Protein Design Part II
Part A: SOD1 Binder Peptide Design
Generate Binders with PepMLM
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence and add the known SOD1-binding peptide FLYRWLPSRRGG for comparison. Record the perplexity scores that indicate PepMLM’s confidence in the binders.
| Peptide Sequence | Perplexity Score |
|---|---|
| WRSYVAALAHWK | 12.24 |
| WHYPAVAAAWKE | 9.54 |
| WRYGVAAAEHKK | 12.30 (Best) |
| WRYYAVAAELWK | 16.22 |
| FLYRWLPSRRGG (Known) | 20.63 |
The perplexity score for the known binder was calculated reusing the function compute_pseudo_perplexity() with the next code snippet:
Evaluate Binders with AlphaFold3
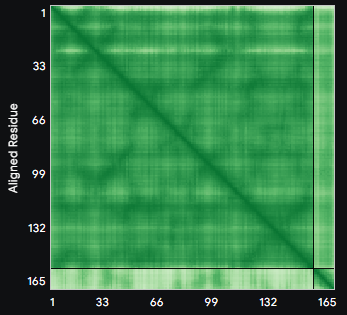
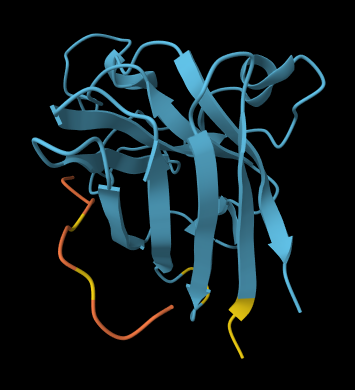
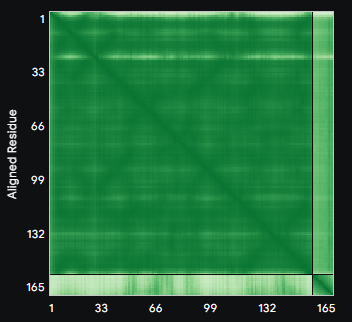
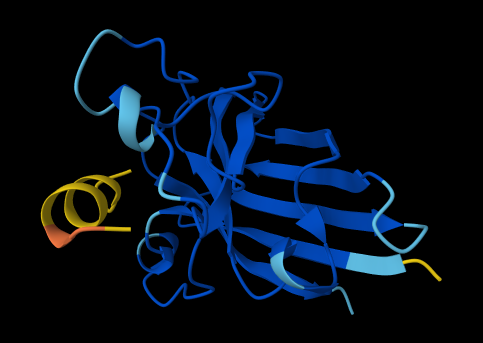
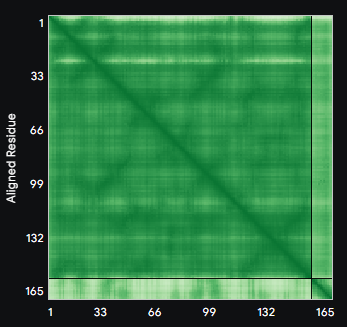
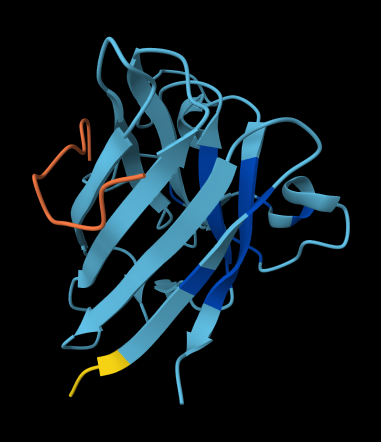
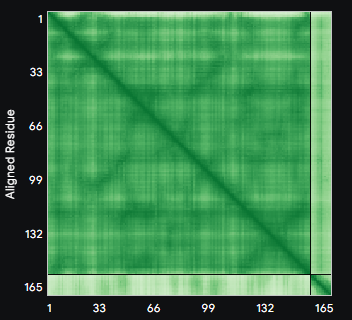
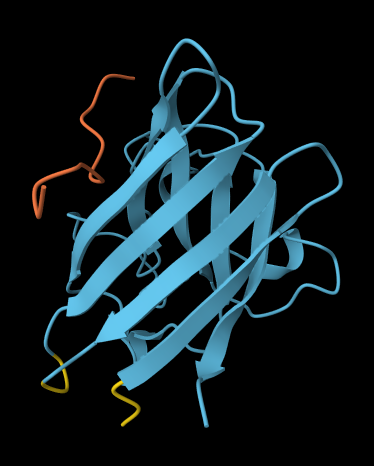
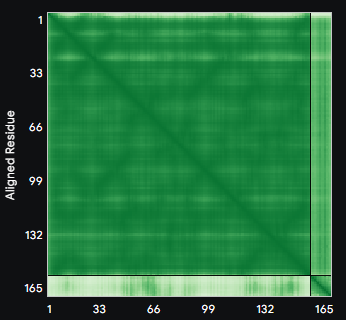
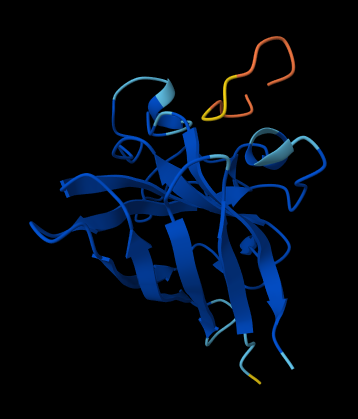
ALL ipTM scores ranged from 0.21 to 0.37, indicating a low overall confidence in protein-peptide interfaces across all models. The peptide WRYYAVAAELWK shows the lowest ipTM score of all models, while peptides WRSYVAALAHWK, WHYPAVAAAWKE, and WRYGVAAAEHKK all exceed the score of the known binder, where WRYGVAAAEHKK stands out as the interface prediction with the best confidence.
Evaluate Properties in the PeptiVerse
| Peptide Focus | Property | Prediction | Value | Unit |
|---|---|---|---|---|
| WRYYAVAAELWK | Solubility | Soluble | 1.000 | Probability |
| Hemolysis | Non-hemolytic | 0.101 | Probability | |
| Binding Affinity | Medium binding | 7.116 | pKd/pKi | |
| Length | - | 12 | aa | |
| Molecular Weight | - | 1555.8 | Da | |
| Net Charge (pH 7) | - | 0.76 | - | |
| Isoelectric Point | - | 8.50 | pH | |
| Hydrophobicity | Medium binding | -0.24 | GRAVY | |
| WHYPAVAAAWKE Selected Candidate | Solubility | Soluble | 1.000 | Probability |
| Hemolysis | Non-hemolytic | 0.025 | Probability | |
| Binding Affinity | Weak binding | 5.140 | pKd/pKi | |
| Length | - | 12 | aa | |
| Molecular Weight | - | 1428.6 | Da | |
| Net Charge (pH 7) | - | -0.15 | - | |
| Isoelectric Point | - | 6.76 | pH | |
| Hydrophobicity | - | -0.32 | GRAVY | |
| WRYGVAAAEHKK | Solubility | Soluble | 1.000 | Probability |
| Hemolysis | Non-hemolytic | 0.017 | Probability | |
| Binding Affinity | Weak binding | 5.487 | pKd/pKi | |
| Length | - | 12 | aa | |
| Molecular Weight | - | 1415.6 | Da | |
| Net Charge (pH 7) | - | 1.85 | - | |
| Isoelectric Point | - | 9.70 | pH | |
| Hydrophobicity | - | -1.00 | GRAVY |
It looks like there is no direct correlation between structural confidence and binding affinity, as the peptide with the highest ipTM score (WRYGVAAAEHKK) shows the weakest predicted affinity, while the one with the strongest affinity has the lowest confidence. All peptides show potentially good toxicity profiles, as they all are predicted as soluble an non-hemolytic.
I would choose a peptide to move forward by balancing structural confidence, binding affinity and therapeutic properites. In this context, WHYPAVAAAWKE emerges as a very good candidate: It has a solid structural confidence (ipTM 0.33), a reasonable binding affinity (5.14 pKd/pki), a low hemolysis probabilty (0.025) and is perfectly soluble.
Generate Optimized Peptides with moPPIt
| Peptide | Hemolysis | Solubility | Affinity | Motif |
|---|---|---|---|---|
| RCQRKEFTNLAA | 0.94 | 0.67 | 5.96 | 0.83 |
| GGTQCEVKKISW | 0.96 | 0.75 | 6.69 | 0.79 |
| ETYAPEYTDINA | 0.94 | 0.67 | 5.78 | 0.81 |
The peptides generated with moPPit are optimized for multiple therapeutic objectives, such as low hemolysis, high solubility, strong binding affinity, and motif presence. The moPPit peptides differ from the PepMLM-generated ones in the next ways:
- They exhibit very high hemolysis probabilities (~0.94), making them potentially toxic, whereas all PepMLM peptides are non-hemolytic (≤0.101).
- Their solubility is lower (~0.67 vs. ~1.0), and binding affinities are similar (5.78–5.96 pKd/pKi) except for one weak binder.
- moPPit peptides were optimized for motif presence, achieving high motif scores in two cases.
Next Steps: To evaluate these peptides before clinical studies, I would first perform in vitro hemolysis and cytotoxicity assays on relevant cell lines, followed by binding affinity measurements and structural validation to ensure specific target engagement and exclude any non desired effects. Stability and solubility tests would also be essential. Finaly, we would take into account the delivery method to assess how effectively the peptide reaches the target [1].
BRD4 Drug Discovery Platform Tutorial
Has not received credits for Boltz platform
Final Project: L-Protein Mutants
Comparison between experimental results and language model predictions.
Observation: There exists very little correlation, and the language model often predicts combinations that yielded a Lysis=0 experimentally, indicating poor functional capture.
Table 1 – Experimental Results on Mutagenesis
| Position | Mutation | Lysis | Protein Levels |
|---|---|---|---|
| 1 | M->I | 0 | 0 |
| 1 | M->I | 0 | 0 |
| 1 | M->T | 0 | 0 |
| 2 | E->Stop | 0 | N.D. |
| 3 | T->I | 0 | 0 |
| 3 | T->S | 0 | 0 |
| 6 | P->L | 0 | 0 |
| 8 | Q->Stop | 0 | N.D. |
| 8 | Q->L | 0 | 0 |
| 8 | Q->L | 0 | 0 |
| 10 | Q->Stop | 0 | N.D. |
| 11 | Q->Stop | 0 | N.D. |
| 13 | P->L | 1 | 1 |
| 13 | P->L | 1 | 1 |
| 15 | S->A | 1 | 1 |
| 18 | R->G | 1 | 1 |
| 18 | R->I | 1 | 1 |
| 18 | R->Stop | 0 | N.D. |
| 19 | R->S | 1 | 0 |
| 19 | R->H | 1 | 0 |
| 20 | R->W | 1 | 0 |
| 20 | R->W | 1 | 0 |
| 20 | R->L | 1 | 0 |
| 23 | K->E | 1 | 0 |
| 23 | K->E | 1 | 0 |
| 23 | K->Stop | 0 | N.D. |
| 23 | K->E | 1 | 0 |
| 23 | K->Stop | 0 | N.D. |
| 25 | E->V | 1 | 0 |
| 25 | E->G | 1 | 0 |
| 25 | E->D | 1 | 0 |
| 25 | E->G | 1 | 0 |
| 25 | E->G | 1 | 0 |
| 25 | E->G | 1 | 0 |
| 25 | E->G | 1 | 0 |
| 25 | E->G | 1 | 0 |
| 25 | E->G | 1 | 0 |
| 25 | E->G | 1 | 0 |
| 25 | E->G | 1 | 0 |
| 25 | E->G | 1 | 0 |
| 25 | E->G | 1 | 0 |
| 26 | D->G | 1 | 0 |
| 27 | Y->Stop | 0 | N.D. |
| 29 | C->Stop | 0 | N.D. |
| 29 | C->R | 0 | 0 |
| 29 | C->Stop | 0 | N.D. |
| 30 | R->Q | 1 | 1 |
| 30 | R->L | 1 | 1 |
| 30 | R->Stop | 0 | N.D. |
| 30 | R->Stop | 0 | N.D. |
| 31 | R->Stop | 0 | N.D. |
| 31 | R->Stop | 0 | N.D. |
| 31 | R->I | 1 | 1 |
| 32 | Q->Stop | 0 | N.D. |
| 33 | Q->H | 0 | 1 |
| 33 | Q->H | 0 | 1 |
| 33 | Q->Stop | 0 | N.D. |
| 33 | Q->Stop | 0 | N.D. |
| 34 | R->Stop | 0 | N.D. |
| 34 | R->Stop | 0 | N.D. |
| 36 | S->Stop | 0 | N.D. |
| 36 | S->Stop | 0 | N.D. |
| 39 | Y->H | 0 | 0 |
| 39 | Y->Stop | 0 | N.D. |
| 39 | Y->Stop | 0 | N.D. |
| 40 | V->E | 0 | 0 |
| 41 | L->Stop | 0 | N.D. |
| 41 | L->Stop | 0 | N.D. |
| 41 | L->Stop | 0 | N.D. |
| 42 | I->N | 0 | 0 |
| 43 | F->L | 0 | 1 |
| 44 | L->V | 0 | 1 |
| 44 | L->P | 1 | 1 |
| 44 | L->P | 1 | 1 |
| 45 | A->P | 1 | 1 |
| 46 | I->N | 0 | 0 |
| 46 | I->F | 1 | 1 |
| 46 | I->N | 0 | 0 |
| 47 | F->Y | 0 | 1 |
| 47 | F->Y | 0 | 1 |
| 47 | F->Y | 0 | 1 |
| 48 | L->P | 0 | 1 |
| 49 | S->L | 0 | 1 |
| 49 | S->Stop | 0 | N.D. |
| 49 | S->L | 0 | 1 |
| 49 | S->T | 0 | 1 |
| 49 | S->Stop | 0 | N.D. |
| 49 | S->T | 0 | 1 |
| 50 | K->E | 0 | 1 |
| 50 | K->N | 0 | 1 |
| 50 | K->N | 0 | 1 |
| 50 | K->Stop | 0 | N.D. |
| 50 | K->I | 0 | 1 |
| 50 | K->N | 0 | 1 |
| 50 | K->Q | 0 | 0 |
| 50 | K->I | 0 | 1 |
| 50 | K->E | 0 | 1 |
| 50 | K->N | 0 | 1 |
| 50 | K->Stop | 0 | N.D. |
| 50 | K->N | 0 | 1 |
| 50 | K->Stop | 0 | N.D. |
| 51 | F->S | 0 | 1 |
| 51 | F->S | 0 | 1 |
| 52 | T->N | 0 | 0 |
| 53 | N->S | 0 | 1 |
| 53 | N->S | 0 | 1 |
| 53 | N->D | 0 | 1 |
| 53 | N->H | 0 | 1 |
| 53 | N->S | 0 | 1 |
| 53 | N->I | 0 | 0 |
| 53 | N->Q | 0 | 0 |
| 53 | N->K | 0 | 0 |
| 53 | N->Q | 0 | 0 |
| 54 | Q->Stop | 0 | 0 |
| 55 | L->Stop | 0 | N.D. |
| 55 | L->Stop | 0 | N.D. |
| 56 | L->H | 0 | 1 |
| 56 | L->H | 0 | 1 |
| 56 | L->H | 0 | 1 |
| 56 | L->P | 0 | 0 |
| 56 | L->P | 0 | 0 |
| 56 | L->H | 0 | 1 |
| 57 | L->P | 0 | 0 |
| 60 | L->P | 0 | 0 |
| 60 | L->V | 0 | 0 |
| 60 | L->Q | 0 | 0 |
| 60 | L->P | 0 | 0 |
| 60 | L->Q | 0 | 0 |
| 63 | V->E | 0 | 1 |
| 63 | V->E | 0 | 1 |
| 66 | T->K | 0 | 1 |
| 66 | T->R | 0 | 0 |
| 69 | T->S | 0 | 0 |
| 71 | Q->Stop | 0 | N.D. |
| 72 | Q->Stop | 0 | N.D. |
| 73 | L->Stop | 0 | N.D. |
| 73 | L->Stop | 0 | N.D. |
| 73 | L->Stop | 0 | N.D. |
Table 2 – Predicted scores via Language Models
| Position | Initial AA | Mut AA | LLR Score |
|---|---|---|---|
| 50 | K | L | 2.56 |
| 29 | C | R | 2.40 |
| 39 | Y | L | 2.24 |
| 29 | C | S | 2.04 |
| 9 | S | Q | 2.01 |
| 29 | C | Q | 2.00 |
| 29 | C | P | 1.97 |
| 29 | C | L | 1.96 |
| 50 | K | I | 1.93 |
| 53 | N | L | 1.86 |
| 61 | E | L | 1.82 |
| 52 | T | L | 1.81 |
| 50 | K | F | 1.80 |
| 29 | C | T | 1.80 |
| 29 | C | K | 1.80 |
| 5 | F | Q | 1.80 |
| 5 | F | R | 1.66 |
| 29 | C | A | 1.65 |
| 27 | Y | R | 1.63 |
| 22 | F | R | 1.60 |
| 5 | F | P | 1.60 |
| 50 | K | V | 1.59 |
| 50 | K | S | 1.57 |
| 5 | F | T | 1.56 |
| 5 | F | S | 1.56 |
| 45 | A | L | 1.54 |
| 39 | Y | S | 1.52 |
| 27 | Y | S | 1.50 |
| 40 | V | L | 1.48 |
| 27 | Y | L | 1.47 |
| 22 | F | S | 1.42 |
| 29 | C | E | 1.38 |
| 39 | Y | A | 1.36 |
| 29 | C | N | 1.36 |
| 50 | K | A | 1.36 |
| 29 | C | I | 1.34 |
| 5 | F | L | 1.33 |
| 17 | N | R | 1.32 |
| 39 | Y | I | 1.32 |
| 39 | Y | T | 1.30 |
| 26 | D | R | 1.27 |
| 29 | C | H | 1.25 |
| 39 | Y | F | 1.25 |
| 39 | Y | V | 1.24 |
| 23 | K | R | 1.24 |
| 25 | E | R | 1.23 |
| 24 | H | R | 1.23 |
| 50 | K | T | 1.22 |
| 27 | Y | Q | 1.22 |
| 27 | Y | T | 1.22 |
Original Protein
Original
Original + DnaJ
Predicted Mutant Structures
Structures predicted via MF2 Multimer for selected L-protein mutants. Each mutation targets either the transmembrane region (AA 41–75) or the soluble region (AA 1–40).
Mutations in Transmembrane Region (AA 41–75)
L44P: METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFPAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
L44P
L44P + DnaJ
A45P: METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLPIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
A45P
A45P + DnaJ
V63E: METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAEIRTVTTLQQLLT
V63E
V63E + DnaJ
Mutations in Soluble Region (AA 1–40)
R30Q: METRFPQQSQQTPASTNRRRPFKHEDYPCRQQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
R30Q
R30Q + DnaJ
C29R: METRFPQQSQQTPASTNRRRPFKHEDYPRRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
C29R
C29R + DnaJ
References
- [1] WuXi AppTec, "A Strategic Roadmap for Peptide Preclinical Studies: 3 Key Stages," labtesting.wuxiapptec.com, Oct. 30, 2025. [Online]. Available: https://labtesting.wuxiapptec.com/2025/10/30/a-strategic-roadmap-for-peptide-preclinical-studies-3-key-stages/. [Accessed: Mar. 16, 2026].