Subsections of Homework
Week 1 HW: Principles and Practices




 For the raw text:
https://docs.google.com/document/d/1u3VepFZVKRcZfB8QUowPQvQGaEuspPL8mM99Tzm679Y/edit?usp=sharing
For the raw text:
https://docs.google.com/document/d/1u3VepFZVKRcZfB8QUowPQvQGaEuspPL8mM99Tzm679Y/edit?usp=sharing
Week 2 HW: DNA Read Write and Edit
Homework week 2 - DNA READ, WRITE & EDIT
Part 1: Benchling & In-silico Gel Art
Part 3: DNA Design Challenge
3.1. Choose your protein.
Cocoonase, an enzyme capable of degrading silk protein sericin-2.
GenBank: AB604648.1
Rodbumrer P, Arthan D, Uyen U, Yuvaniyama J, Svasti J, Wongsaengchantra PY. Functional expression of a Bombyx mori cocoonase: potential application for silk degumming. Acta Biochim Biophys Sin (Shanghai). 2012 Dec;44(12):974-83. doi: 10.1093/abbs/gms090. PMID: 23169343.
Amino acid sequence:
MIVGGEEISINKVPYQAYLLLQKDNEYFQCGGSIISKRHILTAA
HCIEGISKVTVRIGSSNSNKGGTVYTAKSKVAHPKYNSKTKNNDFAIVTVNKDMAIDG
KTTKIITLAKEGSSVPDKTKLLVSGWGATSEGGSSSTTLRAVHVQAHSDDECKKYFRS
LTSNMFCAGPPEGGKDSCQGDSGGPAVKGNVQLGVVSFGVGCARKNNPGIYAKVSAAAKWIKSTAGL
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
cDNA:
atgattgtgggcggcgaagaaattagcattaacaaagtgccgtatcaggcgtatctgctg
ctgcagaaagataacgaatattttcagtgcggcggcagcattattagcaaacgccatatt
ctgaccgcggcgcattgcattgaaggcattagcaaagtgaccgtgcgcattggcagcagc
aacagcaacaaaggcggcaccgtgtataccgcgaaaagcaaagtggcgcatccgaaatat
aacagcaaaaccaaaaacaacgattttgcgattgtgaccgtgaacaaagatatggcgatt
gatggcaaaaccaccaaaattattaccctggcgaaagaaggcagcagcgtgccggataaa
accaaactgctggtgagcggctggggcgcgaccagcgaaggcggcagcagcagcaccacc
ctgcgcgcggtgcatgtgcaggcgcatagcgatgatgaatgcaaaaaatattttcgcagc
ctgaccagcaacatgttttgcgcgggcccgccggaaggcggcaaagatagctgccagggc
gatagcggcggcccggcggtgaaaggcaacgtgcagctgggcgtggtgagctttggcgtg
ggctgcgcgcgcaaaaacaacccgggcatttatgcgaaagtgagcgcggcggcgaaatgg
attaaaagcaccgcgggcctg
The chosen chassis is e.coli, as it is really good for genetic circuit implementation
3.3. Codon optimization.
atgattgtgggcggcgaagaaattagcattaacaaagtgccgtatcaggcgtatctgctgctgcagaaagataacgaatattttcagtgcggcggcagcattattagcaaacgccatattctgaccgcggcgcattgcattgaaggcattagcaaagtgaccgtgcgcattggcagcagcaacagcaacaaaggcggcaccgtgtataccgcgaaaagcaaagtggcgcatccgaaatataacagcaaaaccaaaaacaacgattttgcgattgtgaccgtgaacaaagatatggcgattgatggcaaaaccaccaaaattattaccctggcgaaagaaggcagcagcgtgccggataaaaccaaactgctggtgagcggctggggcgcgaccagcgaaggcggcagcagcagcaccaccctgcgcgcggtgcatgtgcaggcgcatagcgatgatgaatgcaaaaaatattttcgcagcctgaccagcaacatgttttgcgcgggcccgccggaaggcggcaaagatagctgccagggcgatagcggcggcccggcggtgaaaggcaacgtgcagctgggcgtggtgagctttggcgtgggctgcgcgcgcaaaaacaacccgggcatttatgcgaaagtgagcgcggcggcgaaatggattaaaagcaccgcgggcctg
Part 4: Prepare a DNA Twist Order
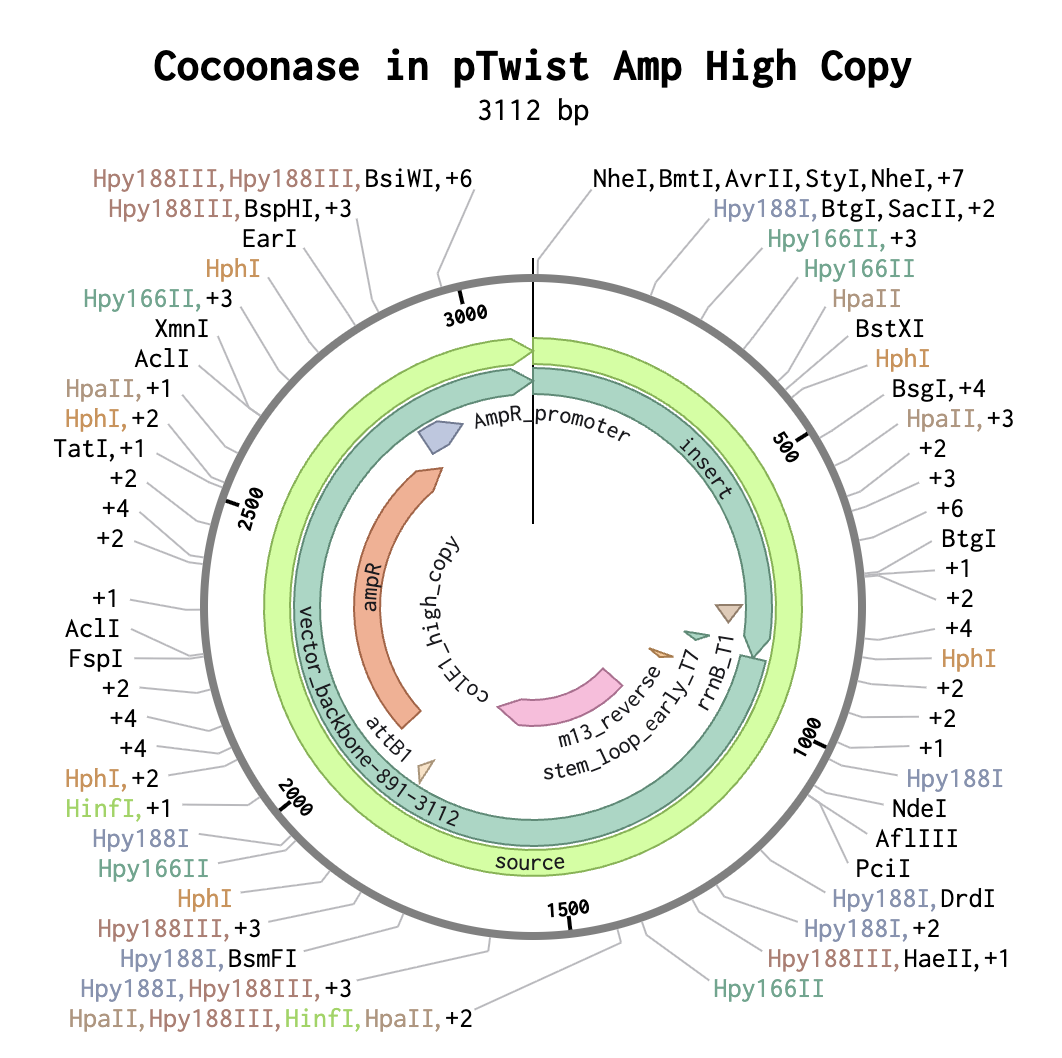
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
The DNA that I would like to sequence is related to my initial project proposal described in last week’s homework. In it I describe L. portucalensis F11; a strain of bacteria that shows evidence to be able to chain-shorten PFAS particles. I would like to sequence this bacteria’s DNA as a first step into isolating the sequence that corresponds to the enzyme that’s able to break down these ‘forever chemicals’.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Also answer the following questions:
The DNA I want to sequence comes from a bacterium whose genome has not been sequenced previously. Initially I figured shotgun sequencing would potentially be a good approach as prokaryotic DNA is simpler in structure and therefore less prone to error than eukaryotic organisms whose genome contains many repetitive sequences. If I simultaneously find and isolate the enzyme responsible for degradation of PFAS, I would be able to cross reference the amino acid chains with the genome, to localise the sequence that codes for it.
However, this is a highly inefficient approach, as it takes a lot of time, resources and money.
L. portucalensis F11, could be cultivated in vitro by only giving it PFAS as potential nutrients. This would mean that the only degenerative enzymatic activity would be by the targeted enzyme, indicating the presence of mRNA coding for the aminoacid-chain of the prospective protein.
Grow bacteria (L. portucalensis F11) on PFAS as the sole carbon source to induce pathway-specific enzymes, ensuring upregulated mRNA dominates the transcriptome.
Lyse cells and extract total RNA using standard kits (e.g., TRIzol), capturing the active enzymatic snapshot.
Remove 80-90% ribosomal RNA through rRNA depletion, with probe-based methods to enrich messenger RNA and avoid wasted sequencing.
Directly sequence the depleted RNA using Oxford Nanopore Sequencing for full-length native transcripts.
Prepare the library: Fix damaged ends on your rRNA-depleted RNA, then attach adapters (a motor protein and anchor) to each strand using ligation.
Load the flow cell: Prime the MinION chip's tiny holes (nanopores) with buffer, then add your RNA library via the SpotON port.
Start sequencing: Apply voltage—the motor pulls RNA through nanopores one-by-one, changing electrical current as each base passes.
Basecall live: Software like Guppy converts the current "squiggles" into RNA sequence reads (A, C, G, U) in real-time.
Assemble reads (e.g., FLAIR/Trinity), quantify expression, and identify degradative enzymes via pathway analysis (KEGG/BLAST for haloacid dehalogenases, monooxygenases).
Computational Reverse Translation: From assembled mRNA, computationally translate to amino acids then back-translate to the precise genomic DNA sequence using codon bias tools (e.g., Backtranseq), accounting for degeneracy.
Oxford Nanopore sequencing is a third-generation sequencing because it reads single DNA/RNA molecules directly in real-time without amplification or short-read assembly
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond.
I would like to synthesize a repressilator-based genetic circuit that could be applied to bacteria that are implemented into the silk structure.
The circuit would be based on three genetic components:
A PFAS detecting biosensor (Mann, et al. 2023)
A gene that encodes for an enzyme that breaks down Sericin 2 (Rodbumrer, et al. 2012)
A gene that encodes for an enzyme that breaks down PFAS particles (Wijayahena et al., 2025)
If the PFAS-detecting biosensor senses the presence of PFAS particles, it would repress the Sericin 2-enzyme gene. This would pressure the bacteria to degrade the PFAS particles. Once the particles are no longer present, the detector would no longer repress the Sericin 2-enzyme gene and the bacteria could continue to survive on metabolising Sericin 2.
Mann, M.M., Berger, B.W. A genetically-encoded biosensor for direct detection of perfluorooctanoic acid. Sci Rep 13, 15186 (2023). https://doi.org/10.1038/s41598-023-41953-1
Rodbumrer P, Arthan D, Uyen U, Yuvaniyama J, Svasti J, Wongsaengchantra PY. Functional expression of a Bombyx mori cocoonase: potential application for silk degumming. Acta Biochim Biophys Sin (Shanghai). 2012 Dec;44(12):974-83. doi: 10.1093/abbs/gms090. PMID: 23169343.
Wijayahena, M.K., Moreira, I.S., Castro, P.M.L., Dowd, S., Marciesky, M.I., Ng, C. and Aga, D.S., 2025. PFAS biodegradation by Labrys portucalensis F11: Evidence of chain shortening and identification of metabolites of PFOS, 6:2 FTS, and 5:3 FTCA. Science of The Total Environment, 959, p.178348.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I would utilize phosphoramidite oligo synthesis to synthesize the separate sequences, to then combine them using Gibson assembly.
Essential steps phosphoramidite oligo synthesis:
Core Synthesis Cycle
The cycle attaches protected phosphoramidite monomers to a solid support (like controlled-pore glass, CPG) bound to the first 3' nucleoside.
Detritylation (Deblocking): Acid (e.g., trichloroacetic acid) removes the 5'-dimethoxytrityl (DMT) protecting group from the growing chain's 5'-OH, exposing it for the next coupling.
Coupling: A phosphoramidite monomer (with DMT-protected 5'-OH, base protections, and reactive 3'-phosphoramidite) plus activator (e.g., tetrazole) reacts with the free 5'-OH to form an unstable phosphite triester linkage (~99% efficiency per step).
Capping: Acetic anhydride caps unreacted 5'-OH groups (as acetyl esters) to prevent truncated failure sequences from proceeding, ensuring high purity.
Oxidation: Iodine in water/pyridine converts the fragile phosphite triester to a stable phosphate triester, mimicking the natural DNA backbone.
Post-Synthesis Processing
After the final cycle and detritylation, the oligo is cleaved from the support (e.g., via ammonia), and all protecting groups are removed (bases, phosphates). Final purification uses HPLC or PAGE.
Essential steps Gibson Assembly:
Gibson assembly joins DNA pieces seamlessly in one simple mix using enzymes. It's great for building genetic circuits like the repressilator from overlapping fragments.
Prep Your DNA
Make or get linear DNA fragments (like PCR products or synthesized genes) with 20-40 base matching ends (overlaps). Use equal amounts, clean them up to remove junk.
One-Pot Reaction
Mix fragments with Gibson master mix (has exonuclease, polymerase, ligase) in a tube on ice. Incubate at 50°C for 15-60 minutes.
Chew-back: Enzyme nibbles 5' ends to make sticky single-strand overlaps.
Anneal and fill: Polymerase glues matching overlaps and fills gaps.
Seal: Ligase connects the nicks for a full circle.
Cool on ice when done.
Oligonucleotide synthesis is limited due to the fact that the longer the chain becomes, the more prone it becomes to error in accuracy. This can be mitigated by only constructing the separate genes described in the previous question.
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
Within the context of my HTGAA project, I would like to make an edit in the genome of my chosen chassis, e.coli k12, by deleting the gene that encodes the tolC protein. The tolC protein is an outer membrane channel, which is required for the function of several efflux systems. As it’s responsible for toxin removal from the cell, it would flush out PFAS particles to the extent that the biosensor would barely be able to detect any particles.
(ii) What technology or technologies would you use to perform these DNA edits and why?
Also answer the following questions:
How does your technology of choice edit DNA? What are the essential steps?
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
I would choose to perform these DNA utilising a CRISPR/CAS9 system, which edits DNA by performing double strand cuts.
It consists of CRISPR and Cas9:
Cas9 is an endonuclease that recognizes protospacer adjacent motifs (PAM) in target DNA. PAM = a short DNA sequence (2-6 base pairs) found next to the target site (protospacer) that a CRISPR-Cas system recognizes and cuts, acting as a crucial "self vs. non-self" marker. Single guide RNA (sgRNA) directs it to target DNA
sgRNA = CRISPR RNA + trans-activating RNA (tracrRNA)
CRISPR RNA is a small, specialized RNA molecule derived from the bacterial CRISPR locus that guides Cas nucleases (like Cas9) to complementary target DNA, enabling precise cutting.
tracrRNA = a small, non-coding RNA essential for the maturation of CRISPR RNA (crRNA) and the activation of the Cas9 nuclease in Type II CRISPR-Cas immune systems. It binds to pre-crRNA, facilitating its cleavage, and forms a dual-RNA complex that guides Cas9 to specific DNA sequences for cleavage. This allows for the sequence to be cut at a specific site, which would then trigger ligase cellular function NHEJ or HDR.
1. Target Design & Guide RNA (gRNA) Selection: Identify the target DNA sequence within the gene to be knocked out. Design a 20-nucleotide sequence complementary to the target, adjacent to a Protospacer Adjacent Motif (PAM, typically 5'-NGG-3' for S. pyogenes Cas9).
2. Assembly of CRISPR Components: The gRNA and Cas9 enzyme are combined to form a ribonucleoprotein (RNP) complex, or plasmids encoding these components are created.
3. Delivery into Cells: The CRISPR components are delivered into target cells (via transfection, electroporation, or viral vectors).
4. Target Recognition and Cleavage: The Cas9-gRNA complex scans the genome for the PAM sequence. Once found, the gRNA hybridizes with the target DNA, activating the Cas9 nuclease to create a double-strand break (DSB) ~3 base pairs upstream of the PAM.
5. DNA Repair and Mutation Induction: The cell attempts to repair the DSB, predominantly via Non-Homologous End Joining (NHEJ). This error-prone process often introduces random insertions or deletions (indels) at the cleavage site.
6. Gene Knockout Confirmation: Indels within the open reading frame (ORF) typically cause frameshift mutations, leading to premature stop codons and nonfunctional proteins (knockout). The resulting mutations are verified via DNA sequencing or mismatch detection assays.
Week 3 HW: Lab Automation
Opentrons Artwork
Post-Lab Questions
Question 1.
My chosen article, entitled ‘AssemblyTron: flexible automation of DNA assembly with Opentrons OT-2 lab robots’, details novel development within the field of synthetic biology and automated lab operations in regards to DNA assembly.
Compared to other steps in the DBTL cycle of synbio, building has progressed the least in terms of automation, optimization and algorithmical performance. This step often involves DNA assembly, which continues to be done manually, with low throughput and often unreliable results.
The article presents AssemblyTron; an open-source Python package to integrate j5 DNA assembly design software outputs with build implementations in Opentrons liquid handling robotics with minimal human intervention.
It describes how AssemblyTron is capable of the following:
Several scarless, multipart DNA assemblies, beginning from fragment amplification.
Polymerase chain reactions across a range of fragment lengths and annealing temperatures by utilising an optimal annealing gradient calculation algorithm.
Golden Gate and homology dependent in vivo assemblies.
Site-directed mutagenesis reactions via homology-dependent IVA
John A Bryant, Mason Kellinger, Cameron Longmire, Ryan Miller, R Clay Wright, AssemblyTron: flexible automation of DNA assembly with Opentrons OT-2 lab robots, Synthetic Biology, Volume 8, Issue 1, 2023, ysac032, https://doi.org/10.1093/synbio/ysac032
Question 2.
For this assignment I decided to focus on one of my three final graduation project, investigating the possibilities to utilize Opentrons to safely and efficiently conduct lab research:
Final project option 2 - Isolating the DNA sequence that codes for the enzyme responsible for PFAS degradation in L. portucalensis F11
For this option, if I could get access to F11, I would use opentrons to inoculate 96-well plates with the bacteria in a medium solution that contains PFAS as the sole carbon source. I would then measure OD600 timepoints to determine cell density (Myers, et al. 2020) to find the optimal timing for RNA extraction.
When the optimal timing for RNA extraction is reached, I could then automate the cell lysing process.
For the extraction of excessive rRNA, I would automate the pipetting process of the hybridisation of DNA probes to the rRNA. Then, using the Opentrons magnetic module (Opentrons, 2026), I could remove the rRNA by using streptavidin-coated magnetic beads.
Finally, I would utilise opentrons automation to library prep for Oxford Nanopore Sequencing.
References:
Myers, J.A., Kitajima, M., Dai, L.C., & Wright, M.S. (2020) 'Robust estimation of bacterial cell count from optical density', Communications Biology, 3(1), p. 446. Available at: https://doi.org/10.1038/s42003-020-01127-5 (Accessed: 24 February 2026).
Opentrons (2026). Opentrons Flex Magnetic Block. [online] Opentrons.com. Available at: https://opentrons.com/products/opentrons-flex-magnetic-block-gen1?utm_source=google&utm_medium=cpc&utm_campaign={campaignname}&utm_term=&hsa_cam=21988025885&hsa_grp=&hsa_mt=&hsa_src=x&hsa_ad=&hsa_acc=2303351826&hsa_net=adwords&hsa_kw =&hsa_tgt=&hsa_ver=3&gad_source=1&gad_campaignid=21981643080&gbraid=0AAAAADemOsWv5oA02ZBRucCPg3Hfq31a8&gclid=Cj0KCQiAtfXMBhDzARIsAJ0jp3DelnoEGaaIAaFZozVi7Rp9gGhAOWiMezpLkMCDgPqzlIMIj1niM3oaAvkeEALw_wcB
Oxford Nanopore Technologies (2026) Platform technology. Available at:
https://nanoporetech.com/platform/technology (Accessed: 24 February 2026)
Final Project Ideas


Week 4 HW: Protein Design Part 1
Part A: Conceptual Questions
Part A
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons?
A piece of meat weighing 500 grams contains approximately 100 to 160 grams of protein. 100 Daltons is equivalent to 1.66053907 × 10-22 grams If we divide the amount of protein by this number, it would mean that a piece of 500 grams of meat would contain between 6.0221407e+23 and 9.6354252e+23 amino acids.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
The digestion system breaks down any genetic material, moreover since the cells are not endogenous to our body they do not get “access” to our bodily system.
Why are there only 20 natural amino acids?
“The 20 standard amino acids encoded by the Genetic Code were adopted during the RNA World, around 4 billion years ago. This amino acid set could be regarded as a frozen accident, implying that other possible structures could equally well have been chosen to use in proteins. Amino acids were not primarily selected for their ability to support catalysis, as the RNA World already had highly effective cofactors to perform reactions, such as oxidation, reduction and transfer of small molecules. Rather, they were selected to enable the formation of soluble structures with close-packed cores, allowing the presence of ordered binding pockets. Factors to take into account when assessing why a particular amino acid might be used include its component atoms, functional groups, biosynthetic cost, use in a protein core or on the surface, solubility and stability. Applying these criteria to the 20 standard amino acids, and considering some other simple alternatives that are not used, we find that there are excellent reasons for the selection of every amino acid. Rather than being a frozen accident, the set of amino acids selected appears to be near ideal.”
Directly quoted from:
Can you make other non-natural amino acids? Design some new amino acids.
Yes it is possible to make other non-natural amino acids, as amino acids have the same structure: a central alpha carbon, an amino group on one side of it and a carboxyl group on the other, with a unique variable side chain.
Where did amino acids come from before enzymes that make them, and before life started?In 1952, the Miller-Urey experiment proved that complex carbon based amino acids can form spontaneously from inorganic precursors under conditions simulating early Earth. This indicates that abiotic synthesis happened before enzymes existed that would make them.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
In opposition to L-amino acids that make right handed α-helices, I would expect α-helices made up of D-amino acids to be left handed.
Can you discover additional helices in proteins?
Yes one can discover additional helices in proteins. While traditional methods like DSSP only take strong hydrogen bonds into consideration, it disregards smaller structures and fringe residues of alpha helices. Methods like STRIDE and geometry tools do detect these residues as alpha helix structures by analysing hydrogen bonds as well as twist angles.
Why are most molecular helices right-handed?
Most molecular helices are right-handed, because amino acids in nature use L-amino acids. Their left-handed chirality forces helices to form in right-handed coils for stability.
Why do β-sheets tend to aggregate?
β-sheets tend to aggregate due to their sheet-like nature; their edges are exposed and therefore have the tendency to latch onto other sheets via hydrogen bonds and hydrophobic contacts. The open strand edges of β-sheets are primed for edge-to-edge zipping with neighbouring sheets.
Part B.
Briefly describe the protein you selected and why you selected it.
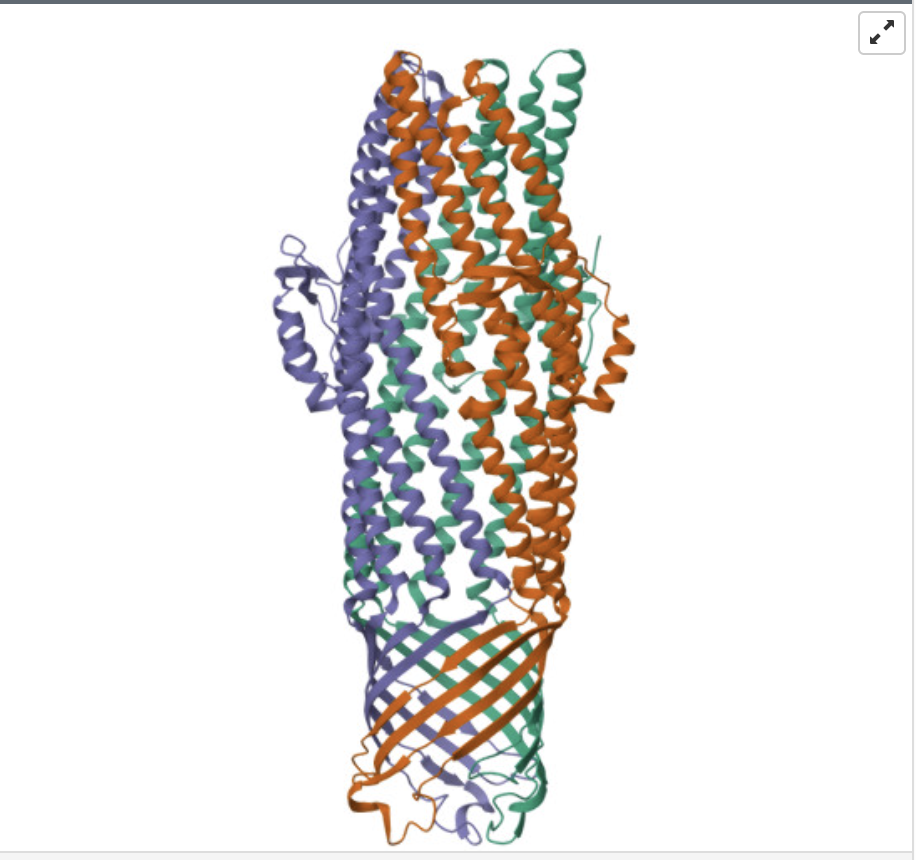
The protein I selected is tolC, a protein in the outer membrane of E.coli K12 that’s responsible for the afflux of various toxins. I selected it for my project as I intend to build a genetic circuit that represses this protein’s function.
Identify the amino acid sequence of your protein.
The amino acid sequence is: MKKLLPILIGLSLSGFSSLSQAENLMQVYQQARLSNPELRKSAADRDAAFEKINEARSPLLPQLGLGADYTYSNGYRDANGINSNATSASLQLTQSIFDMSKWRALTLQEKAAGIQDVTYQTDQQTLILNTATAYFNVLNAIDVLSYTQAQKEAIYRQLDQTTQRFNVGLVAITDVQNARAQYDTVLANEVTARNNLDNAVEQLRQITGNYYPELAALNVENFKTDKPQPVNALLKEAEKRNLSLLQARLSQDLAREQIRQAQDGHLPTLDLTASTGISDTSYSGSKTRGAAGTQYDDSNMGQNKVGLSFSLPIYQGGMVNSQVKQAQYNFVGASEQLESAHRSVVQTVRSSFNNINASISSINAYKQAVVSAQSSLDAMEAGYSVGTRTIVDVLDATTTLYNAKQELANARYNYLINQLNIKSALGTLNEQDLLALNNALSKPVSTNPENVAPQTPEQNAIADGYAPDSPAPVVQQTSARTTTSNGHNPFRN
The sequence is 493 amino acids in length and the most common amino acid is alanine which appears 57 times.
The protein has various homologous proteins, at 100% shared identity, 34 variants of e.coli share the same protein. At 50% similarity there are 1252 entries.
My protein belongs to the outer membrane factor (omf) family.
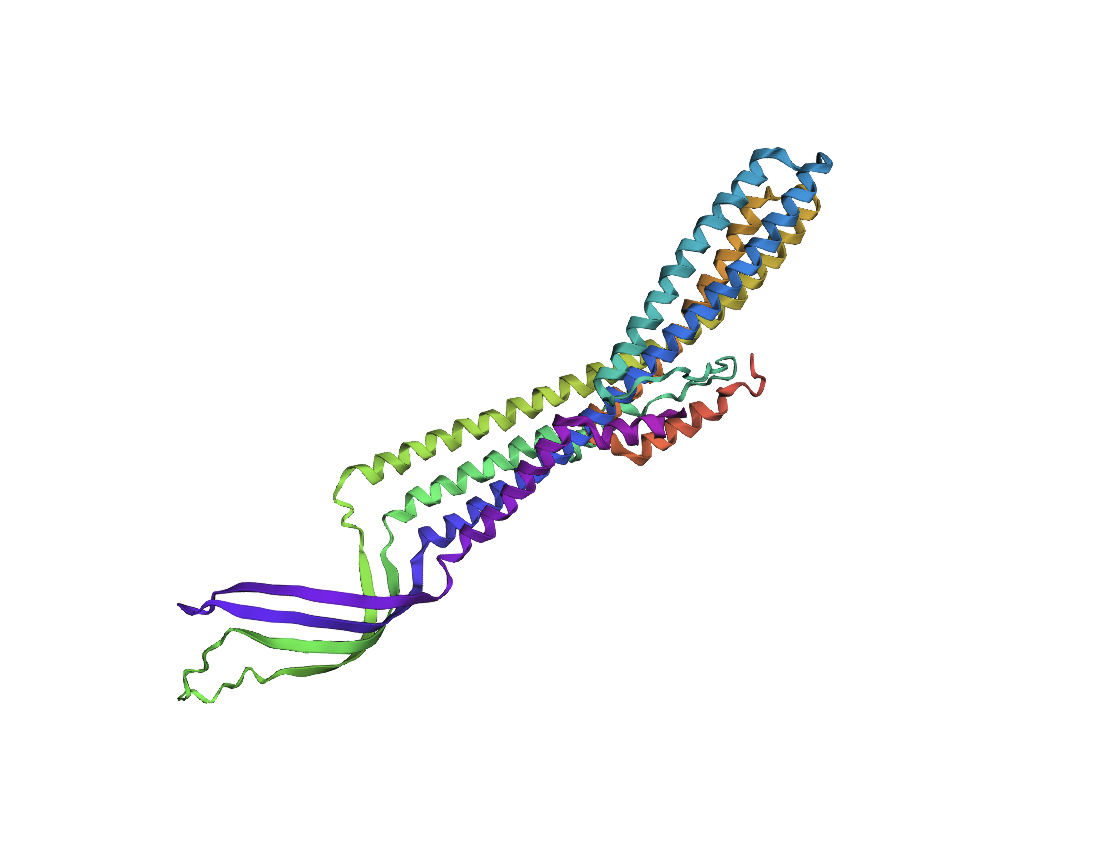
Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
The structure of my protein was solved in 2004, with a resolution of 2.75 Å. This indicates that it’s a good quality structure.
Are there any other molecules in the solved structure apart from protein?
The ligand Cobalt Hexammine(III)
Does your protein belong to any structure classification family?
Yes it belongs to Multidrug efflux transporter AcrB TolC docking domain (DN and DC subdomains)
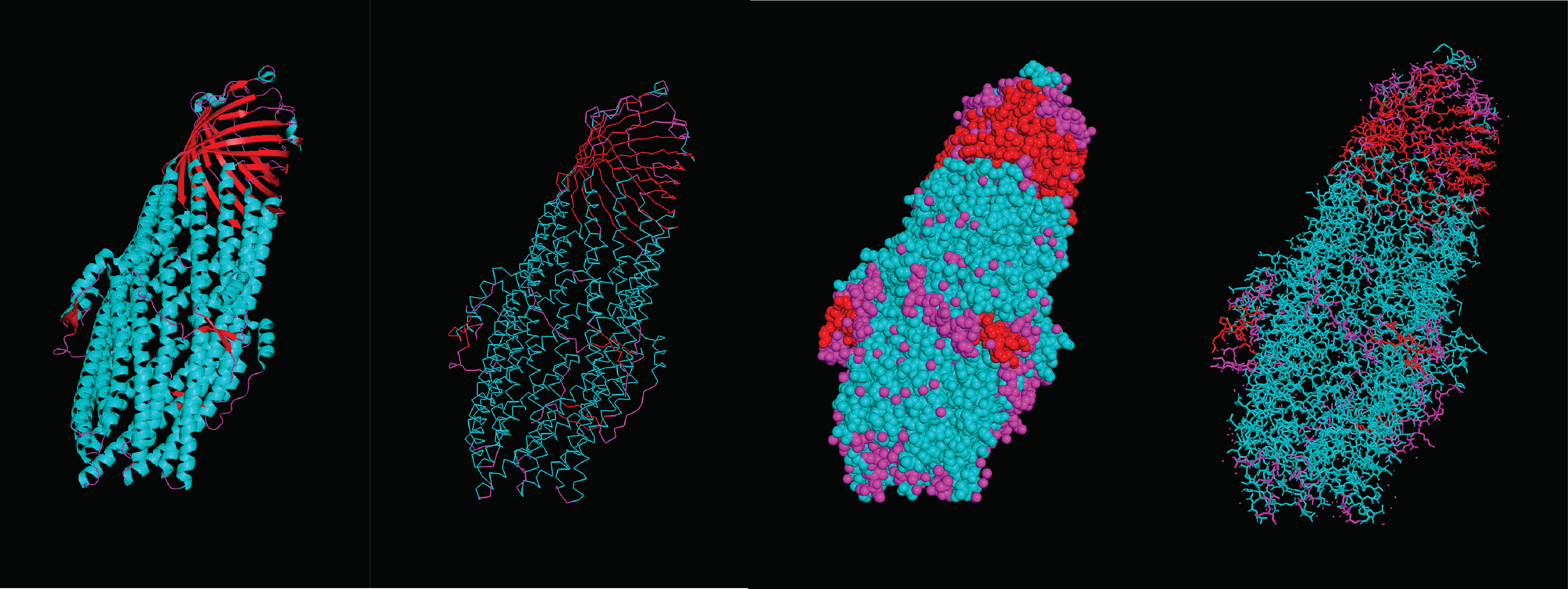
When coloring the secondary structures, it is revealed that the protein contains both helices and sheets, with more helices than sheets.
What can you tell about the distribution of hydrophobic vs hydrophilic residues? There seems to be a pretty homogenous distribution of hydrophobic and hydrophilic residues throughout the protein, this makes sense considering its function as an efflux system that connects the watery inner cell and the lipid outer membrane. Therefore it has to be compatible with both environments. Moreover, this distribution allows for molecules to pass through the structure without getting stuck on any specific spot.
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)
Due to the nature of the protein being an efflux gate in the cell membrane of E.coli k12, it does not contain specified binding pockets. However, it does have larger areas that compounds can bind to
Deep Mutational Scans
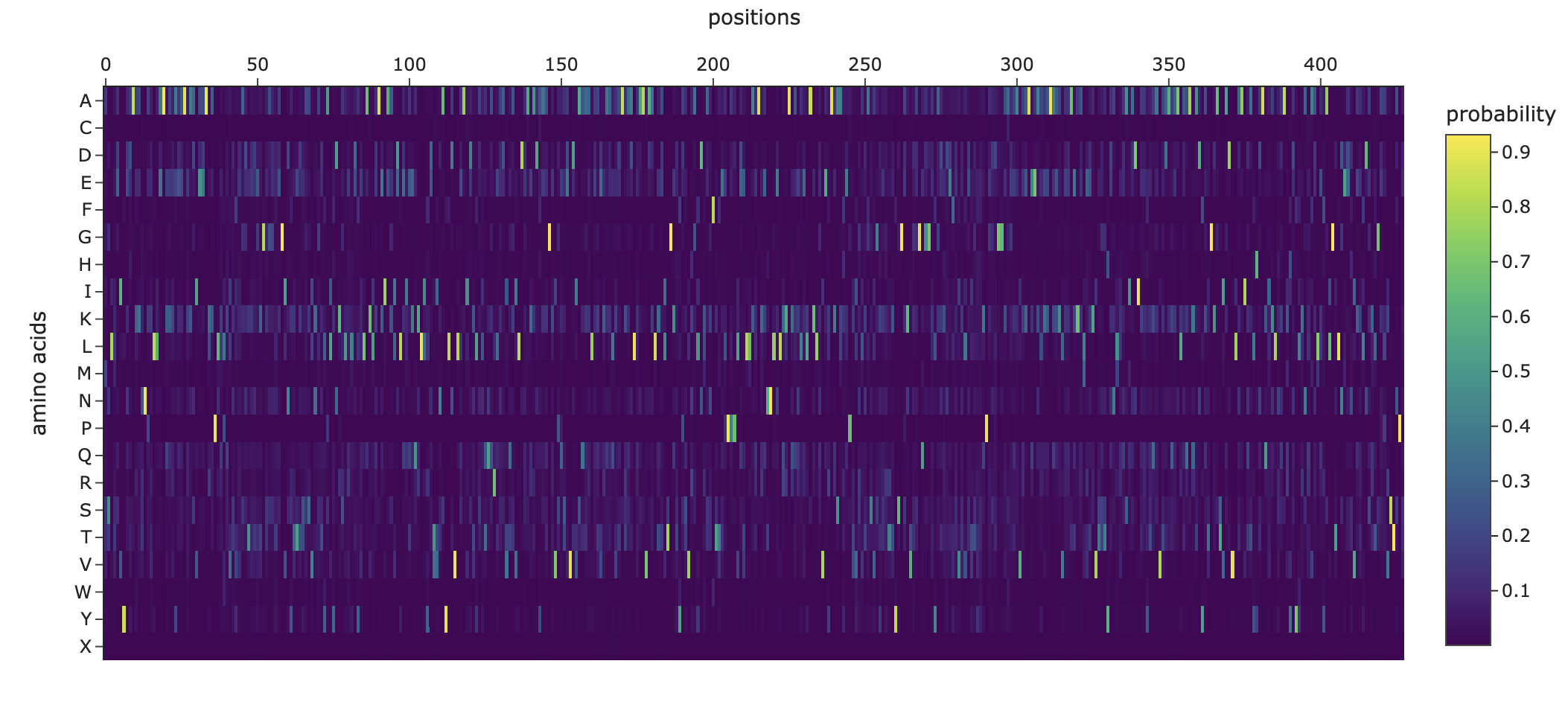
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
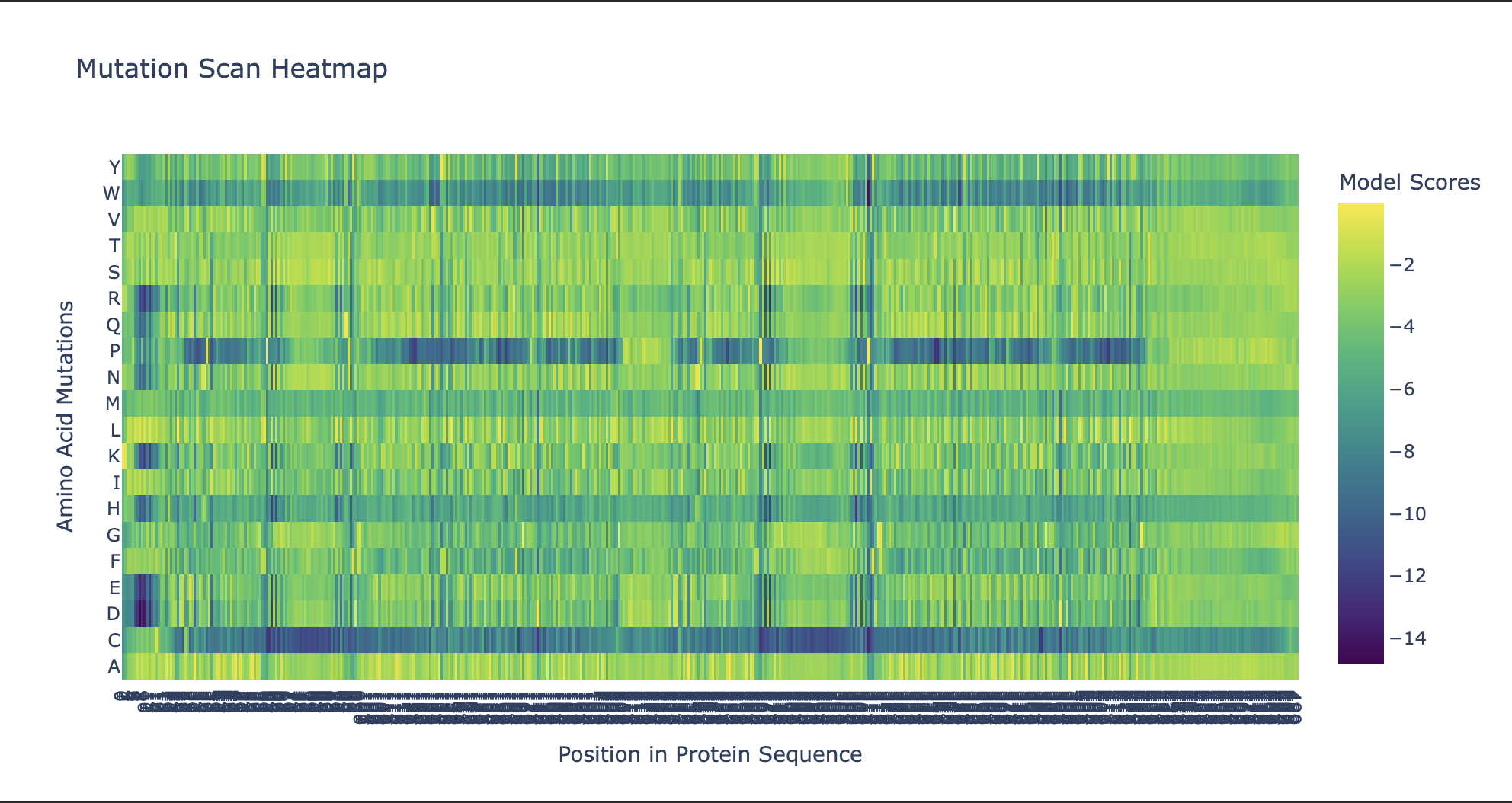
Noteworthy in the deep mutation analysis is Proline's presence seeming to be essential at positions 266 311 60 35 central to the sequence. Proline is known to induce bends, turns and loops to break alpha helices. Knowing what we know about tolC's structural composition as an efflux system within the cell membrane being made up of helices on one side and sheets on the other, this indicates that proline could be positioned at transitional points within the protein structure. When highlighting proline in our PyMol model with yellow, it can be seen positioned throughout the protein at transitional points between secondary structures thus confirming this notion.
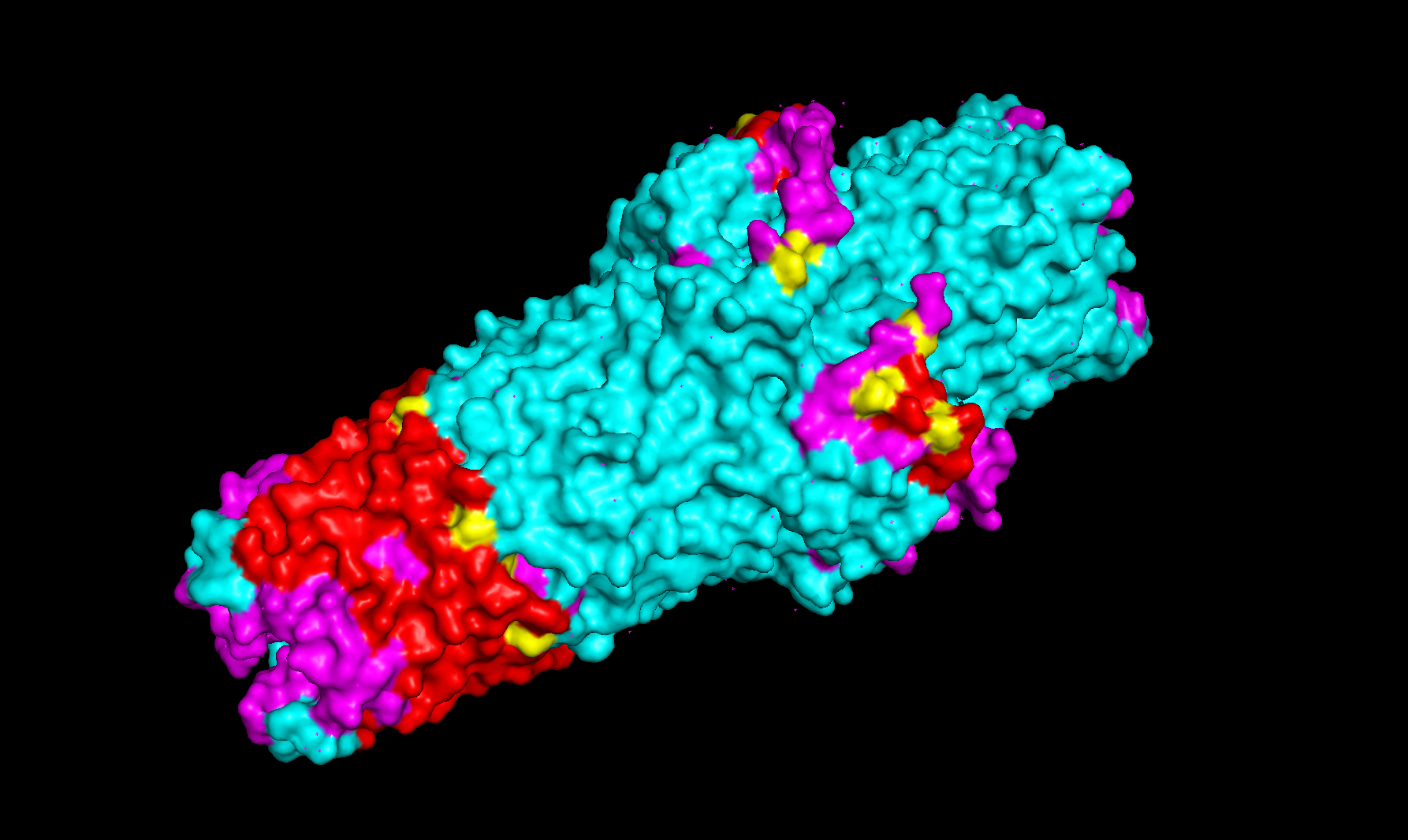
Cysteine is strongly disfavoured at most positions in TolC because it is chemically reactive and can disrupt folding, disulfide patterns, and smooth channel function in the oxidizing periplasmic/extracellular environment. TolC’s role is to provide a robust, relatively passive exit duct for many substrates, so the protein is generally selected to avoid cysteine except in a few tolerant regions, rather than to create specific internal cysteine-based toxin binding pockets. The enrichment or tolerance of Cys at position 14 is more likely due to local structural flexibility or low functional constraint there, not a dedicated heavy-metal binding site.
2. Latent Space Analysis
After a lot of trial and error, I finally got my protein secured in the latent space map highlighted in red.
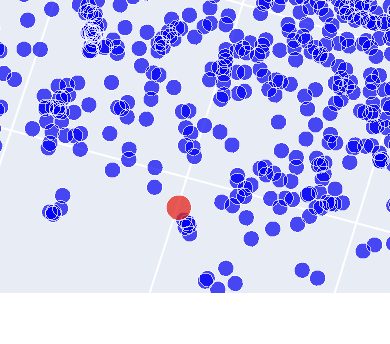
As shown on this image there is a number of proteins that are closely related. While further investigation denoted that two of these were likely to be tolC in e.coli itself, one of the others was derived from Vibrio cholerae, a bacterium that causes cholera in humans. The last one within the family was a protein derived from Pseudomonas aeruginosa. Considering all three of these are gram negative bacteria, it makes sense that these proteins are grouped together.
Folding a protein
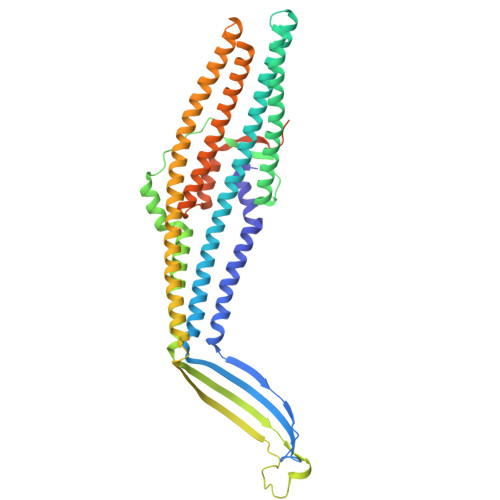
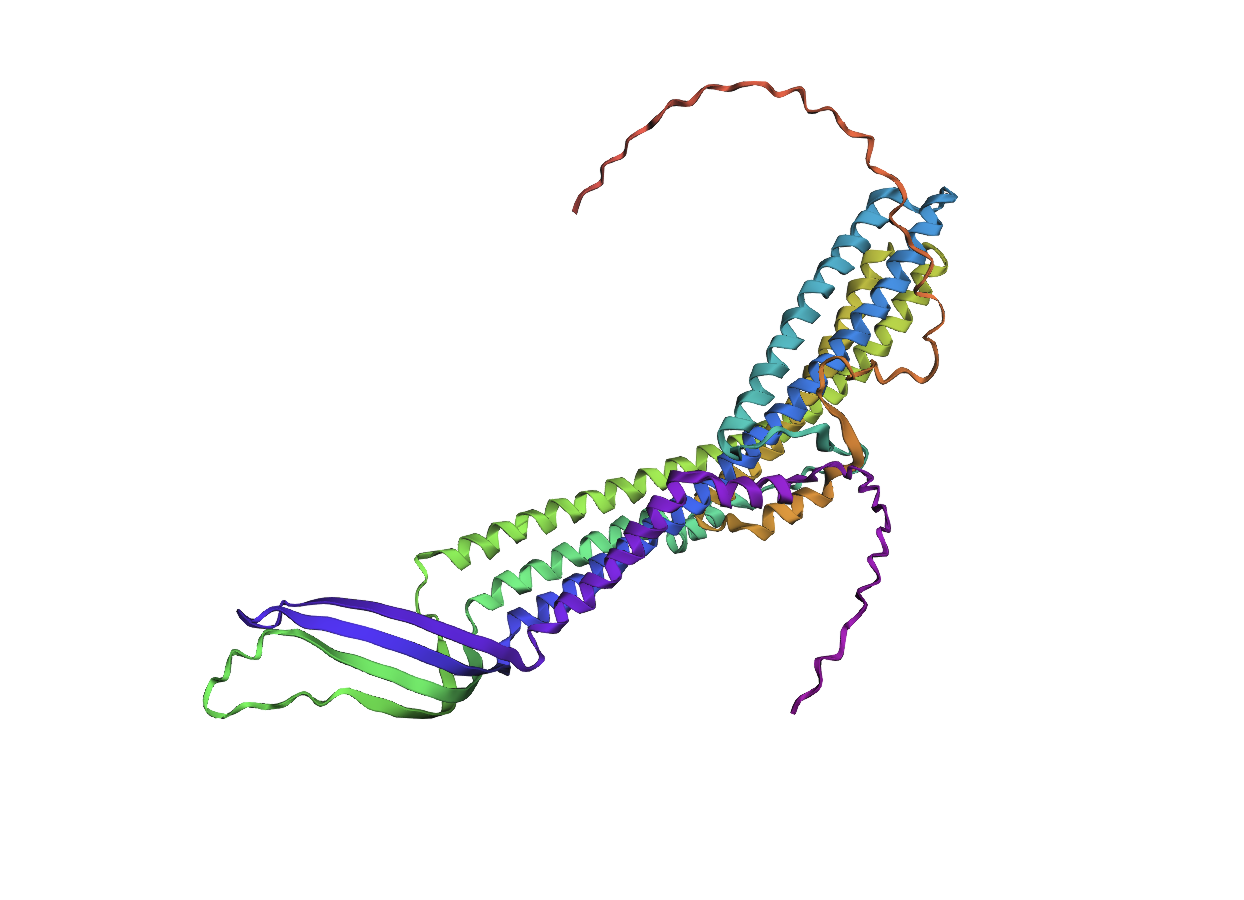
Comparing the ESMFold folded protein to the original structure, the predicted coordination seems to mostly match the original structure in terms of secondary structures. However, the placement of the helices in the ESMFold’ed protein seems to be much more tangled. Moreover, the edges of the protein seem to be completely out of place as well.
Based on prior inquiry into the importance of proline and the absence of cysteine in the sequence, I replaced each proline with a cystine in the sequence, resulting in the following fold:
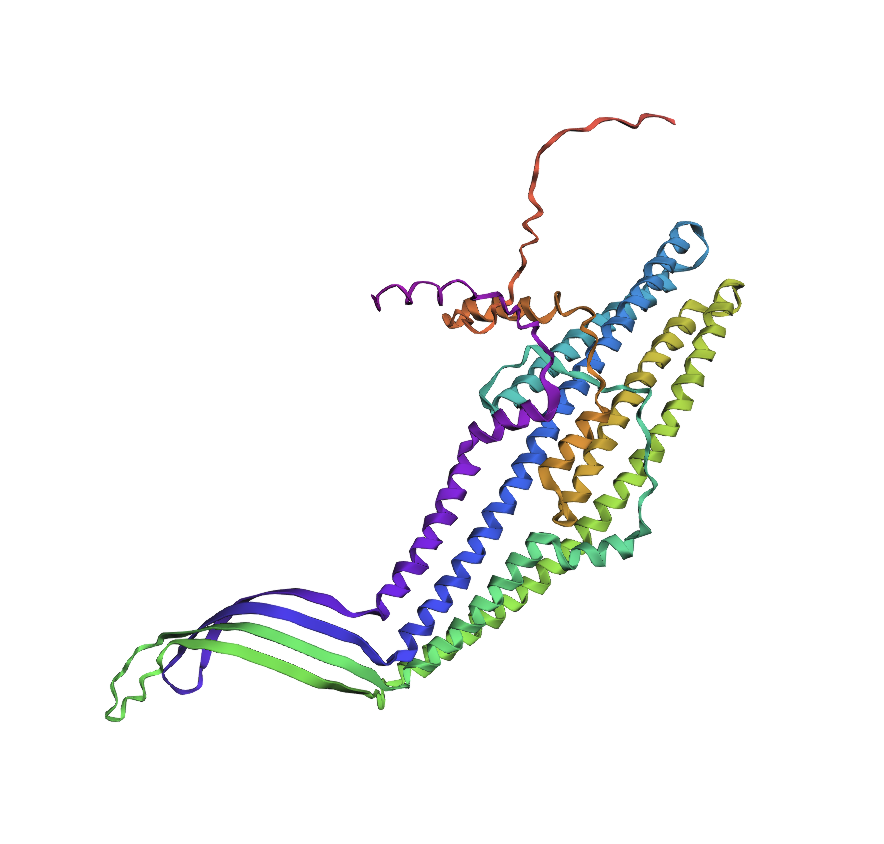
While the positioning and some of the placement of the alpha helices seems to be quite distorted, the effects are not as dramatic as I originally expected them to be.
This prompted me to try replacing another amino acid, in this case alanine with proline to see if this would result in drastically different formations. Once again, the protein seems to be misfolded, but not in a drastic manner that I was expecting:
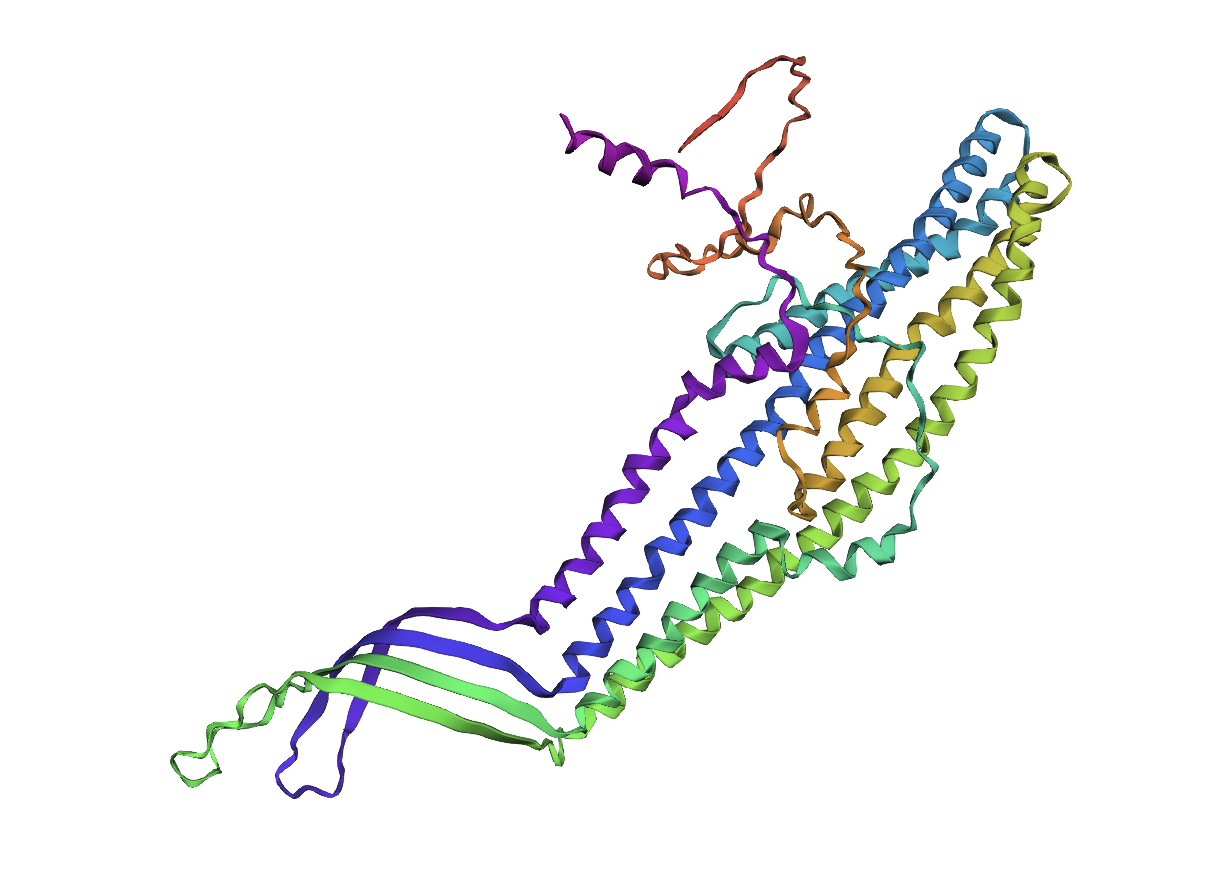
Lastly, I took a chunk in the middle of the sequence and replaced it with a random replacement sequence, which resulted in the following:
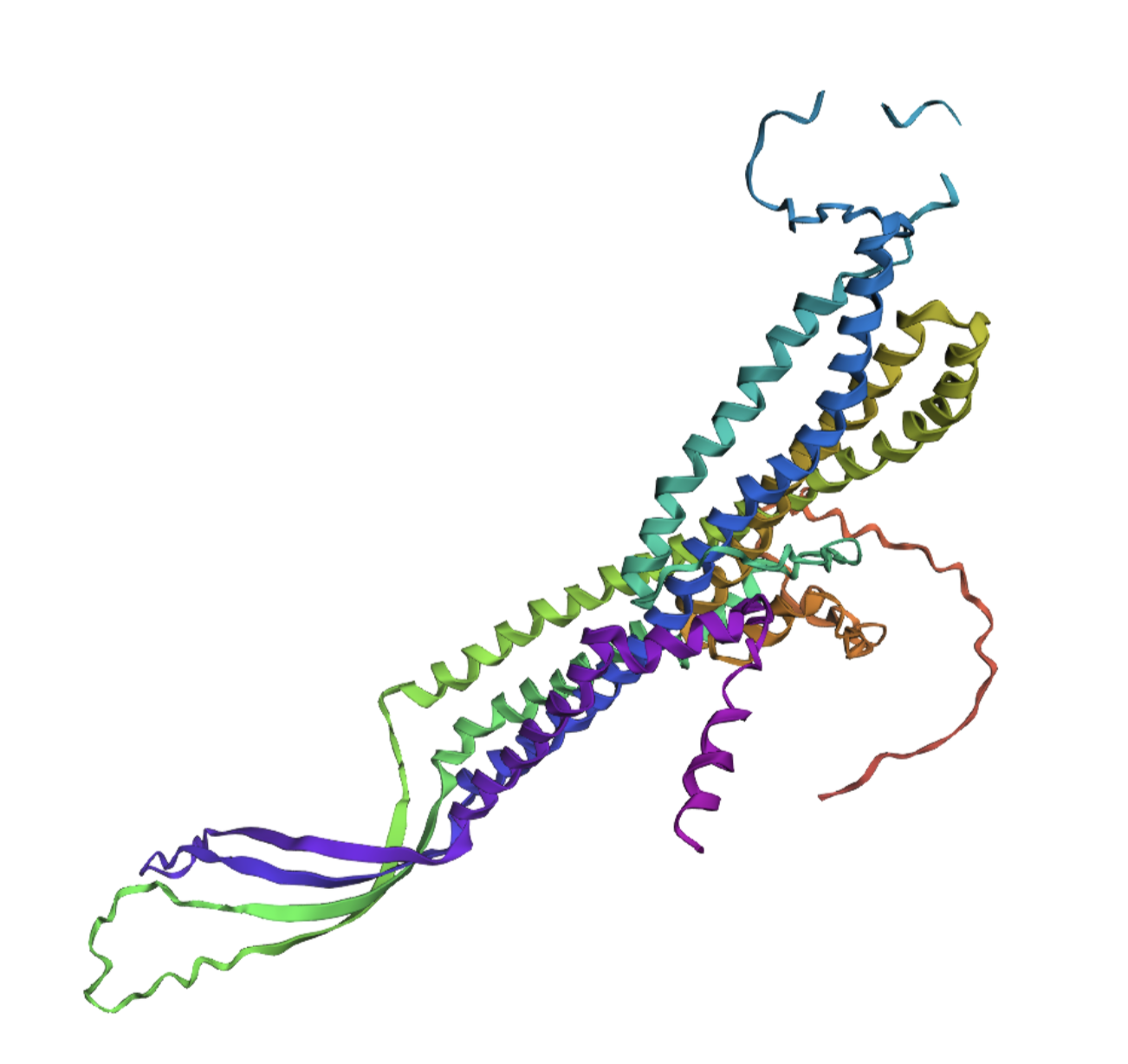
This seemed to drastically alter the structural integrity of the alpha helices within the protein.
Part D. Group Brainstorm on Bacteriophage Engineering
Group of Commited Listeners LifeFabs: Sara Gaviria Escobar, Ruben Janssen, Justine de Riedmatten
Proposal: Engineering the MS2 Lysis Protein L to Enhance Stability Background
The MS2 bacteriophage lysis (L) is a 75 amino acid long-protein, and it is responsible for triggering host cell lysis; this is why it is also called a toxin from the group of bacteriophages (Mezhyrova, 2023). It is a powerful protein that has been widely used in studies where researchers seek to control cell death, but it is difficult to do so due to its instability (Mylon, 2010).
Objectives
To use computational protein design tools to engineer possible stable variants of the MS2 L-protein. To identify variants where structure is preserved but higher stability is shown Analyse if the variants can fold like the original protein and if they interact correctly with DnaJ, its chaperone Methods
Obtain an initial protein backbone for MS2 L-protein using ESMFold.
Obtain alternative sequences to the protein using ProteinMPNN.
Mutate the alternative sequences to the protein using ESM-2.
Model the variants using ESMFold and analyse if the folding is maintained in comparison to the initial protein (MS2 L-protein). Create a ranking based on the confidence metric.s
Assess the top 3 variants’ interactions with DnaJ using AlphaFold-Multimer to predict 3D structures of protein complexes (co-folding multiple chains)
Expected Outcomes
Hopefully, these methods are able to identify some stabilized MS2 L-protein variants with correct folding and interaction with its chaperone, DnaJ. If successful, these designs could serve as templates for further experimental testing in E. coli and provide a methodology adaptable to other phage‑derived membrane proteins that also show decreased stability.
Potential Challenges
The main concern would be generating variants with correct folding, but incorrect interaction with DnaJ, as computational models sometimes can not predict the dynamics of those interactions (Chamakura et al., 2017 & Mondal et al., 2024), thus disrupting the lysis mechanism.
References
Mezhyrova, J., Martin, J., Börnsen, C., Dötsch, V., Frangakis, A. S., Morgner, N., & Bernhard, F. (2023). In vitro characterization of the phage lysis protein MS2-L. Microbiome Research Reports, 2(4), 28.
Mylon, S. E., Rinciog, C. I., Schmidt, N., Gutierrez, L., Wong, G. C., & Nguyen, T. H. (2010). Influence of salts and natural organic matter on the stability of bacteriophage MS2. Langmuir, 26(2), 1035-1042.
Chamakura, K. R., Tran, J. S., & Young, R. (2017). MS2 lysis of Escherichia coli depends on host chaperone DnaJ. Journal of Bacteriology, 199(12), 10-1128.
Mondal, A., Singh, B., Felkner, R. H., De Falco, A., Swapna, G. V. T., Montelione, G. T., … & Perez, A. (2024). A Computational Pipeline for Accurate Prioritization of Protein‐Protein Binding Candidates in High‐Throughput Protein Libraries.Angewandte Chemie International Edition, 63(24), e202405767.
Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
1. Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Original:
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS
AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV
HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Mutated:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS
AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV
HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
2. Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence, add the known SOD1-binding peptide FLYRWLPRSRRGG and record the perplexity scores of your generated binders.
Binder | Pseudo Perplexity |
WRYPVTVLRHKX | 11.329218109003818 |
WRYYAVVLEHWX | 14.262868870651067 |
WRYPVAALAHGX | 7.885580275805914 |
WLYYATGAAWKK | 15.833767982778536 |
FLYRWLPSRRGG |
|
The perplexity scores of my polypeptides are relatively low, as all except one are under 15. This indicates a high confidence in the generated samples.
3. Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
Binder | ipTM scores | Binding description |
WRYPVTVLRHKA | 0.46 | The polypeptide appears to bind to the length of the beta barrel, at approximately a third of the diameter of the beta barrel away from the N-terminus and dimer interface. It appears to be surface bound. |
WRYYAVVLEHWA | 0.19 | It seems to superficially bind to the protein at the furthest distance away from the beta-barrel, with low proximity to the N-terminus. Perhaps one end could be described as close to the dimer interface. |
WRYPVAALAHGA | 0.36 | The polypeptide seems to bind far away from the N-terminus and the dimer interface, it approaches the beta barrel region somewhat, but still keeps a relatively large distance from it as well. It appears to be surface bound. |
WLYYATGAAWKK | 0.26 | The polypeptide appears to superficially bind at the edge of the beta-barrel, with very low proximity to both the N-terminus and the dimer interface. |
FLYRWLPSRRGG | 0.31 | The polypeptide appears to be partially buried at the c-terminus, this terminus appears to be the closest part to the beta-barrel even though overall there seems to be very little interaction with that specific structure. There seems to be very low proximity to both the N-terminus and the dimer interface. |
4. In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
All of my ipTM are below the threshold of acceptable probability, the lowest being 0.19. However, two of my PepMLM-generated peptides do exceed the known binder’s score with a score of 0.36 and 0.46.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
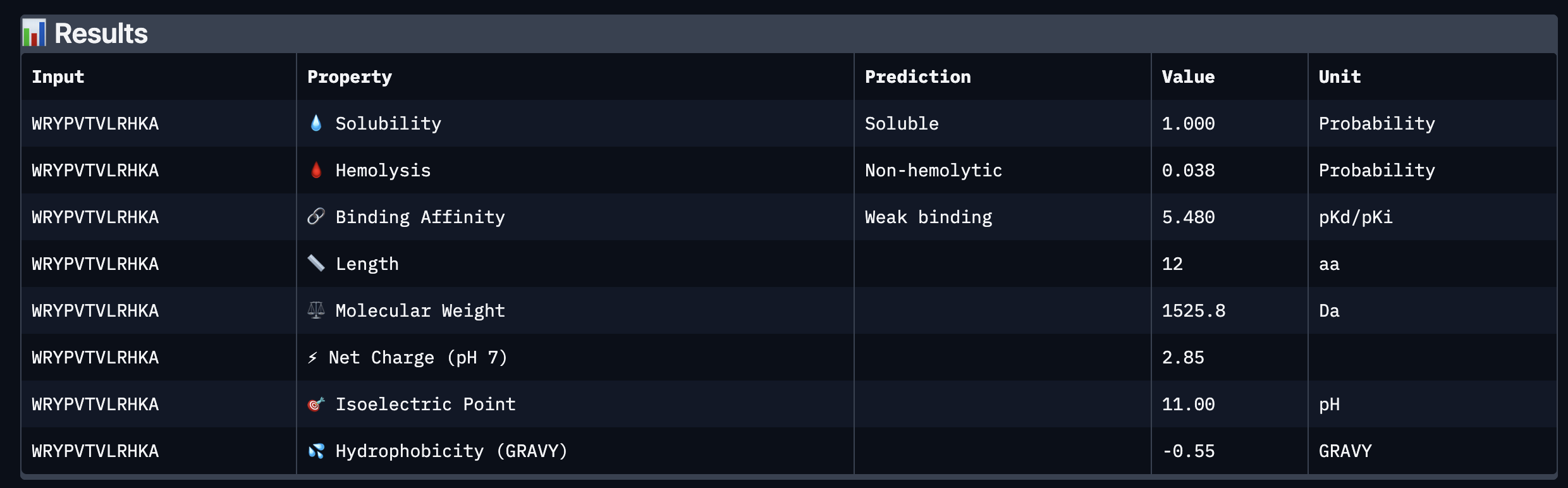
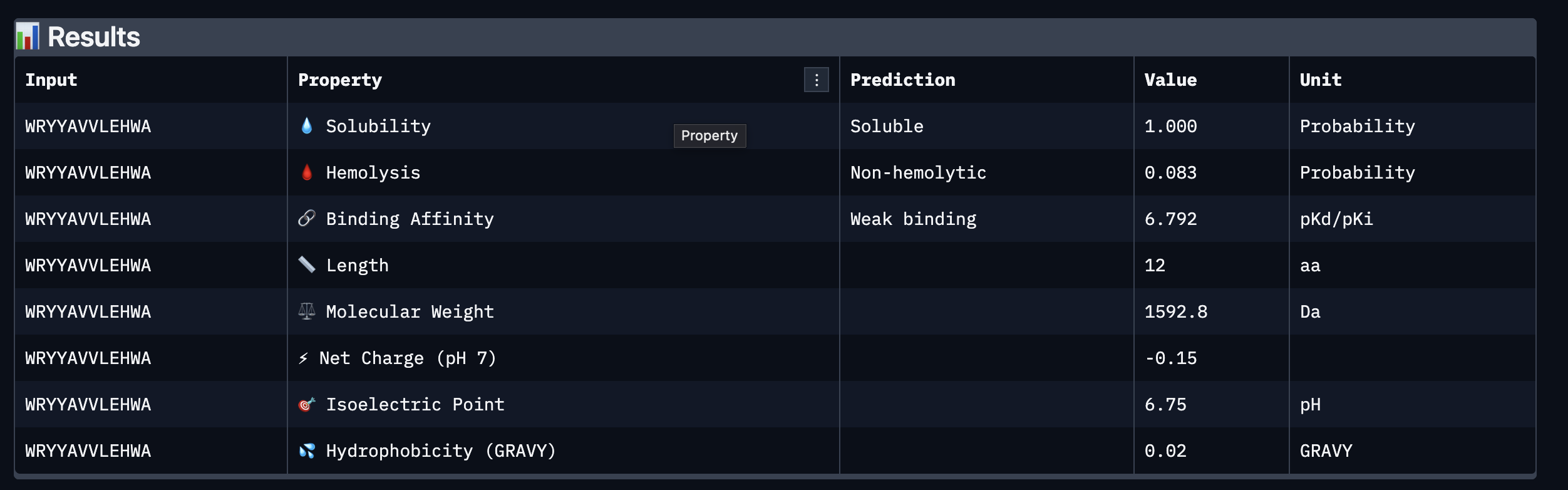
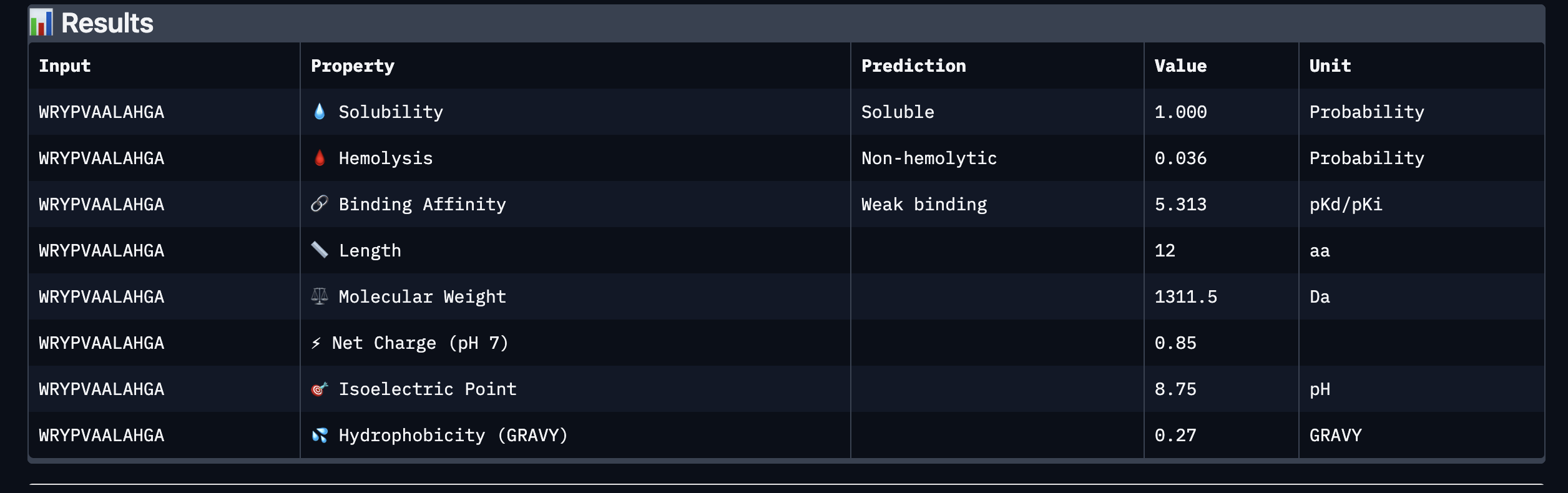
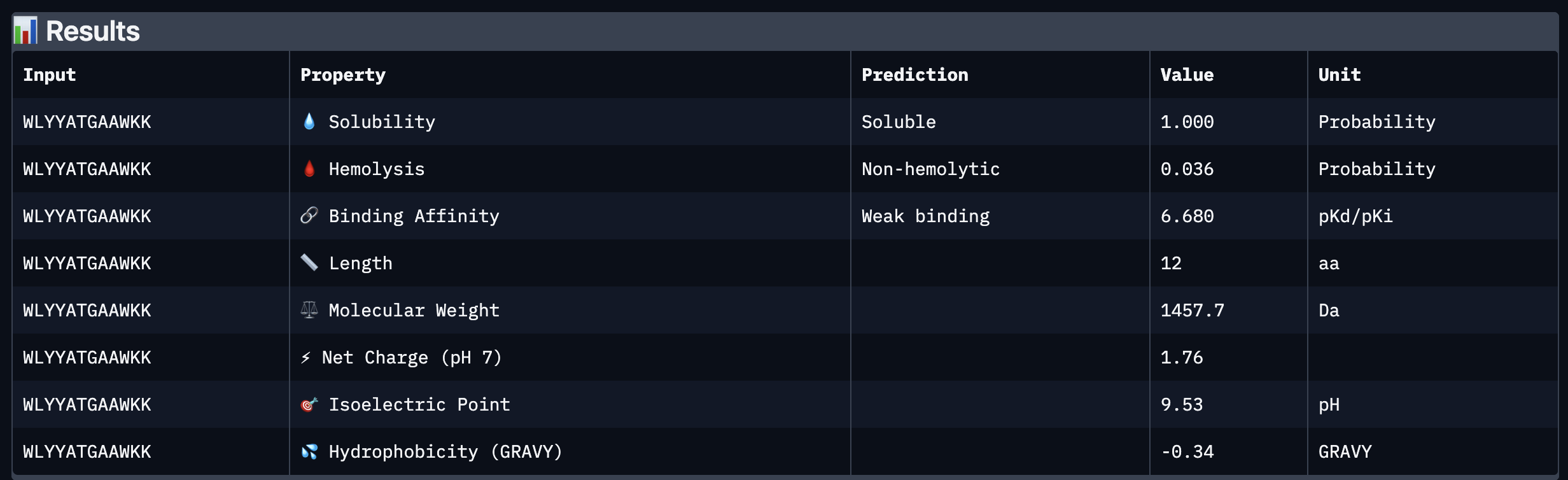
Based on the property prediction by PeptiVerse, it can be stated that all of the polypeptides are water soluble and non-hemolytic. These are highly necessary factors for potential application as they could prove to be detrimental if otherwise.
All polypeptides are predicted to be weak binding, which matches the ipTM scores of the AlphaFold models. This weak binding was also clearly seen in the visualisation of the polypeptides’ binding to the target protein within the AlphaFold models.
The molecular weight of all pp’s ranges between 1311 and 1592 Da, which is within a low range. This could indicate a lower aggregation rate, which would be good.
The SOD-1 protein is negatively charged (-6 per monomer) at physiological pH (7.4) (Shi, 2014), the binders have a small positive charge with the exclusion of WRYVAVVLEHWA having a minute negative charge of -0.15. A strongly positively charged binder would be more prone to non-specific binding to the protein and would result in a higher toxicity, therefore it is good that they are only slightly positively charged.
WRYPVTVLRHKA is the peptide that I would advance as it has the highest ipTM score, it also binds to the protein over the length of the beta-barrel. It has a very low hemolytic score of 0.038. It does however have a relatively weak binding score, however, so do all the other options.
Reference:
Shi Y, Abdolvahabi A, Shaw BF. Protein charge ladders reveal that the net charge of ALS-linked superoxide dismutase can be different in sign and magnitude from predicted values. Protein Sci. 2014 Oct;23(10):1417-33. doi: 10.1002/pro.2526. Epub 2014 Aug 7. PMID: 25052939; PMCID: PMC4287002.
Part C: Final Project: L-Protein Mutants
Information about Lysis protein in Bacteriophage MS2
Amino acid sequence of Lysis
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
DNA Sequence of Lysis
atggaaacccgctttccgcagcagagccagcagaccccggcgagcaccaaccgccgccgcccgtttaaacatgaagattatccgtgccgccgccagcagcgcagcagcaccctgtatgtgctgatttttctggcgatttttctgagcaaatttaccaaccagctgctgctgagcctgctggaagcggtgattcgcaccgtgaccaccctgcagcagctgctgacc
DNAj Sequence
tcgacgctga atttgaagaa gtcaaagaca aaaaataatc gccctataaa cgggtaatta 60
tactgacacg ggcgaagggg aatttcctct ccgcccgtgc attcatctag gggcaattta 120
aaaaagatgg ctaagcaaga ttattacgag attttaggcg tttccaaaac agcggaagag 180
cgtgaaatca gaaaggccta caaacgcctg gccatgaaat accacccgga ccgtaaccag 240
ggtgacaaag aggccgaggc gaaatttaaa gagatcaagg aagcttatga agttctgacc 300
gactcgcaaa aacgtgcggc atacgatcag tatggtcatg ctgcgtttga gcaaggtggc 360
atgggcggcg gcggttttgg cggcggcgca gacttcagcg atatttttgg tgacgttttc 420
ggcgatattt ttggcggcgg acgtggtcgt caacgtgcgg cgcgcggtgc tgatttacgc 480
tataacatgg agctcaccct cgaagaagct gtacgtggcg tgaccaaaga gatccgcatt 540
ccgactctgg aagagtgtga cgtttgccac ggtagcggtg caaaaccagg tacacagccg 600
cagacttgtc cgacctgtca tggttctggt caggtgcaga tgcgccaggg attcttcgct 660
gtacagcaga cctgtccaca ctgtcagggc cgcggtacgc tgatcaaaga tccgtgcaac 720
aaatgtcatg gtcatggtcg tgttgagcgc agcaaaacgc tgtccgttaa aatcccggca 780
ggggtggaca ctggagaccg catccgtctt gcgggcgaag gtgaagcggg cgagcatggc 840
gcaccggcag gcgatctgta cgttcaggtt caggttaaac agcacccgat tttcgagcgt 900
gaaggcaaca acctgtattg cgaagtcccg atcaacttcg ctatggcggc gctgggtggc 960
gaaatcgaag taccgaccct tgatggtcgc gtcaaactga aagtgcctgg cgaaacccag 1020
accggtaagc tattccgtat gcgcggtaaa ggcgtcaagt ctgtccgcgg tggcgcacag 1080
ggtgatttgc tgtgccgcgt tgtcgtcgaa acaccggtag gcctgaacga aaggcagaaa 1140
cagctgctgc aagagctgca agaaagcttc ggtggcccaa ccggcgagca caacagcccg 1200
cgctcaaaga gcttctttga tggtgtgaag aagttttttg acgacctgac ccgctaacct 1260
ccccaaaagc ctgcccgtgg gcaggcctgg gtaaaaatag ggtgcgttga agatatgcga 1320
gcacctgtaa agtggcgggg atcactccca taagcgct 1358
Conserved sites
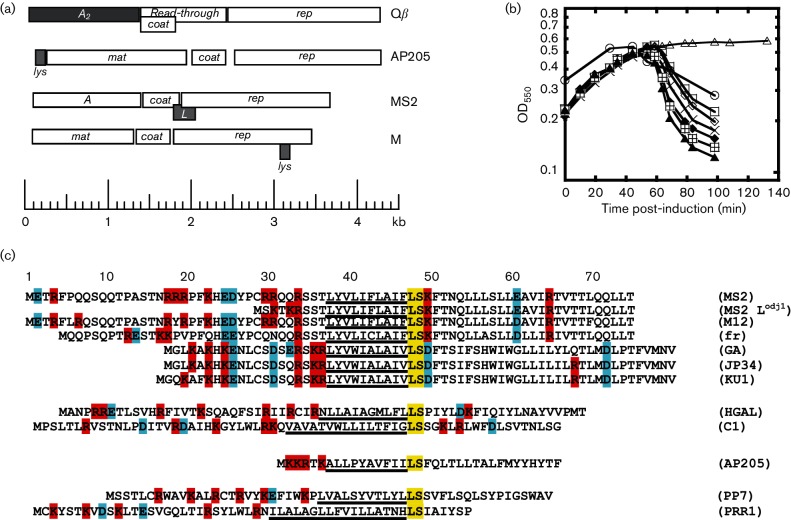
The provided article “Mutational analysis of the MS2 lysis protein L. Microbiology” describes how throughout homologs, position 48 and 49 are consistently conserved sites (LEU en SER). In the central domain there is a high conservation level across most homologs in position 38 39 40, (Leu, Tyr and Val) (Chamakura, 2017)
Known mutational effects from research.
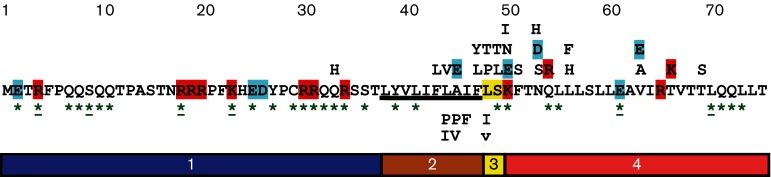
The above figure obtained from the aforementioned paper shows an overview of a mutational analysis of MS2 L. The analysis shows that certain missense mutations in the second domain do not affect lytic function (indicated below the L sequence). In the second, third and fourth domain, numerous mutations did negatively affect lysis function but did not result in protein accumulation (indicated above the L sequence). Green asterisks represent positions where a single nucleotide mutation would result in a nonsense mutation.
References:
Chamakura, K. R., Edwards, G. B., & Young, R. (2017). Mutational analysis of the MS2 lysis protein L. Microbiology (Reading, England), 163(7), 961–969. https://doi.org/10.1099/mic.0.000485
My selected approach to determining sequence variations is cross referencing the provided experimental data on L-protein mutants with generated point mutations and accompanying success scores from the provided Protein Language Models.
As the assignment dictates, 2 mutations should be located in the soluble N-terminal domain (position 1 - 39) and 2 in the transmembrane domain ( position 40 - 75)
Top 10 of colab
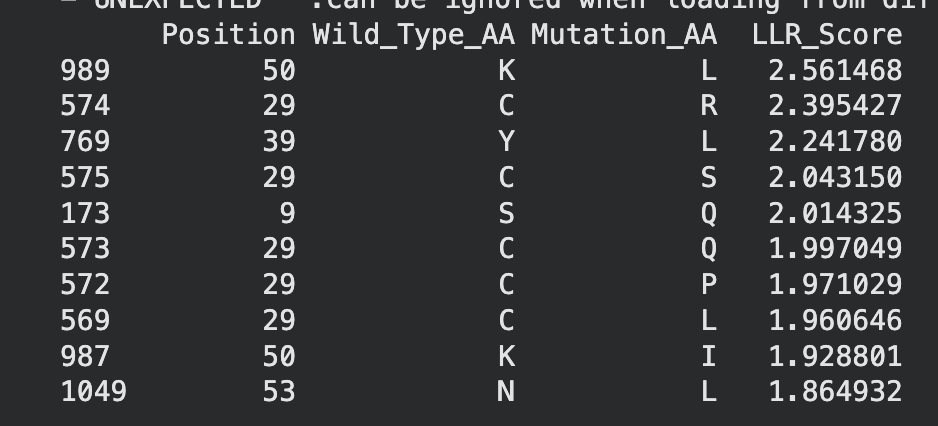
When crossreferencing this list with the original experimental mutations, two mutations, namely C29R and K50I appeared to result in neither lysis or protein formation. Therefore they will be excluded from potential mutation candidates.
This leaves the following mutations:
K50L - While this mutation has the highest LLR score, further research indicates that Lysine (K) and Leucine (L) are structurally very different residues. This could indicate the potential for problematic protein formation. However, due to its high score it should still be considered.
Y39L - Tyrosine and Leucine
S9Q - Serine and glutamine
C29Q - Cystine and glutamine
C29P - Cystine and proline
C29L - cystine and leucine
N53L - Asparagine and leucine
I asked Perplexity to compare the structural properties of the wild-type amino acids to their mutated amino acids.
Mutation | Amino Acids | Structural Similarity? | Reason |
Y39L | Tyrosine → Leucine | No | Aromatic ring vs. branched alkyl chain; different bulk and polarity. www2.chemistry.msu |
S9Q | Serine → Glutamine | Low | Short hydroxyl vs. longer amide; size/volume mismatch despite some polarity. benchchem |
C29Q | Cysteine → Glutamine | No | Thiol (-SH) vs. amide; polarity similar but chain length and reactivity differ. wikipedia |
C29P | Cysteine → Proline | No | Linear thiol vs. rigid cyclic; proline disrupts helices unlike cysteine. youtubeweb02.gonzaga |
C29L | Cysteine → Leucine | No | Small polar thiol vs. large hydrophobic branch; major size/chemistry shift. www2.chemistry.msu |
N53L | Asparagine → Leucine | No | Polar amide vs. hydrophobic branch; polarity and H-bonding lost. www2.chemistry.msu |
However, I will continue the assignment with the following five mutations:
K50L
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT
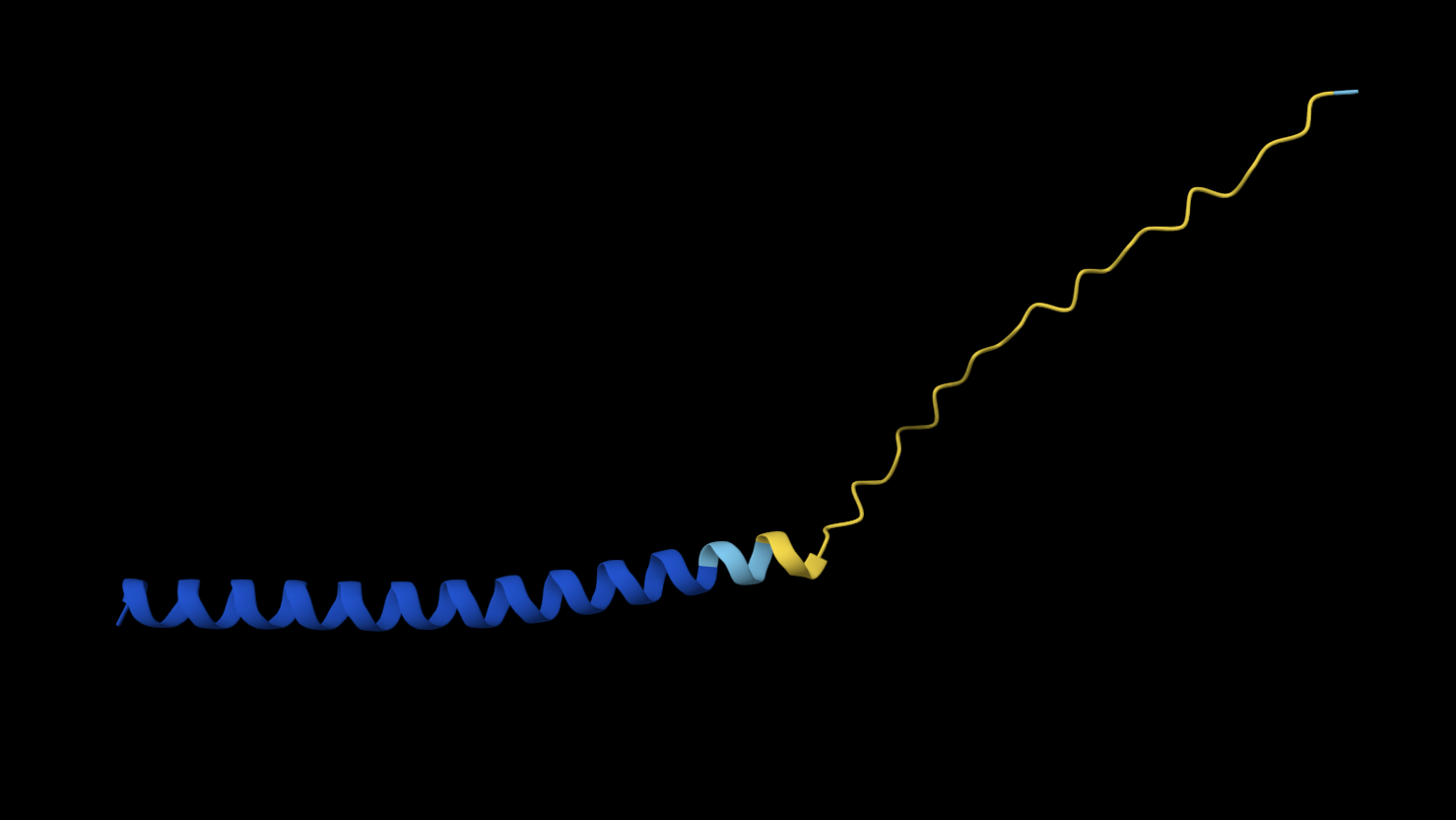
N53L
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT
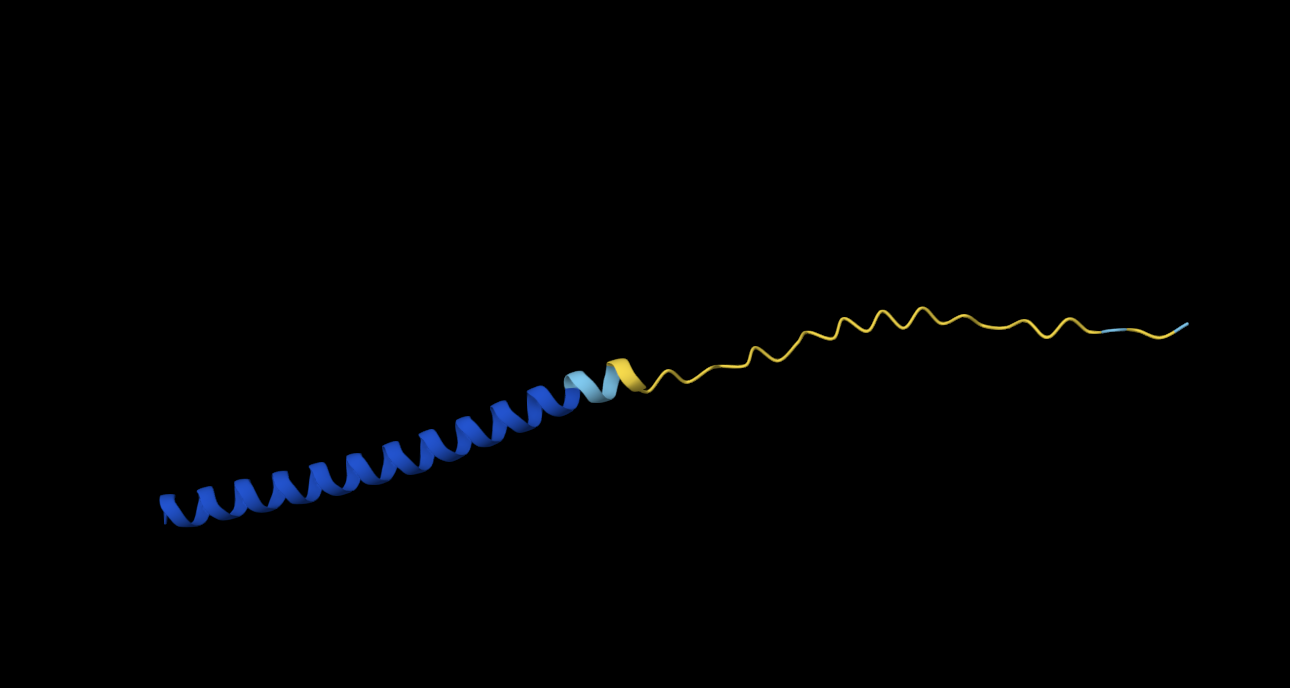
S9Q
METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
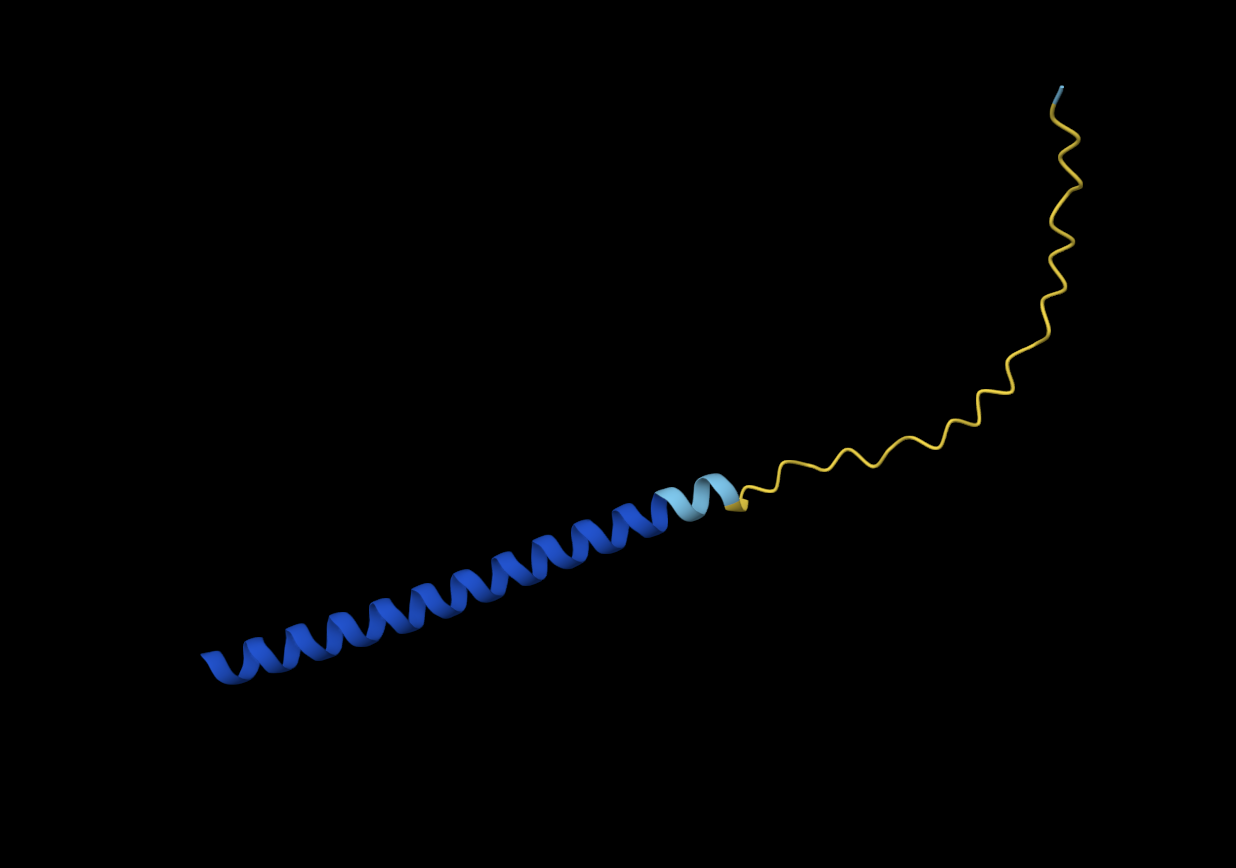
Y39L
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
C29L
METRFPQQSQQTPASTNRRRPFKHEDYPLRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT

Week 6 HW: Genetic Circuits Part I
Assignment: DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion High-Fidelity PCR Master Mix contains Phusion DNA Polymerase, nucleotides, and optimized reaction buffer including MgCl2.
The DNA Polymerase synthesizes new strands of DNA by assembling nucleotides based on a template strand, the nucleotides are necessary for assembly and the optimized reaction buffers assure variable factors are optimised for PCR.
What are some factors that determine primer annealing temperature during PCR?
The main factor that determines primer annealing during PCR is the melting temperature of the primer.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Polymerase Chain Reaction (PCR) is a widely used, highly sensitive laboratory technique invented by Kary Mullis in 1983 to amplify specific DNA sequences, producing millions to billions of copies in just a few hours. UPCR is a nucleic acid amplification technique involving denaturation, annealing, extension, and amplification of short DNA or RNA segments. The reaction employs DNA polymerase derived from Thermus aquaticus, known as Taq polymerase. Using a thermal cycler, DNA polymerase (like Taq polymerase), and primers, it enables rapid diagnosis of diseases, forensic analysis, and genetic research.
The thermostability of Taq polymerase preserves the physical and chemical integrity of nucleic acids throughout repeated high-temperature cycles, making it suitable for the PCR-based detection of bacterial and viral pathogens and for screening genetic disorders.
During denaturation, prepared samples are heated to 94 to 96 degrees Celsius for 30 seconds, this separates the double stranded DNA in single strands. Next, during annealing the sample temperature is lowered to 50 to 65 degrees Celsius for 30 seconds. This allows for the primers (short, single-stranded nucleic acid sequences (oligonucleotide) that serves as a necessary starting point for DNA synthesis and replication) to bind to the target sites on the ssDNA. Then the samples are heated to 72 degrees for at least another 30 seconds. This allows for the Taq polymerase to bind to the primers and extend a new DNA strand onto the ssDNA.
The cycle is then restarted.
Restriction enzyme digests use bacterial enzymes to cut DNA at specific sites. Restriction enzymes recognize short, palindromic DNA sequences (typically 4-8 base pairs) and cleave the phosphodiester backbone. Type II enzymes, most common in labs, cut within or near the recognition site, producing either blunt ends (straight cuts) or sticky ends (overhanging single strands that facilitate ligation).
PCR is a simple method to replicate DNA sequences exponentially in a short amount of time with a large variety of applications from diagnostics to genetics research. Restriction enzyme digests are used to cut (out) specific strands of DNA that would for example be used for genetic engineering. Relationally, RED could be done to then perform PCR cloning, this would allow one to isolate a sequence and then amplify it for further
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
An important step to ensure the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning is purifying your samples. This would remove any contaminants, such as reagents from prior steps, that interfere with further processing.
Moreover, one could perform gel electrophoresis to establish that the target sequence length is present in the sample.
How does the plasmid DNA enter the E. coli cells during transformation?
There are two methods through which plasmid DNA enters E.coli cells during transformation:
Chemical competence method - E. coli cells are grown to log phase, chilled on ice, and treated with cold calcium chloride (CaCl₂), which binds to the cell membrane's phospholipids and DNA phosphates. This neutralises charges, making the negatively charged plasmid DNA less repelled by the membrane. The DNA-cell mix is then heat-shocked (42°C for 30-90 seconds), temporarily fluidising the membrane to form pores that let the plasmid slip inside; cells are quickly iced again to reseal the membrane.
Reference:
Froger, A., & Hall, J. E. (2007). Transformation of plasmid DNA into E. coli using the heat shock method. Journal of visualized experiments : JoVE, (6), 253. https://doi.org/10.3791/253
Electroporation
Cells are mixed with DNA and zapped with a high-voltage pulse (e.g., 2.5 kV), creating transient membrane holes via dielectric breakdown. The electric field drives charged DNA into the cell before pores close.
Biology LibreTexts (2021) Transforming E. coli. Available at: https://bio.libretexts.org/Bookshelves/Biotechnology/Lab_Manual:_Introduction_to_Biotechnology/01:_Techniques/1.13:_Transformation
Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Model this assembly method with Benchling or Asimov Kernel!
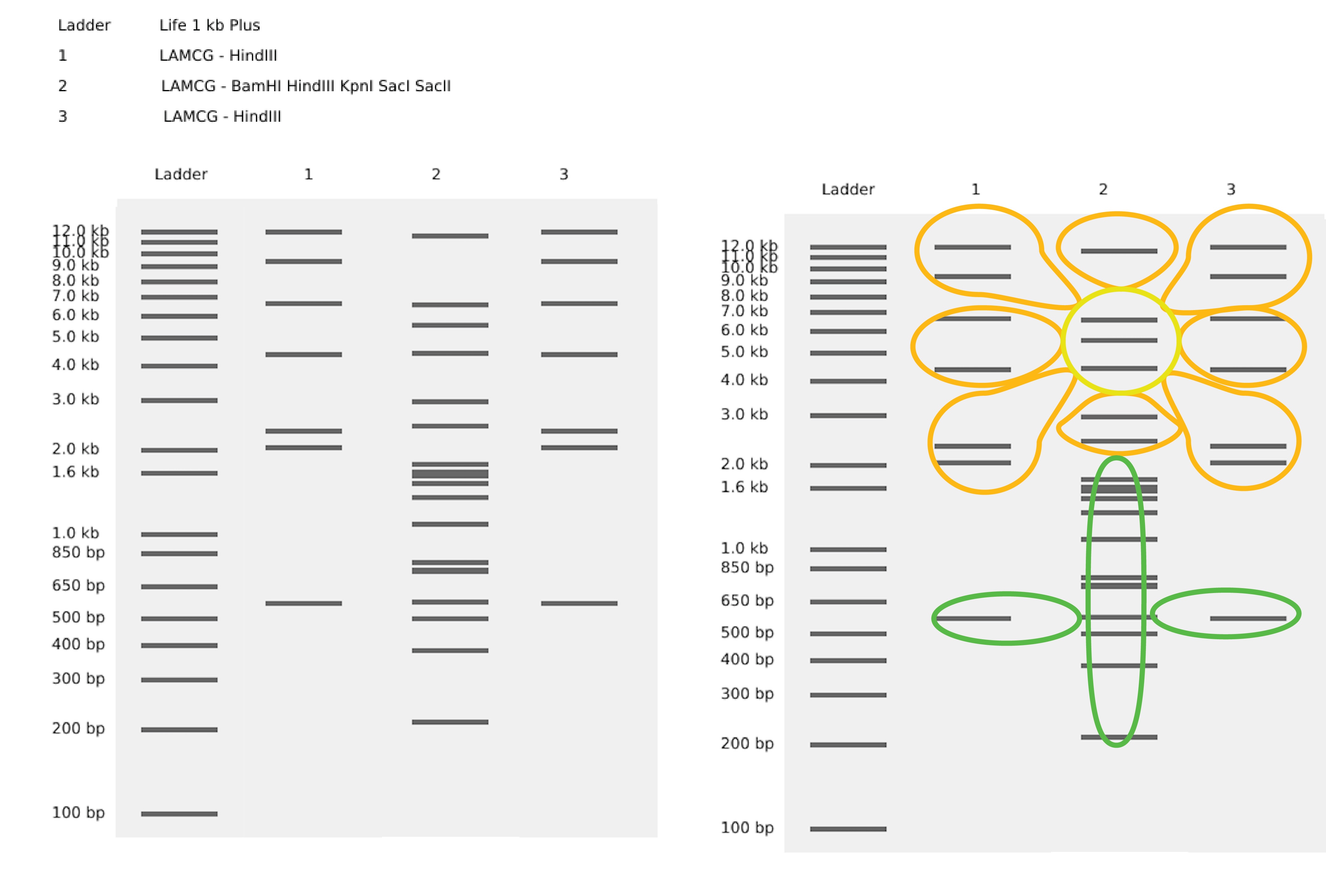
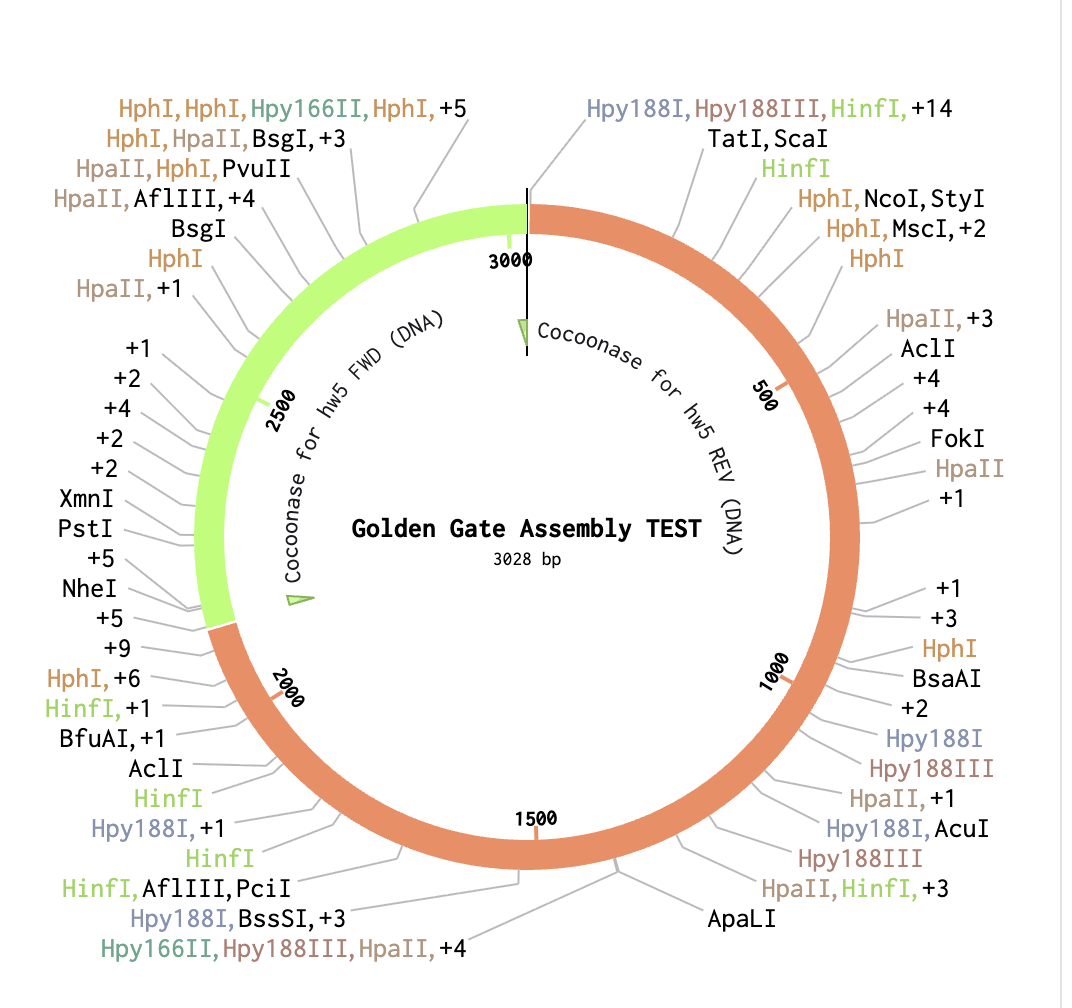
Golden Gate Assembly is a modular DNA cloning method for joining multiple fragments in a precise, scarless manner. It employs Type IIS restriction enzymes like BsaI, which cut outside their recognition sites to produce custom 4-base sticky overhangs. These overhangs are designed via PCR primers or pre-made parts to ensure fragments only ligate in the intended order. A one-pot reaction cycles between enzyme digestion at 37°C and T4 ligase joining at 16°C, self-correcting mismatches. Domesticated sequences—lacking internal cut sites—prevent unwanted breaks, resulting in clean final constructs without residual enzyme motifs. This approach outperforms traditional cloning by enabling efficient, hierarchical assembly of complex genetic circuits.
Bird, J.E. et al. (2022) 'A User's Guide to Golden Gate Cloning Methods and Standards', ACS Synthetic Biology, 11(12), pp. 3895–3909. Available at: https://pubs.acs.org/doi/10.1021/acssynbio.2c00355
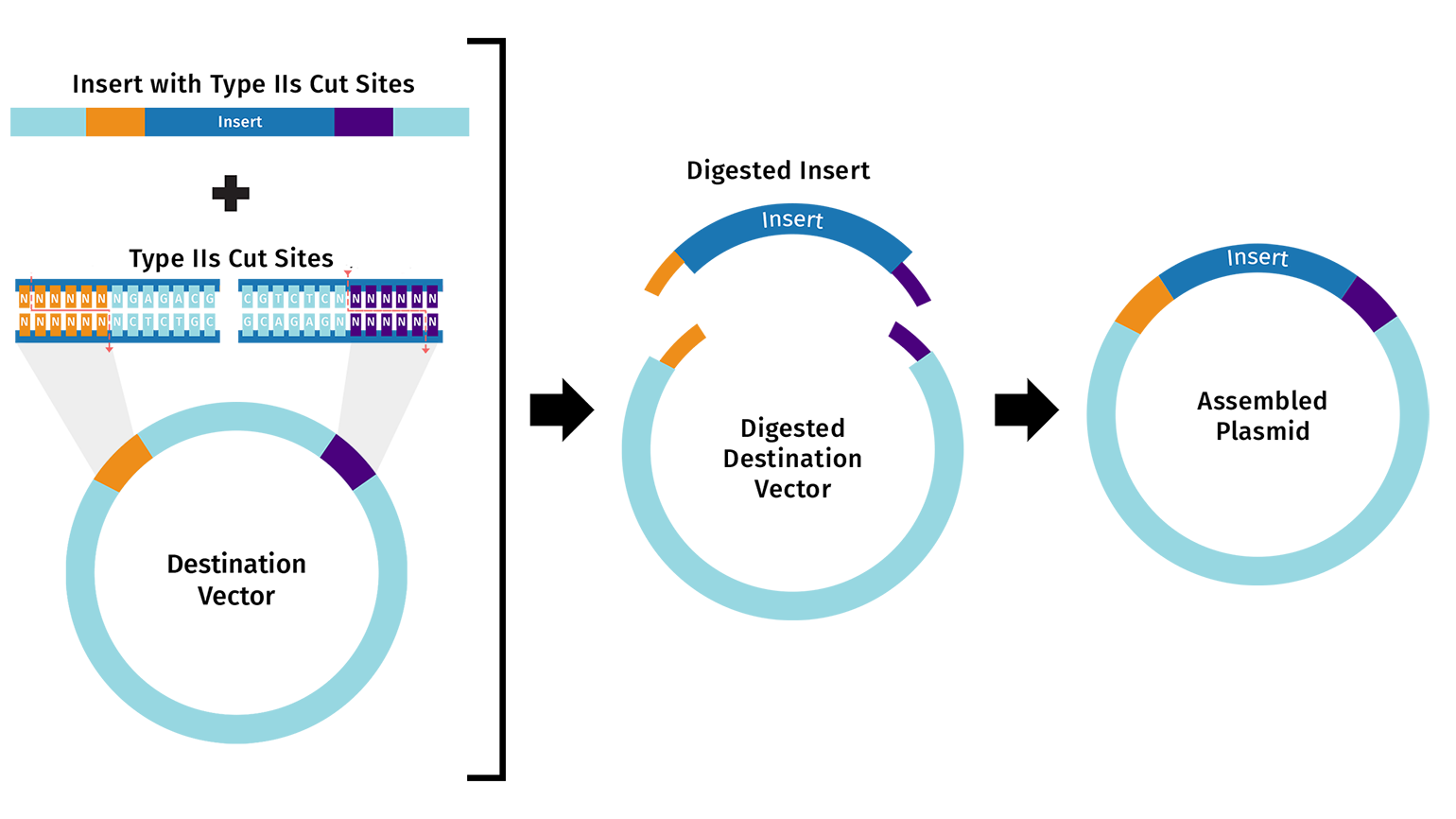
I chose to model Golden Gate Assembly using Benchling.
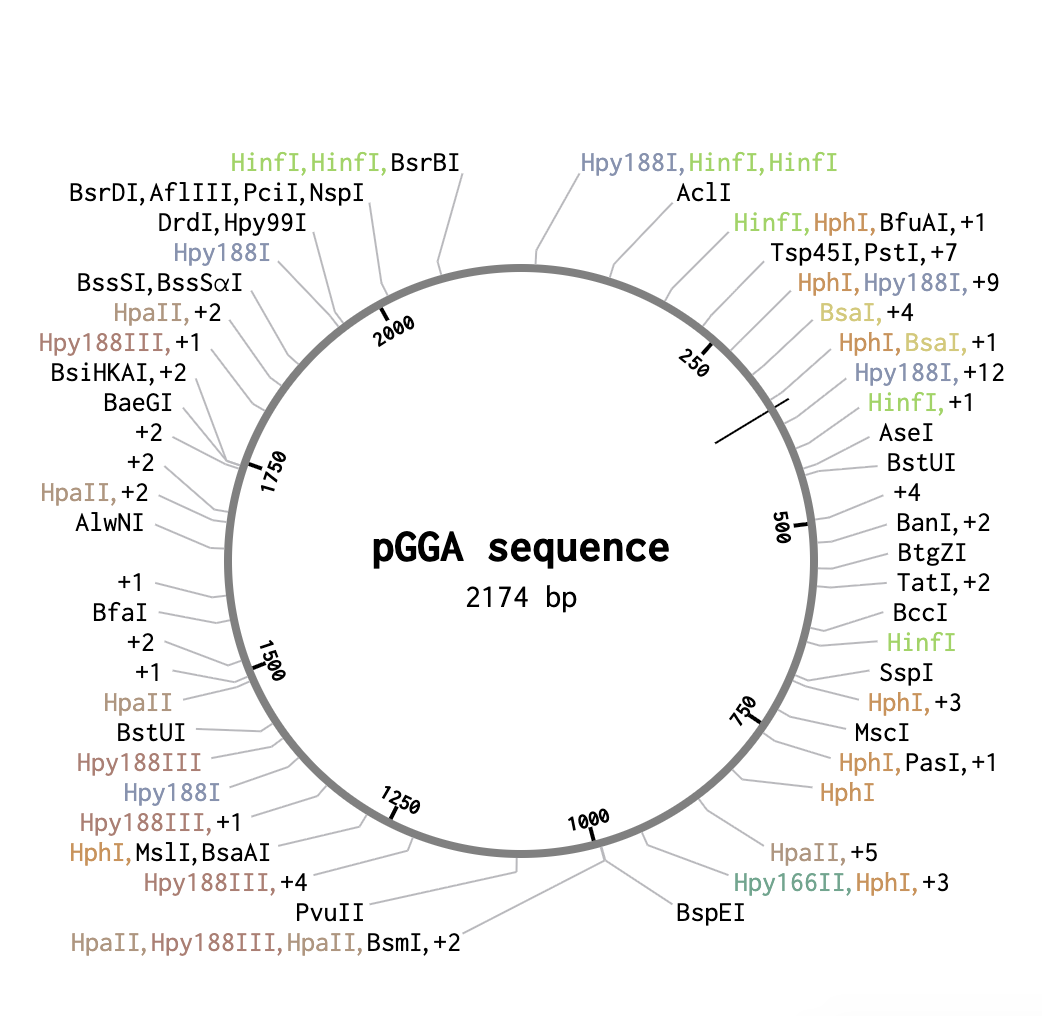
First, I looked up a suitable backbone vector. Through quick research I found that for golden gate assembly has domesticated vectors like pGGA which I ended up selecting for this exercise. I then opened the sequence in Benchling.
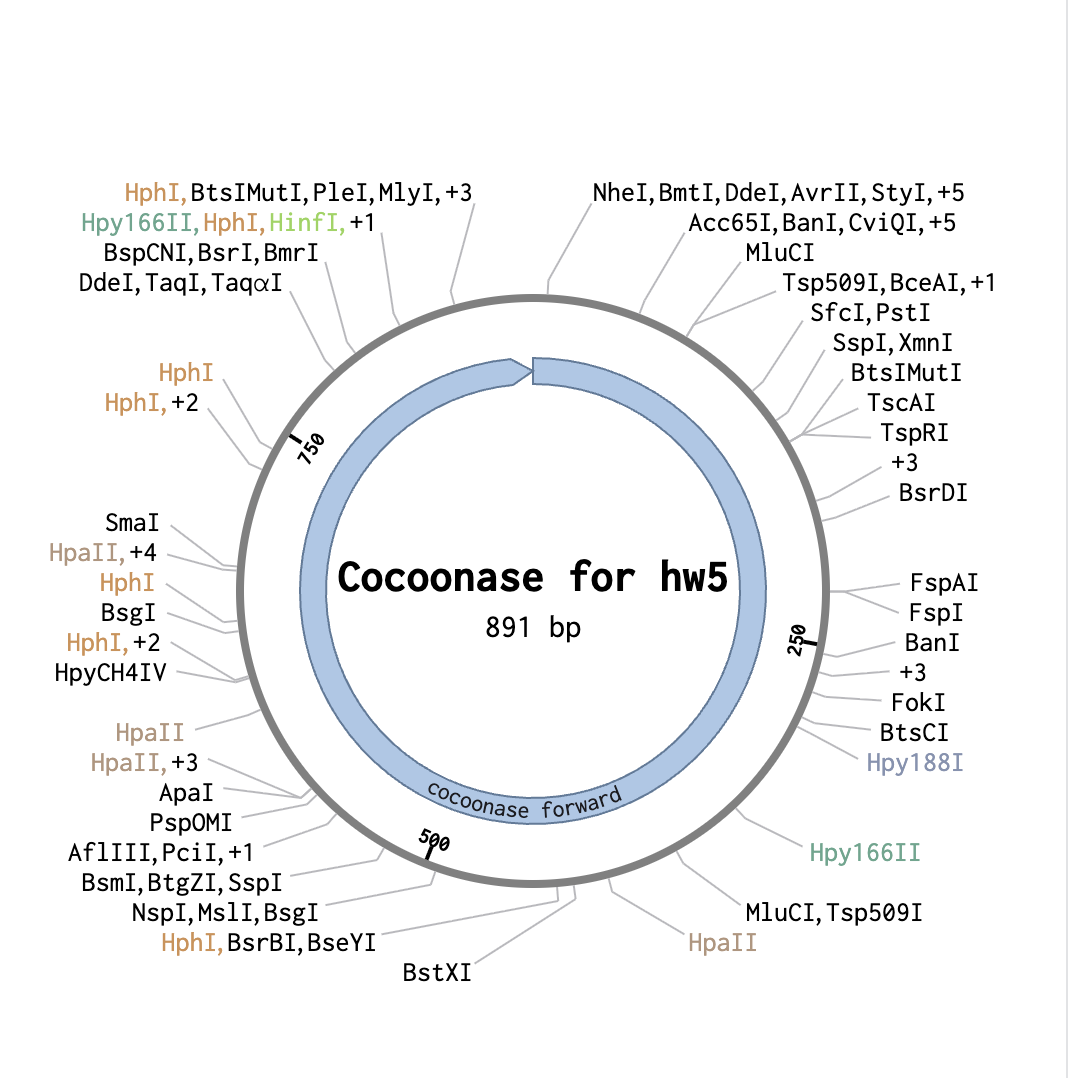
From a previous homework assignment I selected the sequence for cocoonase as my insert sequence.
With both tabs open, I started the assembly wizard for Golden Gate Assembly.
As a backbone I selected pGGA, with BsaI cut sites. For the Insert selection, I selected the entire cocoonase sequence and pressed insert. This resulted in the following circular sequence:
Week 7 HW: Genetic Circuits Part II
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
One of the core advantages of IANNs over traditional genetic circuits is that IANNs can be used for complex computations whereas traditional genetic circuits are restricted to simpler digital operations. Moreover, IANNs have a higher predictability than traditional genetic circuits and can be modified to perform precise therapeutic functions.
Traditional genetic circuits also struggle with scalability, as stochastic fluctuation of genetic expression and molecular concentrations cause noise. Similar to ANNs, IANNs smooth out input perturbations through by distributing these stochastic fluctuations across nodes and pathways,
Sources:
Ameen Eetemadi, Ilias Tagkopoulos, Genetic Neural Networks: an artificial neural network architecture for capturing gene expression relationships, Bioinformatics, Volume 35, Issue 13, July 2019, Pages 2226–2234, https://doi.org/10.1093/bioinformatics/bty945
Next-generation biocomputing: mimicking artificial neural network with genetic circuits Leo Chi U Seak, Owen Lok In Lo, Wade Chun-Wai Suen, Ming-Tsung Wu bioRxiv 2021.03.12.435120; doi: https://doi.org/10.1101/2021.03.12.435120
2. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
For my final project aim 1 I intend to develop a genetic circuit implemented in e.coli: a
For aim 1 of my final project, I propose implementing an intracellular artificial neural network (IANN) as a genetic circuit in E. coli chassis cells applied to real-world ermine moth silk, which serves as the substrate source for sericin-2; BmCoc enzymatic degradation of this silk-derived sericin-2 triggers GFP fluorescence readout.
Inputs are spatially localized BmCoc activity levels on the silk (sensed via cleavage products or substrate modifications contacting the chassis), feeding into layer 1 sensor modules that compute a weighted sum through regulatory proteins or RNA processors, with nonlinearity from thresholded promoter logic.
The layer 1 output (e.g., an endoribonuclease) regulates a layer 2 promoter driving GFP, yielding low fluorescence without degradation and high GFP above a tunable threshold when silk sericin-2 is broken down, enabling binary or analog reporting via microscopy or flow cytometry of silk-colonizing cells. This allows live quantification of enzymatic silk processing directly on the natural substrate.
Limitations include poor specificity (crosstalk from moth silk contaminants or host pathways), gene expression noise blurring single-cell outputs on heterogeneous silk fibers, fixed genetic "weights" limiting adaptability, metabolic burden under silk-adherent growth conditions, and kinetic mismatches between rapid proteolysis and slow transcription/translation. Despite these, the IANN provides a modular platform for screening BmCoc variants on authentic ermine moth silk and multi-input enzymatic classification in a substrate-embedded context.
3. Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
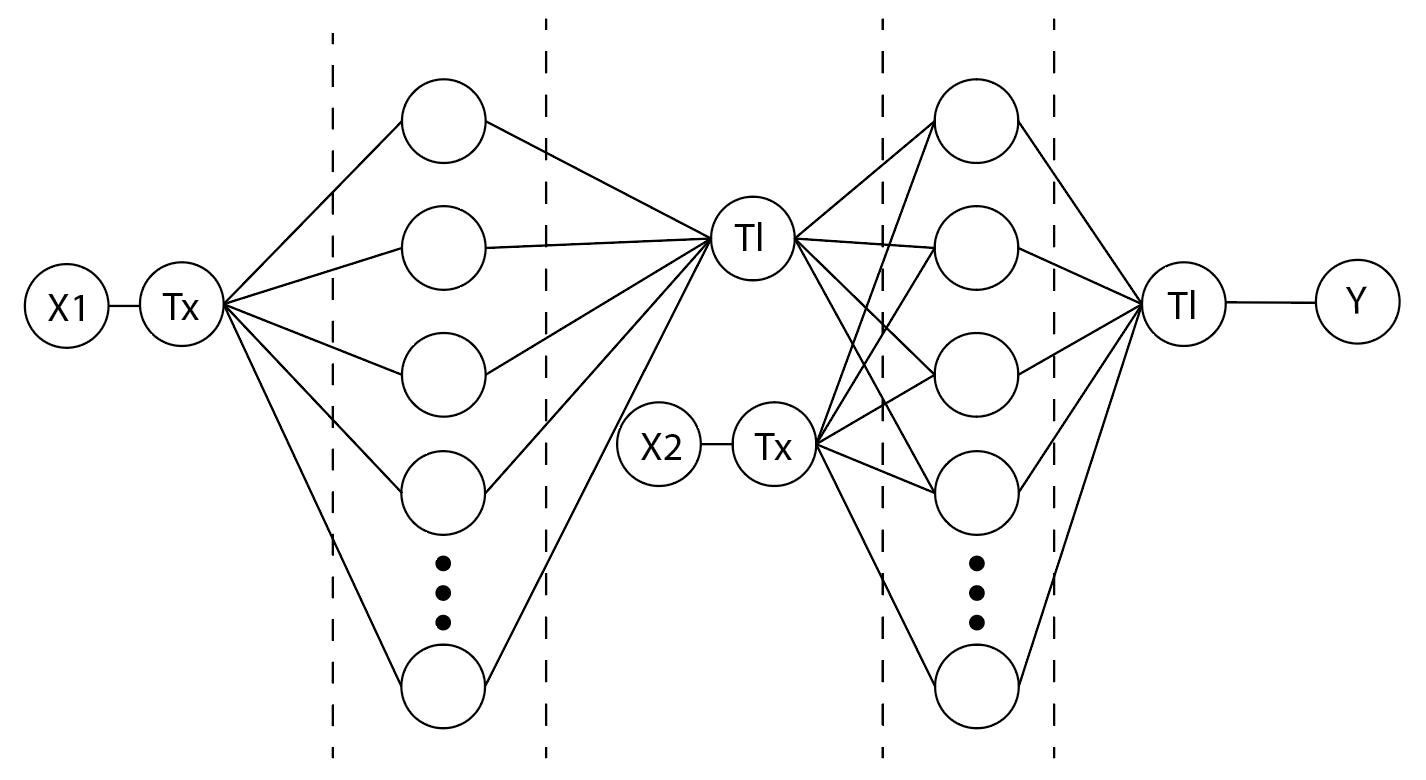
In the first layer the transcription rate of the Cys4 DNA forms the weighted value of the Cys4 translation rate, then in layer two the transcription of the GFP DNA sequence as well as the output of Cys4 translation inform the weighted value that dictates the translation of the GFP protein
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Over the past decade, a growing interest in fungal materials as a sustainable substitute for traditional, often plastic-based, materials. One example of fungal materials is mycelium packaging as a replacement for polystyrene packaging. This replacement material is completely biodegradable, shock absorbent and has a negative carbon footprint. However it is much more costly to produce and it takes a lot more time to develop.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Fungi are increasingly becoming a domain of interest in regards to bioremediation strategies, showing promising potential in the remediation of heavy metals, synthetic dyes, hydrocarbons and more. (Dinakarkumar, et al., 2024).
The reality of bioremediative initiatives however, is the possibility that these efforts are not a one-time solution. As long as pollutive industries continue to operate, it is not possible to say with full certainty that bioremediative efforts can be stopped, still leaving local communities vulnerable to the ramifications of exposure to these pollutants. Quick, accessible communication of ecological safety is therefore an important public health concern. I propose to genetically engineer fungi to produce differently coloured fruiting bodies when degrading pollutants in the soil. This would give a direct indicator of soil health and safety.

When comparing the application of synthetic biology to fungi to that of bacteria, a key advantage in favour of fungi is the fact that filamentous fungi excel in protein secretion and are particularly suited for producing complex enzymes due to their ability to perform post-translational modifications. (Garg, 2026)
References
Yuvaraj Dinakarkumar, Gnanasekaran Ramakrishnan, Koteswara Reddy Gujjula, Vishali Vasu, Priyadharishini Balamurugan, Gayathri Murali,
Fungal bioremediation: An overview of the mechanisms, applications and future perspectives, Environmental Chemistry and Ecotoxicology, Volume 6, 2024, Pages 293-302,
ISSN 2590-1826, https://doi.org/10.1016/j.enceco.2024.07.002.
Shilpa Garg, The importance of fungal biotechnology for sustainable applications, Trends in Biotechnology, Volume 44, Issue 1, 2026, Pages 79-91, ISSN 0167-7799, https://doi.org/10.1016/j.tibtech.2025.06.010.
Week 9 HW: Cell Free Systems
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis offers a number of advantages over traditional in vivo methods. An important advantage is the fact that cell-free systems are much more time efficient in comparison to their in-vivo counterparts. Cell-free systems also can operate using linear DNA fragments, whereas in-vivo systems necessitate the use of plasmid DNA.
Genetic expression in cell-free systems is directly proportional to the concentration of DNA that is present, this allows for highly specific protein production quantities.
Cases where cell-free expression is more beneficial than cell production:
Cell-free systems allow for the production of proteins that would otherwise be toxic to cellular systems.
Cell-free systems could potentially result in higher throughput for biofuel production as compared to in-vivo mechanisms.
Nico J Claassens, Simon Burgener, Bastian Vögeli, Tobias J Erb, Arren Bar-Even,
A critical comparison of cellular and cell-free bioproduction systems,Current Opinion in Biotechnology, Volume 60, 2019, Pages 221-229, ISSN 0958-1669, https://doi.org/10.1016/j.copbio.2019.05.003. (https://www.sciencedirect.com/science/article/pii/S0958166918301861)
Rollin, J.A., Tam T. K. and Zhang Y.-H. P. , (2014) ‘New biotechnology paradigm: cell-free biosystems for biomanufacturing’, Green Chemistry, 16(9), pp. 3248–3255.
https://pubs.rsc.org/en/content/articlelanding/2013/gc/c3gc40625c#!divCitation
Describe the main components of a cell-free expression system and explain the role of each component.
The core components of the cell-free expression system include cell-free extracts, template DNA, and energy sources (such as ATP, GTP, etc.).
Cell extracts form the basis of the in vitro expression system, containing all the necessary cellular machinery, such as ribosomes, transcription factors, and modifying enzymes. Template DNA encodes the target protein and can be obtained through PCR amplification or synthesis.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy provision regeneration is critical in cell-free systems, as there is no cellular metabolic function to continuously provide the necessary energy for any cellular expression.
A method to ensure continuous ATP supply in cell-free experiments is the addition
A common strategy for achieving phosphorylation of ADP is using pairs of compounds with high-energy phosphate bonds, along with their cognate kinases such as acetylpjosphate/acetate kinase, phospoenolpyruvate/pyruvate kinase or creatine phosphate/xreatine kinase. However, use of these compounds inevitably results in accumulation of inorganic phosphate in the reaction mixture.
Lee, Kyung-Ho & Kim, Dong-Myung. (2018). Recent advances in development of cell-free protein synthesis systems for fast and efficient production of recombinant proteins. FEMS microbiology letters. 365. 10.1093/femsle/fny174.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic cell-free expression systems often based on E. coli extract, have a high protein yield, simple cultivation and cell growth and lysate preparation. They’re cost efficient and easy to genetically engineer. The disadvantages of these systems, however, are that they have limited posttranslational modifications, no endogenous membrane structures for the synthesis of integral membrane proteins and only native prokaryotic chaperones are available. It might not be able to fold eukaryotic proteins correctly. (Zemella, 2015)
Eukaryotic cell-free systems offer a wider variety in extract sources. The main disadvantages that often arise with these cell free systems are high cultivation costs and low protein yield in mammalian and yeast cells. Plant-based extracts like wheat germ and tobacco BY-2 do seem to have a higher protein yield, but relative to prokaryotic systems it is still low. However, eukaryotic CFSs have the ability to handle complex post-translational modifications. (Zemella, 2015)
In prokaryotic cell-free expression systems I would choose to produce a protein that incorporates non-canonical amino acids, as these systems make this feasible. A protein I would produce in a eukaryotic cell-free expression system would be GFP.
Zemella, A., Thoring, L., Hoffmeister, C., & Kubick, S. (2015). Cell-Free Protein Synthesis: Pros and Cons of Prokaryotic and Eukaryotic Systems. Chembiochem : a European journal of chemical biology, 16(17), 2420–2431. https://doi.org/10.1002/cbic.201500340
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Cell-free expression of membrane proteins struggles with hydrophobic aggregation and ribosome stalling during nascent chain synthesis.
Setup: Use PURExpress or E. coli lysate with a GFP-fused bacterial membrane protein. Add liposomes (PC:PE mix) or nanodiscs (0.1-1 μM) at t=0. Test lipid variants and N-terminal modifications in 20 μL reactions at 25°C for 4h.
Readouts: Total GFP fluorescence; centrifugation for soluble/insoluble fractions; protease protection for insertion.
Solutions: Nanodiscs promote co-translational insertion; optimized lipids aid folding. Select condition maximizing functional, membrane-associated yield. Controls: soluble GFP, no-lipid baseline. Boosts often 5-10x.
Roos C, Kai L, Haberstock S, Proverbio D, Ghoshdastider U, Ma Y, Filipek S, Wang X, Dötsch V, Bernhard F. High-level cell-free production of membrane proteins with nanodiscs. Methods Mol Biol. 2014;1118:109-30. doi: 10.1007/978-1-62703-782-2_7. PMID: 24395412.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Three possible reasons for a low yield in a cell-free system:
Chosen prokaryotic or eukaryotic extract is not suitable for high yield protein output, a troubleshooting strategy for this would be to shift to a prokaryotic extract to then compare the resulting protein yield.
Initial DNA concentration and necessary components are too low, resulting in low output, alter the concentrations of components within the set-up to maximise ratios for highest protein yield.
The chosen experimental set-up is a batch system, resulting in lower protein yield. If possible, switch toa CECF reaction system. (Schwarz, 2007)
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
Pick a function and describe it.
What would your synthetic cell do? What is the input and what is the output?
In line with my final project research, my proposed synthetic cell would hold the capacity to degrade PFAS particles, based on the discovery of enzymatic degradation of PFAS by soil bacteria found in Portugal. The input therefore would be various compounds within the category of PFAS particles. The output would be chain-shortened, non-toxic molecules.
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
It would be very interesting to have this function be realized by cell-free Tx/Tl alone, as the enzymatic degradation system would be deployable much faster and much safer than an in-vivo counterpart.
Could this function be realized by genetically modified natural cell?
This function could be realized by genetically modified natural cells, as the envisioned functionality was first observed in L. portucalensis F11. However, this strain of bacteria takes a significantly long time to metabolise these compounds. Transplanting this enzymatic function into a different chassis like E.coli K12 would also come with its own challenges, as efflux systems would have to be neutralised due to the toxic nature of the PFAS particles.
Describe the desired outcome of your synthetic cell operation.
The desired outcome of my synthetic cell operation would be the degradation of PFAS in its surrounding
Design all components that would need to be part of your synthetic cell.
What would be the membrane made of?
The membrane would be a zwitterionic phospholipid membrane consisting of DOPC and DOPG.
Insights into adenosine A2A receptor activation through cooperative modulation of agonist and allosteric lipid interactions - Scientific Figure on ResearchGate. Available from: https://www.researchgate.net/figure/Structural-comparison-of-DOPG-and-DOPC-lipid-headgroups-DOPG-A-headgroup-is-composed-of_fig1_340707406 [accessed 6 Apr 2026]
What would you encapsulate inside? Enzymes, small molecules.
Inside of the membrane I would encapsulate the Tx/Tl system for the PFAS degrading enzyme, as well as all necessary resources for polypeptide creation.
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
The organism my Tx/Tl system would come from would be bacterial, as described prior in this exercise.
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
The PFAS particle substrates are permeable, therefore there would not be a need for additional membrane channels.
Sangwoo Ryu, Emi Yamaguchi, Seyed Mohamad Sadegh Modaresi, Juliana Agudelo, Chester Costales, Mark A. West, Fabian Fischer, Angela L. Slitt, Evaluation of 14 PFAS for permeability and organic anion transporter interactions: Implications for renal clearance in humans, Chemosphere, Volume 361, 2024, 142390, ISSN 0045-6535,
https://doi.org/10.1016/j.chemosphere.2024.142390.
Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
Lipids: DOPC and DOPG
Genes: Yet to be fully documented sequences for the enzymatic degradation of PFAS particles.
How will you measure the function of your system?
I would utilize mass spectrometry to measure the degradation of the initial molecules.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
As described in my answers to Kate Amala’s homework questions, I would propose a cell-free system that is able to degrade PFAS particles, which can be adapted to function as a freeze-dried cell-free system.
PFAS pollution is one of the great pollutive issues we face today. Recent scientific evidence shows the potential of certain bacteria to biodegrade these particles. By utilizing lyophilization technology, cell-free versions of this enzymatic system can be distributed world wide. Freeze-dried systems are activated by water; this component can be utilised to our advantage when the system is deployed in PFAS contaminated waters.
This will address the need to remediate against the presence of PFAS particles in our environment.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
While cell-free reactions depend on water to activate, this can be used to our advantage when dealing with contaminated waters. While one-time use has its downsides, it could prove to be advantageous when deploying the system into the environment. We cannot predict what uncontrolled accumulation of these components would do in the environment in the long run, therefore their temporary nature would be less hazardous.
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
It is well-known that exposure to cosmic radiation is a great problem in space exploration. An important possible health risk is the development of cancer. However, cancer diagnostics are
more difficult due to environmental and spatial factors in space.
Carcinoembryonic antigen (CEA) is a nonspecific serum biomarker that is elevated in many malignancies, including colorectal cancer, medullary thyroid cancer, breast cancer, and mucinous ovarian cancer. The ELISA method detects antigen-antibody interactions by using enzyme-labelled conjugates and enzyme substrates that generate colour changes; it is often used to assess CEA-values in blood.
Outside of cancer diagnostics, the availability of antibodies is vital for various fields of research.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
The main molecular target I propose to study is the anti-CEA antibody, which is a receptor protein used in ELISA detection of CEA concentration in blood.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
Currently, production of antibodies is predominantly done through Polyclonal Antibody Production, a process that often involves significant animal cruelty, is relatively inefficient and can often take months. Besides a matter of optimisation, moving towards a cell-free synthesizing methodology is an important matter of animal welfare.
While animal rights are still incredibly important in a space setting, it is obvious to state that this technology would not be available in the first place. Besides moral and ethical objections, taking animals into space for this purpose would additionally come with significant additional challenges in terms of resources, time and spatial capacities.
Therefore ensuring that cell-free synthesis of antibodies is functional in space is vital in the development of sustainable space exploration.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
Goal:
The research goal of this project is understanding the possibilities of synthesizing anti-CEA antibodies in outer space for disease diagnostics, which would be indicative of the synthesizing possibilities of other antibodies.
Reasoning:
As stated prior, the synthesis of antibodies in space is an important technology to hone in the development of space adapted diagnostics. Anti-CEA antibodies are selected for this experiment as their CEA counterpart is a key indicator for various cancers in the body.
Moreover, the BioBits® cell-free protein expression system could be a suitable candidate for this experiment as its lyophilised system would be applicable for experiment reproduction in space.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
The samples I would test in my experiment would be synthetic genetic pathways for protein production, specifically for anti-CEA antibodies. These pathways would be developed based on sequencing data and antibody synthesis precedents obtained from literature review.
The DNA would be obtained from in-vivo specimens and amplified using PCR.
Type of data measured: In line with the BioBits® cell-free protein expression’s functionality, the measured data would be a GFP readout that would be in proportion to the expression of the antibodies.
Week 10 HW: Advanced Imaging and Measurement Technology
Homework: Final Project
For your final project:
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
Aspect | Measurement Goal | Scientific Rationale |
Silk Substrate | Percent weight loss | Quantifies the effectiveness of BmCoc in selectively degrading sericin. |
Bacterial Growth | Colony Forming Units (CFU) | Determines if E. coli proliferates by utilizing degraded sericin as a nutrient source. |
Enzyme Output | Concentration of His-tagged BmCoc | Verifies production, secretion, and stability of the functional enzyme in the supernatant. |
|
|
|
Technologies and Methodologies
1. Gravimetric Analysis (Mass Measurement)
I will perform precise weighing of silk fibers before and after incubation. By using a high-precision analytical balance and drying samples in a desiccator/oven to a constant mass, I will establish a reliable "dry mass" baseline. This is the most direct metric for determining the total degradation of the sericin-fibroin matrix.
2. Microbial Quantification (CFU Assay)
To measure proliferation, I will use serial dilution plating on selective media (LB + Ampicillin). By vortexing the silk fibers with a detergent like Tween20, you detach biofilm-associated cells, allowing for an accurate count of viable bacteria. This confirms that the engineered cells are not just surviving but actively utilizing the silk-derived nutrients.
3. Protein Quantification and Verification
Bradford Assay: This colorimetric assay will be used for the rapid, high-throughput quantification of the total His-tagged BmCoc protein recovered from the culture supernatant. It provides a standard estimate of the enzyme titer.
Ni-NTA Affinity Chromatography: Before quantification, I will use Ni-NTA resin to specifically capture the His-tagged BmCoc. This serves as a vital purification step, removing background E. coli proteins and allowing for a more accurate concentration measurement of your enzyme of interest.
A280 Absorbance: Using a NanoDrop spectrophotometer, I will perform direct UV absorption measurements at 280 nm, utilizing the calculated extinction coefficient of BmCoc to provide a secondary confirmation of the protein concentration.
Homework: Waters Part I — Molecular Weight
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
eGFP Sequence:
MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH
Note: This contains a His-purification tag (HHHHHH) and a linker (the LE before it).
Theoretical pI/Mw: 5.90 / 28006.60
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
Determine for each adjacent pair of peaks using:
n = 875,4421
n+1 = 903,7148
z = -31,9638
2. Mw = (m/z * z) = (875,4421*-31.9638) = -27.982,4562 Da
3. Accuracy = (MWexperiment - MWtheory) / MWtheory
= (27982,4562 - 28006,60) / 28006,60
= -0,0008
3. Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
You cannot observe the charge state of the zoomed-in peak in the mass spectrum for the intact eGFP, as the protein is unfolded and in its denatured state. Therefore the protein is in a larger charged state, thus it can not be observed.
Homework: Waters Part III — Peptide Mapping - primary structure
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH
There are 20 Lysines and 6 Arganines in eGFP
How many peptides will be generated from tryptic digestion of eGFP?
Based on the Expasy calculator, 21 peptides generated when using trypsin to perform the digest.
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
I count 21 peaks in the generated peptide map.
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
When comparing the generated peptides from the expasy calculator and the LC-MS data for the Peptide Map data generated in lab, they both indicate that 21 peptides have been created.
Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide (M+H^+) based on its m/z and z.
n =521.7612
n+1 = 526.25918
z = (m/zn+1) / ( (m/zn) – (m/zn+1) )
= 526.25918 / (521.7612 – 526.25918)
= 526.25918 / -4,49798
= -116,999004 = 117
m = m / z * z
= 526.25918 / 117
= 4,49794171
What is the percentage of the sequence that is confirmed by peptide mapping? (see Figure 6)
The percentage of the sequence that is confirmed by peptide mapping is 88%
Homework: Waters Part IV — Did I make GFP?
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
7FU Decamer = 10 subunits 340 * 10 = 3400 kDa = 3.4 MDa ≈
8FU Didecamer = 20 subunits 400 * 20 = 8000 kDa = 8.0 MDa ≈ 8.33 MDa
8FU 3-Decamer = 30 subunits 400 * 30 = 1200 kDA = 12.0 MDa ≈ 12.67 MDA
8FU 4-Decamer = 40 subunits 400 * 40 = 1600 kDA = 16.0 MDa
The calculated weights somewhat correspond to peaks in the CDMS chart, taking possible extraneous digits into consideration when looking at the provided data.
Week 11 HW: Bioproduction and Cloud Labs

Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
My contribution to the Pixel Artwork Canvas is the red pixel circled in pink, located on position Q2K7 and was the 1298th contribution overall. What I like about the project is that it showcases fundamental aspects of human collaboration, by having decisionmaking simplified and visualised in the artwork. The participant is asked to question prioritising their own agency or the agency of another contributor: do you reject what was made before you or do you honour its legacy?
What could make the experience better next year, would be to make the canvas bigger. I think it would be interesting to see what emerges when people have enough space to draw without having to compete for space as much. Would it create smaller islands? Would it still be completely covered?
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
Lysate components
E. coli lysate. This provides the core machinery needed for protein synthesis, including ribosomes, enzymes, and other factors that carry out transcription and translation.
BL21 (DE3) Star lysate (includes T7 RNA polymerase). This is a more specialized extract that contains T7 RNA polymerase, which helps transcribe the DNA template into mRNA efficiently.
Salts and buffer
Potassium glutamate. Helps maintain the right ionic conditions in the reaction so the protein synthesis machinery can work properly.
HEPES-KOH pH 7.5. Acts as a buffer to keep the reaction at a stable pH.
Magnesium glutamate. Supplies magnesium, which is important for ribosome function and many enzyme activities.
Potassium phosphate monobasic. Helps with buffering and maintaining the correct chemical environment.
Potassium phosphate dibasic. Works together with the monobasic form to keep the pH balanced.
Energy / nucleotide system
Ribose. Supports nucleotide-related metabolism and helps with maintaining the reaction chemistry.
Glucose. Serves as an energy source that can help regenerate ATP during the reaction.
AMP. Helps support the energy pool in the reaction.
CMP. Supplies pyrimidine nucleotide building blocks.
GMP. Supplies guanine-related nucleotide building blocks.
UMP. Supplies uracil-related nucleotide building blocks.
Guanine. Helps replenish guanine nucleotide pools for RNA synthesis.
Translation mix
17 amino acid mix. Provides most of the amino acids needed to build the protein.
Tyrosine. Added separately to make sure there is enough of this amino acid in the reaction.
Cysteine. Added separately for the same reason, since it can sometimes be limiting.
Additives
Backfill
Backfill. Used to make up the remaining volume in the reaction without changing the chemistry too much.
Nuclease-free water. Adjusts the final reaction volume while avoiding contamination that could break down DNA or RNA.
References:
Gregorio, N. E., Levine, M. Z., & Oza, J. P. (2019). A User's Guide to Cell-Free Protein Synthesis. Methods and protocols, 2(1), 24. https://doi.org/10.3390/mps2010024
Krinsky N, Kaduri M, Shainsky-Roitman J, Goldfeder M, Ivanir E, Benhar I, et al. (2016) A Simple and Rapid Method for Preparing a Cell-Free Bacterial Lysate for Protein Synthesis. PLoS ONE 11(10): e0165137. https://doi.org/10.1371/journal.pone.0165137
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The 1-hour optimized PEP-NTP master mix is designed for faster, short-term protein production, while the 20-hour NMP-Ribose-Glucose master mix is set up to support longer reactions over a much longer incubation time. In simple terms, the PEP-NTP mix is more geared toward quick energy use and fast output, whereas the NMP-Ribose-Glucose mix seems designed to sustain protein synthesis more gradually over time.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
sfGFP is a superfolder GFP variant with very fast maturation, so it becomes fluorescent quickly after translation and is usually a strong readout in cell-free systems. Its good folding robustness also helps it fluoresce even when expression conditions are not ideal.
mRFP1 is a red fluorescent protein with relatively slow maturation compared with sfGFP, so fluorescence can lag behind protein production in short cell-free runs. It also has moderate acid sensitivity, which can affect signal if the reaction conditions drift in pH.
mKO2 is an orange fluorescent protein that is strongly oxygen dependent for chromophore maturation, so low-oxygen conditions can reduce or delay its fluorescence. It also has moderate acid sensitivity, which can influence readout in reactions where pH is not tightly controlled.
mTurquoise2 is a cyan FP with very low acid sensitivity, which makes its fluorescence more reliable if the reaction environment changes slightly in pH. It is also reported to mature quickly, so it is useful when you want a relatively fast signal after expression.
mScarlet-I is a very bright red FP with relatively fast maturation, so it gives a strong fluorescence signal once enough protein has been made. Its moderate acid sensitivity means low pH could still reduce the observed fluorescence a bit.
Electra2 is a blue fluorescent protein and one important practical point is that blue FPs often tend to be less bright than many green or red FPs, which can make the readout harder in cell-free assays. It also depends on proper folding and chromophore formation, so expression conditions can affect how much signal you actually see. Electra2, published in 2022, is considerable brighter than other bFPs.
2. Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
As stated above, mKO2 is an orange fluorescent protein that is strongly oxygen dependent for chromophore maturation, so low-oxygen conditions can reduce or delay its fluorescence. It also has moderate acid sensitivity, which can influence readout in reactions where pH is not tightly controlled. Therefore, maximizing fluorescence over a 36-hour incubation period would only be feasible through higher-oxygen conditions and a controlled pH that stays near neutral to slightly basic.
I therefore hypothesise that an increase in HEPES-KOH pH 7.5 buffer would allow for the necessary stability of the pH of the mastermix. Oxygen availability on the other hand, does not necessarily relate to any specific reagent, but rather the aeration of the reaction. This includes allowing for sufficient headspace, not too much total volume and potentially aeration through continuous stirring.
3. The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
Based on my answer to question 2, I decided to increase the amount of HEPES-KOH pH 7.5 in the master mix composition with 5.000 mM to test whether my hypothesis was correct. Due to the fact that I do not have access to the lab, I would unfortunately not be able to test it in vitro.