Conceptual Questions
We have that one aminoacid is approximately 100 Daltons (100g/mol). For 130g of protein, we have 130g / 100g/mol = 1.3 moles. Using Avogadro's number: 1.3 * 6.022*10^23 = 7.826*10^23 molecules.
Driving Force: The hydrogen bonds are polar. When two sheets bond, they dehydrate. The release of bound water molecules provides an entropic force for aggregation.
Structural Analysis
Selected Protein:
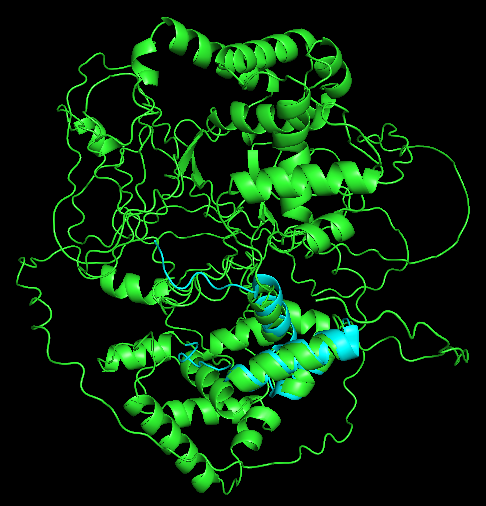
DMTF1
(Cyclin D-binding Myb-like transcription factor 1) is a transcription factor regulating cell growth and survival. It acts as a tumor suppressor by activating the p53 pathway. Restoring DMTF1 levels can reverse neural stem cell dormancy, potentially "reversing" brain aging.

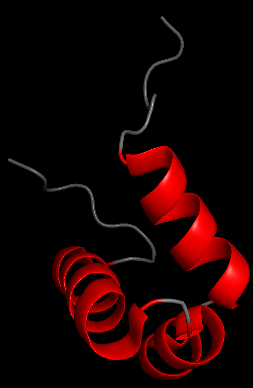
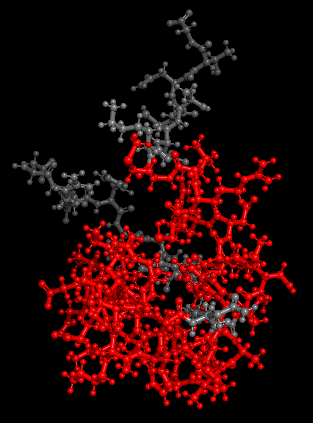
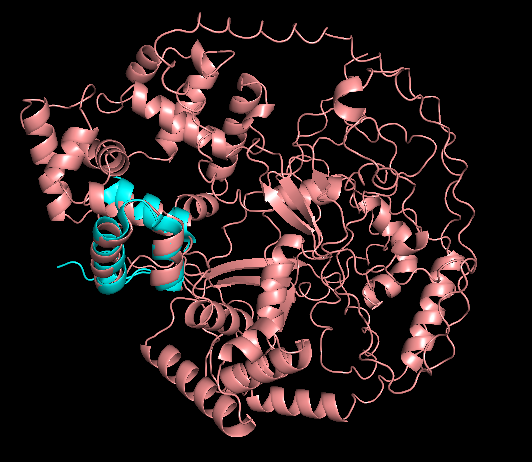
Dynamic Overlays


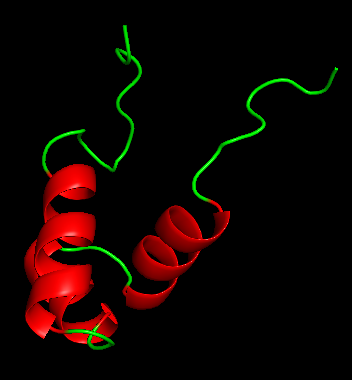
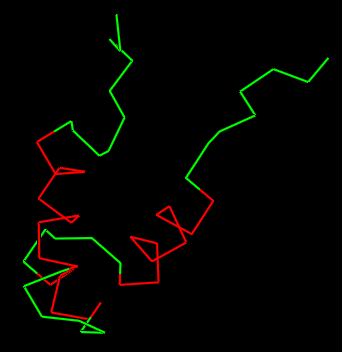
Secondary Structure
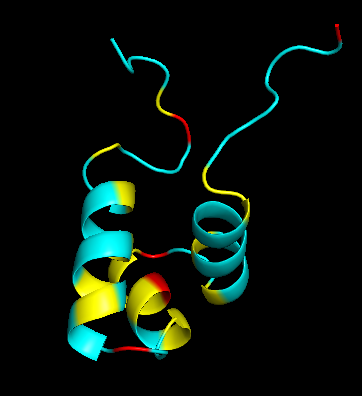
Does it have more helices or sheets?
It has more helices than sheets.
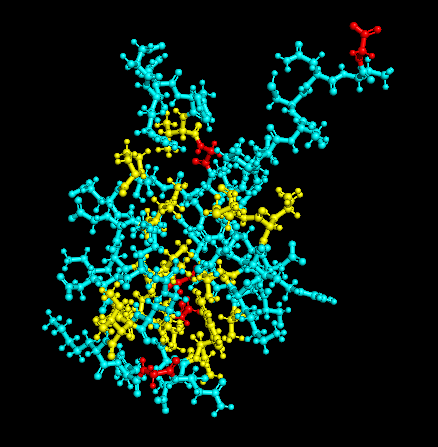
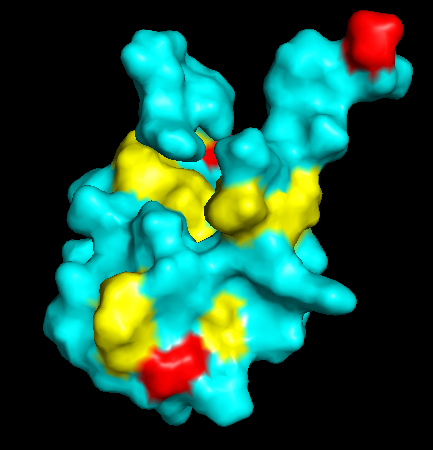
Residue Type
Distribution of hydrophobic vs hydrophilic residues?
The surface is dominated by hydrophilic residues (blue), necessary for this nuclear transcription factor to bind DNA.
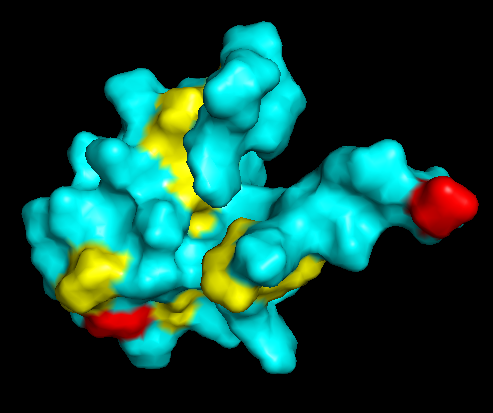
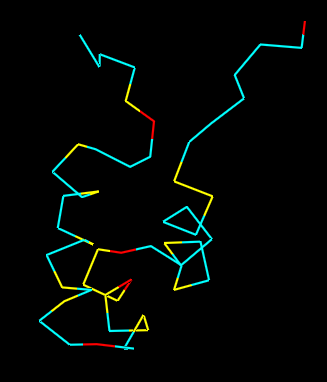
Surface Topology
Does it have any “holes”?
No deep holes. Instead, it contains shallow depressions and elongated surfaces, typical for protein-DNA interactions.
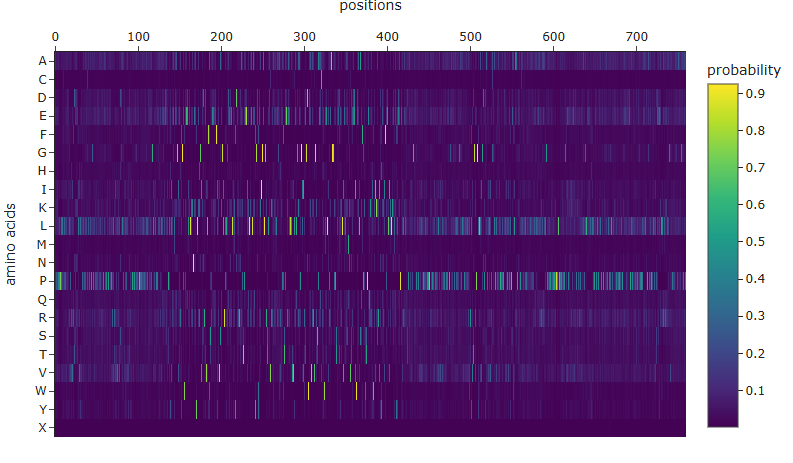
ML-Based Generation
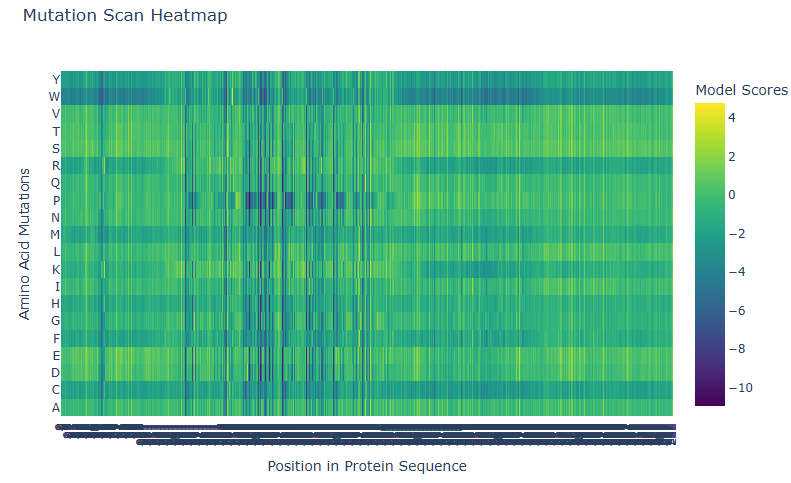
- Positions 230-385 feature frequent dark blue columns. Almost no mutation is well-tolerated here; a.a. are highly specific.
- Outer edges are greener, representing greatest tolerance.
- Amino acids like Tryptophan (W), R, M, H, F, C are almost always bad tolerated.
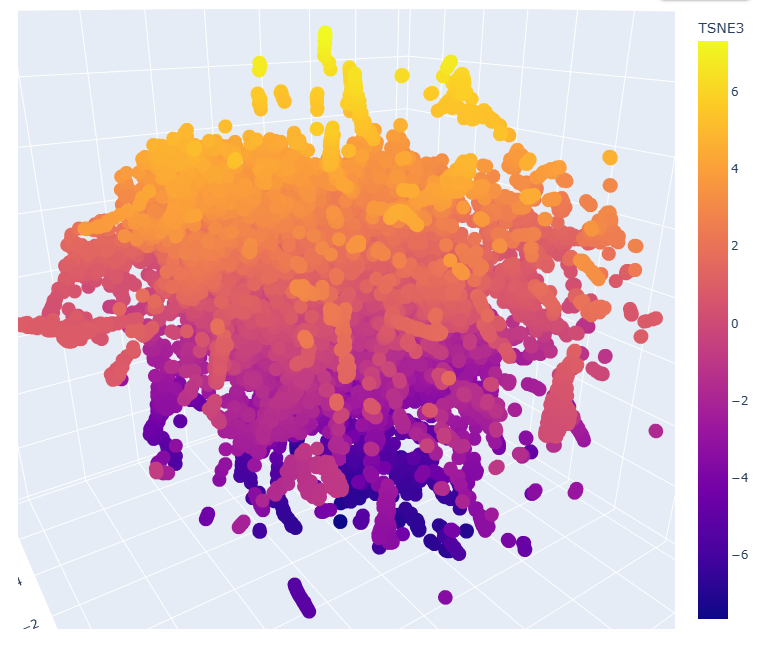
Proteins close in space often share organism, classification, and function.
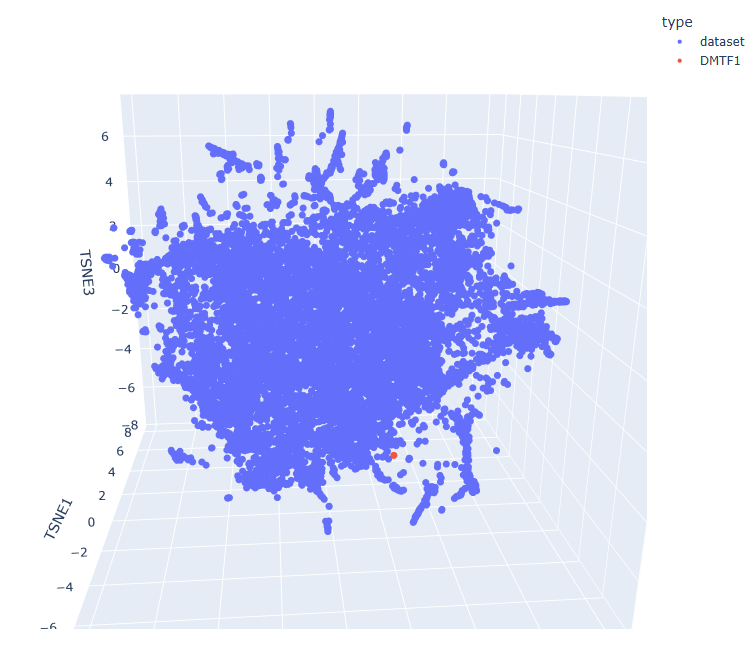
- d2g77a2 a.69.2.1 Ypt/Rab-GAP domain
- d4c3hd_ g.98.1.1 (D:) RNA polymerase I
- d2v8qa1 d.129.6.2 AMPK1
Part D. Group Brainstorm on Bacteriophage Engineering
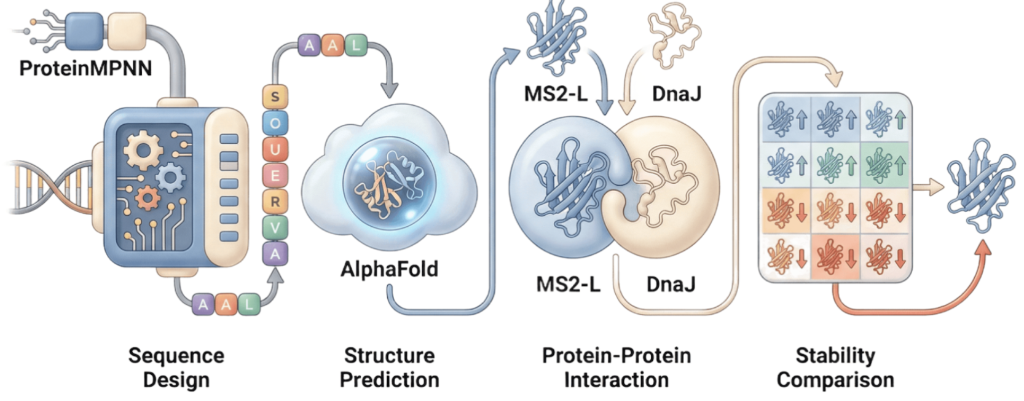
As discussed in “Phage Therapy: Past, Present and Future”, phage therapy represents an interesting alternative to antibiotic treatments, especially as recent developments allow researchers to engineer bacteriophages and their proteins. Our final group project for HTGAA Spring 2026 focuses on improving the bacteriophage MS2’s ability to kill its host bacteria E. coli by engineering its lysis protein MS2-L.
As an interdisciplinary team with different levels of experience in biotechnology, we propose increasing the stability of MS2-L. The lysis protein relies on the chaperone DnaJ for proper protein folding, a process E. coli can disrupt. However, it has been previously demonstrated that mutations deleting the N-terminal half of the MS2-L remove its dependence on DnaJ while also accelerating bacterial lysis. We believe this direction is promising for discovering variants that have structural stability within its host.
Our proposed approach begins with ProteinMPNN to look for alternative amino acid sequences that will improve the stability of MS2-L, then the sequences can be evaluated using AlphaFold and AlphaFold-Multimer to verify compatibility with their biological function and their interaction with DnaJ, with Alphafold specialized to model oligomeric complexes like MS2 and AlphaFold-Multimer tailored to predict protein-protein interactions like the one between MS2 and DnaJ.
Lastly, we must identify promising sequences for experimentation. We can do this by comparing variants quantitatively, e.g. using a deep mutational scan to see how each variant holds up when introduced to point mutations. This will narrow our candidate list to the most promising candidates for synthesis and experimental validation, reducing costs and promoting data-informed decision-making.
Any pitfalls are tied to the reliability of our tools; computational predictions of stability may not fully reflect protein behavior. For example, AlphaFold-Multimer has a systematic bias toward interactions between ordered protein regions, with a reduced accuracy for disordered regions and transient interactions like that of a chaperone and its complex.
We are also held back with a narrow scope. Phage therapy depends on several biological variables beyond a single protein, and there is currently a lack of pharmacokinetic and pharmacodynamic studies on phage therapy. This means that we can make MS2-L more stable, but other factors could limit the effectiveness of the bacteriophage.