DNA
Playground
From in-silico gel electrophoresis art to codon-optimized gene design and CRISPR editing.
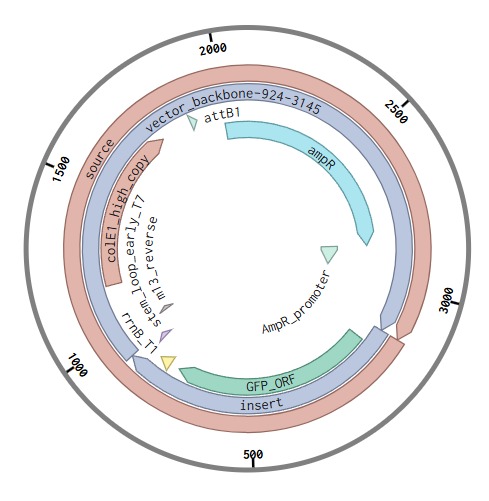
Benchling & In-silico Gel Art
Simulate Restriction Enzyme Digestion with the following Enzymes:

Create a pattern/image in the style of Paul Vanouse's Latent Figure Protocol artworks.
I tried creating a heart for Saint Valentine's Day. I focused on enzymes that would create a small number of cuts because that way I had more control over the "noise" that generates when there are lots of cuts.
I also tried selecting a specific fragment of the sequence to try to get the cuts at the kbp I wanted.
That way (and with lots of trial and error), I managed to find enzymes that worked even though they were not at Ronan's website.



DNA Design Challenge
The protein I chose is: DMTF1 (Cyclin D Binding Myb-like Transcription Factor 1)
The reason I find this protein interesting is because it is thought to act as a tumor suppressor in cancers like lung, lymphoma, and breast cancer by Arf activation. But the most interesting part for me is that recently it was discovered that it has an important role in regulating neural stem cell (NSC) function during aging.
Researchers from the National University of Singapore reported that DMTF1 expression is significantly down-regulated in NSCs with telomere dysfunction, but more importantly: Restoring DMTF1 levels was sufficient to rescue the impaired proliferation of these NSCs.
Obtaining from UniProt:
Obtaining the reverse translation from the sequence with a tool I found on Google:
Finally, performing codon optimization on a Google tool, avoiding cleavage sites of enzyme BbsI, I obtained the following sequence:
Codon optimization is important because different organisms have different preferences for codon usage. Even though multiple codons may encode the same amino acid, the abundance of tRNA may differ between organisms, so the "preference" of codons in a given organism depends on its tRNA abundance. If you take a gene from one organism and introduce it into another, the codon usage of the introduced gene may differ from the codon usage of the organism.
In the end, that could lead to inefficient translation of the gene into proteins, reducing the expression of that particular protein, or leading to production of non-functional proteins. I chose to optimize DMTF1 for human expression to get that tumor suppression effect and NSC proliferation.
Cell-Dependent Method
Using mammalian cell lines such as HEK293 (Human Embryonic Kidney 293), the gene relevant to DMTF1 is introduced in the cells, and whilst culturing, they produce the protein with very similar properties and folding to the real ones found in nature.
Cell-Free Method
In vitro translation of proteins is a technique that produces proteins using extracted transcriptional and translational resources from cells. The system comprises all the necessary components for the translation, the gene relevant to DMTF1, and the necessary nucleotides.
Prepare a Twist Synthesis Order
Following the tutorial from the homework I got the next results:

DNA Read / Write / Edit
DNA Read
I would like to sequence the APOE gene in human patients to see the version of the gene they have. The APOE gene is the most important genetic risk factor associated with Alzheimer's disease. It has three major variants: ε2, ε3, and ε4, being defined by just 2 specific points in the DNA sequence. People who carry the ε4 version have a significantly increased risk of Alzheimer's, while the ε2 version of the gene is protective and associated with a slower rate of cognitive decline. By sequencing this single gene we can get a lot of information about the risk of the disease in patients and start thinking about prevention strategies.
I would use the first-generation sequencing technology Sanger sequencing, a technique developed in 1977 that is still the gold standard for sequencing of individual genes. For APOE genotyping, I only need to determine the bases at two specific positions, so I don't need high-throughput capabilities.
The input for this process would be the genomic DNA extracted from a patient's saliva or blood sample. The part of the APOE gene that codes for the different versions is then amplified by PCR to produce enough DNA for analysis. After purification to eliminate excess primers and nucleotides, a Sanger sequencing reaction is started using:
- The PCR product
- A sequencing primer
- DNA Polymerase
- Standard deoxynucleotides (dNTPs)
- Fluorescently labeled dideoxynucleotides (ddNTPs)
The fluorescent ddNTPs work as fluorescent terminators that are occasionally incorporated into the growing DNA strand. As these ddNTPs lack a 3'-OH group, when they are incorporated into the strand, they terminate the strand elongation, producing fragments of varying lengths ending at specific nucleotides. This process is known as Chain Termination.
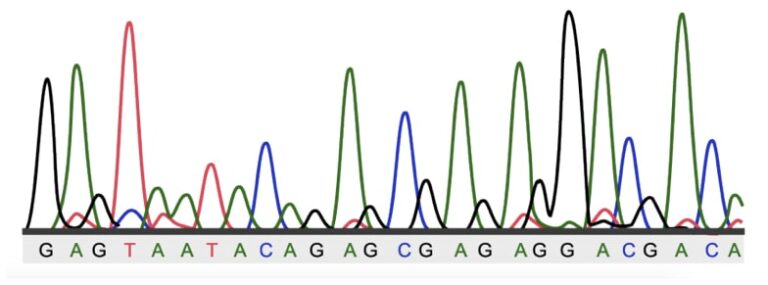
The fragments are then separated by size via capillary electrophoresis. As the fragments pass a detection window, a laser excites fluorescent dyes bound to the terminal ddNTPs, and a detector measures the fluorescence, where each of the four nucleotides is stained with a different dye color, and a computer interprets the data as a DNA sequence represented graphically as an electropherogram with colored peaks. By analyzing the nucleotide calls at positions 112 and 158 of the APOE gene, the genotype of the patient can be inferred.
DNA Write
I would like to synthesize (if possible and on-scope) the human gene SIRT6 and investigate its function in protecting neurons against aging-related injury. SIRT6 is an intriguing protein that has been shown to be directly involved in mammalian longevity. Studies have demonstrated that mice with the SIRT6 gene knocked out exhibit signs of aging and die prematurely, while mice overexpressing SIRT6 live longer (up to 15.8 percent longer in males). Biologically, SIRT6 is involved in the repair of DNA damage and the stabilization of telomeres. Both of these processes are essential for neurons, which must survive and maintain their function for our entire lifetime without dividing. By synthesizing this gene, I could introduce it into human neurons grown in the lab and determine if overexpressing SIRT6 makes them more resilient against stress, DNA damage, or other aging-related perturbations.
Enzymatic DNA synthesis employs the use of engineered enzymes to synthesize DNA strands one base at a time in mild aqueous conditions, as if it were done in a cell. The current platforms are derived from the terminal deoxynucleotidyl transferase (TdT) enzyme or its engineered versions, which have the ability to add one nucleotide to an existing strand without the need for a template. In controlled synthesis, each nucleotide is added with a temporary blocking group that prevents further additions until the blocking group is removed, thus allowing for the stepwise addition of bases to form a desired sequence. This cycle of adding a base, removing its blocker, and repeating is continued until the target DNA segment is synthesized. The reactions are carried out in water at room temperature as opposed to the harsh conditions of organic solvents, and for this reason, the DNA strand is less damaged and can be extended to a much longer length than in chemical synthesis.
The cycle starts with a DNA initiator strand that is already fixed to a solid support, which enables the repeated addition of a single base. For each cycle, a specific blocked nucleotide is added by the enzyme, which, after the removal of the blocking group by a chemical reaction, is ready for the addition of the next base. This precise addition of bases by the enzyme can reach very high coupling efficiencies, often reported to be above 99% for each step, and enables the preparation of longer contiguous sequences that would otherwise need to be assembled from many smaller fragments. In contrast, chemical synthesis is not very efficient beyond 150–200 bases, while enzymatic synthesis is increasingly being used for the direct synthesis of hundreds of bases in a single fragment, which would otherwise require a lot of assembly steps for a 1,000-base gene.
There are other advantages to enzymatic synthesis as well: It does not use toxic solvents, produces less dangerous waste, and it can be more environmentally friendly than traditional chemical synthesis. It may be faster too, as each step of nucleotide addition is very fast and does not involve multiple chemical reactions per step. On the other hand, because the technology is relatively young and still being developed, there are still some challenges to be overcome, such as providing absolute fidelity of sequence, dealing with unwanted additions, and working with certain modified nucleotides, so the costs and products may vary depending on the application. Nevertheless, for applications involving longer, high-fidelity sequences such as a gene construct, enzymatic DNA synthesis is a promising new alternative.
DNA Edit
I would like to make an edit to the APOE gene in human cells to change the version of the gene that increases the risk of Alzheimer's (ε4) to the version that protects against it (ε2). This would involve changing one or two letters of the DNA code at particular points in the gene. The APOE ε4 is the largest genetic risk factor for Alzheimer's disease, while the ε2 version actually protects against the disease. There has already been some success in changing APOE ε4 to APOE ε2 in mice to improve problems associated with Alzheimer's disease in the brain, such as metabolism and function. If we could make the same edit in human neurons or glial cells, it would be possible to investigate whether this would protect them from the pathologies of Alzheimer's disease. If it works in cells, it could potentially be developed into a therapy for individuals who carry the high-risk allele.
I would use CRISPR-Cas9 genome editing because of its accuracy and established use for targeted DNA editing. The CRISPR-Cas9 system relies on a guide RNA (gRNA), which is usually 20 nucleotides long and consists of a target-specific sequence in a scaffold of around 100 nucleotides in total, to guide the Cas9 nuclease to a specific genomic location in the genome adjacent to a PAM sequence. To convert the APOE ε4 to APOE ε2, I would design a gRNA that targets the region around the relevant codons and supply a single-stranded or double-stranded DNA repair template (around 100–200 nucleotides) that contains the ε2 sequence flanked by homology arms. The basic elements of the CRISPR-Cas9 system are:
Once it is inside the cell, the Cas9 protein combines with the gRNA and searches the genome for the complementary sequence that is located adjacent to a PAM site. As a result of binding, a double-strand break is introduced by the Cas9 protein. The cell then repairs the break in the DNA by either nonhomologous end joining, which is error-prone, or by homology-directed repair (HDR). If a template is provided, the HDR pathway can introduce the ε2 sequence into the genome at the site of the break. Cells are then expanded, and clones are isolated to verify the modification by sequencing the APOE gene site.
This method has several limitations. HDR can be inefficient, and the presence of off-target cleavage can cause unwanted mutations in other parts of the genome. Double-strand breaks can also result in bigger deletions or chromosomal rearrangements at the target site, and certain cell types, like primary neurons, are hard to transfect efficiently. Obtaining and characterizing properly edited clones can take weeks to months. More recent tools, like base editing, prime editing, and CRISPR-associated transposases, are being developed to minimize the use of double-strand breaks, but they are still being optimized for general use in mammalian cells.