Week 2 HW: DNA Read Write and Edit
Homework week 2 - DNA READ, WRITE & EDIT
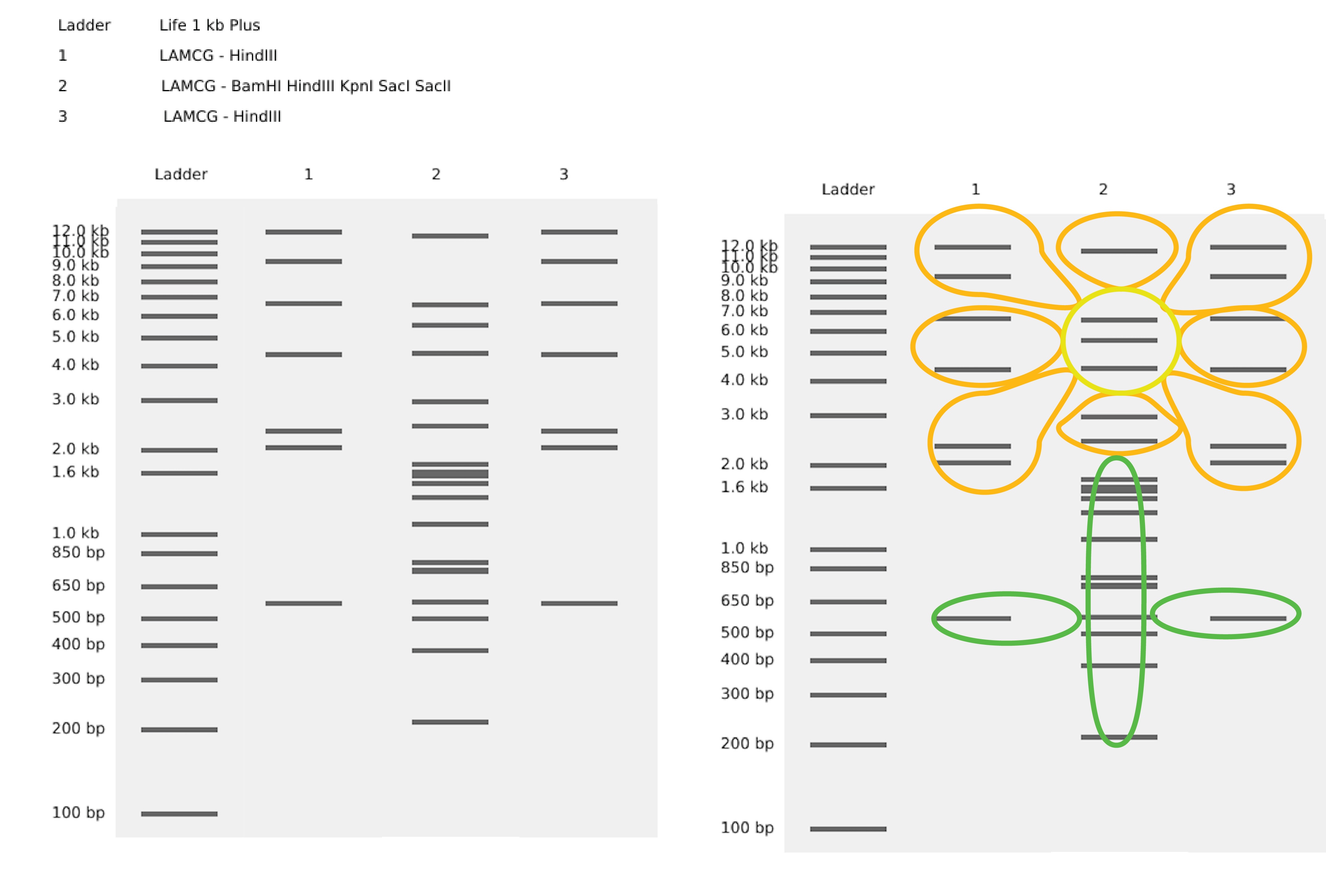
Part 1: Benchling & In-silico Gel Art
Part 3: DNA Design Challenge
3.1. Choose your protein.
Cocoonase, an enzyme capable of degrading silk protein sericin-2.
GenBank: AB604648.1
Rodbumrer P, Arthan D, Uyen U, Yuvaniyama J, Svasti J, Wongsaengchantra PY. Functional expression of a Bombyx mori cocoonase: potential application for silk degumming. Acta Biochim Biophys Sin (Shanghai). 2012 Dec;44(12):974-83. doi: 10.1093/abbs/gms090. PMID: 23169343.
Amino acid sequence:
MIVGGEEISINKVPYQAYLLLQKDNEYFQCGGSIISKRHILTAA
HCIEGISKVTVRIGSSNSNKGGTVYTAKSKVAHPKYNSKTKNNDFAIVTVNKDMAIDG
KTTKIITLAKEGSSVPDKTKLLVSGWGATSEGGSSSTTLRAVHVQAHSDDECKKYFRS
LTSNMFCAGPPEGGKDSCQGDSGGPAVKGNVQLGVVSFGVGCARKNNPGIYAKVSAAAKWIKSTAGL
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
cDNA:
atgattgtgggcggcgaagaaattagcattaacaaagtgccgtatcaggcgtatctgctg
ctgcagaaagataacgaatattttcagtgcggcggcagcattattagcaaacgccatatt
ctgaccgcggcgcattgcattgaaggcattagcaaagtgaccgtgcgcattggcagcagc
aacagcaacaaaggcggcaccgtgtataccgcgaaaagcaaagtggcgcatccgaaatat
aacagcaaaaccaaaaacaacgattttgcgattgtgaccgtgaacaaagatatggcgatt
gatggcaaaaccaccaaaattattaccctggcgaaagaaggcagcagcgtgccggataaa
accaaactgctggtgagcggctggggcgcgaccagcgaaggcggcagcagcagcaccacc
ctgcgcgcggtgcatgtgcaggcgcatagcgatgatgaatgcaaaaaatattttcgcagc
ctgaccagcaacatgttttgcgcgggcccgccggaaggcggcaaagatagctgccagggc
gatagcggcggcccggcggtgaaaggcaacgtgcagctgggcgtggtgagctttggcgtg
ggctgcgcgcgcaaaaacaacccgggcatttatgcgaaagtgagcgcggcggcgaaatgg
attaaaagcaccgcgggcctg
The chosen chassis is e.coli, as it is really good for genetic circuit implementation
3.3. Codon optimization.
atgattgtgggcggcgaagaaattagcattaacaaagtgccgtatcaggcgtatctgctgctgcagaaagataacgaatattttcagtgcggcggcagcattattagcaaacgccatattctgaccgcggcgcattgcattgaaggcattagcaaagtgaccgtgcgcattggcagcagcaacagcaacaaaggcggcaccgtgtataccgcgaaaagcaaagtggcgcatccgaaatataacagcaaaaccaaaaacaacgattttgcgattgtgaccgtgaacaaagatatggcgattgatggcaaaaccaccaaaattattaccctggcgaaagaaggcagcagcgtgccggataaaaccaaactgctggtgagcggctggggcgcgaccagcgaaggcggcagcagcagcaccaccctgcgcgcggtgcatgtgcaggcgcatagcgatgatgaatgcaaaaaatattttcgcagcctgaccagcaacatgttttgcgcgggcccgccggaaggcggcaaagatagctgccagggcgatagcggcggcccggcggtgaaaggcaacgtgcagctgggcgtggtgagctttggcgtgggctgcgcgcgcaaaaacaacccgggcatttatgcgaaagtgagcgcggcggcgaaatggattaaaagcaccgcgggcctg
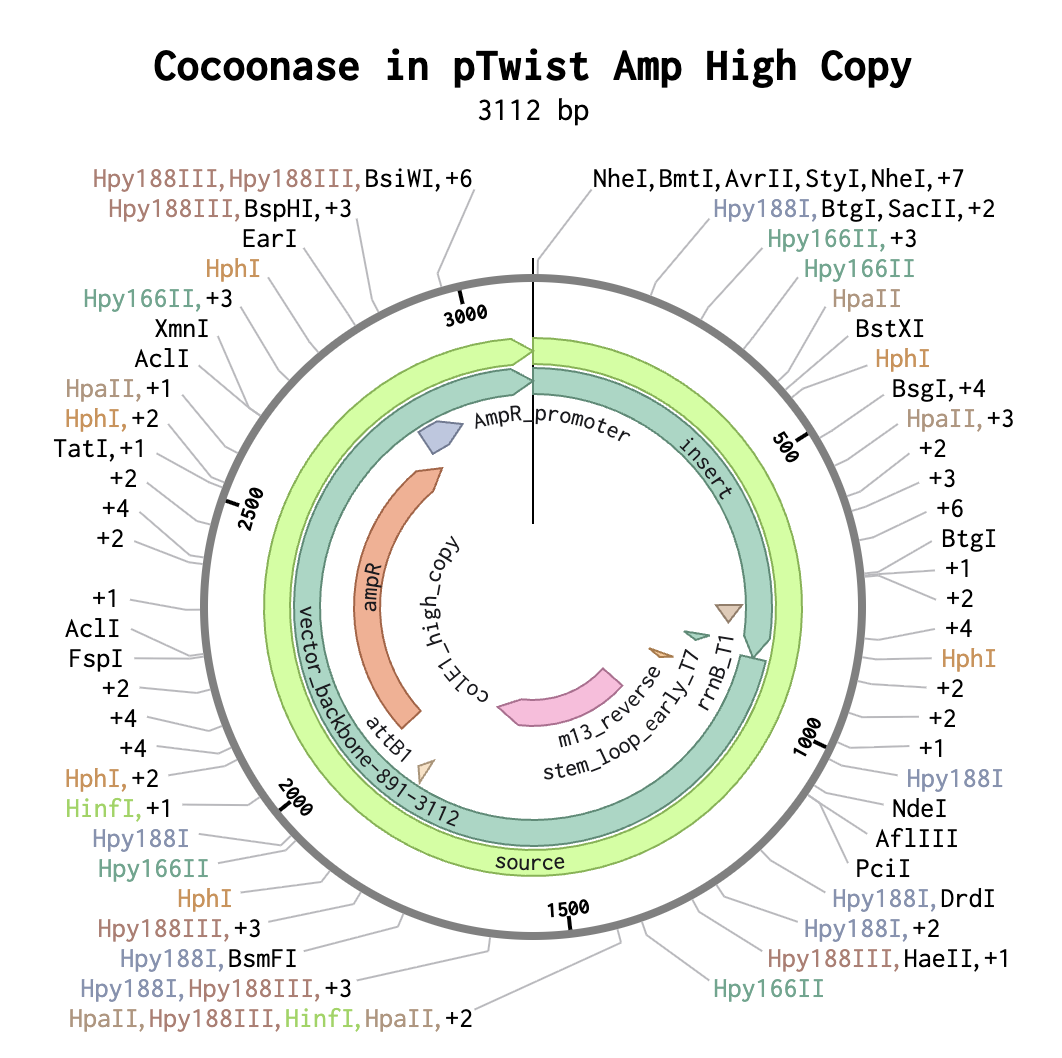
Part 4: Prepare a DNA Twist Order
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
The DNA that I would like to sequence is related to my initial project proposal described in last week’s homework. In it I describe L. portucalensis F11; a strain of bacteria that shows evidence to be able to chain-shorten PFAS particles. I would like to sequence this bacteria’s DNA as a first step into isolating the sequence that corresponds to the enzyme that’s able to break down these ‘forever chemicals’.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Also answer the following questions:
The DNA I want to sequence comes from a bacterium whose genome has not been sequenced previously. Initially I figured shotgun sequencing would potentially be a good approach as prokaryotic DNA is simpler in structure and therefore less prone to error than eukaryotic organisms whose genome contains many repetitive sequences. If I simultaneously find and isolate the enzyme responsible for degradation of PFAS, I would be able to cross reference the amino acid chains with the genome, to localise the sequence that codes for it.
However, this is a highly inefficient approach, as it takes a lot of time, resources and money.
L. portucalensis F11, could be cultivated in vitro by only giving it PFAS as potential nutrients. This would mean that the only degenerative enzymatic activity would be by the targeted enzyme, indicating the presence of mRNA coding for the aminoacid-chain of the prospective protein.
Grow bacteria (L. portucalensis F11) on PFAS as the sole carbon source to induce pathway-specific enzymes, ensuring upregulated mRNA dominates the transcriptome.
Lyse cells and extract total RNA using standard kits (e.g., TRIzol), capturing the active enzymatic snapshot.
Remove 80-90% ribosomal RNA through rRNA depletion, with probe-based methods to enrich messenger RNA and avoid wasted sequencing.
Directly sequence the depleted RNA using Oxford Nanopore Sequencing for full-length native transcripts.
Prepare the library: Fix damaged ends on your rRNA-depleted RNA, then attach adapters (a motor protein and anchor) to each strand using ligation.
Load the flow cell: Prime the MinION chip's tiny holes (nanopores) with buffer, then add your RNA library via the SpotON port.
Start sequencing: Apply voltage—the motor pulls RNA through nanopores one-by-one, changing electrical current as each base passes.
Basecall live: Software like Guppy converts the current "squiggles" into RNA sequence reads (A, C, G, U) in real-time.
Assemble reads (e.g., FLAIR/Trinity), quantify expression, and identify degradative enzymes via pathway analysis (KEGG/BLAST for haloacid dehalogenases, monooxygenases).
Computational Reverse Translation: From assembled mRNA, computationally translate to amino acids then back-translate to the precise genomic DNA sequence using codon bias tools (e.g., Backtranseq), accounting for degeneracy.
Oxford Nanopore sequencing is a third-generation sequencing because it reads single DNA/RNA molecules directly in real-time without amplification or short-read assembly
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond.
I would like to synthesize a repressilator-based genetic circuit that could be applied to bacteria that are implemented into the silk structure.
The circuit would be based on three genetic components:
A PFAS detecting biosensor (Mann, et al. 2023)
A gene that encodes for an enzyme that breaks down Sericin 2 (Rodbumrer, et al. 2012)
A gene that encodes for an enzyme that breaks down PFAS particles (Wijayahena et al., 2025)
If the PFAS-detecting biosensor senses the presence of PFAS particles, it would repress the Sericin 2-enzyme gene. This would pressure the bacteria to degrade the PFAS particles. Once the particles are no longer present, the detector would no longer repress the Sericin 2-enzyme gene and the bacteria could continue to survive on metabolising Sericin 2.
Mann, M.M., Berger, B.W. A genetically-encoded biosensor for direct detection of perfluorooctanoic acid. Sci Rep 13, 15186 (2023). https://doi.org/10.1038/s41598-023-41953-1
Rodbumrer P, Arthan D, Uyen U, Yuvaniyama J, Svasti J, Wongsaengchantra PY. Functional expression of a Bombyx mori cocoonase: potential application for silk degumming. Acta Biochim Biophys Sin (Shanghai). 2012 Dec;44(12):974-83. doi: 10.1093/abbs/gms090. PMID: 23169343.
Wijayahena, M.K., Moreira, I.S., Castro, P.M.L., Dowd, S., Marciesky, M.I., Ng, C. and Aga, D.S., 2025. PFAS biodegradation by Labrys portucalensis F11: Evidence of chain shortening and identification of metabolites of PFOS, 6:2 FTS, and 5:3 FTCA. Science of The Total Environment, 959, p.178348.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I would utilize phosphoramidite oligo synthesis to synthesize the separate sequences, to then combine them using Gibson assembly.
Essential steps phosphoramidite oligo synthesis:
Core Synthesis Cycle
The cycle attaches protected phosphoramidite monomers to a solid support (like controlled-pore glass, CPG) bound to the first 3' nucleoside.
Detritylation (Deblocking): Acid (e.g., trichloroacetic acid) removes the 5'-dimethoxytrityl (DMT) protecting group from the growing chain's 5'-OH, exposing it for the next coupling.
Coupling: A phosphoramidite monomer (with DMT-protected 5'-OH, base protections, and reactive 3'-phosphoramidite) plus activator (e.g., tetrazole) reacts with the free 5'-OH to form an unstable phosphite triester linkage (~99% efficiency per step).
Capping: Acetic anhydride caps unreacted 5'-OH groups (as acetyl esters) to prevent truncated failure sequences from proceeding, ensuring high purity.
Oxidation: Iodine in water/pyridine converts the fragile phosphite triester to a stable phosphate triester, mimicking the natural DNA backbone.
Post-Synthesis Processing
After the final cycle and detritylation, the oligo is cleaved from the support (e.g., via ammonia), and all protecting groups are removed (bases, phosphates). Final purification uses HPLC or PAGE.
Essential steps Gibson Assembly:
Gibson assembly joins DNA pieces seamlessly in one simple mix using enzymes. It's great for building genetic circuits like the repressilator from overlapping fragments.
Prep Your DNA
Make or get linear DNA fragments (like PCR products or synthesized genes) with 20-40 base matching ends (overlaps). Use equal amounts, clean them up to remove junk.
One-Pot Reaction
Mix fragments with Gibson master mix (has exonuclease, polymerase, ligase) in a tube on ice. Incubate at 50°C for 15-60 minutes.
Chew-back: Enzyme nibbles 5' ends to make sticky single-strand overlaps.
Anneal and fill: Polymerase glues matching overlaps and fills gaps.
Seal: Ligase connects the nicks for a full circle.
Cool on ice when done.
Oligonucleotide synthesis is limited due to the fact that the longer the chain becomes, the more prone it becomes to error in accuracy. This can be mitigated by only constructing the separate genes described in the previous question.
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
Within the context of my HTGAA project, I would like to make an edit in the genome of my chosen chassis, e.coli k12, by deleting the gene that encodes the tolC protein. The tolC protein is an outer membrane channel, which is required for the function of several efflux systems. As it’s responsible for toxin removal from the cell, it would flush out PFAS particles to the extent that the biosensor would barely be able to detect any particles.
(ii) What technology or technologies would you use to perform these DNA edits and why?
Also answer the following questions:
How does your technology of choice edit DNA? What are the essential steps?
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
I would choose to perform these DNA utilising a CRISPR/CAS9 system, which edits DNA by performing double strand cuts.
It consists of CRISPR and Cas9:
Cas9 is an endonuclease that recognizes protospacer adjacent motifs (PAM) in target DNA. PAM = a short DNA sequence (2-6 base pairs) found next to the target site (protospacer) that a CRISPR-Cas system recognizes and cuts, acting as a crucial "self vs. non-self" marker. Single guide RNA (sgRNA) directs it to target DNA
sgRNA = CRISPR RNA + trans-activating RNA (tracrRNA)
CRISPR RNA is a small, specialized RNA molecule derived from the bacterial CRISPR locus that guides Cas nucleases (like Cas9) to complementary target DNA, enabling precise cutting.
tracrRNA = a small, non-coding RNA essential for the maturation of CRISPR RNA (crRNA) and the activation of the Cas9 nuclease in Type II CRISPR-Cas immune systems. It binds to pre-crRNA, facilitating its cleavage, and forms a dual-RNA complex that guides Cas9 to specific DNA sequences for cleavage. This allows for the sequence to be cut at a specific site, which would then trigger ligase cellular function NHEJ or HDR.
1. Target Design & Guide RNA (gRNA) Selection: Identify the target DNA sequence within the gene to be knocked out. Design a 20-nucleotide sequence complementary to the target, adjacent to a Protospacer Adjacent Motif (PAM, typically 5'-NGG-3' for S. pyogenes Cas9).
2. Assembly of CRISPR Components: The gRNA and Cas9 enzyme are combined to form a ribonucleoprotein (RNP) complex, or plasmids encoding these components are created.
3. Delivery into Cells: The CRISPR components are delivered into target cells (via transfection, electroporation, or viral vectors).
4. Target Recognition and Cleavage: The Cas9-gRNA complex scans the genome for the PAM sequence. Once found, the gRNA hybridizes with the target DNA, activating the Cas9 nuclease to create a double-strand break (DSB) ~3 base pairs upstream of the PAM.
5. DNA Repair and Mutation Induction: The cell attempts to repair the DSB, predominantly via Non-Homologous End Joining (NHEJ). This error-prone process often introduces random insertions or deletions (indels) at the cleavage site.
6. Gene Knockout Confirmation: Indels within the open reading frame (ORF) typically cause frameshift mutations, leading to premature stop codons and nonfunctional proteins (knockout). The resulting mutations are verified via DNA sequencing or mismatch detection assays.