1 Sequence
I retrieved the human SOD1 canonical sequence from UniProt (P00441). I introduced the disease-associated A4V mutation by changing the alanine in the original N-terminal sequence MATKAV… to valine, resulting in MATKVV…. Because I included the initiating methionine in the AlphaFold3 input sequence, this disease-associated A4V position appears as VAL5 in the AlphaFold viewer.
A4V mutant SOD1 sequence used for all computational submissions:
Note: I used VAL5 as the visual reference for the A4V site when inspecting AlphaFold3 outputs.
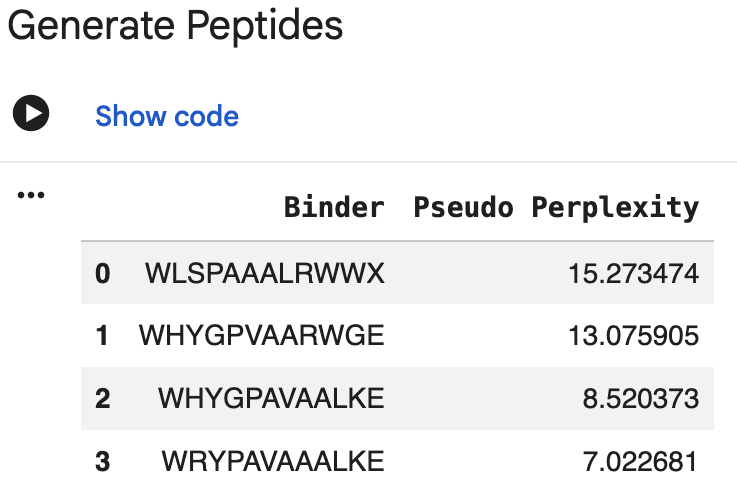
3. PepMLM Peptide Generation
I generated four 12-aa peptide binders using PepMLM conditioned on the A4V mutant SOD1 sequence. One initial output contained an unknown amino acid symbol (X), so I discarded it and regenerated an additional candidate. I also included the known SOD1-binding peptide FLYRWLPSRRGG as a reference control.
| Peptide ID | Sequence | Source | Length | PepMLM pseudo perplexity |
|---|---|---|---|---|
| P1 | WHSGATAAAWKE | PepMLM | 12 aa | 7.160 |
| P2 | WHYGPVAARWGE | PepMLM | 12 aa | 13.076 (highest; least plausible) |
| P3 | WHYGPAVAALKE | PepMLM | 12 aa | 8.520 |
| P4 | WRYPAVAAALKE | PepMLM | 12 aa | 7.023 (lowest; most plausible) |
| Known binder | FLYRWLPSRRGG | Literature control | 12 aa | N/A |
Interpretation: Lower pseudo perplexity indicates that the model views the sequence as more plausible given the SOD1 target. Based on this metric, P4 and P1 ranked as the strongest PepMLM outputs, while P2 ranked as the weakest. However, pseudo perplexity reflects sequence plausibility, not physical binding affinity or interface confidence. I therefore used AlphaFold3 as the next structural evaluation step.
4. AlphaFold3 Structural Evaluation
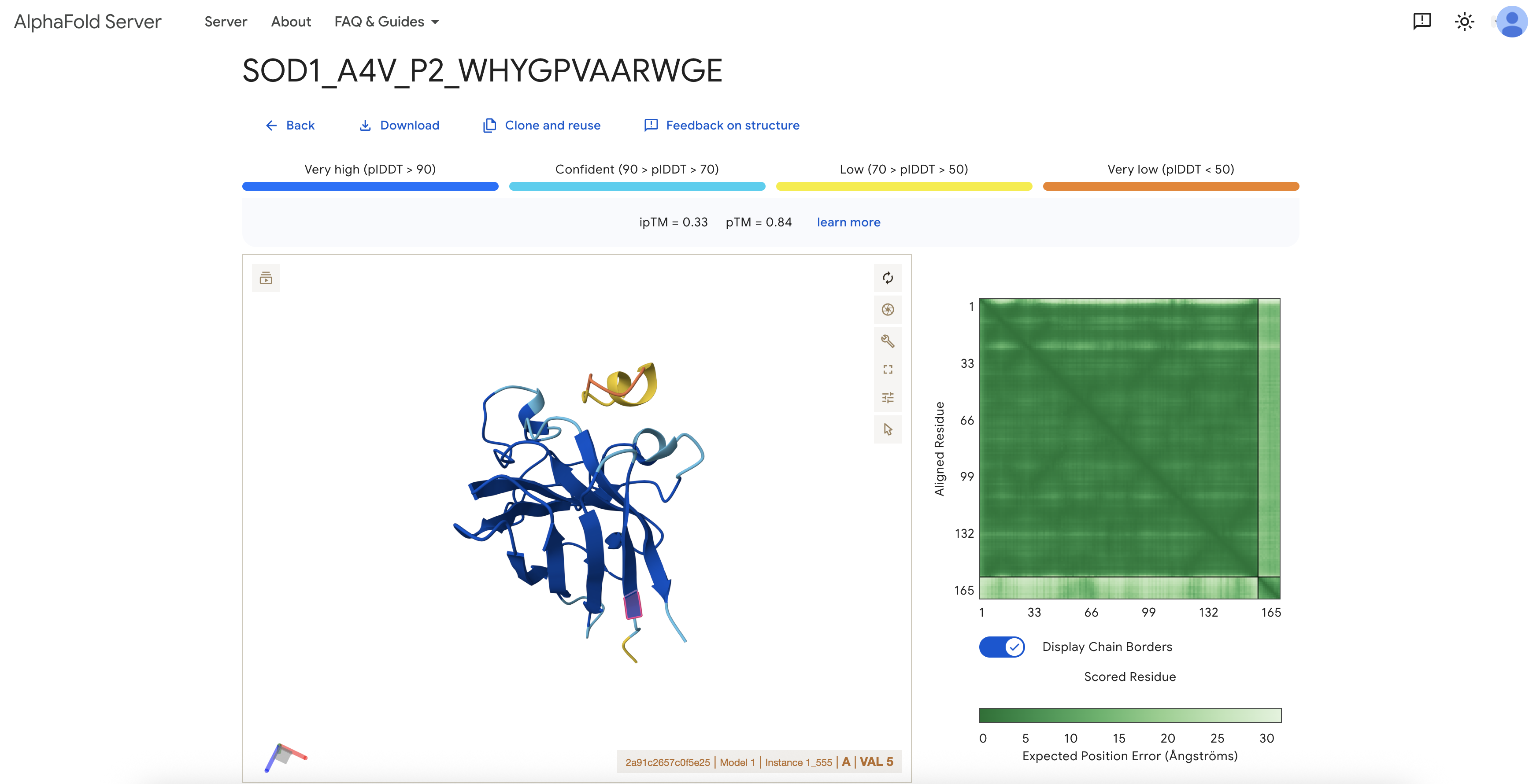
I modeled each peptide as a separate chain alongside A4V mutant SOD1 using the AlphaFold3 server. The SOD1 chain was submitted as Chain A and the peptide as Chain B; no fusion was introduced. I used AlphaFold3 to test whether each peptide could form a plausible protein-peptide complex with SOD1.
Note on VAL5: Because the AlphaFold input includes the initiating methionine, the A4V mutation site corresponds to residue VAL5 in the output structure. All localization descriptions below use this numbering.
| Peptide ID | Sequence | ipTM | pTM | Near VAL5 / A4V? | Structural interpretation |
|---|---|---|---|---|---|
| P1 | WHSGATAAAWKE | — | — | Not assessed | AlphaFold3 result was not available in this run; evaluated via PeptiVerse only. |
| P2 | WHYGPVAARWGE | 0.33 | 0.84 | Not clearly near VAL5 | Low-confidence interface; SOD1 structure itself was reasonably predicted. |
| P3 | WHYGPAVAALKE | 0.34 | 0.80 | No | Highest available ipTM in my completed runs, but still low; peptide appeared as a low-confidence yellow/orange segment outside the SOD1 beta-barrel and did not engage VAL5. |
| P4 | WRYPAVAAALKE | 0.29 | 0.74 | Not clearly near VAL5 | Very low interface confidence; lowest pTM of the completed set. |
| Known binder | FLYRWLPSRRGG | 0.29 | 0.78 | Not clearly near VAL5 | Also low ipTM in this setup, suggesting that AlphaFold3 interface prediction was uncertain for these short peptides. |
Structural interpretation: All available ipTM scores were low (0.29–0.34). The SOD1 monomer itself was modeled with reasonable confidence (pTM 0.74–0.84), but the protein-peptide interfaces were not predicted with high confidence in any case. Inspection of the P3 model around VAL5 showed that the peptide appeared as a yellow/orange low-confidence segment outside the main SOD1 beta-barrel, with no clear engagement of the A4V site. The known SOD1-binding peptide also produced a low ipTM in this setup, suggesting that low ipTM should not be interpreted as definitive evidence against binding; rather, this workflow did not produce high-confidence SOD1-peptide interfaces for any tested sequence.
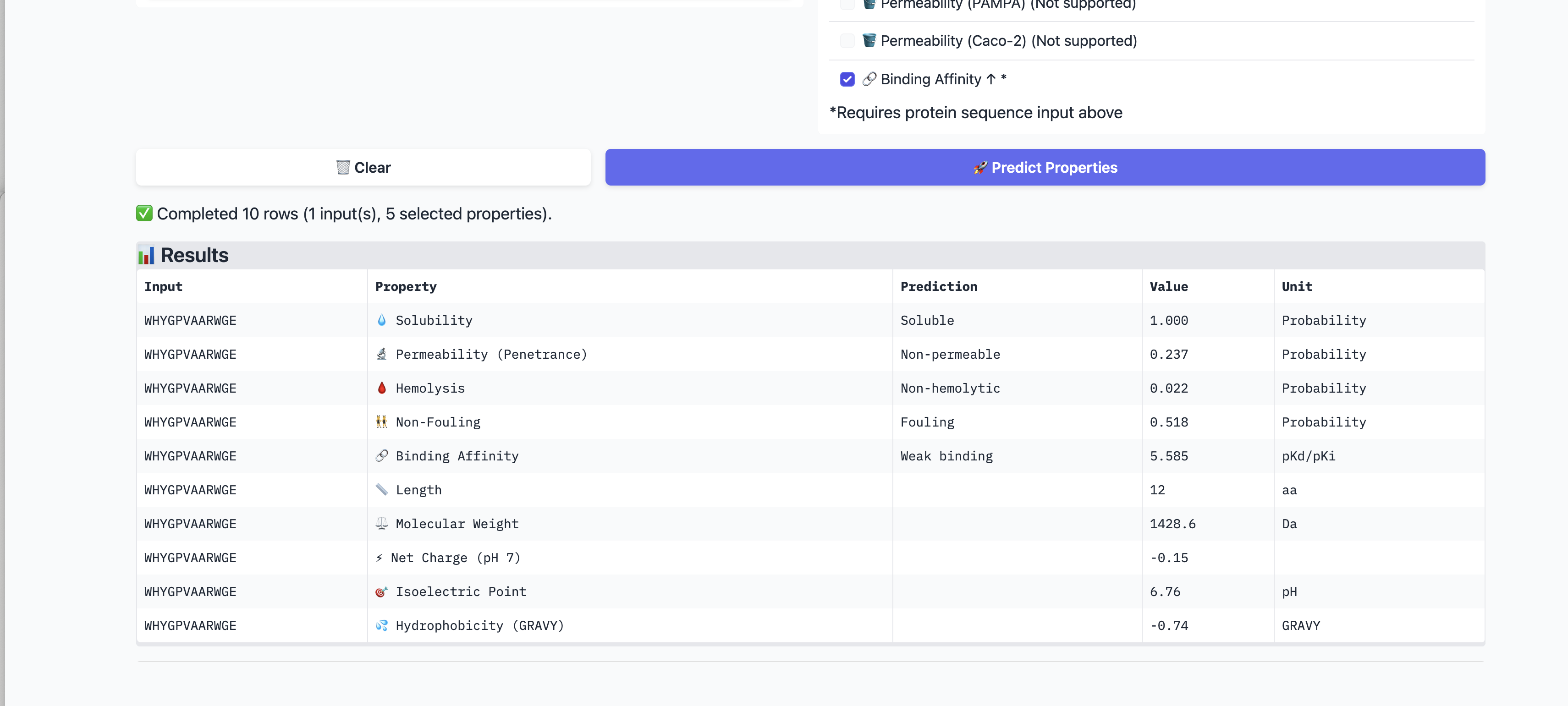
5. PeptiVerse Therapeutic Property Evaluation
I evaluated all four PepMLM-generated peptides and the known binder using PeptiVerse. I supplied the A4V mutant SOD1 sequence as the target for binding affinity prediction. I assessed solubility, hemolysis probability, binding affinity (pKd/pKi), molecular weight, net charge at pH 7, permeability, non-fouling behavior, and GRAVY index.
| ID | Sequence | Solubility | Hemolysis | Binding affinity | MW | Charge pH7 | Perm. | Non-fouling | GRAVY |
|---|---|---|---|---|---|---|---|---|---|
| P1 | WHSGATAAAWKE | 1.000 | 0.013 | Weak 5.360 pKd | 1314 Da | -0.15 | 0.528 | 0.593 | -0.59 |
| P2 | WHYGPVAARWGE | 1.000 | 0.022 | Weak 5.585 pKd | 1429 Da | -0.15 | 0.237 | Fouling | -0.74 |
| P3 | WHYGPAVAALKE | 1.000 | 0.031 | Weak 5.408 pKd | 1342 Da | -0.15 | 0.099 | 0.647 | -0.12 |
| P4 | WRYPAVAAALKE | 1.000 | 0.039 | Weak 5.462 pKd | 1375 Da | +0.77 | 0.331 | 0.697 | -0.04 |
| Known | FLYRWLPSRRGG | 1.000 | 0.047 | Weak 5.550 pKd | 1508 Da | +2.76 | 0.862 | 0.652 | -0.71 |
PeptiVerse findings: All peptides were predicted to be soluble (1.000) and had low hemolysis probabilities (0.013–0.047), which is a favorable developability signal. PeptiVerse classified every candidate, including the known binder, as a weak binder in a narrow 5.36–5.59 pKd range.
P1 had the cleanest overall property profile: lowest hemolysis (0.013), near-neutral charge (-0.15), and no fouling flag. Its binding affinity prediction was not strong, but none of the candidates showed convincing affinity in PeptiVerse.
P2 showed the highest predicted binding affinity among the PepMLM peptides, but it had the worst PepMLM pseudo perplexity and was flagged as fouling, making it less attractive overall.
P4 had the best PepMLM pseudo perplexity and acceptable PeptiVerse properties, but its AlphaFold3 ipTM was very low (0.29).
P3 had the highest available AlphaFold3 ipTM (0.34) among the completed runs, but this is still a low-confidence interface. Its PeptiVerse profile was acceptable but not outstanding.
Comparison with the known binder: FLYRWLPSRRGG had a slightly higher
predicted affinity than several generated peptides and the highest
predicted permeability, but it also had the highest hemolysis
probability among this small set. Because PeptiVerse also classified the
known binder as weak, I treated these predictions as useful screening
signals rather than definitive binding evidence.
6. moPPIt Targeted Design Attempt
Following the PepMLM and AlphaFold3 analysis, I attempted a more targeted design step using moPPIt. The goal was to bias peptide generation toward residues 5–10 of the A4V SOD1 sequence, which include VAL5 / the A4V site and adjacent N-terminal residues, rather than allowing the peptide to bind anywhere on the protein surface.
| Parameter | Value |
|---|---|
| Target protein | A4V mutant SOD1 (154 aa) |
| Peptide length | 12 aa |
| Motif positions | Residues 5–10 (VVCVLK) |
| Main objectives | Affinity + Motif |
| Additional objectives attempted | Hemolysis, Solubility |
| Number of samples | 10 |
Runtime failure: The public moPPIt Colab notebook failed during model initialization due to a missing PeptiVerse artifact. The error message was:
This error indicates that the Non-Fouling classifier, which is part of the PeptiVerse suite used internally by moPPIt, was not present in the public notebook environment. Because the failure occurred at model loading, no moPPIt peptides were generated. I therefore documented the intended targeted-design setup but did not include moPPIt-generated candidates in the final results. If the notebook is updated or a local installation is available, running moPPIt with motif guidance on residues 5–10 would be my recommended next step.
7. Final Candidate Interpretation
No peptide in this workflow emerged as a high-confidence SOD1 binder. The key reasons are:
AlphaFold3 ipTM scores were low across all completed runs (0.29–0.34), below what I would expect for a confident protein-peptide interface.
None of the inspected peptides clearly bound near VAL5 / A4V, the biologically relevant region of the mutant protein.
PeptiVerse classified all candidates, including the known literature binder, as weak binders. Solubility and hemolysis profiles were uniformly good, but binding affinity predictions were not convincing.
moPPIt could not be completed due to a missing model artifact in the public Colab environment.
Provisional candidate selection: If one peptide must be selected from the completed AlphaFold3 runs, P3 (WHYGPAVAALKE) is the provisional structural candidate only because it had the highest available ipTM score (0.34). However, the difference between candidates is small and the absolute ipTM value remains low, so this should not be interpreted as strong binding evidence.
An alternative choice is P1 (WHSGATAAAWKE), which had the cleanest PeptiVerse property profile: the lowest hemolysis probability, near-neutral charge, and no fouling flag. If P1 is later submitted to AlphaFold3 and produces a substantially higher ipTM than the other candidates, it would become a more reasonable candidate for additional screening.
My honest conclusion at this stage is that the current PepMLM peptides are plausible 12-aa sequences with good basic developability properties, but they do not yet show strong computational evidence of specific binding to the A4V SOD1 region. The most productive next step would be a targeted redesign using a functioning moPPIt setup, or an equivalent motif-guided method, focused on residues 5–10 around VAL5.
8. Conclusion
I generated four 12-aa peptide candidates with PepMLM and evaluated them alongside the known SOD1-binding peptide FLYRWLPSRRGG. PepMLM pseudo perplexity scores ranked P4 and P1 as the most plausible sequences. PeptiVerse predicted that all candidates were soluble and had low hemolysis risk, but classified all as weak binders. AlphaFold3 did not produce high-confidence SOD1-peptide interfaces (ipTM 0.29–0.34), and no peptide clearly engaged the VAL5/A4V region. I attempted a targeted moPPIt design run targeting residues 5–10, but the public notebook could not complete generation due to a missing model artifact.
This workflow demonstrates both the utility and the limitations of computational peptide design. PepMLM provides rapid plausible sequences; PeptiVerse filters for developability properties; AlphaFold3 tests structural plausibility of interfaces. However, none of these tools alone constitutes proof of binding. In this case, the combined evidence indicates that none of the current peptides should be advanced as a therapeutic candidate without further targeted design and experimental binding validation.
Part C: Final Project — L-Protein Mutants
1. Background and Objective
The MS2 bacteriophage L-protein is a small lysis protein that contributes to bacterial lysis by disrupting the E. coli inner membrane. This mechanism is relevant to phage-based strategies against antibiotic-resistant infections. DnaJ, an E. coli chaperone, appears to support normal L-protein processing, so mutations that make L-protein more stable or more capable of auto-folding could help reduce dependence on host chaperone assistance.
Because my Part C analysis is less complete than Part A, I treat the following section as a provisional design proposal. Any specific mutation choice should be checked against the course mutagenesis-score table and experimental lysis dataset before final experimental submission.
2. L-Protein Sequence and Region Logic
L-protein full-length sequence (75 aa):
| Region | Residues | Functional relevance |
|---|---|---|
| Soluble N-terminal domain | 1–40 | Likely related to DnaJ interaction, folding initiation, and processing. |
| Transmembrane region | 41–75 | Likely related to membrane insertion, pore formation, and lysis activity. |
Design logic: I would avoid known essential or highly conserved residues, especially LS motifs and strongly conserved hydrophobic positions in the transmembrane region. I would prioritize positions that are less conserved, experimentally tolerated, and mechanistically likely to improve folding or membrane insertion.
3. Provisional Mutant Design Strategy
I would use three filters to select L-protein variants: (1) mutagenesis score, (2) experimental lysis behavior relative to wild type, and (3) conservation status from sequence alignment. Since my current notes do not fully verify every score and lysis percentage, I avoid treating the specific candidates below as final. They are proposed directions to check against the assignment dataset.
| Provisional variant | Region | Why it is interesting | Uncertainty / check needed |
|---|---|---|---|
| Q8K | Soluble region | May alter surface charge in the N-terminal region and test whether DnaJ-related recognition can be changed without touching the transmembrane lysis domain. | Verify score, conservation, and lysis data before final use. |
| F22L | Soluble region | A conservative hydrophobic substitution that may reduce aromatic recognition while preserving general hydrophobic character. | Verify that the position is not strongly conserved and that lysis remains acceptable. |
| A45G | Transmembrane edge | Could introduce local flexibility near the start of the TM region and test whether flexibility helps membrane insertion. | Higher-risk candidate; verify that lysis is not strongly reduced. |
| N53A | Transmembrane core | A polar-to-nonpolar change that may increase TM hydrophobicity and potentially improve autonomous membrane insertion. | Strong candidate only if supported by positive score and maintained/improved lysis. |
| Q54A | Transmembrane core | Similar logic to N53A; may improve hydrophobic compatibility of the TM region. Could also be tested later as part of an N53A/Q54A double-mutant hypothesis. | Verify single-mutant performance before considering double mutant. |
4. Tentative Priority
My tentative priority would be to first validate one soluble-domain mutant and one transmembrane-region mutant rather than immediately making a complex multi-mutant design. If the assignment dataset supports that N53A and/or Q54A maintain or improve lysis, I would prioritize them as the most mechanistically plausible transmembrane candidates. If Q8K maintains lysis, I would prioritize it as the most useful soluble-domain candidate for probing DnaJ-dependence effects.
5. Structural Validation Plan
As a qualitative check, I would use AlphaFold2 Multimer or a similar structure-prediction workflow to compare wild-type and mutant L-protein assemblies. For soluble-domain candidates, I would also consider co-folding residues 1–40 with the DnaJ J-domain to see whether the predicted interaction surface changes. I would treat these models as qualitative support only, because membrane proteins and phage lysis assemblies are difficult for structure predictors. The decisive test would still be an experimental lysis assay, ideally including conditions that test DnaJ dependence.
6. Definition of a Successful Mutant
A successful L-protein mutant should:
retain overall L-protein structural integrity, especially in the transmembrane region;
avoid disrupting LS motifs or other known essential residues;
maintain or improve lysis activity relative to wild type;
show evidence of improved stability, auto-folding, or reduced DnaJ dependence;
be practical to synthesize, express, and test within the course workflow.
In short, I would not define a good mutant only by the highest computational score. I would define it as a mutation that balances model support, experimental lysis behavior, conservation logic, and a clear biological mechanism.