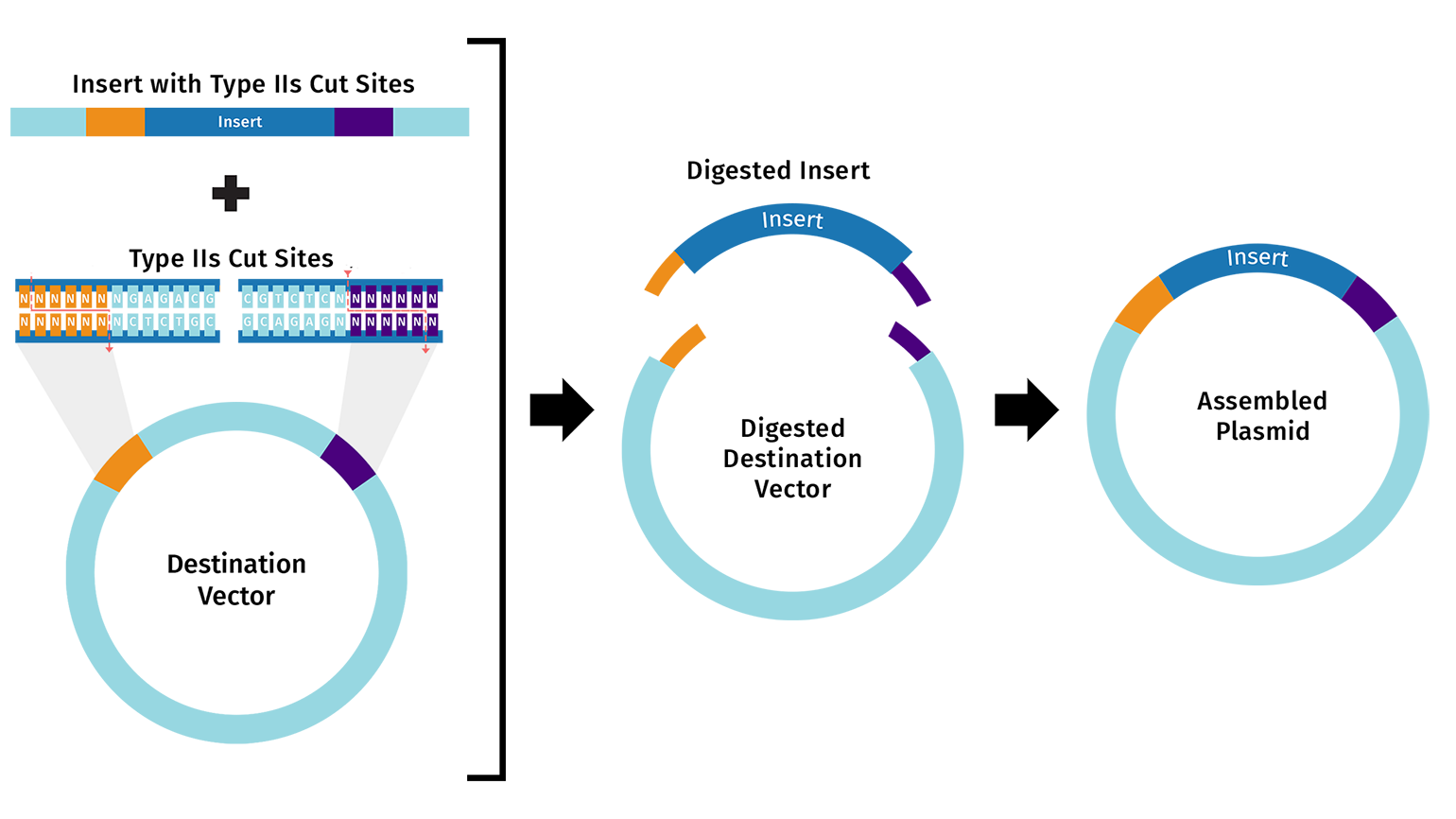I’m a graduate student working at the intersection of Business, design and Technology. This site documents my coursework, lab work, and projects for How To Grow (Almost) Anything, Spring 2026.
My interests include biological fabrication, genetic code engineering, and the social and ethical implications of programmable life.
AI Cite Prompts were directly based on the homework questions provided.
Homework Questions from Professor Jacobson 1. What is the error rate of polymerase? How does this compare to the human genome length, and how does biology address the discrepancy? Polymerase error rate:
~10⁻⁵ per base without proofreading; ~10⁻⁷–10⁻⁸ with proofreading; ~10⁻⁹–10⁻¹⁰ with mismatch repair.
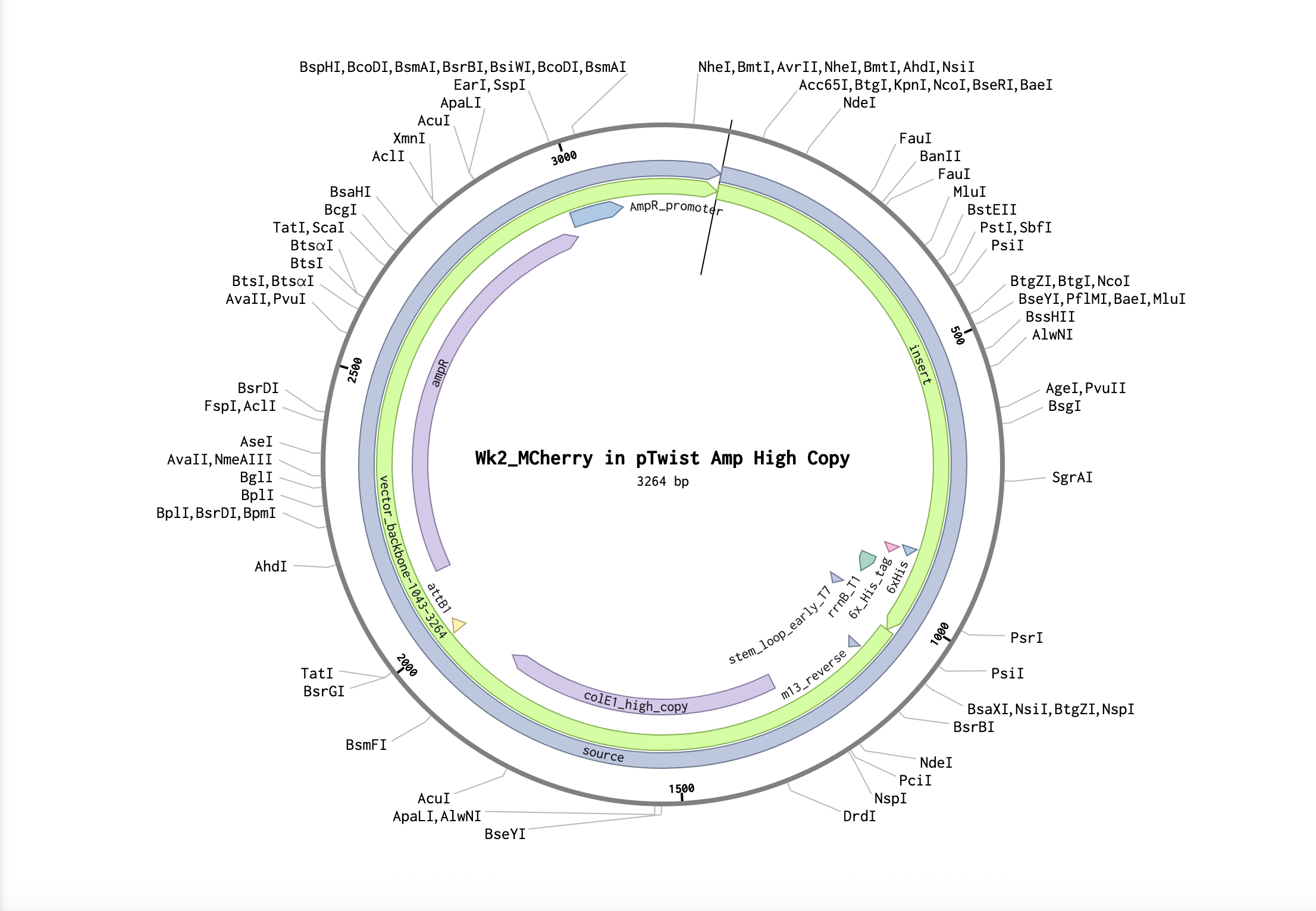
3.1 mCherry I was thinking about observation under UV illumination — red fluorescence seems easier to detect compared to blue or green light. It also has stronger visual impact.
Protein Sequence MVSKGEEDNMAIIKEFMRFKVHMEGSVNGHEFEIEGEGEGRPYEGTQTAKLKVTKGGPLPFAWDILSPQFMYGSKAYVKHPADIPDYLKLSFPEGFKWERVMNFEDGGVVTVTQDSSLQDGEFIYKVKLRGTNFPSDGPVMQKKTMGWEASSERMYPEDGALKGEIKQRLKLKDGGHYDAEVKTTYKAKKPVQLPGAYNVNIKLDITSHNEDYTIVEQYERAEGRHSTGGMDELYK Source: https://www.fpbase.org/protein/mcherry/
3.2 Original DNA Sequence atggtgagcaaaggcgaagaagataacatggcgattattaaagaatttatgcgctttaaa gtgcatatggaaggcagcgtgaacggccatgaatttgaaattgaaggcgaaggcgaaggc cgcccgtatgaaggcacccagaccgcgaaactgaaagtgaccaaaggcggcccgctgccg tttgcgtgggatattctgagcccgcagtttatgtatggcagcaaagcgtatgtgaaacat ccggcggatattccggattatctgaaactgagctttccggaaggctttaaatgggaacgc gtgatgaactttgaagatggcggcgtggtgaccgtgacccaggatagcagcctgcaggat ggcgaatttatttataaagtgaaactgcgcggcaccaactttccgagcgatggcccggtg atgcagaaaaaaaccatgggctgggaagcgagcagcgaacgcatgtatccggaagatggc gcgctgaaaggcgaaattaaacagcgcctgaaactgaaagatggcggccattatgatgcg gaagtgaaaaccacctataaagcgaaaaaaccggtgcagctgccgggcgcgtataacgtg aacattaaactggatattaccagccataacgaagattataccattgtggaacagtatgaa cgcgcggaaggccgccatagcaccggcggcatggatgaactgtataaa 3.3 Codon Optimization Why Optimize? The same amino acid can be encoded by multiple codons, but different organisms have different codon usage preferences (tRNA abundance, translation efficiency, mRNA structure, etc.). To allow the host to express the protein more efficiently, codon optimization is necessary.
Published paper Villanueva-Cañas et al., PLOS ONE (2021) built a multi-station SARS-CoV-2 RT-qPCR testing workflow using Opentrons OT-2 robots. The core novelty is a reusable software + station architecture that makes a complex diagnostic pipeline programmable, modular, and reproducible across setups.
Final project automation plan Project: “Living Ice Cream” A temperature-responsive dessert system with:
Protein & Amino Acid Questions 1) How many molecules of amino acids are in 500 g of meat? (Assume average amino acid ≈ 100 Da ≈ 100 g/mol)
Upper-bound estimate: treat the 500 g as entirely amino acids.
Moles of amino acids ≈ 500 g / (100 g/mol) = 5 mol Number of molecules ≈ 5 mol × 6.02 × 10²³ molecules/mol ≈ 3.0 × 10²⁴ amino acid molecules 2) Why do humans eat beef but do not become a cow? Dietary proteins are not incorporated intact into our bodies.
Part A: SOD1 Binder Peptide Design 1. Background and Design Goal Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, SOD1 forms a stable homodimer and binds copper and zinc cofactors. Mutations in SOD1 are associated with familial ALS; among them, the A4V variant is one of the aggressive disease-associated variants and has been linked to SOD1 destabilization, altered folding behavior, and toxic aggregation.
DNA Assembly 1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? Phusion High-Fidelity PCR Master Mix contains Phusion DNA Polymerase, dNTPs, reaction buffer, and MgCl₂. The DNA polymerase copies the DNA template during PCR. dNTPs are the building blocks used to synthesize the new DNA strand. The buffer keeps the reaction at the correct pH and salt condition. MgCl₂ is an important cofactor that helps the polymerase function properly. Since Phusion is a high-fidelity polymerase, it is useful when the amplified DNA sequence needs to be accurate for cloning or assembly.
Part 1: Intracellular Artificial Neural Networks (IANNs) Question 1 - Advantages of IANNs Over Traditional Genetic Circuits Traditional genetic circuits usually work through Boolean logic. An input is treated as either ON or OFF, and the circuit produces an output based on that discrete state. This is useful for simple decisions, but it is limited when the cell needs to respond to gradual or mixed signals.
General homework questions 1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell-free protein synthesis has two major advantages over traditional in vivo expression: flexibility and experimental control.
Homework: Final Project For my final project, Glowing Ice Cream, the main things I would measure are the identity, quantity, activity, and release behavior of the NanoLuc/substrate system.
Protein Identity and Purity I need to confirm whether I successfully made the right protein, NanoLuc-GSGS-His₆.
Part 1: The 1,536 Pixel Artwork Canvas | Collective Artwork For the collective bioart project, I contributed a small bacteriophage drawing in the upper-right corner of the canvas. (Later seemed to have been modified by someone else)
What I liked about this project is that it made the artwork feel genuinely collective. Instead of everyone making a separate image, the final result became a shared and evolving biological canvas. The fact that my pixel contribution could be changed by someone else was a little unexpected, but it also made the project feel more alive.
Error accumulation during synthesis and replication
Homework Questions from Dr. LeProust
3. What is the most commonly used method for oligo synthesis today?
Phosphoramidite solid-phase synthesis.
4. Why is it difficult to synthesize oligos longer than ~200 nt directly?
Each coupling step is less than 100% efficient
Errors accumulate linearly with length
Yield and purity drop exponentially
5. Why can’t a 2000 bp gene be made by direct oligo synthesis?
Error rates become prohibitive
Full-length product yield approaches zero
Long genes must be assembled from shorter oligos (e.g., Gibson assembly, PCA)
Homework Question from George Church
What are the 10 essential amino acids in all animals?
The ten essential amino acids that animals cannot synthesize de novo and must obtain from diet are:
Histidine, Isoleucine, Leucine, Lysine, Methionine, Phenylalanine, Threonine, Tryptophan, Valine, and Arginine (Arginine is conditionally essential in adults but universally essential during growth.)
How does this affect the “Lysine Contingency”?
Lysine’s essentiality reflects a deep evolutionary constraint: animals universally lost lysine biosynthesis pathways, making them metabolically dependent on external sources. This supports the “lysine contingency” as a system-level lock-in rather than an arbitrary biochemical choice. Once lysine synthesis was abandoned, translational machinery, diet, and ecological dependencies co-evolved around its availability, making reversal highly unlikely. Thus, lysine exemplifies how early metabolic decisions constrain future evolutionary trajectories.
Week 2 HW: DNA Read, Write, & Edit
3.1 mCherry
I was thinking about observation under UV illumination — red fluorescence seems easier to detect compared to blue or green light. It also has stronger visual impact.
The same amino acid can be encoded by multiple codons, but different organisms have different codon usage preferences (tRNA abundance, translation efficiency, mRNA structure, etc.). To allow the host to express the protein more efficiently, codon optimization is necessary.
Organism Selected
E. coli K-12.
It is one of the most commonly used host strains. The technical maturity and widespread adoption of this system make it highly suitable for experimental work.
(There’s unknown error for the optimization version,,,)
5.1 DNA Read
5.1(i) What DNA Would I Sequence?
A synthetic DNA library used for digital DNA data storage.
Artificially designed DNA fragments encoding digital information (text or images).
Why?
DNA here serves as an information storage medium rather than biological genetic material.
Sequencing verifies:
Whether the written digital information is preserved
Whether errors occurred during storage or amplification
The error rate (substitutions, insertions, deletions)
This is effectively a biotechnology-based data integrity check.
5.1(ii) Sequencing Technology
First-generation sequencing: Sanger sequencing.
Why?
High accuracy
Suitable for validating single fragments
Characteristics
Reads one DNA template at a time
Read length ~700–900 bp
Very high accuracy
Inputs
Template DNA
Primer
DNA polymerase
dNTPs
Fluorescently labeled ddNTPs
Core Principle
DNA synthesis is terminated at random positions using ddNTPs.
Process:
Polymerase copies the template
ddNTP incorporation stops elongation
Fragments of different lengths are produced
Fragments separated by size
Fluorescent signal read to reconstruct sequence
Output
DNA sequence
Chromatogram
Limitations
Cannot sequence thousands of fragments simultaneously
5.2 DNA Write
5.2(i) What DNA Would I Synthesize?
An expression cassette expressing mCherry in E. coli.
Includes:
Promoter
RBS
Codon-optimized mCherry CDS
His tag
Terminator
Reason:
Produces visible red fluorescence
Strong contrast under blue light
5.2(ii) Synthesis Technology
Solid-phase chemical DNA synthesis (phosphoramidite method) + gene assembly.
Steps
Design sequence computationally
Split into short oligos
Chemically synthesize oligos
Assemble via PCR or Gibson Assembly
Clone into vector
Sequence verify
Limitations
Error rate increases with length
Assembly required
Sequencing verification required
Cost scales with length
5.3 DNA Edit
5.3(i) What DNA Would I Edit?
A single-base mutation (e.g., disease-causing point mutation).
Reason:
Represents the most precise editing scenario
Relevant for therapeutic research
5.3(ii) Editing Technology
CRISPR-Cas9 + HDR repair template.
Principle
Design gRNA
Cas9 creates double-strand break
Provide donor DNA
HDR replaces base precisely
Required Inputs
gRNA
Cas9 protein or plasmid
Donor DNA
Target cells
Limitations
Low HDR efficiency
Possible off-target effects
Complex delivery
Cell-type dependent precision
Week 3 HW: AUTOMATION
1) Published paper
Villanueva-Cañas et al., PLOS ONE (2021) built a multi-station SARS-CoV-2 RT-qPCR testing workflow using Opentrons OT-2 robots. The core novelty is a reusable software + station architecture that makes a complex diagnostic pipeline programmable, modular, and reproducible across setups.
Visual shift (color / glow) near melt-adjacent temperatures
Why Ginkgo automation
I’m using Ginkgo’s autonomous / cloud-lab framing as an iteration engine for high-throughput DOE: stable automation backbone, fast experimental loops, and standardized readouts for repeated screening rounds.
What I will automate
A) “Breathing” kinetics screening (high-throughput DOE)
Goal: Find enzyme/substrate + formulation conditions that yield slow, non-violent micro-gas behavior around ~15–25°C.
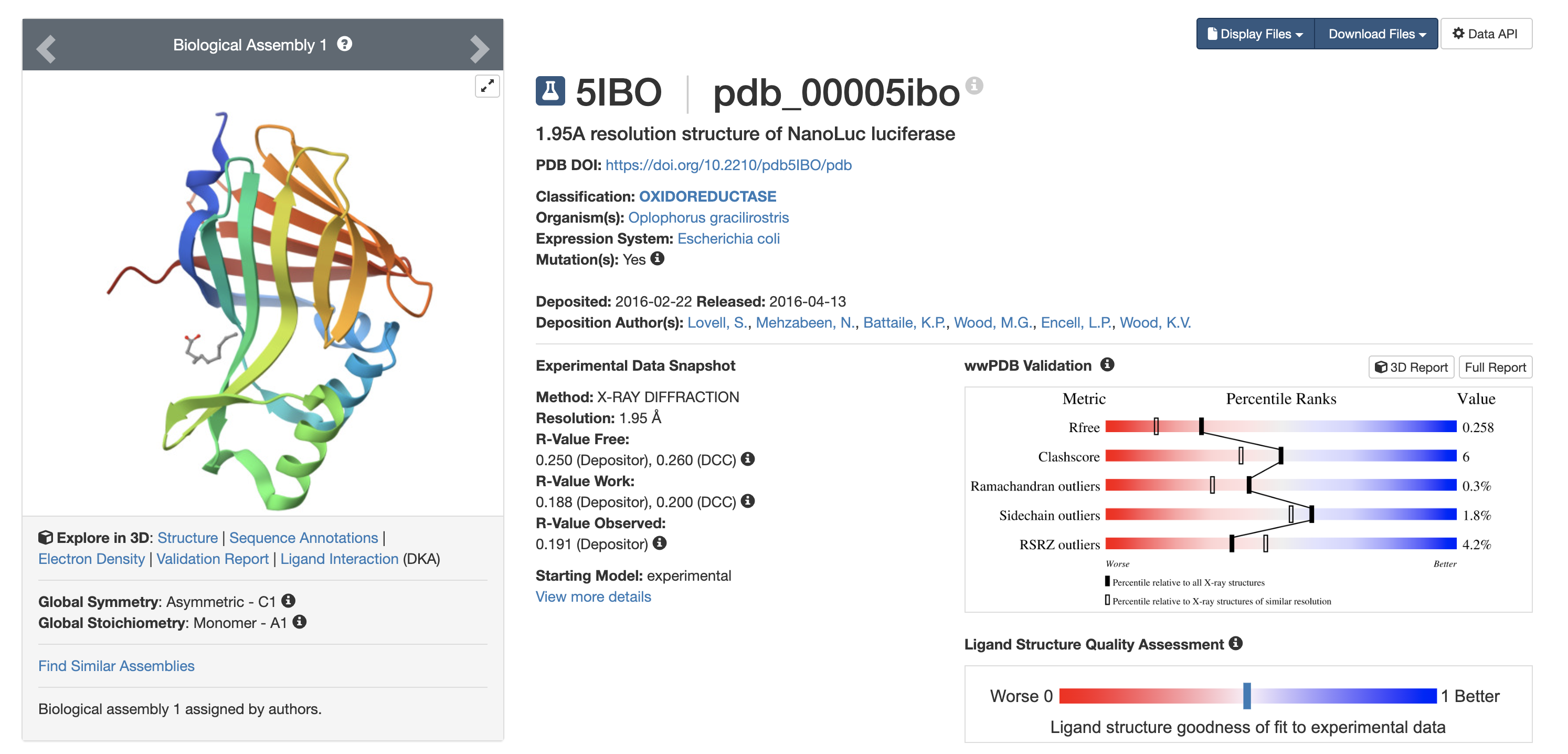
Resolution < 2.5 Å = high-quality structure 1.95 Å is considered reliable.
8️ Other Molecules in Structure
Yes. Bound ligand: Decanoic acid
Why this matters:
Indicates defined binding pocket
Active site accommodates hydrophobic molecules
Design implications for ice cream:
Does luciferin need micro-encapsulation?
Does fat content affect substrate diffusion?
Could dairy fat interact with hydrophobic pocket?
Structural data → formulation constraints.
PyMOL Visualization
Observation:
Beta-sheet dominant structure
Consistent with beta-barrel fold
Interpretation:
Beta-rich cores = tightly packed
Likely structurally stable
Implication:
May retain activity under:
Slight warming
Partial melting
Oxygen exposure
11 Residue Surface Analysis
Hydrophobic residues:
Cluster in interior
Polar/charged residues:
Dominant on surface
Interpretation:
Hydrophobic core → structural stability
Hydrophilic surface → water compatibility
From ice cream perspective:
Ice cream = water + fat + sugar
Hydrophilic exterior = favorable
Hydrophobic core remains shielded
Encouraging for enzyme stability in food matrix.
Week 5 HW: Protein Design II
Part A: SOD1 Binder Peptide Design
1. Background and Design Goal
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that
converts superoxide radicals into hydrogen peroxide and oxygen. In its
native state, SOD1 forms a stable homodimer and binds copper and zinc
cofactors. Mutations in SOD1 are associated with familial ALS; among
them, the A4V variant is one of the aggressive disease-associated
variants and has been linked to SOD1 destabilization, altered folding
behavior, and toxic aggregation.
In this section, I designed 12-amino-acid peptide binders against A4V
mutant SOD1 and evaluated which candidates showed enough computational
evidence to justify experimental validation. I treated this as an
early-stage computational screening exercise rather than proof of
therapeutic activity.
2. A4V Mutant SOD1 Sequence
I retrieved the human SOD1 canonical sequence from UniProt (P00441). I
introduced the disease-associated A4V mutation by changing the alanine
in the original N-terminal sequence MATKAV… to valine, resulting in
MATKVV…. Because I included the initiating methionine in the
AlphaFold3 input sequence, this disease-associated A4V position appears
as VAL5 in the AlphaFold viewer.
A4V mutant SOD1 sequence used for all computational submissions:
Note: I used VAL5 as the visual reference for the A4V site when
inspecting AlphaFold3 outputs.
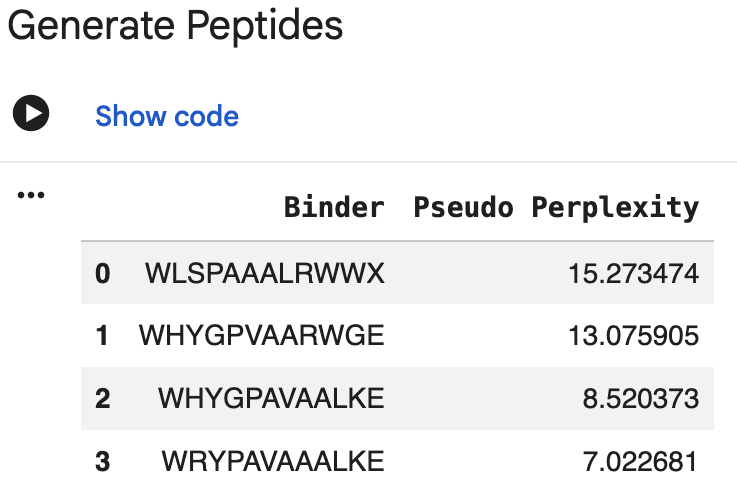
3. PepMLM Peptide Generation
I generated four 12-aa peptide binders using PepMLM conditioned on the
A4V mutant SOD1 sequence. One initial output contained an unknown amino
acid symbol (X), so I discarded it and regenerated an additional
candidate. I also included the known SOD1-binding peptide FLYRWLPSRRGG
as a reference control.
Peptide ID
Sequence
Source
Length
PepMLM pseudo perplexity
P1
WHSGATAAAWKE
PepMLM
12 aa
7.160
P2
WHYGPVAARWGE
PepMLM
12 aa
13.076 (highest; least plausible)
P3
WHYGPAVAALKE
PepMLM
12 aa
8.520
P4
WRYPAVAAALKE
PepMLM
12 aa
7.023 (lowest; most plausible)
Known binder
FLYRWLPSRRGG
Literature control
12 aa
N/A
Interpretation: Lower pseudo perplexity indicates that the model views
the sequence as more plausible given the SOD1 target. Based on this
metric, P4 and P1 ranked as the strongest PepMLM outputs, while P2
ranked as the weakest. However, pseudo perplexity reflects sequence
plausibility, not physical binding affinity or interface confidence. I
therefore used AlphaFold3 as the next structural evaluation step.
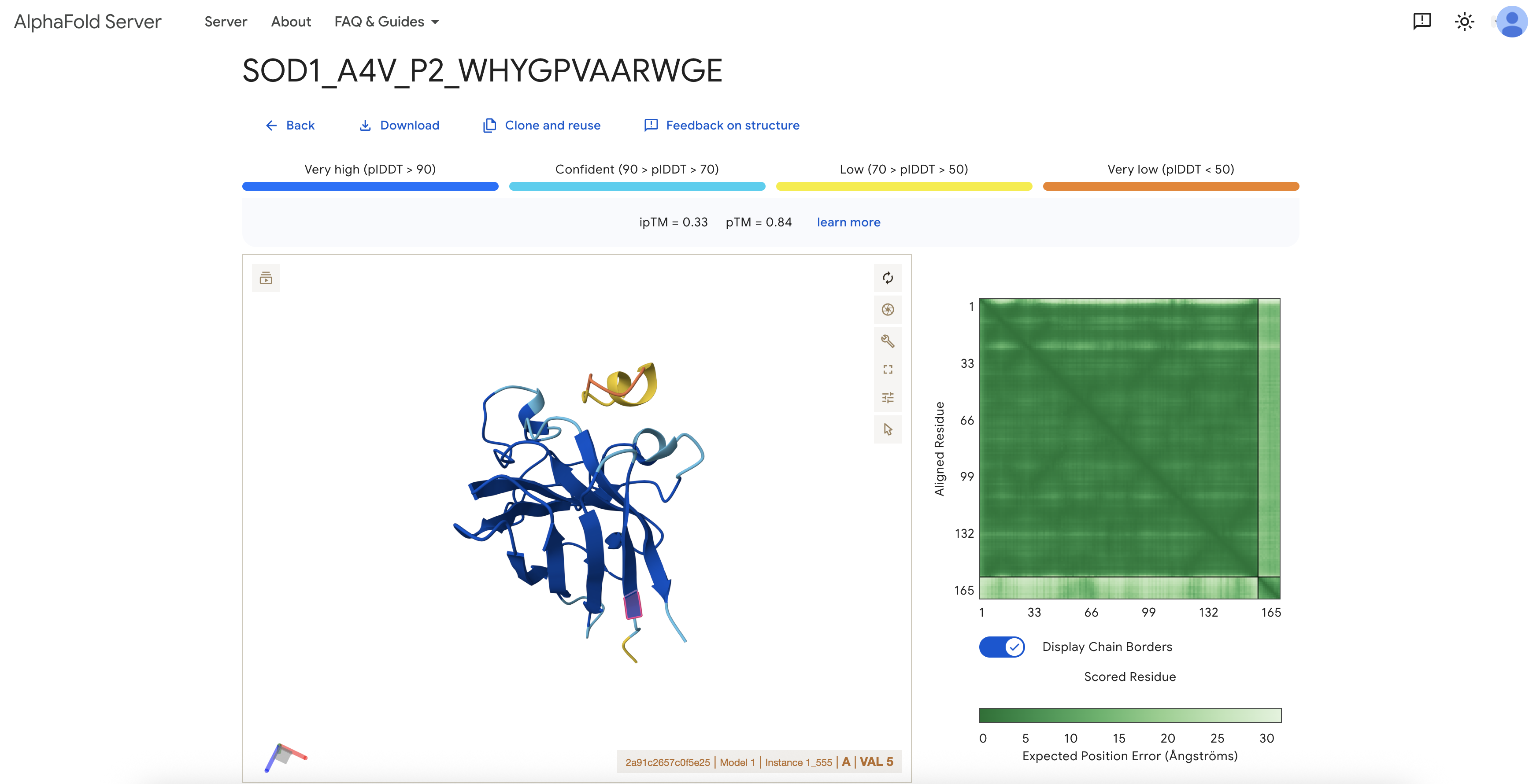
4. AlphaFold3 Structural Evaluation
I modeled each peptide as a separate chain alongside A4V mutant SOD1
using the AlphaFold3 server. The SOD1 chain was submitted as Chain A and
the peptide as Chain B; no fusion was introduced. I used AlphaFold3 to
test whether each peptide could form a plausible protein-peptide complex
with SOD1.
Note on VAL5: Because the AlphaFold input includes the initiating
methionine, the A4V mutation site corresponds to residue VAL5 in the
output structure. All localization descriptions below use this
numbering.
Peptide ID
Sequence
ipTM
pTM
Near VAL5 / A4V?
Structural interpretation
P1
WHSGATAAAWKE
—
—
Not assessed
AlphaFold3 result was not available in this run; evaluated via PeptiVerse only.
P2
WHYGPVAARWGE
0.33
0.84
Not clearly near VAL5
Low-confidence interface; SOD1 structure itself was reasonably predicted.
P3
WHYGPAVAALKE
0.34
0.80
No
Highest available ipTM in my completed runs, but still low; peptide appeared as a low-confidence yellow/orange segment outside the SOD1 beta-barrel and did not engage VAL5.
P4
WRYPAVAAALKE
0.29
0.74
Not clearly near VAL5
Very low interface confidence; lowest pTM of the completed set.
Known binder
FLYRWLPSRRGG
0.29
0.78
Not clearly near VAL5
Also low ipTM in this setup, suggesting that AlphaFold3 interface prediction was uncertain for these short peptides.
Structural interpretation: All available ipTM scores were low
(0.29–0.34). The SOD1 monomer itself was modeled with reasonable
confidence (pTM 0.74–0.84), but the protein-peptide interfaces were not
predicted with high confidence in any case. Inspection of the P3 model
around VAL5 showed that the peptide appeared as a yellow/orange
low-confidence segment outside the main SOD1 beta-barrel, with no clear
engagement of the A4V site. The known SOD1-binding peptide also produced
a low ipTM in this setup, suggesting that low ipTM should not be
interpreted as definitive evidence against binding; rather, this
workflow did not produce high-confidence SOD1-peptide interfaces for any
tested sequence.
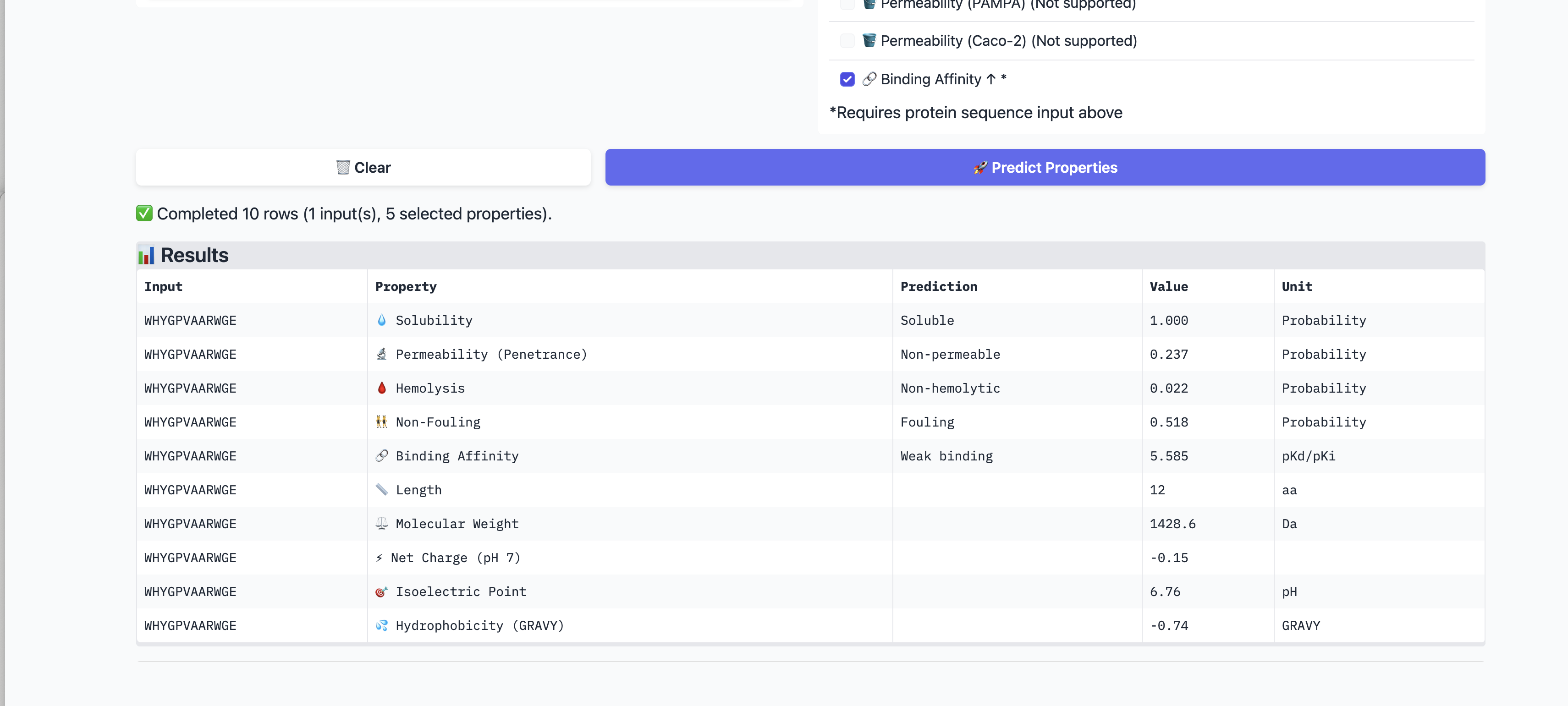
5. PeptiVerse Therapeutic Property Evaluation
I evaluated all four PepMLM-generated peptides and the known binder
using PeptiVerse. I supplied the A4V mutant SOD1 sequence as the target
for binding affinity prediction. I assessed solubility, hemolysis
probability, binding affinity (pKd/pKi), molecular weight, net charge at
pH 7, permeability, non-fouling behavior, and GRAVY index.
ID
Sequence
Solubility
Hemolysis
Binding affinity
MW
Charge pH7
Perm.
Non-fouling
GRAVY
P1
WHSGATAAAWKE
1.000
0.013
Weak 5.360 pKd
1314 Da
-0.15
0.528
0.593
-0.59
P2
WHYGPVAARWGE
1.000
0.022
Weak 5.585 pKd
1429 Da
-0.15
0.237
Fouling
-0.74
P3
WHYGPAVAALKE
1.000
0.031
Weak 5.408 pKd
1342 Da
-0.15
0.099
0.647
-0.12
P4
WRYPAVAAALKE
1.000
0.039
Weak 5.462 pKd
1375 Da
+0.77
0.331
0.697
-0.04
Known
FLYRWLPSRRGG
1.000
0.047
Weak 5.550 pKd
1508 Da
+2.76
0.862
0.652
-0.71
PeptiVerse findings: All peptides were predicted to be soluble (1.000)
and had low hemolysis probabilities (0.013–0.047), which is a favorable
developability signal. PeptiVerse classified every candidate, including
the known binder, as a weak binder in a narrow 5.36–5.59 pKd range.
P1 had the cleanest overall property profile: lowest hemolysis (0.013),
near-neutral charge (-0.15), and no fouling flag. Its binding affinity
prediction was not strong, but none of the candidates showed convincing
affinity in PeptiVerse.
P2 showed the highest predicted binding affinity among the PepMLM
peptides, but it had the worst PepMLM pseudo perplexity and was flagged
as fouling, making it less attractive overall.
P4 had the best PepMLM pseudo perplexity and acceptable PeptiVerse
properties, but its AlphaFold3 ipTM was very low (0.29).
P3 had the highest available AlphaFold3 ipTM (0.34) among the completed
runs, but this is still a low-confidence interface. Its PeptiVerse
profile was acceptable but not outstanding.
Comparison with the known binder: FLYRWLPSRRGG had a slightly higher
predicted affinity than several generated peptides and the highest
predicted permeability, but it also had the highest hemolysis
probability among this small set. Because PeptiVerse also classified the
known binder as weak, I treated these predictions as useful screening
signals rather than definitive binding evidence.
6. moPPIt Targeted Design Attempt
Following the PepMLM and AlphaFold3 analysis, I attempted a more
targeted design step using moPPIt. The goal was to bias peptide
generation toward residues 5–10 of the A4V SOD1 sequence, which include
VAL5 / the A4V site and adjacent N-terminal residues, rather than
allowing the peptide to bind anywhere on the protein surface.
Parameter
Value
Target protein
A4V mutant SOD1 (154 aa)
Peptide length
12 aa
Motif positions
Residues 5–10 (VVCVLK)
Main objectives
Affinity + Motif
Additional objectives attempted
Hemolysis, Solubility
Number of samples
10
Runtime failure: The public moPPIt Colab notebook failed during model
initialization due to a missing PeptiVerse artifact. The error message
was:
FileNotFoundError: No best_model artifact found in
PeptiVerse/training_classifiers/nf/enet_gpu_smiles
This error indicates that the Non-Fouling classifier, which is part of
the PeptiVerse suite used internally by moPPIt, was not present in the
public notebook environment. Because the failure occurred at model
loading, no moPPIt peptides were generated. I therefore documented the
intended targeted-design setup but did not include moPPIt-generated
candidates in the final results. If the notebook is updated or a local
installation is available, running moPPIt with motif guidance on
residues 5–10 would be my recommended next step.
7. Final Candidate Interpretation
No peptide in this workflow emerged as a high-confidence SOD1 binder.
The key reasons are:
AlphaFold3 ipTM scores were low across all completed runs (0.29–0.34),
below what I would expect for a confident protein-peptide interface.
None of the inspected peptides clearly bound near VAL5 / A4V, the
biologically relevant region of the mutant protein.
PeptiVerse classified all candidates, including the known literature
binder, as weak binders. Solubility and hemolysis profiles were
uniformly good, but binding affinity predictions were not convincing.
moPPIt could not be completed due to a missing model artifact in the
public Colab environment.
Provisional candidate selection: If one peptide must be selected from
the completed AlphaFold3 runs, P3 (WHYGPAVAALKE) is the provisional
structural candidate only because it had the highest available ipTM
score (0.34). However, the difference between candidates is small and
the absolute ipTM value remains low, so this should not be interpreted
as strong binding evidence.
An alternative choice is P1 (WHSGATAAAWKE), which had the cleanest
PeptiVerse property profile: the lowest hemolysis probability,
near-neutral charge, and no fouling flag. If P1 is later submitted to
AlphaFold3 and produces a substantially higher ipTM than the other
candidates, it would become a more reasonable candidate for additional
screening.
My honest conclusion at this stage is that the current PepMLM peptides
are plausible 12-aa sequences with good basic developability properties,
but they do not yet show strong computational evidence of specific
binding to the A4V SOD1 region. The most productive next step would be a
targeted redesign using a functioning moPPIt setup, or an equivalent
motif-guided method, focused on residues 5–10 around VAL5.
8. Conclusion
I generated four 12-aa peptide candidates with PepMLM and evaluated them
alongside the known SOD1-binding peptide FLYRWLPSRRGG. PepMLM pseudo
perplexity scores ranked P4 and P1 as the most plausible sequences.
PeptiVerse predicted that all candidates were soluble and had low
hemolysis risk, but classified all as weak binders. AlphaFold3 did not
produce high-confidence SOD1-peptide interfaces (ipTM 0.29–0.34), and no
peptide clearly engaged the VAL5/A4V region. I attempted a targeted
moPPIt design run targeting residues 5–10, but the public notebook could
not complete generation due to a missing model artifact.
This workflow demonstrates both the utility and the limitations of
computational peptide design. PepMLM provides rapid plausible sequences;
PeptiVerse filters for developability properties; AlphaFold3 tests
structural plausibility of interfaces. However, none of these tools
alone constitutes proof of binding. In this case, the combined evidence
indicates that none of the current peptides should be advanced as a
therapeutic candidate without further targeted design and experimental
binding validation.
Part C: Final Project — L-Protein Mutants
1. Background and Objective
The MS2 bacteriophage L-protein is a small lysis protein that
contributes to bacterial lysis by disrupting the E. coli inner membrane.
This mechanism is relevant to phage-based strategies against
antibiotic-resistant infections. DnaJ, an E. coli chaperone, appears to
support normal L-protein processing, so mutations that make L-protein
more stable or more capable of auto-folding could help reduce dependence
on host chaperone assistance.
Because my Part C analysis is less complete than Part A, I treat the
following section as a provisional design proposal. Any specific
mutation choice should be checked against the course mutagenesis-score
table and experimental lysis dataset before final experimental
submission.
Likely related to DnaJ interaction, folding initiation, and processing.
Transmembrane region
41–75
Likely related to membrane insertion, pore formation, and lysis activity.
Design logic: I would avoid known essential or highly conserved
residues, especially LS motifs and strongly conserved hydrophobic
positions in the transmembrane region. I would prioritize positions that
are less conserved, experimentally tolerated, and mechanistically likely
to improve folding or membrane insertion.
3. Provisional Mutant Design Strategy
I would use three filters to select L-protein variants: (1) mutagenesis
score, (2) experimental lysis behavior relative to wild type, and (3)
conservation status from sequence alignment. Since my current notes do
not fully verify every score and lysis percentage, I avoid treating the
specific candidates below as final. They are proposed directions to
check against the assignment dataset.
Provisional variant
Region
Why it is interesting
Uncertainty / check needed
Q8K
Soluble region
May alter surface charge in the N-terminal region and test whether DnaJ-related recognition can be changed without touching the transmembrane lysis domain.
Verify score, conservation, and lysis data before final use.
F22L
Soluble region
A conservative hydrophobic substitution that may reduce aromatic recognition while preserving general hydrophobic character.
Verify that the position is not strongly conserved and that lysis remains acceptable.
A45G
Transmembrane edge
Could introduce local flexibility near the start of the TM region and test whether flexibility helps membrane insertion.
Higher-risk candidate; verify that lysis is not strongly reduced.
N53A
Transmembrane core
A polar-to-nonpolar change that may increase TM hydrophobicity and potentially improve autonomous membrane insertion.
Strong candidate only if supported by positive score and maintained/improved lysis.
Q54A
Transmembrane core
Similar logic to N53A; may improve hydrophobic compatibility of the TM region. Could also be tested later as part of an N53A/Q54A double-mutant hypothesis.
Verify single-mutant performance before considering double mutant.
4. Tentative Priority
My tentative priority would be to first validate one soluble-domain
mutant and one transmembrane-region mutant rather than immediately
making a complex multi-mutant design. If the assignment dataset supports
that N53A and/or Q54A maintain or improve lysis, I would prioritize them
as the most mechanistically plausible transmembrane candidates. If Q8K
maintains lysis, I would prioritize it as the most useful soluble-domain
candidate for probing DnaJ-dependence effects.
5. Structural Validation Plan
As a qualitative check, I would use AlphaFold2 Multimer or a similar
structure-prediction workflow to compare wild-type and mutant L-protein
assemblies. For soluble-domain candidates, I would also consider
co-folding residues 1–40 with the DnaJ J-domain to see whether the
predicted interaction surface changes. I would treat these models as
qualitative support only, because membrane proteins and phage lysis
assemblies are difficult for structure predictors. The decisive test
would still be an experimental lysis assay, ideally including conditions
that test DnaJ dependence.
6. Definition of a Successful Mutant
A successful L-protein mutant should:
retain overall L-protein structural integrity, especially in the
transmembrane region;
avoid disrupting LS motifs or other known essential residues;
maintain or improve lysis activity relative to wild type;
show evidence of improved stability, auto-folding, or reduced DnaJ
dependence;
be practical to synthesize, express, and test within the course
workflow.
In short, I would not define a good mutant only by the highest
computational score. I would define it as a mutation that balances model
support, experimental lysis behavior, conservation logic, and a clear
biological mechanism.
Week 6 HW: DNA Assembly
DNA Assembly
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion High-Fidelity PCR Master Mix contains Phusion DNA Polymerase, dNTPs, reaction buffer, and MgCl₂. The DNA polymerase copies the DNA template during PCR. dNTPs are the building blocks used to synthesize the new DNA strand. The buffer keeps the reaction at the correct pH and salt condition. MgCl₂ is an important cofactor that helps the polymerase function properly. Since Phusion is a high-fidelity polymerase, it is useful when the amplified DNA sequence needs to be accurate for cloning or assembly.
2. What are some factors that determine primer annealing temperature during PCR?
Primer annealing temperature mainly depends on the primer’s melting temperature, or Tm. Tm is affected by primer length, GC content, sequence composition, and whether the primer has mismatches with the template. Longer primers and higher GC content usually increase the Tm. Salt concentration and buffer conditions can also affect annealing. In practice, the annealing temperature should be high enough to avoid nonspecific binding, but not so high that the primer cannot bind efficiently.
3. Compare PCR and restriction enzyme digests as ways to create linear DNA fragments.
PCR creates linear DNA fragments by amplifying a selected region from a DNA template using primers. It is flexible because primers can be designed to add extra sequences, such as Gibson overlaps, mutations, or restriction sites. Restriction enzyme digest creates linear fragments by cutting DNA at specific recognition sites. It is simpler when useful cut sites already exist in the plasmid or insert, but it is limited by where those enzyme sites are located. PCR is preferable when custom fragment boundaries or added overlap sequences are needed. Restriction digest is preferable when the construct already has clean enzyme sites and a more direct cut-and-paste workflow is possible.
4. How can you ensure that digested or PCR-ed DNA is appropriate for Gibson cloning?
For Gibson cloning, each neighboring DNA fragment needs compatible overlapping ends. Primers can be designed so that PCR products contain overlap regions matching the adjacent fragment or vector. The overlap should be long enough for efficient assembly, usually around 15–30 bp depending on the protocol and sequence composition. The fragments also need to be in the correct order and orientation, without unwanted mutations. The vector should be linearized only once. After PCR or digestion, fragment size can be checked by gel electrophoresis, and the final assembled construct should ideally be confirmed by sequencing.
5. How does plasmid DNA enter the E. coli cells during transformation?
During transformation, plasmid DNA enters chemically competent or electrocompetent E. coli cells. In heat-shock transformation, calcium chloride treatment helps make the cell membrane more permissive to DNA, and the short heat shock creates a temporary condition that allows plasmid DNA to pass into the cells. In electroporation, an electrical pulse briefly opens pores in the bacterial membrane, allowing DNA to enter. After transformation, the cells recover in growth medium and are plated on antibiotic plates. Only the cells that successfully took up the plasmid with the antibiotic resistance marker should grow.
6. Golden Gate Assembly.
Golden Gate Assembly is a DNA assembly method that uses Type IIS restriction enzymes and DNA ligase in the same reaction. Type IIS enzymes, such as BsaI or BsmBI, recognize a specific DNA sequence but cut outside of that recognition site. This allows the designer to create custom sticky ends, or overhangs, that determine the order of DNA fragments. After the enzyme cuts the fragments, T4 DNA ligase joins compatible overhangs together. Because the recognition sites can be designed to disappear from the final product, Golden Gate can produce scarless assemblies. It is especially useful for modular assembly of multiple DNA parts, such as promoter, RBS, coding sequence, and terminator. Unlike Gibson Assembly, which relies on longer homologous overlaps, Golden Gate relies on short designed overhangs produced by restriction enzyme cutting.
Golden Gate Assembly Diagram
Week 7: Genetic Circuits Part II: Neuromorphic Circuits
Part 1: Intracellular Artificial Neural Networks (IANNs)
Question 1 - Advantages of IANNs Over Traditional Genetic Circuits
Traditional genetic circuits usually work through Boolean logic. An input is treated as either ON or OFF, and the circuit produces an output based on that discrete state. This is useful for simple decisions, but it is limited when the cell needs to respond to gradual or mixed signals.
IANNs have several advantages over that kind of circuit.
Continuous input processing
IANNs can respond to graded signals, such as different concentrations of transcription factors, proteins, or small molecules. Instead of only detecting whether a signal is present or absent, the circuit can respond differently to low, medium, and high input levels.
Weighted signal integration
In an IANN, each input can contribute with a different strength. For example, one marker could have a strong positive effect on the output, while another marker has a weaker or inhibitory effect. This makes the circuit more flexible than a simple AND/OR gate, where inputs are usually treated more equally.
More complex classification
Because IANNs can combine several continuous inputs, they can classify cellular states that are hard to define with simple Boolean gates. A cell state may depend on the relative levels of multiple markers rather than one clear signal. An IANN is better suited for this kind of decision.
Potential adaptability
If the strength of regulatory connections can be tuned over time, an IANN could also have some capacity for adaptation or memory. This would be difficult to achieve with a fixed Boolean circuit.
Overall, IANNs are useful because they allow genetic circuits to behave more like analog decision-making systems rather than simple switches.
Question 2 - A Useful Application for an IANN
Concept: Smart CAR-T Cells for Solid Tumor Targeting
One application I would be interested in is a CAR-T cell that uses an IANN to decide whether a target cell is likely to be a tumor cell. A major problem in CAR-T therapy, especially for solid tumors, is off-target toxicity. Many tumor-associated antigens are also present at lower levels in healthy tissue, so a single-input circuit can be too blunt.
In this design, the engineered T cell would read several continuous inputs at the same time:
X1: surface density of a tumor-associated antigen, such as HER2
X2: level of an immune checkpoint ligand, such as PD-L1
X3: level of a hypoxia-related signal, such as HIF-1alpha, which is often higher in a tumor microenvironment
Each input would feed into a regulatory layer with a different weight. The final output would control a killing response, such as expression or activation of a cytotoxic program. The cell would only respond strongly when the weighted sum of the inputs passes a threshold.
This would be useful because the circuit would not rely on a single marker. A healthy cell might express a small amount of HER2, but it may not also show the same PD-L1 and hypoxia-related profile as a tumor cell. The IANN would allow the CAR-T cell to respond to a combined pattern instead of one isolated signal.
There are also clear limitations. Gene expression is noisy, so the same circuit may behave differently across individual cells. It would also be hard to tune the weights precisely, because biological regulatory parts are not as predictable as electronic components. Another issue is timing. Transcription and translation are relatively slow, so the circuit may not respond quickly enough in every therapeutic setting. Finally, patient-to-patient and cell-to-cell variability could make the behavior of the engineered T-cell population difficult to predict.
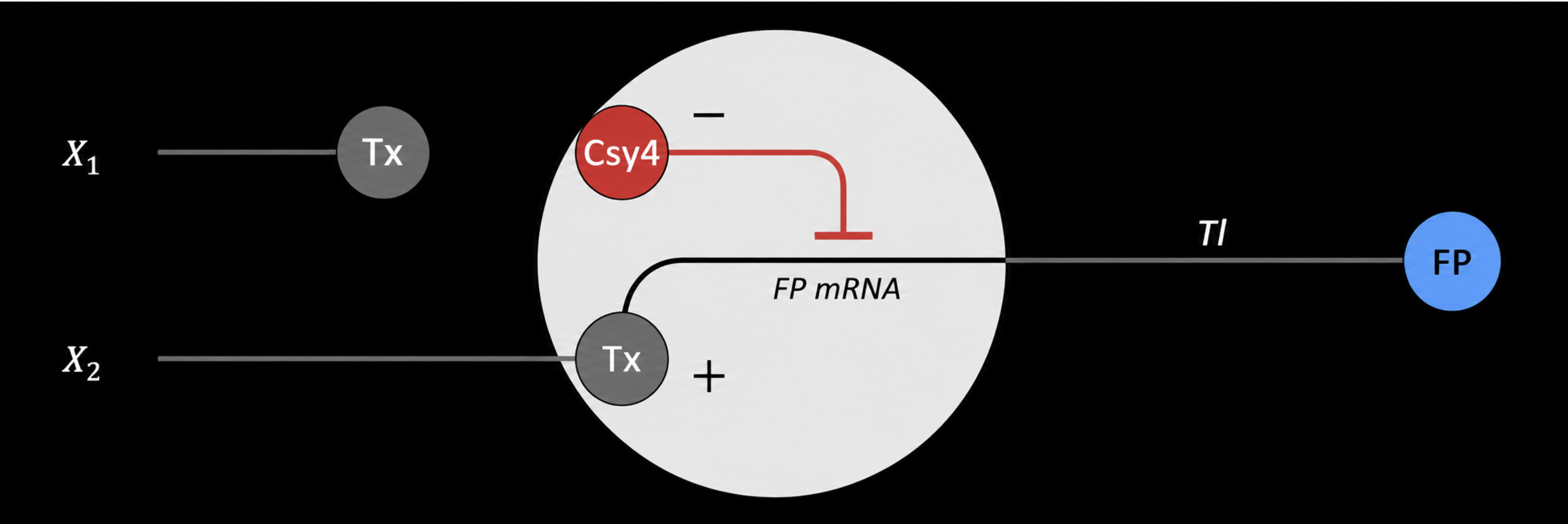
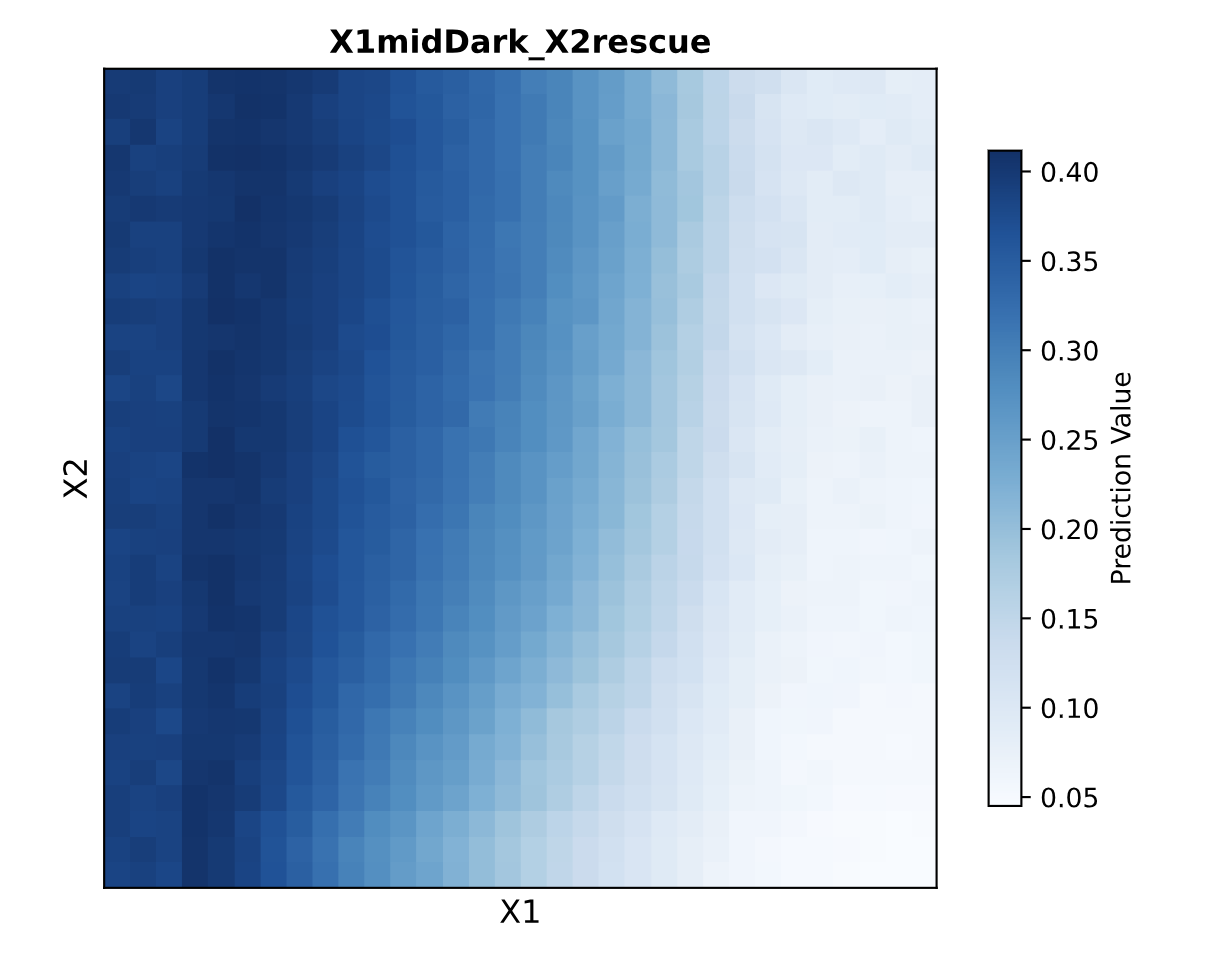
In this design, X1 drives the production of an endoribonuclease, such as Csy4. This creates the intermediate regulatory signal in the first layer.
X2 drives the production of fluorescent protein mRNA. This mRNA contains a recognition site for the endoribonuclease made from X1. When Csy4 is present, it can cleave or regulate the FP mRNA, changing how much fluorescent protein is translated.
The final fluorescent output depends on both inputs. X2 provides the transcript for the fluorescent protein, while X1 controls a regulator that changes how efficiently that transcript becomes protein. In that sense, the system behaves like a simple multilayer perceptron: the first layer converts one input into a regulatory molecule, and the second layer combines that regulatory signal with another input to control the final output.
Part 2: Fungal Materials
Question 1 - Existing Fungal Materials: Examples, Advantages, and Disadvantages
Fungal materials are often based on mycelium, the root-like network of fungal hyphae. These materials are already being explored in packaging, fashion, food coatings, and building materials.
Mycelium-Based Packaging
One common example is mycelium packaging. Companies such as Ecovative grow mycelium on agricultural waste, such as hemp hurd or corn stalks, inside molds. After the material grows into the desired shape, it is dried or heat-treated so it becomes a stable packaging block. It can be used as an alternative to expanded polystyrene foam.
Feature
Mycelium Packaging
EPS / Styrofoam
Biodegradability
Compostable under the right conditions
Persists for a very long time
Feedstock
Can use agricultural waste
Petroleum-derived
Shape control
Can be grown in molds
Easily molded through industrial processing
Cushioning
Good enough for many packaging uses
Very strong and lightweight
Moisture resistance
Usually weaker unless coated or treated
Strong moisture resistance
Cost and scale
Improving, but still less mature
Very cheap and highly scaled
The main advantage of mycelium packaging is that it can use waste biomass and break down after use. The main disadvantages are moisture sensitivity, slower production, and the difficulty of competing with the cost and performance of mature plastic foams.
Mycelium Leather Alternatives
Another example is mycelium-based leather alternatives. Bolt Threads developed Mylo, which was used in limited products and prototypes by brands such as Stella McCartney, Adidas, and lululemon. MycoWorks makes Reishi, another mycelium-based material aimed at leather-like applications.
Compared with animal leather, mycelium leather alternatives can avoid animal use and may require less land and water. They can also be grown into sheets with some control over thickness, texture, and surface quality.
The disadvantages are still important. These materials are expensive to produce, and their durability, abrasion resistance, and long-term aging are still being tested against traditional leather. Some products may also require finishing layers, backing materials, or coatings, which can make end-of-life composting more complicated.
Other Examples
Fungal chitosan can be used for films, food coatings, and biodegradable packaging. These materials are interesting because chitosan has film-forming and antimicrobial properties.
Mycelium biocomposites are also being explored for insulation panels and acoustic materials. In those cases, mycelium is useful because it can bind plant fibers together into a lightweight structure. The challenge is that building materials need to meet strict requirements for strength, fire behavior, moisture resistance, and long-term stability.
Question 2 - Genetically Engineering Fungi: What and Why
If I were engineering fungi for materials, I would focus on improving mechanical strength and moisture resistance. Those are two major limits for many mycelium-based products. Native mycelium networks are useful because they grow into a shape and bind biomass together, but they can still be brittle or sensitive to water.
One possible direction would be to engineer fungi to produce stronger structural proteins as they grow. For example, a fungus could be designed to secrete silk-like proteins into the mycelium matrix. This could make the final material tougher and more flexible while keeping the basic advantage of mycelium: it grows into the structure itself.
Another direction would be controlling degradation rate. For packaging, fast composting is useful. For furniture, shoes, or building materials, the material needs to last much longer during normal use. By tuning genes involved in cell wall formation and breakdown, such as chitin synthases or glucanases, it may be possible to make fungal materials that are stable in use but degrade under specific composting conditions.
Fungi are also a strong chassis for synthetic biology compared with bacteria for several reasons.
First, fungi are eukaryotes, so they are often better at folding and secreting complex proteins than bacteria such as E. coli. This matters if the engineered material depends on secreted enzymes or structural proteins.
Second, filamentous fungi naturally form a physical network. With bacteria, the cells usually need to be harvested and processed into a material after growth. With mycelium, the organism is already building a material as it grows. That makes genetic engineering directly connected to the material-forming process.
Third, many fungi can grow on lignocellulosic feedstocks, including agricultural waste. This is useful for biomaterials because the feedstock is cheap and renewable.
Finally, some industrial fungi, such as Aspergillus and Trichoderma species, are already used for high-level protein secretion. That makes them attractive if the goal is to produce and export useful proteins directly into a growing material.
Overall, I would use fungal synthetic biology to make mycelium materials stronger, more water-resistant, and more controllable after disposal. The strongest reason to use fungi is that the growth process and the material-making process are closely linked.
Week 9 HW: Cell-Free Systems
General homework questions
1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis has two major advantages over traditional in vivo expression: flexibility and experimental control.
First, it is more flexible because it does not require maintaining living cells. That means researchers can rapidly test different DNA templates, reaction conditions, cofactors, salts, temperatures, and additives without worrying about whether the host cell survives. This is especially useful when the protein of interest is toxic, unstable, or difficult for living cells to produce.
Second, it offers much tighter control over experimental variables. In a cell-free system, the researcher can directly define the reaction environment, including magnesium concentration, pH, energy source, template concentration, and molecular chaperones. In contrast, in vivo systems are influenced by the cell’s own metabolism, stress responses, membrane transport, and growth state, which makes it harder to isolate specific causes of success or failure.
At least two cases where cell-free expression is more beneficial than cell production are:
When expressing toxic proteins that would harm or kill living cells.
When doing rapid prototyping or screening, because many conditions can be tested quickly without cloning and culturing cells.
Additional useful cases include membrane protein optimization, incorporation of noncanonical amino acids, and applications where living cells are undesirable for safety or regulatory reasons.
2. Describe the main components of a cell-free expression system and explain the role of each component.
A cell-free expression system typically contains the following major components:
1. Cell lysate or extract This is the core biological machinery taken from broken cells. It contains ribosomes, translation factors, tRNAs, aminoacyl-tRNA synthetases, and many enzymes needed for transcription and translation.
2. Genetic template (DNA or RNA) This provides the instructions for the protein to be produced. In many systems, plasmid DNA or linear DNA is used as the template for transcription and translation.
3. Amino acids These are the building blocks of proteins. They are required for translation.
4. Nucleotides ATP, GTP, CTP, and UTP are needed for transcription, and ATP/GTP are also consumed during translation and other reaction steps.
5. Energy regeneration system Because protein synthesis uses large amounts of ATP and GTP, the reaction needs a system to regenerate usable energy and keep the reaction going for longer.
6. Salts and buffer These maintain the chemical environment needed for the reaction. Magnesium and potassium are especially important because they affect ribosome function and overall expression efficiency.
7. Optional additives Depending on the application, the system may also include chaperones, disulfide bond helpers, detergents, liposomes, nanodiscs, crowding agents, or protease inhibitors. These help support difficult proteins or special reaction goals.
Together, these parts recreate the minimum biochemical environment needed for gene expression outside a living cell.
3. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy regeneration is critical in cell-free systems because transcription and translation consume large amounts of ATP and GTP. Unlike living cells, cell-free systems do not have a full self-sustaining metabolism, so the reaction can quickly stall if energy molecules are depleted. Without continuous energy supply, protein yield drops, reaction time shortens, and the system becomes inefficient.
One method to ensure continuous ATP supply is to use an energy regeneration substrate such as phosphoenolpyruvate (PEP). In this setup, PEP donates phosphate groups that help recycle ATP during the reaction. This extends the active lifetime of the cell-free system and improves protein yield.
Other energy systems, such as creatine phosphate or 3-phosphoglycerate, can also be used, but PEP is a standard and effective example.
4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic cell-free systems, such as E. coli-based lysates, are generally faster, cheaper, and easier to optimize. They often provide high yields and are excellent for bacterial proteins or simple reporters. However, they are less suitable for proteins that require complex post-translational modifications or advanced folding machinery.
Eukaryotic cell-free systems, such as wheat germ, rabbit reticulocyte, insect, or mammalian extracts, are better for proteins that need more complex folding environments or eukaryotic-specific processing. These systems are usually more expensive and can be harder to optimize, but they are better suited for many eukaryotic proteins.
A good example for a prokaryotic system would be GFP. GFP is relatively straightforward to express, does not require complicated post-translational modification, and works well in E. coli-based cell-free systems.
A good example for a eukaryotic system would be a mammalian membrane receptor, such as a GPCR fragment or another receptor protein. This type of protein often requires a more eukaryotic-like folding environment and is harder to produce correctly in bacterial systems.
In short, prokaryotic systems are better for speed and simplicity, while eukaryotic systems are better for proteins that need more sophisticated cellular processing.
5. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
To optimize the expression of a membrane protein in a cell-free system, I would design the experiment so that the protein has access to a membrane-like environment during synthesis. I would not express it in a plain aqueous reaction alone, because membrane proteins contain hydrophobic regions that tend to misfold or aggregate in water.
My setup would include:
A cell-free expression system
The DNA template encoding the membrane protein
A membrane-mimicking support such as liposomes, nanodiscs, or suitable detergents
A series of optimization conditions, such as different magnesium concentrations, temperatures, template concentrations, and lipid support levels
The main challenges are:
1. Aggregation Hydrophobic regions may stick together instead of inserting properly into a membrane-like structure.
Response: add liposomes or nanodiscs so the protein has a compatible environment while it is being synthesized.
2. Misfolding Even if the protein is produced, it may not fold into a functional form.
Response: optimize temperature, reaction rate, and possibly include chaperones.
3. Low functionality despite expression A membrane protein may be made, but still not behave properly.
Response: evaluate both yield and functional activity, not just total protein amount.
I would screen multiple conditions in parallel and compare both expression level and functionality to identify the best setup.
6. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
A low protein yield in a cell-free system could result from several factors:
Reason 1: Poor template quality or ineffective construct design The DNA may be degraded, the promoter may be weak, or the sequence may not be optimized for the system.
Troubleshooting strategy: Verify the DNA sequence, check DNA purity, compare plasmid versus linear DNA, and test different promoter or untranslated region designs.
Reason 2: Suboptimal reaction conditions The magnesium concentration, potassium concentration, temperature, or pH may not be suitable.
Troubleshooting strategy: Run a condition matrix varying temperature, Mg²⁺, K⁺, and reaction time to identify a better expression window.
Reason 3: The target protein is intrinsically difficult to express The protein may aggregate, degrade, or require folding support.
Troubleshooting strategy: Lower the temperature, add chaperones or stabilizers, shorten the construct if appropriate, or add membrane mimics if the protein is membrane-associated.
These troubleshooting approaches help distinguish whether the problem comes from the template, the reaction environment, or the protein itself.
Homework question from Kate Adamala
7. Design an example of a useful synthetic minimal cell as follows:
Pick a function and describe it.
I would design a synthetic minimal cell that makes an ice cream surface emit a temporary visible glow when it warms up. The goal is to create a dessert that appears to become active only during melting, producing a controlled “living-like” response without using living cells.
What would your synthetic cell do? What is the input and what is the output?
The synthetic cell would detect a warming event near the ice cream surface.
Input: an increase in temperature and local melting at the surface
Output: a visible luminescent signal
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Only partially. A bulk cell-free Tx/Tl reaction could generate a signal, but without encapsulation the system would be more diluted, less protected, and much harder to localize spatially. Encapsulation is useful because it protects the reaction components and keeps the signal confined to specific regions of the dessert.
Could this function be realized by genetically modified natural cell?
In principle, yes. A genetically modified natural cell could potentially be engineered to respond to temperature and generate light. However, this would be less desirable here because using living engineered cells in an edible dessert raises stronger concerns about safety, regulation, storage stability, and user acceptance. A synthetic minimal cell is more appropriate for a controlled design application like this.
Describe the desired outcome of your synthetic cell operation.
The desired outcome is that the ice cream remains visually inactive while frozen, but begins to emit a faint and localized glow when it starts warming. The signal should be short-lived, noticeable, and limited mainly to the exposed surface.
Design all components that would need to be part of your synthetic cell.
The synthetic minimal cell would need:
A membrane compartment
A cell-free transcription/translation system
A reporter module for light production
A regulatory mechanism that links warming to activation
An energy regeneration module
Protective components to improve freeze-thaw stability
What would be the membrane made of?
The membrane could be made from phospholipids plus cholesterol. For example, a POPC and cholesterol membrane would be a reasonable prototype system because it provides a stable artificial membrane structure.
What would you encapsulate inside? Enzymes, small molecules.
Inside the synthetic cell, I would encapsulate:
A bacterial cell-free Tx/Tl lysate
Amino acids
Nucleotides
Buffer salts
An ATP regeneration system
DNA encoding a luminescent reporter
Any required small-molecule cofactors or substrates
Cryoprotective additives to improve survival during freezing
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason?
I would start with a bacterial cell-free system, most likely E. coli-based. A bacterial system is appropriate because it is easier to use, faster to optimize, and sufficient for a proof-of-concept visible reporter system. I would only switch to a mammalian system if the regulatory logic specifically required mammalian transcription factors or eukaryotic response elements.
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
The synthetic cell would communicate with the environment through a warming-triggered change. Ideally, the system would be designed so that increased temperature or local phase change activates the internal reaction. Depending on the final design, this could happen either through passive thermal sensitivity of the encapsulated system or through a membrane property that changes permeability during warming. If needed, a membrane channel or pore could be included in a more advanced design.
Experimental details
8. List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
Lipids:
POPC
Cholesterol
Genes:
A reporter gene for visible output, such as a luciferase-related construct
If needed, a membrane pore gene such as alpha-hemolysin (aHL) for controlled transport in a more advanced version
If a temperature-sensitive regulatory element is used, the corresponding control sequence would also be included in the construct design
For a concept-stage design, the exact reporter gene can be chosen based on brightness, ease of detection, and compatibility with the cell-free system. For an early proof of concept, a robust reporter is more important than final food compatibility.
9. How will you measure the function of your system?
I would measure the system in stages.
First, I would test the reporter in a standard cell-free reaction and quantify signal output using fluorescence or luminescence measurements. Second, I would test the same system after encapsulation to see whether the synthetic cells still function. Third, I would test the encapsulated system after freeze-thaw exposure to evaluate whether activity is preserved. Finally, I would place the synthetic cells into a model ice cream matrix and monitor signal intensity as the temperature increases.
The key measurements would be:
Signal onset temperature
Signal brightness
Signal duration
Spatial localization of the signal
Retention of function after freezing
Homework question from Peter Nguyen
10. Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
I propose a freeze-dried cell-free architectural coating that becomes visibly activated when water intrusion occurs, allowing walls or building surfaces to function as passive biological leak indicators.
How will the idea work, in more detail? Write 3-4 sentences or more.
The material would contain freeze-dried cell-free reactions embedded in a coating layer or sensing patch. Under normal dry conditions, the system remains inactive and stable. When water enters the material due to leakage or condensation, the reaction becomes hydrated and activates a reporter output, such as a visible color or fluorescence signal. This would allow building managers to detect hidden moisture problems earlier, before severe mold growth or structural damage occurs.
What societal challenge or market need will this address?
This concept addresses the need for early detection of water damage in buildings. Moisture problems are costly, often hidden, and can lead to mold, indoor air quality issues, and expensive repairs. A passive biological leak indicator could reduce inspection delays and improve maintenance efficiency.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
I would address these limitations by designing the system as a replaceable sensing layer rather than a permanent active material. The freeze-dried reactions would remain stable until hydration, and the sensing patches could be modular and periodically replaced during maintenance cycles. I would also explore protective matrices and packaging strategies to improve shelf life and reduce accidental activation from ambient humidity.
Homework question from Ally Huang
11. Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Long-duration spaceflight exposes biological systems to radiation, microgravity, confinement, and limited medical infrastructure. One major challenge is the ability to detect and respond to biological stress quickly without relying on large lab equipment. Developing compact cell-free diagnostic tools for space is significant because it supports crew health, enables on-site biological measurement, and advances low-resource biotechnology relevant both in space and on Earth. Scientifically, it is interesting because it tests how simplified biochemical systems perform under extreme conditions and whether portable synthetic biology can serve as a reliable research and health-monitoring platform.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
A DNA reporter construct responsive to oxidative stress-associated molecular signals, measured through cell-free reporter output.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
Oxidative stress is one of the most important biological effects associated with radiation exposure and altered physiological conditions in space. A reporter construct linked to oxidative stress sensing can serve as a simplified way to detect biologically meaningful environmental or sample-related changes. By monitoring reporter output in a cell-free system, the experiment would test whether portable gene expression platforms can be used to detect molecular stress signatures in space-compatible settings. This directly relates to the challenge of performing biological monitoring in space with limited equipment, low mass, and limited crew time.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
My hypothesis is that a freeze-dried cell-free expression system can be used as a compact and reliable platform to detect oxidative stress-related molecular signals in a constrained spaceflight-like environment. The reasoning is that cell-free systems remove the need to culture living cells, which makes them safer, faster, and easier to deploy in resource-limited settings. If a stress-responsive reporter can be activated reproducibly after rehydration, then this approach could support future low-resource biological monitoring during long-duration missions. The broader goal is not only to detect a signal, but to evaluate whether simplified gene expression tools can function as practical diagnostic or experimental platforms in space, where traditional wet-lab workflows are difficult to implement.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
I would test freeze-dried BioBits cell-free reactions containing a stress-responsive reporter construct. Experimental samples would include reactions exposed to oxidative stress-related input conditions and matched negative controls without the activating condition. Additional controls would include a positive-expression control to confirm the cell-free system is functioning properly. After rehydration and incubation, I would measure reporter output using fluorescence readout with the P51 viewer. The main data would be signal intensity, time to detectable signal, and comparison between activated, negative-control, and positive-control reactions.
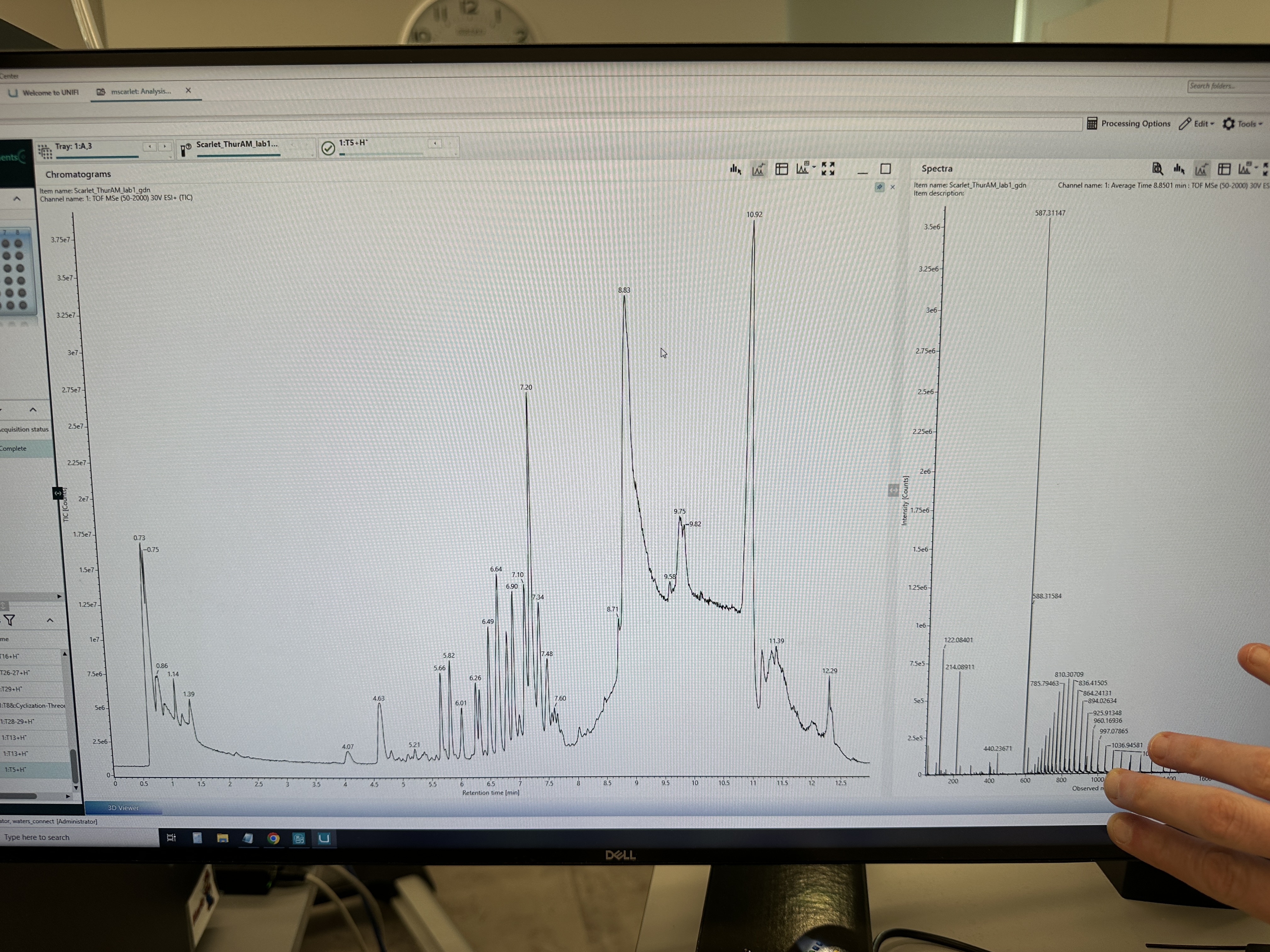
For my final project, Glowing Ice Cream, the main things I would measure are the identity, quantity, activity, and release behavior of the NanoLuc/substrate system.
1. Protein Identity and Purity
I need to confirm whether I successfully made the right protein, NanoLuc-GSGS-His₆.
I would measure:
Protein molecular weight Use intact protein LC-MS or MALDI/ESI-MS to check whether the measured mass matches the expected NanoLuc-GSGS-His₆ molecular weight.
Protein purity Use SDS-PAGE after Ni-NTA purification. A strong band around the expected NanoLuc size would support successful expression and purification.
Sequence confirmation Use DNA sequencing for the plasmid and, if available, peptide mapping by LC-MS/MS to confirm that the expressed protein sequence matches the designed construct.
2. Protein Quantity
I need to know how much active or total protein I have before testing brightness.
I would measure:
Total protein concentration using A280, Bradford, or BCA assay.
Purified fraction concentration after Ni-NTA purification.
Approximate NanoLuc concentration by combining A280 with SDS-PAGE purity estimation.
This matters because the visible glow depends heavily on protein concentration.
3. Bioluminescence Output
The most important functional measurement is whether the system glows visibly and for how long.
I would measure:
Peak brightness
Time to peak
Glow duration
Brightness decay curve
Area under the brightness-time curve
Technologies:
Plate reader or luminometer for quantitative luminescence.
Dark-room phone imaging for the actual visual effect.
ImageJ / Python image analysis to extract brightness from time-lapse videos.
For each protein:substrate condition, I would plot brightness over time and compare which ratio gives the best “lick-to-glow” effect.
4. Substrate Encapsulation and Temperature Release
Because the project uses cocoa butter / lipid encapsulation as a temperature trigger, I would measure:
Whether the substrate stays separated from protein at cold temperature.
Whether release happens after warming.
Whether the glow starts only after the lipid melts.
Technologies:
Cold-room vs warm-room luminescence test.
Microscopy to observe lipid bead size and distribution.
Time-lapse imaging to compare cold stability and warm-triggered release.
If available, LC-MS or HPLC to measure how much substrate is retained or released.
5. Ice Cream Matrix Stability
I would also measure whether the system still works inside or on real ice cream.
I would test:
Glow before freezing.
Glow after frozen storage.
Glow after short warming.
Whether fat, sugar, water, and temperature affect signal.
The goal is not only to prove that NanoLuc works, but to measure whether it can work in the actual food-like prototype environment.
Waters Part I — Molecular Weight
Question 1
Based on the predicted amino acid sequence of eGFP and known modifications, what is the calculated molecular weight?
The sequence given in the assignment includes eGFP, the LE linker, and a His₆ tag.
Experimental MW ≈ 27,983.86 Da
Theoretical mature MW ≈ 27,986.57 Da
Mass error ≈ 0.0097%
Mass error ≈ 97 ppm
Question 3
Can you observe the charge state for the zoomed-in peak in the intact eGFP mass spectrum?
Yes. In the zoomed-in peak around m/z ≈ 1473, the isotope peaks are separated by about:
Δm/z ≈ 0.053
Charge state can be estimated from isotope spacing:
z = 1 / isotope spacing
z = 1 / 0.053
z ≈ 18.9
So the charge state is approximately:
z = 19+
This also matches the molecular weight check:
MW ≈ 19 × (1473 - 1.007)
MW ≈ 27,900–28,000 Da
which is consistent with eGFP.
Answer:
Yes, the zoomed-in intact eGFP peak is approximately 19+.
The isotope spacing is about 1/19 ≈ 0.053 m/z.
Waters Part II — Secondary / Tertiary Structure
Question 1
Explain the difference between native and denatured protein conformations. What changes in the mass spectrum?
A native protein is folded. Its 3D structure is still mostly preserved, so many charged residues are buried inside the folded structure. Because fewer sites are exposed to solvent and protonation, the protein usually carries fewer charges.
A denatured protein is unfolded. When the protein unfolds, more basic and polar residues become exposed. This allows the protein to pick up more protons, so it appears in higher charge states.
In the mass spectrum:
Denatured eGFP:
More charge states
Higher charge numbers
Peaks appear at lower m/z
Broader charge-state distribution
Native eGFP:
Fewer charge states
Lower charge numbers
Peaks appear at higher m/z
More compact charge-state distribution
In Figure 2, the denatured spectrum has many peaks across the lower m/z range, while the native spectrum has fewer, stronger peaks at higher m/z values.
Answer:
The denatured protein unfolds, exposes more chargeable residues, and therefore produces higher charge states at lower m/z. The native protein remains folded, carries fewer charges, and appears at higher m/z with fewer charge states.
Question 2
What is the charge state of the native eGFP peak around m/z 2800?
For mature eGFP:
MW ≈ 27,986.57 Da
The native peak near:
m/z ≈ 2799.4
Charge state:
z ≈ MW / (m/z - proton mass)
z ≈ 27,986.57 / (2799.4 - 1.007)
z ≈ 27,986.57 / 2798.39
z ≈ 10.0
So the peak at around 2800 m/z is:
10+
This is also consistent with the adjacent native peaks around 2333, 2545, and 2799 m/z, which correspond approximately to 12+, 11+, and 10+.
Answer:
The native eGFP peak near 2800 m/z is 10+.
Small note: the actual zoomed inset in the provided figure appears to show the peak around 2545 m/z, which would correspond to 11+. But the peak specifically near 2800 m/z is 10+.
Waters Part III — Peptide Mapping / Primary Structure
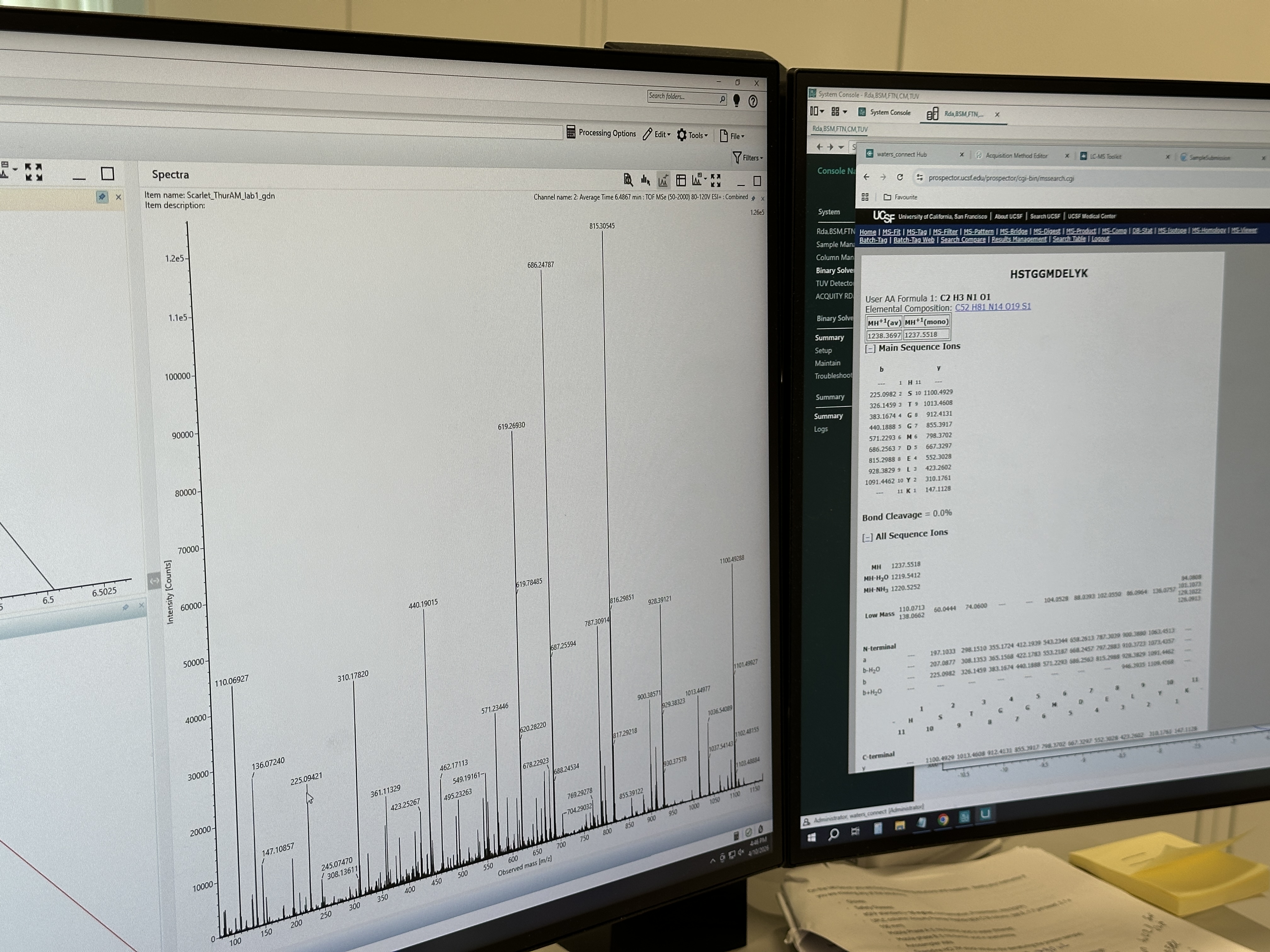
Question 1
How many Lysines and Arginines are in eGFP? Highlight them.
Peptide = FEGDTLVNR
Theoretical [M+H]+ = 1050.52145 Da
Experimental [M+H]+ = 1050.52696 Da
Mass error ≈ 5.25 ppm
If using the directly labeled singly charged peak at 1050.52438, the error is approximately:
2.8 ppm
Question 7
What percentage of the sequence is confirmed by peptide mapping?
Figure 6 reports:
Identified: 88%
Answer:
The peptide map confirms 88% of the eGFP sequence.
Bonus Question 8
What peptide sequence best matches the fragmentation spectrum in Figure 5c?
The peptide identified from the precursor mass is:
FEGDTLVNR
The fragmentation spectrum in Figure 5c is consistent with this peptide assignment.
Answer:
FEGDTLVNR
Bonus Question 9
Does the peptide map data indicate that the protein is eGFP?
Yes.
The data supports that the protein is eGFP because:
The intact protein mass is close to the expected mature eGFP-His₆ mass.
The tryptic peptide map contains peptides that match expected eGFP digestion products.
The peptide at 2.78 min matches the expected peptide FEGDTLVNR.
The peptide map confirms 88% sequence coverage.
Answer:
Yes. The intact mass, peptide masses, fragmentation data, and 88% sequence coverage are all consistent with the protein being the eGFP standard.
Waters Part IV — Oligomers
KLH subunit masses from the homework table:
7FU subunit = 340 kDa
8FU subunit = 400 kDa
Calculations
Oligomeric species
Calculation
Theoretical mass
Where it appears in Figure 7
7FU Decamer
10 × 340 kDa
3,400 kDa = 3.4 MDa
Peak around 3.4 MDa
8FU Didecamer
20 × 400 kDa
8,000 kDa = 8.0 MDa
Major peak around 8.33 MDa
8FU 3-Decamer
30 × 400 kDa
12,000 kDa = 12.0 MDa
Peak around 12.67 MDa
8FU 4-Decamer
40 × 400 kDa
16,000 kDa = 16.0 MDa
Expected around 16 MDa; weak/broad signal region
The figure shows strong peaks around 3.4, 4.013, 8.33, and 12.67 MDa. The 4.013 MDa peak likely corresponds to an 8FU decamer, although that specific species was not one of the four requested answers.
Answer:
7FU Decamer = 3.4 MDa
8FU Didecamer = 8.0 MDa, observed near 8.33 MDa
8FU 3-Decamer = 12.0 MDa, observed near 12.67 MDa
8FU 4-Decamer = 16.0 MDa, expected near 16 MDa but not as clearly resolved in the screenshot
Yes, the intact LC-MS data is consistent with eGFP. The measured molecular weight is close to the theoretical mature eGFP-His₆ molecular weight, and the peptide mapping data further supports the protein identity.
Week 11 HW: Week 11 — Bioproduction & Cloud Labs
Part 1: The 1,536 Pixel Artwork Canvas | Collective Artwork
For the collective bioart project, I contributed a small bacteriophage drawing in the upper-right corner of the canvas. (Later seemed to have been modified by someone else)
What I liked about this project is that it made the artwork feel genuinely collective. Instead of everyone making a separate image, the final result became a shared and evolving biological canvas. The fact that my pixel contribution could be changed by someone else was a little unexpected, but it also made the project feel more alive.
For next year, it could be helpful to have a simple version-history or layer system, so each person can still see what they originally contributed. Another small improvement could be to reserve some collaborative zones and some individual zones, so the artwork can support both collective editing and personal authorship.
Part 2: Cell-Free Protein Synthesis | Cell-Free Reagents
Roles of Each Component
E. coli Lysate / BL21(DE3) Star Lysate
The lysate provides the core biological machinery for transcription and translation, including ribosomes, tRNAs, translation factors, metabolic enzymes, and T7 RNA polymerase. In this reaction, it functions like the “cell” without the cell membrane.
Potassium Glutamate
Potassium glutamate helps set the ionic strength of the reaction and makes the environment more similar to the inside of an E. coli cell. This supports proper ribosome function, enzyme activity, and protein folding.
HEPES-KOH pH 7.5
HEPES-KOH is the main buffer that keeps the reaction near physiological pH. This is important because transcription, translation, and fluorescent protein maturation are all sensitive to pH changes.
Magnesium Glutamate
Magnesium is essential for ribosome stability, RNA structure, and many enzymatic reactions in transcription and translation. However, too much or too little magnesium can strongly reduce cell-free protein expression.
Potassium Phosphate Monobasic / Dibasic
The phosphate buffer pair helps control pH and provides phosphate species that can participate in energy metabolism. Using both monobasic and dibasic forms allows the system to tune the buffer around the desired pH.
Ribose
Ribose provides a sugar backbone source for nucleotide regeneration. In the longer NMP-ribose-glucose system, ribose helps the lysate rebuild nucleotide triphosphates from simpler nucleotide or base precursors.
Glucose
Glucose works as a long-term carbon and energy source. Instead of giving the reaction all of its energy immediately, glucose allows the lysate’s metabolic enzymes to regenerate energy over a longer incubation.
AMP, CMP, GMP, UMP
These nucleotide monophosphates act as lower-cost precursors for the NTPs needed during transcription. The lysate can phosphorylate them into ATP, CTP, GTP, and UTP.
Guanine
Guanine is a nucleobase precursor that can be converted into GMP through salvage pathways in the lysate. This allows the system to produce GTP even when GMP is reduced or omitted.
17 Amino Acid Mix
The amino acid mix supplies most of the building blocks needed for protein synthesis. The ribosome uses these amino acids to translate the fluorescent protein sequence.
Tyrosine
Tyrosine is included separately because it has solubility and stability issues compared with many other amino acids. It is still essential for translation and is also part of the chromophore chemistry in many fluorescent proteins.
Cysteine
Cysteine is also supplied separately because it can be chemically unstable and oxidation-sensitive. Keeping it separate helps maintain a more controlled amino acid composition.
Nicotinamide
Nicotinamide supports cofactor metabolism and can help maintain redox and energy-related activity in the lysate. This is especially useful for longer cell-free reactions.
Nuclease-Free Water
Nuclease-free water is used to bring the reaction to the final volume without adding enzymes that could degrade DNA or RNA.
Difference Between the 1-Hour PEP-NTP Mix and the 20-Hour NMP-Ribose-Glucose Mix
The 1-hour optimized PEP-NTP master mix is designed for fast expression. It directly provides NTPs and uses PEP as an immediate energy source, so transcription and translation can start quickly and produce a strong short-term signal.
The 20-hour NMP-ribose-glucose master mix is designed for longer, more sustainable protein production. Instead of directly providing all NTPs, it uses NMPs, ribose, glucose, and guanine so the lysate can regenerate nucleotides and energy over time. This makes it more suitable for long-incubation fluorescent artwork, where the goal is not only fast expression but sustained fluorescence over many hours.
Bonus: How can transcription occur if GMP is not included but Guanine is?
Transcription can still occur because guanine can be converted into GMP through nucleotide salvage pathways in the E. coli lysate. After guanine is converted to GMP, cellular kinases can phosphorylate GMP into GDP and then GTP, which T7 RNA polymerase can use for RNA synthesis.
Part 3: Planning the Global Experiment | Cell-Free Master Mix Design
Biophysical / Functional Properties of the Six Fluorescent Proteins
sfGFP
sfGFP is useful in cell-free systems because it folds very efficiently and matures quickly, making it a strong positive-control fluorescent protein. Its main advantage is that it is robust, so changes in fluorescence are more likely to reflect the reaction condition rather than complete folding failure.
mRFP1
mRFP1 is a red fluorescent protein, but it matures more slowly and has lower brightness compared with newer red fluorescent proteins. In a cell-free reaction, this means the readout may lag behind actual protein production.
mKO2
mKO2 is an orange fluorescent protein with moderate acid sensitivity and relatively slow maturation. For a long 36-hour incubation, pH stability could strongly affect how much orange fluorescence is visible at the endpoint.
mTurquoise2
mTurquoise2 is a cyan fluorescent protein with high quantum yield, rapid maturation, and very low acid sensitivity. These properties make it a good candidate for stable readout over time, although cyan fluorescence can be closer to background autofluorescence than red/orange channels.
mScarlet-I
mScarlet-I is a bright, rapidly maturing monomeric red fluorescent protein. Its brightness makes it attractive for artwork, but its moderate acid sensitivity means that pH control still matters during long incubation.
Electra2
Electra2 is a blue fluorescent protein with strong brightness and blue emission around 454 nm. Since blue fluorescence can be more sensitive to background and imaging conditions, the final readout may depend strongly on the plate reader settings and optical channel.
Hypothesis for Master Mix Adjustment
For mKO2, I would test whether increasing the buffering capacity of the master mix improves the final fluorescence after 36 hours. Because mKO2 has moderate acid sensitivity and slow maturation, a longer reaction may lose signal if the reaction becomes too acidic over time.
My hypothesis is that adding extra HEPES-KOH pH 7.5 and/or carefully tuning the potassium phosphate buffer would help maintain pH stability during the 36-hour incubation. The expected result would be stronger and more consistent orange fluorescence at the endpoint, especially compared with conditions where the reaction produces protein but the fluorescent signal is reduced by pH drift.
Assigned Wells / Final Project Slide
For the next phase, I would use the assigned wells to test how the custom reagent supplement changes fluorescence over time. My final project slide is focused on Glowing Ice Cream, so I would connect this experiment back to the broader question of how biological light production or fluorescence can be made visible, playful, and stable enough for a designed experience.
Data Analysis Plan
Once the fluorescence data is returned, I would compare not only the final endpoint brightness, but also the shape of the fluorescence curve over time. For artwork, the most useful condition is not necessarily the one with the highest short peak, but the one that gives a strong and stable visible signal across the full incubation window.
This week’s experiment was done late at night. To speed things up, we tried to simplify several steps in the protocol and cut a few corners, such as filtering only once when the protocol called for two rounds of filtration. In the end, we did not get the result we were hoping for. The plates only showed some unidentified white colonies, possibly E. coli, but the issue is that the original E. coli we used was supposed to be purple.
This week, I worked on designing a small genetic circuit system based on RNA endonucleases and fluorescent protein outputs. Instead of starting directly from a final circuit, we first organized the available biological parts and tried to understand how they could be connected into a regulatory network.
Organizing the Basic Parts We started by making a spreadsheet of possible genetic circuit part names. The first group of parts was the RNA endonucleases, or ERNs:
This week’s Lab work is effectively part of this week’s Homework;
Week 5_Lab: Protein Design Part II
This week’s Lab work is effectively part of this week’s Homework;
Week 6_Lab: Gibson Assembly
This week’s experiment was done late at night. To speed things up, we tried to simplify several steps in the protocol and cut a few corners, such as filtering only once when the protocol called for two rounds of filtration. In the end, we did not get the result we were hoping for. The plates only showed some unidentified white colonies, possibly E. coli, but the issue is that the original E. coli we used was supposed to be purple.
So we took some samples from another group and tried again. This time, they grew successfully, which suggests that the problem most likely came from the way we modified or skipped parts of the protocol during the workflow.
Week 7_Lab: Neuromorphic Circuits
This week, I worked on designing a small genetic circuit system based on RNA endonucleases and fluorescent protein outputs. Instead of starting directly from a final circuit, we first organized the available biological parts and tried to understand how they could be connected into a regulatory network.
1. Organizing the Basic Parts
We started by making a spreadsheet of possible genetic circuit part names. The first group of parts was the RNA endonucleases, or ERNs:
CasE
Csy4
PgU
These ERNs can recognize specific RNA sequences and cut or regulate transcripts that contain their recognition sites. This made them useful as regulatory components in the circuit.
We also listed several fluorescent proteins as possible outputs:
mKO2
eBFP2
mMaroon1
mNeonGreen
These fluorescent proteins would allow us to visually read out the behavior of the circuit.
2. Building a Naming System
After listing the basic parts, we created a naming rule to describe how one part regulates another:
ERN_rec_target
In this naming system, rec means that the target transcript contains the recognition site for a specific ERN.
For example:
PgU_rec_Csy4
This means that the Csy4 transcript contains a PgU recognition site. If PgU is expressed, it can recognize and regulate the Csy4 mRNA.
This naming system helped us describe different circuit connections clearly and consistently.
3. Designing ERN-to-ERN Regulation
Next, we used the naming rule to generate possible regulatory connections between the ERNs:
PgU_rec_Csy4
PgU_rec_CasE
Csy4_rec_CasE
CasE_rec_Csy4
These parts describe cases where one ERN controls the expression of another ERN. This creates the possibility of building layered regulatory circuits, where one RNA-processing enzyme affects another regulatory node.
At this stage, I was trying to think of the circuit less as a single linear pathway and more as a small network of interacting regulatory parts.
4. Connecting ERNs to Fluorescent Outputs
After that, we connected the ERNs to fluorescent protein outputs. We listed several possible reporter constructs:
Csy4_rec_mNeonGreen
CasE_rec_mNeonGreen
PgU_rec_mNeonGreen
CasE_rec_Csy4_rec_mKO2
The first three designs are simpler reporter outputs. For example, Csy4_rec_mNeonGreen means that the mNeonGreen transcript contains a Csy4 recognition site, so Csy4 can regulate the final green fluorescence output.
The last design, CasE_rec_Csy4_rec_mKO2, is more complex because it includes more than one regulatory recognition site. This suggests that the final mKO2 output could be controlled by multiple ERN-related inputs.
5. Sketching the Circuit Logic
Once the parts were organized, we began drawing the circuit logic. The goal was to show how the ERNs could regulate each other and how those regulatory relationships could eventually control fluorescent protein expression.
The general logic was:
Input / ERN expression
↓
ERN recognizes specific RNA site
↓
Target transcript is regulated
↓
Fluorescent protein output changes
For example, if Csy4 is present and the mNeonGreen transcript contains a Csy4 recognition site, then Csy4 can affect the amount of mNeonGreen that is produced. The final fluorescence becomes a readout of the regulatory interaction.
6. What I Learned from This Process
Through this process, I learned that designing a genetic circuit is not only about choosing biological parts, but also about defining clear relationships between them. The spreadsheet helped me organize the parts as modular components, while the circuit diagram helped me understand how those components could interact.
I also realized that the naming system is important because it makes the design easier to read, discuss, and modify. Once we had a consistent structure like ERN_rec_target, it became much easier to generate new circuit possibilities and compare them.
Overall, this exercise helped me understand how RNA endonucleases such as Csy4, CasE, and PgU can be used as programmable regulatory elements in a genetic circuit. By connecting them to fluorescent protein reporters, we could design circuits where the output is visible and experimentally testable.
ERN_rec_target
Week 8_Lab:
NO LAB THIS WEEK
Week 9_Lab: Cell Free
Protein Purification (LOA this week, moved lab to final week)
FINAL PROJECT DECK: https://www.figma.com/deck/G1Zvluf6m7BrTVp1HMR95F SECTION 1: ABSTRACT My final project explores whether ice cream can become a visible biological interface instead of a passive frozen dessert. The project began with a simple question: could an ice cream appear to “wake up” when it warms, by producing visible bioluminescence through a designed protein-substrate reaction? For this class, I narrowed the idea into a proof-of-concept experiment using NanoLuc luciferase and its substrate furimazine. NanoLuc is a small engineered luciferase derived from Oplophorus gracilirostris and designed to produce strong glow-type luminescence when paired with furimazine.
My final project explores whether ice cream can become a visible biological interface instead of a passive frozen dessert. The project began with a simple question: could an ice cream appear to “wake up” when it warms, by producing visible bioluminescence through a designed protein-substrate reaction? For this class, I narrowed the idea into a proof-of-concept experiment using NanoLuc luciferase and its substrate furimazine. NanoLuc is a small engineered luciferase derived from Oplophorus gracilirostris and designed to produce strong glow-type luminescence when paired with furimazine.
The broad objective is to test whether purified NanoLuc can produce naked-eye-visible light in or on ice cream when mixed with substrate under controlled conditions. My hypothesis is that if the protein and substrate are kept separated during frozen storage and then brought into contact during warming, the system can generate visible local light spots. During the course, I focused on designing a NanoLuc-GSGS-His6 construct, expressing or preparing NanoLuc through a cell-free or bacterial expression workflow, purifying the His-tagged protein with Ni-NTA resin, and testing protein-to-substrate ratios in a small-volume assay.
I also explored a food-material strategy where furimazine-containing substrate droplets could be physically separated from protein using cocoa butter as a temperature-responsive fat barrier. The current project is a lab demonstration only and should not be consumed, because Nano-Glo and furimazine are research reagents rather than approved food ingredients.
SECTION 2: PROJECT AIMS
Aim 1: Experimental Aim
The first aim of my final project is to produce and test visible NanoLuc-based bioluminescence in an ice cream setting by utilizing DNA construct design, cell-free or bacterial protein expression, Ni-NTA purification of NanoLuc-GSGS-His6, and controlled mixing of purified protein with Nano-Glo substrate.
Relevant methods and resources include:
NanoLuc-GSGS-His6 DNA design
pET-28a expression design
DH5alpha plasmid preparation
BL21(DE3) expression planning
Cell-free protein synthesis
Ni-NTA affinity purification
Promega Nano-Glo substrate assay
96-well plate ratio testing
Dark-room imaging
Image-based brightness analysis
Aim 2: Development Aim
The next development aim is to build a more controlled frozen dessert prototype by encapsulating or physically separating the substrate inside small cocoa-butter-based particles, so the protein and substrate do not fully react during frozen storage but can contact each other when warmed locally or during simulated oral-temperature conditions.
This would move the project beyond simply dropping protein and substrate together. It would test whether a food-compatible fat matrix can act as a physical timing mechanism for a bioluminescent reaction. The main technical questions would be particle size, substrate loading, reaction brightness after storage, and whether the cocoa butter actually melts fast enough at the moment of contact.
Aim 3: Visionary Aim
The long-term vision is to develop a safe, food-grade version of a “living-feeling” ice cream that gives a visible biological response as it warms, melts, or is eaten.
A future version would need to replace the current research-use NanoLuc/furimazine system with food-safe or fully toxicology-tested components. It could also use a food-grade secretion host such as Pichia pastoris for protein production, and a thermally responsive split luciferase design, such as NanoBiT fused to a temperature-sensitive protein interaction pair. The goal would be a dessert that does not simply display color or decoration, but changes through a biological reaction that the eater can see.
SECTION 3: BACKGROUND
Background and Literature Context
Bioluminescence is widely used in biology because it can convert molecular activity into visible or measurable light. NanoLuc is especially useful for this project because it is small, bright, and stable compared with older luciferase reporters. Hall et al. engineered NanoLuc from a deep-sea shrimp luciferase system and paired it with a synthetic imidazopyrazinone substrate called furimazine. Their work reported glow-type luminescence and much higher specific activity than firefly or Renilla luciferase systems, which makes NanoLuc a good candidate for a visual proof-of-concept experiment rather than only a sensitive instrument-based assay.
A second relevant area is cell-free gene expression. Garenne et al. describe cell-free gene expression as a way to produce proteins outside living cells, and they point out that E. coli-based cell-free systems have become useful for synthetic biology, protein synthesis, and prototyping. This matters for my project because I do not need a living organism inside the ice cream. I only need the designed protein product. Cell-free expression gives a faster way to test whether the DNA design can produce functional NanoLuc before investing in a full bacterial expression and purification pipeline.
The safety context is also central to this project. Shipunova et al. studied furimazine toxicity in vitro and in vivo and reported hepatotoxic effects under prolonged intravenous administration in animals. That does not directly describe oral food exposure, but it clearly shows that furimazine cannot be treated casually as a food ingredient. Promega also labels the Nano-Glo Luciferase Assay System as “For Research Use Only. Not for Use in Diagnostic Procedures.” For this reason, the current project should be framed as a lab demonstration and a material-biology prototype, not as an edible product.
Novelty and Innovation
My project uses NanoLuc outside its usual role as a reporter in cells, tissues, or plate-reader assays. Instead of asking NanoLuc to report on gene expression, I am using it as the active component in a visible material experience. The novelty is in combining a designed protein, a substrate timing problem, and an ice cream material system into one prototype.
The project also treats frozen food as a reaction environment. Ice cream is cold, heterogeneous, fatty, and visually familiar, which makes it very different from a normal buffer or cell culture medium. The cocoa butter idea adds another layer: the fat is not just an ingredient, but a possible physical gate that controls when substrate contacts protein.
Why the Project Matters and Possible Impact
I care about this project because synthetic biology often stays invisible to people unless they are looking at a gel, a graph, or a screen. This project asks whether a biological reaction can become something direct and physical, something a person can see happening in a familiar object. Ice cream is a playful format, but the technical question is real: can we control the time, location, and visibility of a biological reaction inside a complex material?
The immediate impact is educational and experimental. A successful prototype could make enzyme-substrate reactions easier to understand because the reaction is visible without special imaging equipment. It could also show how biological tools can be combined with product design and material design, instead of staying only inside lab workflows.
The longer-term impact depends on safety and food compatibility. I do not think the current NanoLuc/furimazine system should be presented as edible. A future food-safe version could contribute to responsive foods, science communication tools, or controlled-release food materials. Even if the final product never becomes a commercial dessert, the project still tests a useful design pattern: separating biological components in a material until temperature, mixing, or physical contact triggers the reaction.
Ethical Implications
The main ethical issue is safety. The current version uses NanoLuc and furimazine through commercial research reagents, and these materials are not approved as food ingredients. Because of that, the project should follow non-maleficence: no one should eat the prototype, and no public demonstration should create the impression that the current sample is safe for consumption. The project also touches responsibility, because the language around “living ice cream” could be misleading. The prototype is not alive. It is a designed biochemical reaction placed in a food-like object.
To keep the project ethical, I would label all samples as non-edible research prototypes, perform tests only in appropriate lab or controlled demo settings, and dispose of materials as research waste. I would also avoid user testing that involves tasting unless every component is replaced with food-grade materials and reviewed under the appropriate food safety process. A possible unintended consequence is that the visual appeal of a glowing dessert could make people underestimate reagent risk. I could also be wrong about the stability of the reaction in ice cream, the effectiveness of cocoa butter encapsulation, or the safety pathway for future ingredients. A safer alternative for public display would be to keep the reaction sealed in a transparent food-shaped shell or use a non-food hydrogel model before moving back to real ice cream.
SECTION 4: EXPERIMENTAL DESIGN, TECHNIQUES, TOOLS, AND TECHNOLOGY
Detailed Experimental Plan
Design the NanoLuc-GSGS-His6 DNA construct. The construct should include NanoLuc, a short flexible GSGS linker, and a C-terminal His6 tag for Ni-NTA purification.
Place the construct into a pET-28a-style expression design so it can be expressed under a T7 promoter in BL21(DE3), while also being compatible with cell-free expression systems that use T7 RNA polymerase.
Use Benchling or a similar DNA design tool to check the coding sequence, reading frame, His tag position, stop codon, and any required cloning or ordering details.
Prepare the DNA template through plasmid preparation or ordered DNA, depending on the available timeline. Expected result: a verified DNA template that can express NanoLuc-GSGS-His6.
Use a small-scale cell-free protein synthesis reaction to produce NanoLuc. Cell-free systems are appropriate here because they can synthesize protein within a few hours and can use plasmid or linear DNA templates.
Incubate the cell-free reaction under the recommended temperature and time conditions. Expected result: functional NanoLuc protein is produced in the reaction mixture.
Purify the His-tagged NanoLuc using loose Ni-NTA resin in a microcentrifuge tube. Ni-NTA resin is designed to purify recombinant proteins containing a polyhistidine tag.
Equilibrate the Ni-NTA resin with binding buffer, bind the cell-free reaction or cleared lysate to the resin, wash away non-binding components, and elute NanoLuc using imidazole-containing elution buffer.
Quantify the purified protein concentration. In the current experiment, the measured protein concentration was approximately 3.48 mg/L. Assuming NanoLuc is approximately 19.1 kDa, this corresponds to roughly 180 nM protein before dilution.
Prepare a protein dilution series. My current planned set is: N1 as stock protein, N2 as 24 uL stock plus 6 uL PBS, N3 as 20 uL stock plus 10 uL PBS, and N4 as 15 uL stock plus 15 uL PBS.
Prepare Nano-Glo reagent conditions. The main comparison should focus on 1x and 0.5x reagent because earlier trials suggested that excessive dilution makes the signal hard to see by eye.
Set up a small plate assay using approximately 10 uL protein solution and 10 uL substrate reagent per well. Expected result: wells with higher protein and substrate concentration should show stronger visible luminescence.
Record the reaction in a dark box or dark room using a phone camera or camera with fixed exposure. Capture images or video every few seconds for the first minute, then continue at longer intervals if the signal remains visible.
Analyze brightness using image analysis. For each well, define a region of interest and extract average pixel brightness over time. Expected result: each condition produces a brightness-time curve rather than only a single visual observation.
Transfer the best-performing condition to a real ice cream test. Because there was not enough time to make ice cream from scratch with protein and substrate built into the production process, the practical test is to make small holes in commercial ice cream, add the material locally, and image the response.
For the first ice cream test, add purified protein and Nano-Glo reagent directly into small holes or surface pockets. Expected result: visible dots should appear if the reaction remains concentrated enough and the camera exposure is controlled.
For the second ice cream test, test a cocoa-butter-based substrate particle or droplet. The substrate would be mixed or physically trapped with cocoa butter, cooled into small particles, and placed into the ice cream. Expected result: the fat barrier should slow premature contact between protein and substrate.
Simulate warming by local heating or by adding warmed purified protein solution. Expected result: warming should soften the cocoa butter and increase protein-substrate contact.
Compare three outcomes: direct mixing in buffer, direct mixing on ice cream, and cocoa-butter-mediated release on ice cream. The comparison will show whether the ice cream matrix and fat encapsulation reduce or preserve visible signal.
Use the results to decide whether the next stage should focus on higher protein yield, better substrate encapsulation, or a different temperature-gated protein design.