Week 4 HW: Protein Design Part 1
Part A: Conceptual Questions
Part A
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons?
A piece of meat weighing 500 grams contains approximately 100 to 160 grams of protein. 100 Daltons is equivalent to 1.66053907 × 10-22 grams If we divide the amount of protein by this number, it would mean that a piece of 500 grams of meat would contain between 6.0221407e+23 and 9.6354252e+23 amino acids.
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
The digestion system breaks down any genetic material, moreover since the cells are not endogenous to our body they do not get “access” to our bodily system.
Why are there only 20 natural amino acids?
“The 20 standard amino acids encoded by the Genetic Code were adopted during the RNA World, around 4 billion years ago. This amino acid set could be regarded as a frozen accident, implying that other possible structures could equally well have been chosen to use in proteins. Amino acids were not primarily selected for their ability to support catalysis, as the RNA World already had highly effective cofactors to perform reactions, such as oxidation, reduction and transfer of small molecules. Rather, they were selected to enable the formation of soluble structures with close-packed cores, allowing the presence of ordered binding pockets. Factors to take into account when assessing why a particular amino acid might be used include its component atoms, functional groups, biosynthetic cost, use in a protein core or on the surface, solubility and stability. Applying these criteria to the 20 standard amino acids, and considering some other simple alternatives that are not used, we find that there are excellent reasons for the selection of every amino acid. Rather than being a frozen accident, the set of amino acids selected appears to be near ideal.”
Directly quoted from:
Doig AJ. Frozen, but no accident - why the 20 standard amino acids were selected. FEBS J. 2017 May;284(9):1296-1305. doi: 10.1111/febs.13982. Epub 2017 Jan 13. PMID: 27926995.
Can you make other non-natural amino acids? Design some new amino acids.
Yes it is possible to make other non-natural amino acids, as amino acids have the same structure: a central alpha carbon, an amino group on one side of it and a carboxyl group on the other, with a unique variable side chain.
Where did amino acids come from before enzymes that make them, and before life started?In 1952, the Miller-Urey experiment proved that complex carbon based amino acids can form spontaneously from inorganic precursors under conditions simulating early Earth. This indicates that abiotic synthesis happened before enzymes existed that would make them.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
In opposition to L-amino acids that make right handed α-helices, I would expect α-helices made up of D-amino acids to be left handed.
Can you discover additional helices in proteins?
Yes one can discover additional helices in proteins. While traditional methods like DSSP only take strong hydrogen bonds into consideration, it disregards smaller structures and fringe residues of alpha helices. Methods like STRIDE and geometry tools do detect these residues as alpha helix structures by analysing hydrogen bonds as well as twist angles.
Why are most molecular helices right-handed?
Most molecular helices are right-handed, because amino acids in nature use L-amino acids. Their left-handed chirality forces helices to form in right-handed coils for stability.Why do β-sheets tend to aggregate?
β-sheets tend to aggregate due to their sheet-like nature; their edges are exposed and therefore have the tendency to latch onto other sheets via hydrogen bonds and hydrophobic contacts. The open strand edges of β-sheets are primed for edge-to-edge zipping with neighbouring sheets.
Part B.
Briefly describe the protein you selected and why you selected it.
The protein I selected is tolC, a protein in the outer membrane of E.coli K12 that’s responsible for the afflux of various toxins. I selected it for my project as I intend to build a genetic circuit that represses this protein’s function.
Identify the amino acid sequence of your protein.
The amino acid sequence is: MKKLLPILIGLSLSGFSSLSQAENLMQVYQQARLSNPELRKSAADRDAAFEKINEARSPLLPQLGLGADYTYSNGYRDANGINSNATSASLQLTQSIFDMSKWRALTLQEKAAGIQDVTYQTDQQTLILNTATAYFNVLNAIDVLSYTQAQKEAIYRQLDQTTQRFNVGLVAITDVQNARAQYDTVLANEVTARNNLDNAVEQLRQITGNYYPELAALNVENFKTDKPQPVNALLKEAEKRNLSLLQARLSQDLAREQIRQAQDGHLPTLDLTASTGISDTSYSGSKTRGAAGTQYDDSNMGQNKVGLSFSLPIYQGGMVNSQVKQAQYNFVGASEQLESAHRSVVQTVRSSFNNINASISSINAYKQAVVSAQSSLDAMEAGYSVGTRTIVDVLDATTTLYNAKQELANARYNYLINQLNIKSALGTLNEQDLLALNNALSKPVSTNPENVAPQTPEQNAIADGYAPDSPAPVVQQTSARTTTSNGHNPFRN
The sequence is 493 amino acids in length and the most common amino acid is alanine which appears 57 times.
The protein has various homologous proteins, at 100% shared identity, 34 variants of e.coli share the same protein. At 50% similarity there are 1252 entries.
My protein belongs to the outer membrane factor (omf) family.
Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
The structure of my protein was solved in 2004, with a resolution of 2.75 Å. This indicates that it’s a good quality structure.
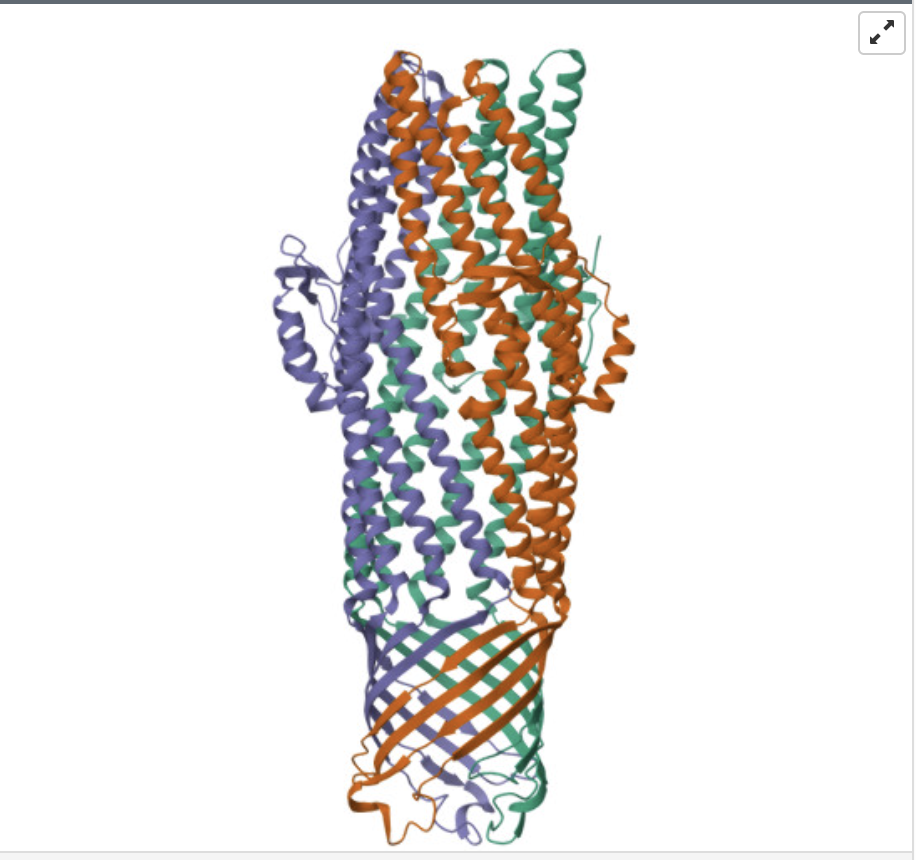
Are there any other molecules in the solved structure apart from protein?
The ligand Cobalt Hexammine(III)
Does your protein belong to any structure classification family?
Yes it belongs to Multidrug efflux transporter AcrB TolC docking domain (DN and DC subdomains)
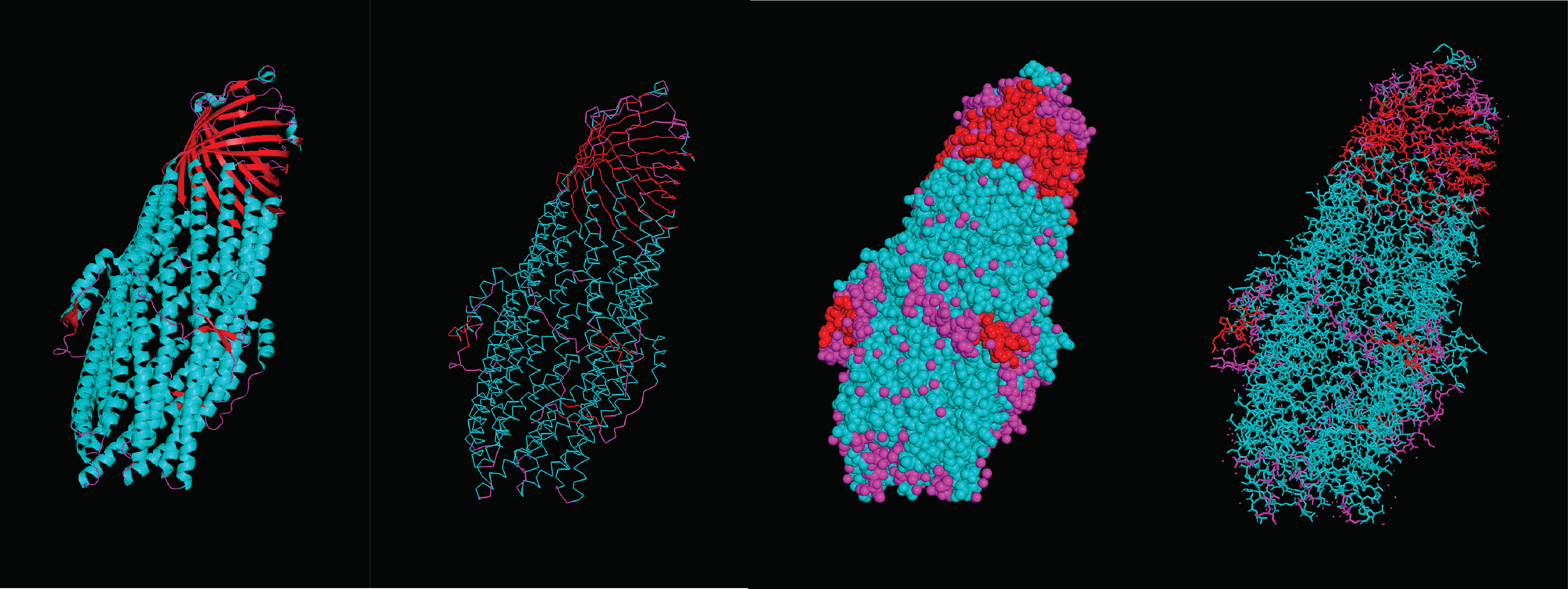
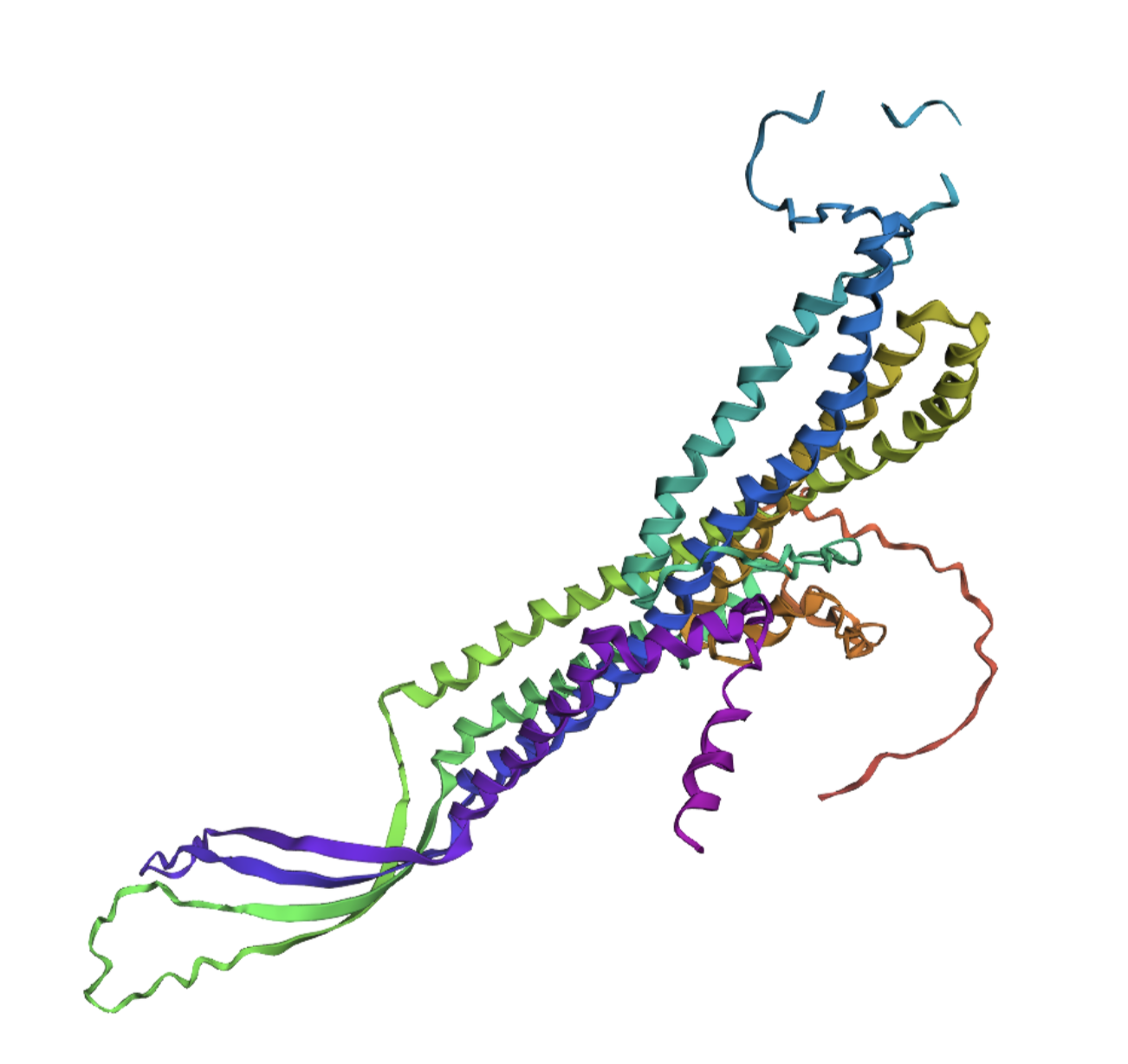
When coloring the secondary structures, it is revealed that the protein contains both helices and sheets, with more helices than sheets.
What can you tell about the distribution of hydrophobic vs hydrophilic residues? There seems to be a pretty homogenous distribution of hydrophobic and hydrophilic residues throughout the protein, this makes sense considering its function as an efflux system that connects the watery inner cell and the lipid outer membrane. Therefore it has to be compatible with both environments. Moreover, this distribution allows for molecules to pass through the structure without getting stuck on any specific spot.
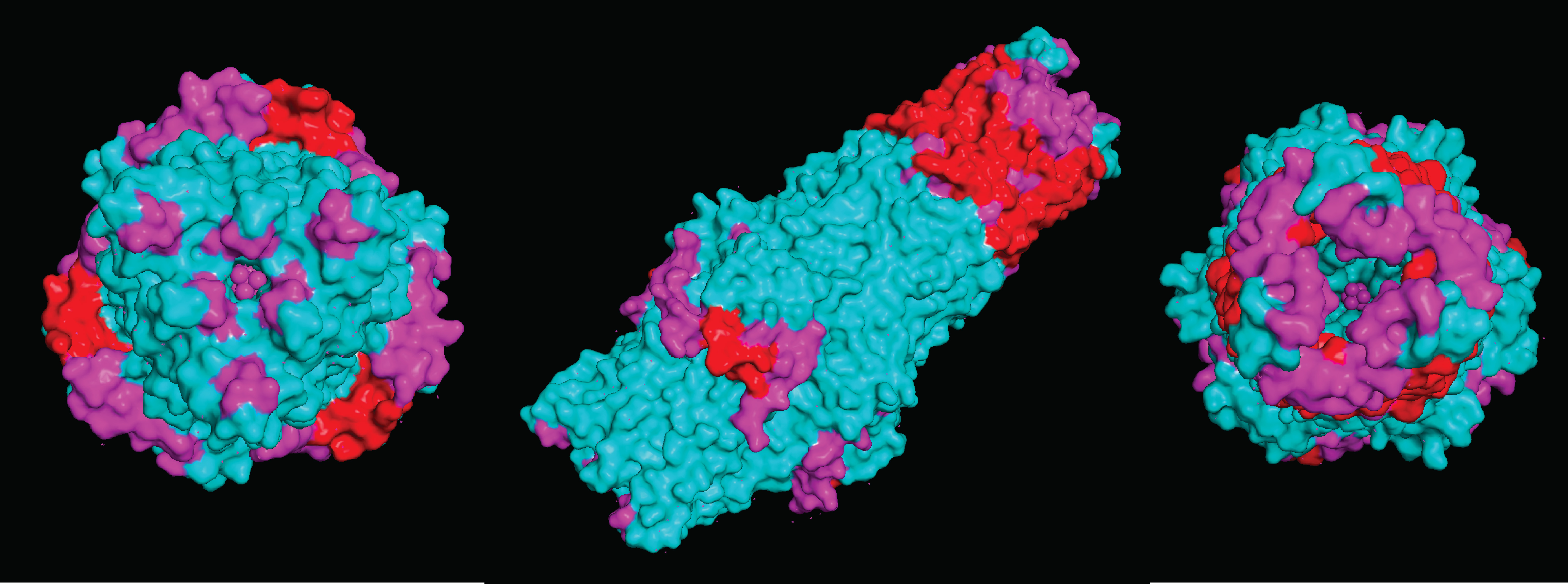
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)
Due to the nature of the protein being an efflux gate in the cell membrane of E.coli k12, it does not contain specified binding pockets. However, it does have larger areas that compounds can bind to
Deep Mutational Scans
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
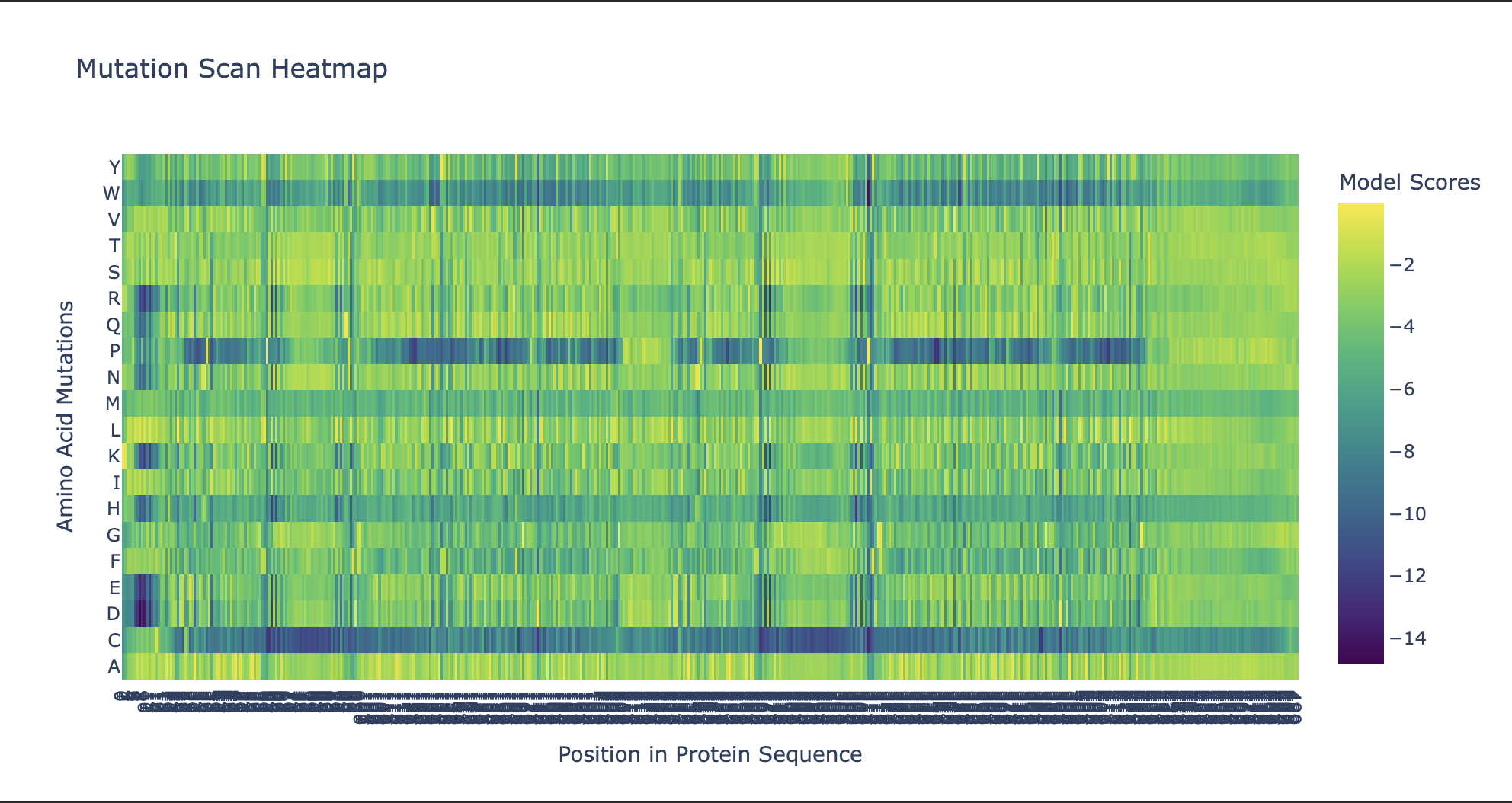
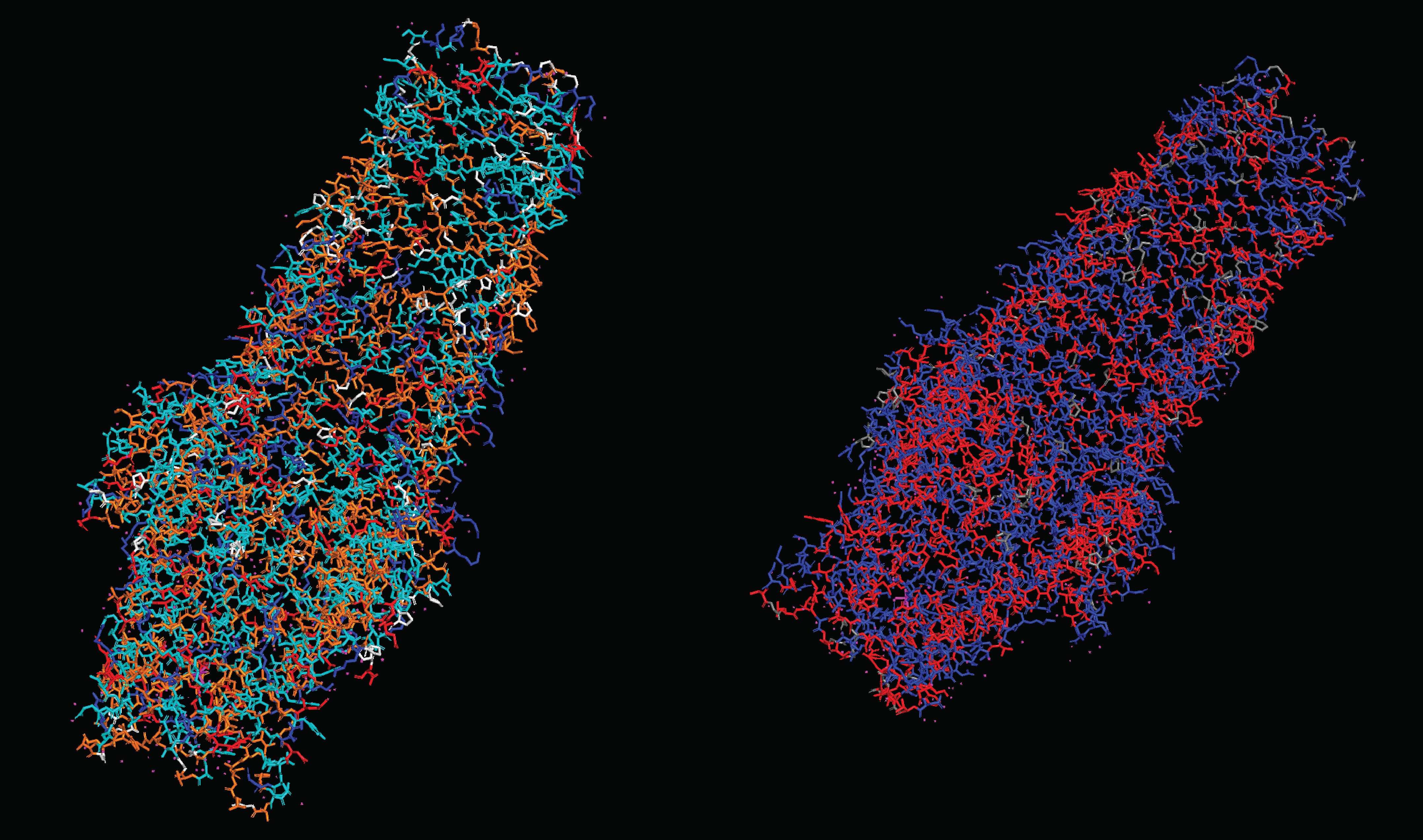
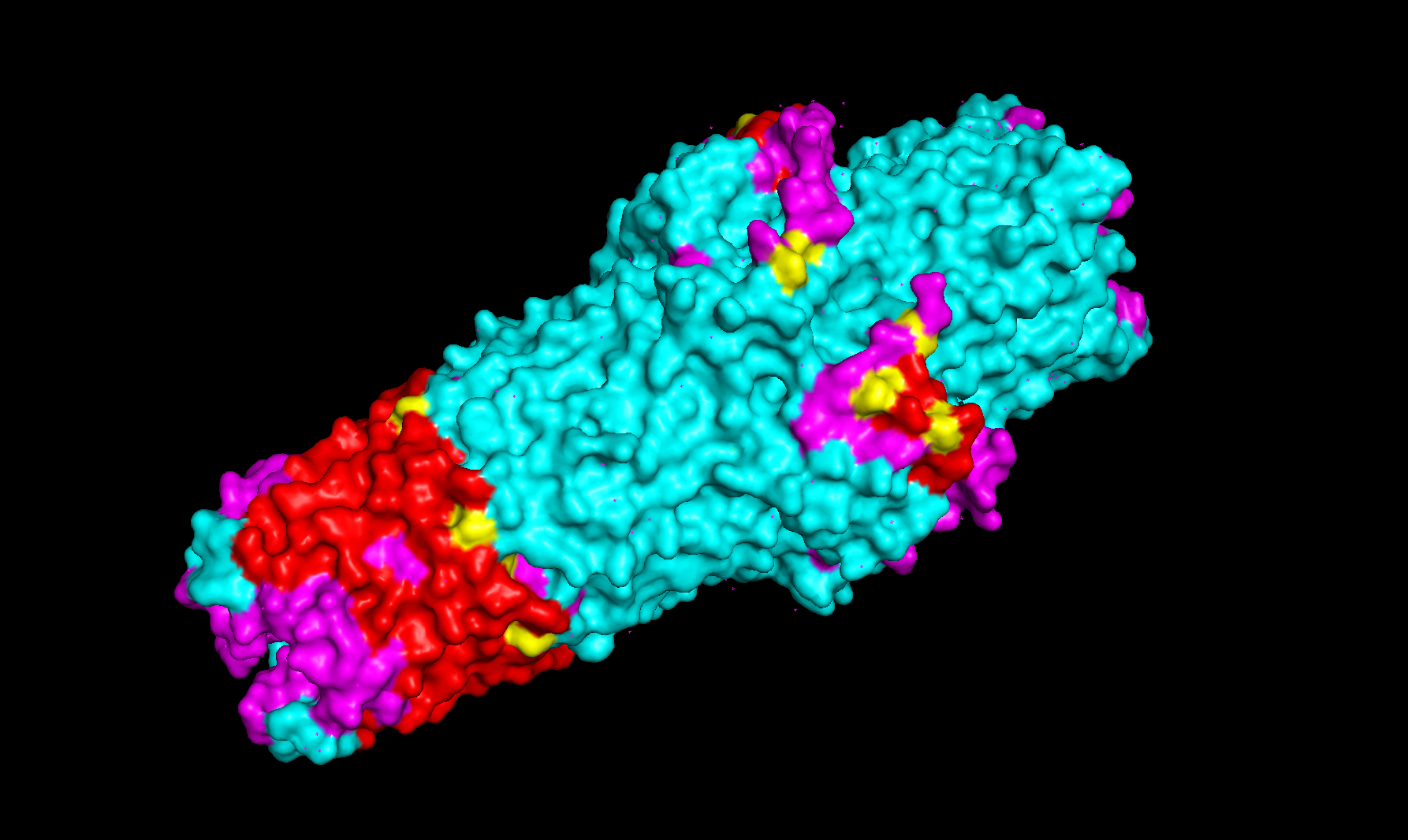
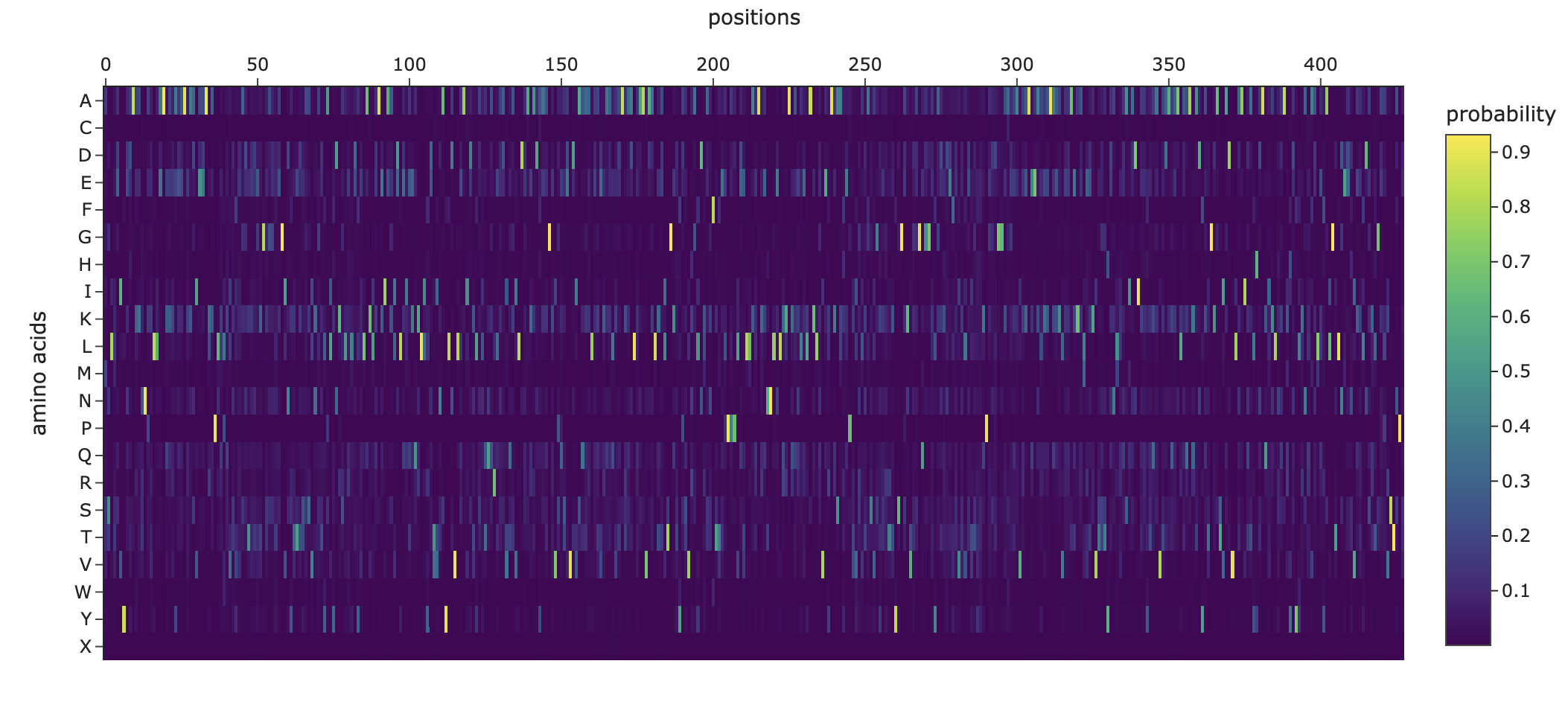
Noteworthy in the deep mutation analysis is Proline's presence seeming to be essential at positions 266 311 60 35 central to the sequence. Proline is known to induce bends, turns and loops to break alpha helices. Knowing what we know about tolC's structural composition as an efflux system within the cell membrane being made up of helices on one side and sheets on the other, this indicates that proline could be positioned at transitional points within the protein structure. When highlighting proline in our PyMol model with yellow, it can be seen positioned throughout the protein at transitional points between secondary structures thus confirming this notion.
Cysteine is strongly disfavoured at most positions in TolC because it is chemically reactive and can disrupt folding, disulfide patterns, and smooth channel function in the oxidizing periplasmic/extracellular environment. TolC’s role is to provide a robust, relatively passive exit duct for many substrates, so the protein is generally selected to avoid cysteine except in a few tolerant regions, rather than to create specific internal cysteine-based toxin binding pockets. The enrichment or tolerance of Cys at position 14 is more likely due to local structural flexibility or low functional constraint there, not a dedicated heavy-metal binding site.
2. Latent Space Analysis
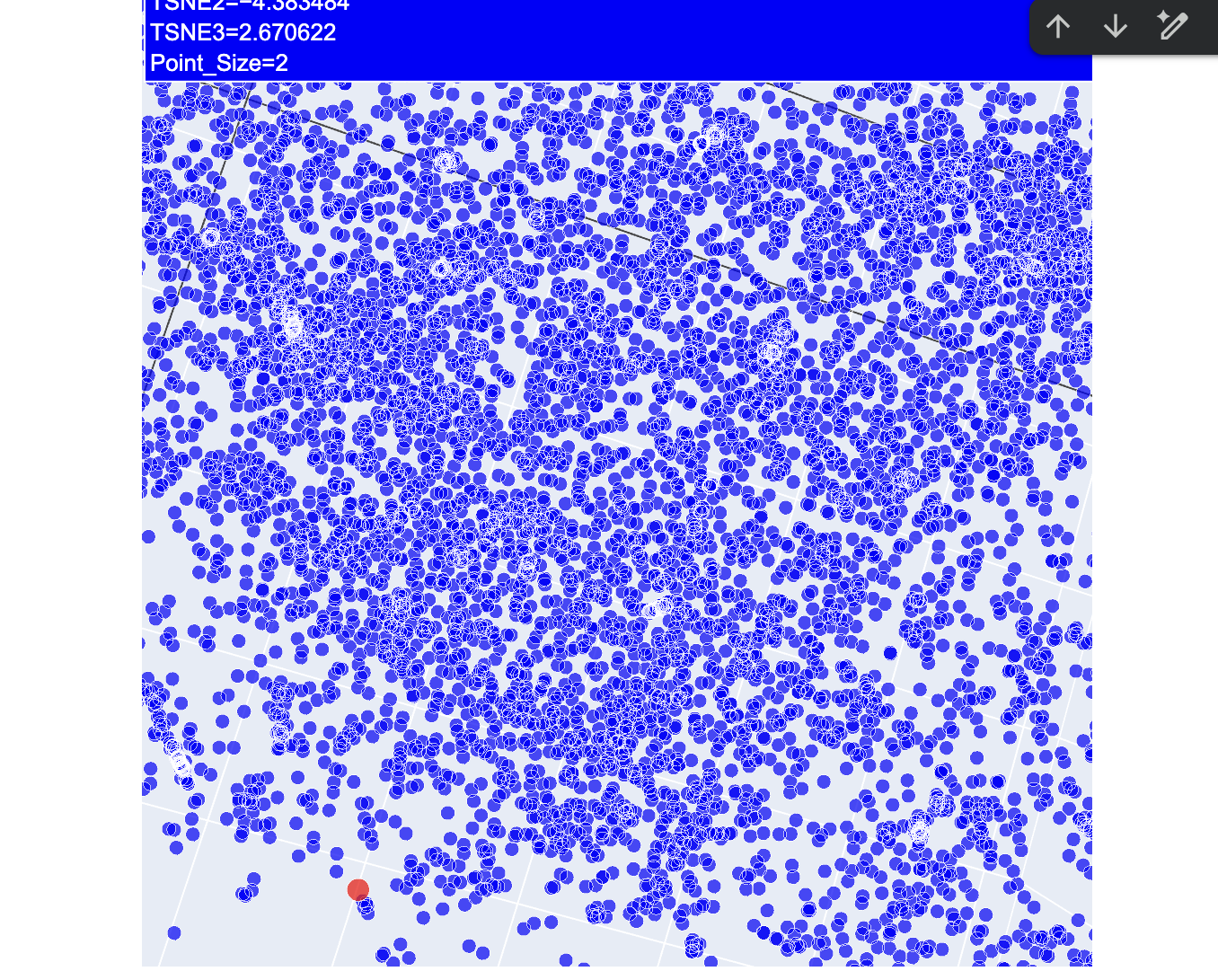
After a lot of trial and error, I finally got my protein secured in the latent space map highlighted in red.
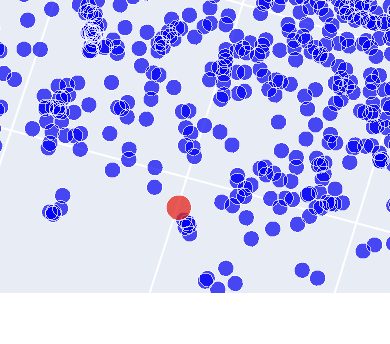
As shown on this image there is a number of proteins that are closely related. While further investigation denoted that two of these were likely to be tolC in e.coli itself, one of the others was derived from Vibrio cholerae, a bacterium that causes cholera in humans. The last one within the family was a protein derived from Pseudomonas aeruginosa. Considering all three of these are gram negative bacteria, it makes sense that these proteins are grouped together.
Folding a protein
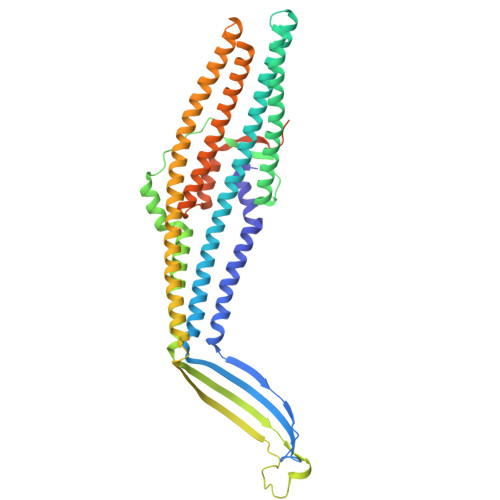
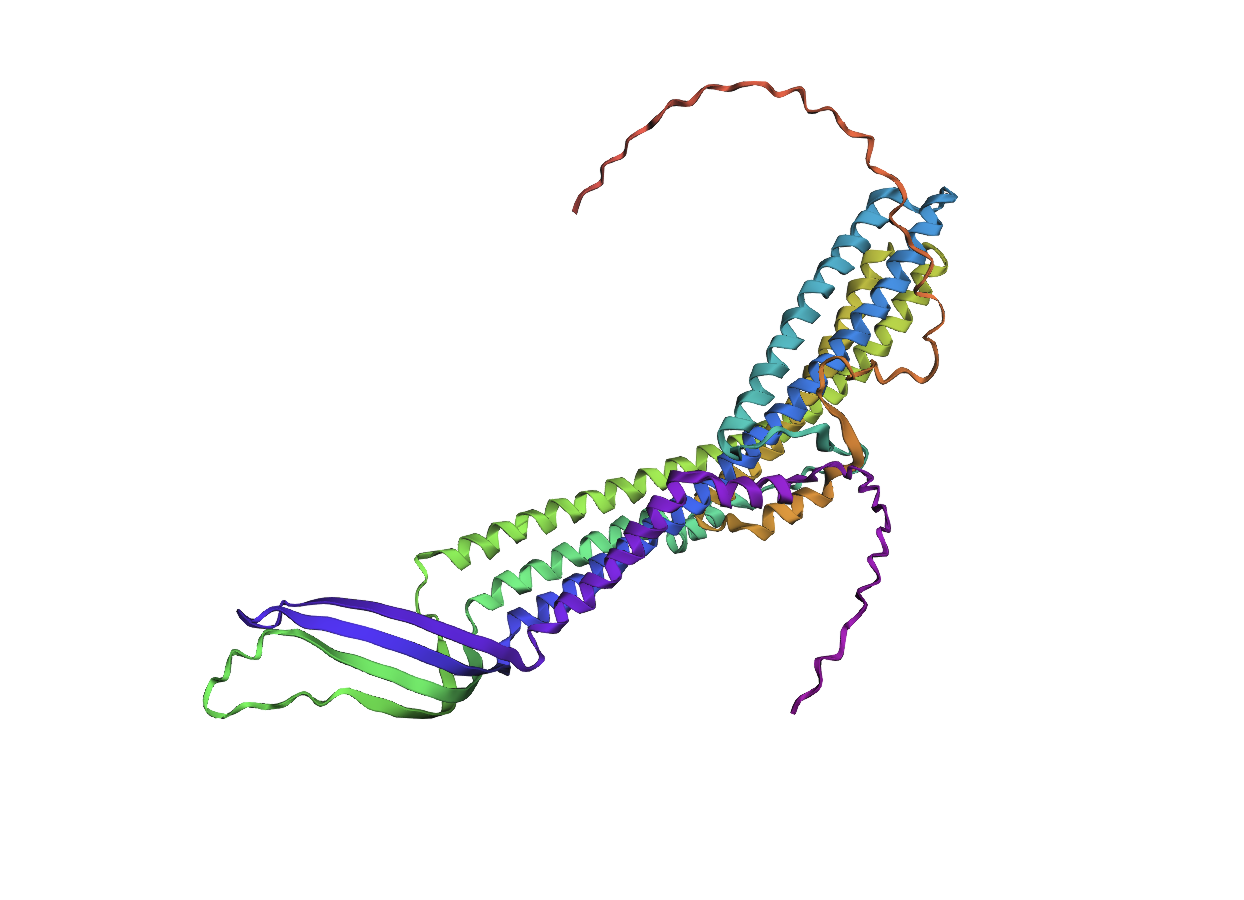
Comparing the ESMFold folded protein to the original structure, the predicted coordination seems to mostly match the original structure in terms of secondary structures. However, the placement of the helices in the ESMFold’ed protein seems to be much more tangled. Moreover, the edges of the protein seem to be completely out of place as well.
Based on prior inquiry into the importance of proline and the absence of cysteine in the sequence, I replaced each proline with a cystine in the sequence, resulting in the following fold:
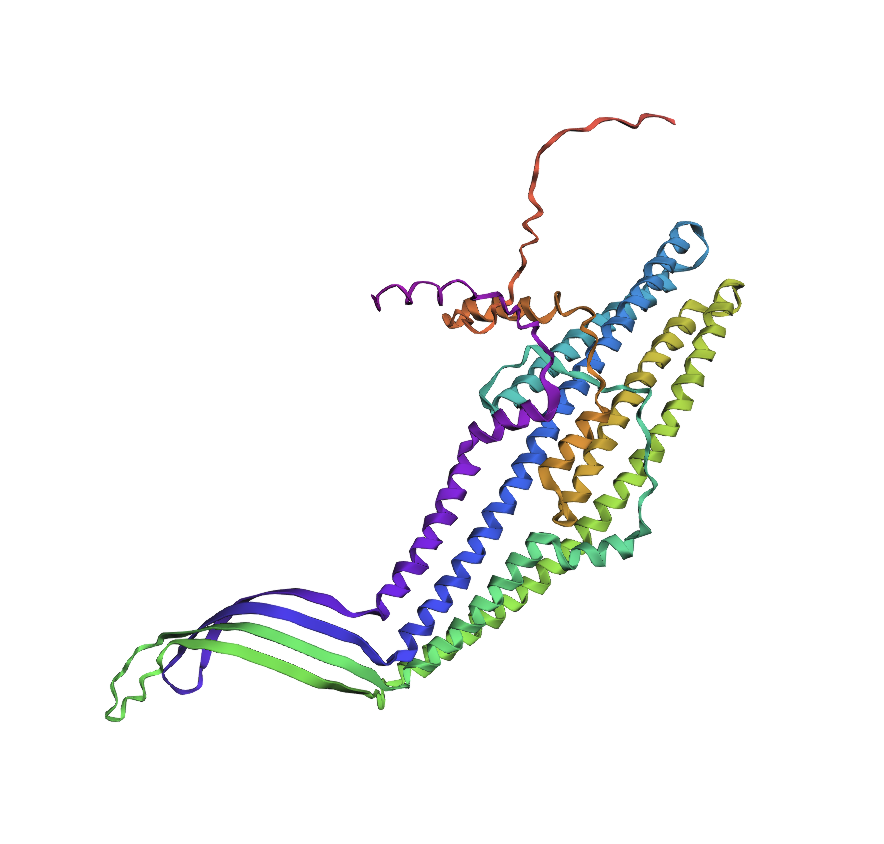
While the positioning and some of the placement of the alpha helices seems to be quite distorted, the effects are not as dramatic as I originally expected them to be.
This prompted me to try replacing another amino acid, in this case alanine with proline to see if this would result in drastically different formations. Once again, the protein seems to be misfolded, but not in a drastic manner that I was expecting:
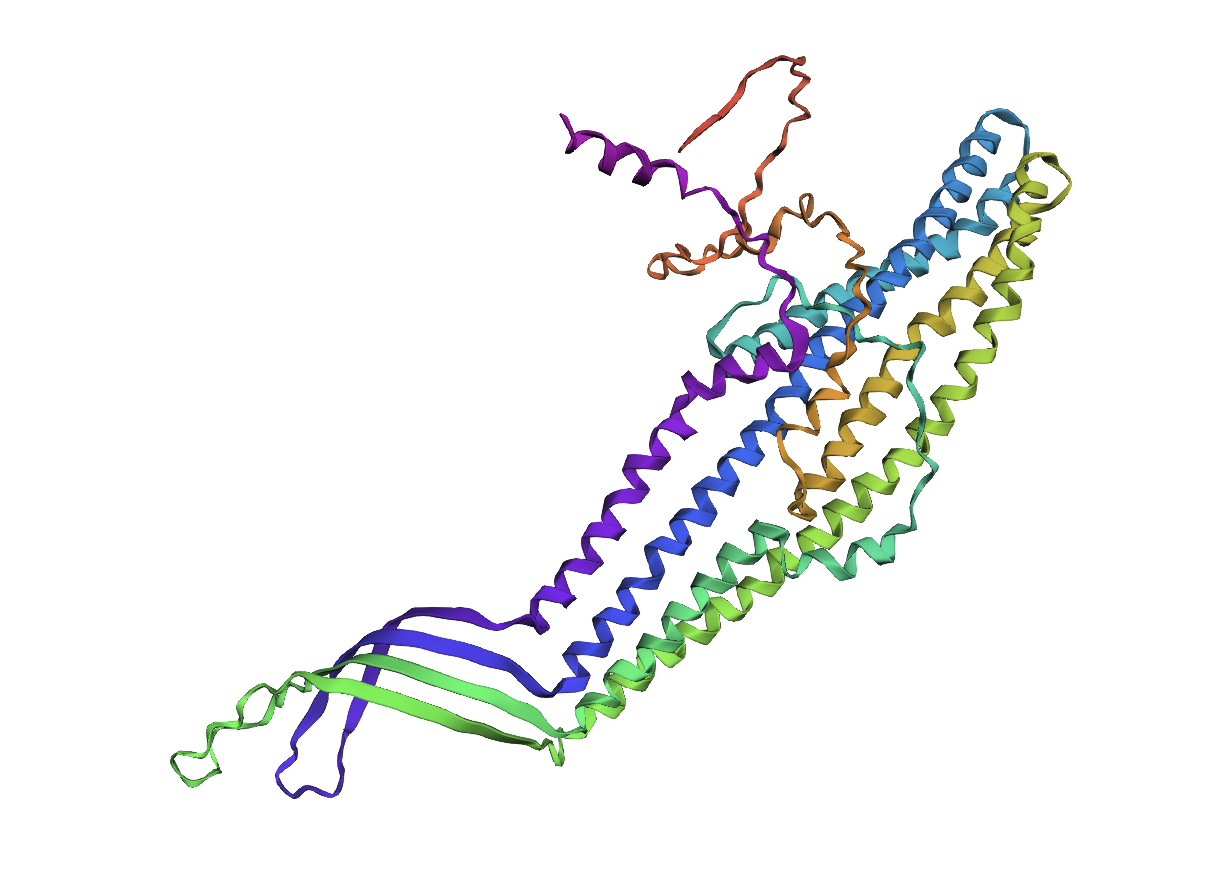
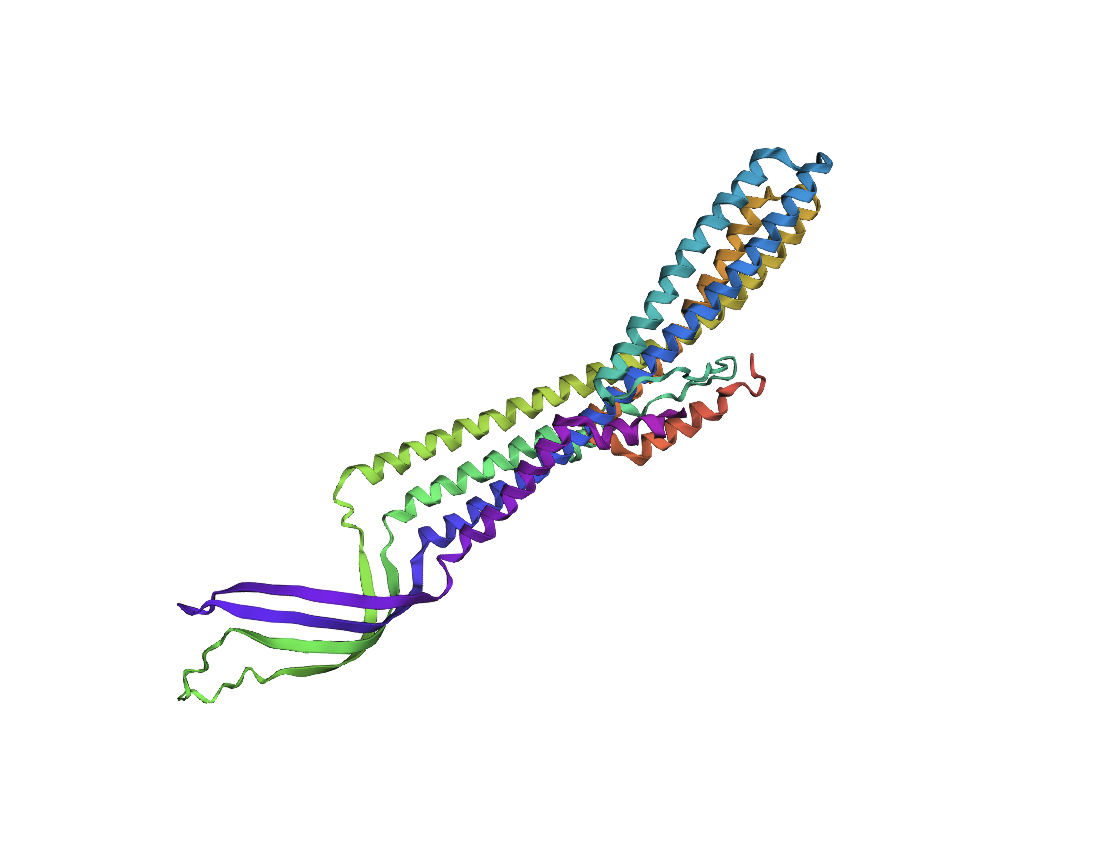
Lastly, I took a chunk in the middle of the sequence and replaced it with a random replacement sequence, which resulted in the following:
This seemed to drastically alter the structural integrity of the alpha helices within the protein.
Part D. Group Brainstorm on Bacteriophage Engineering
Group of Commited Listeners LifeFabs: Sara Gaviria Escobar, Ruben Janssen, Justine de Riedmatten
Proposal: Engineering the MS2 Lysis Protein L to Enhance Stability Background
The MS2 bacteriophage lysis (L) is a 75 amino acid long-protein, and it is responsible for triggering host cell lysis; this is why it is also called a toxin from the group of bacteriophages (Mezhyrova, 2023). It is a powerful protein that has been widely used in studies where researchers seek to control cell death, but it is difficult to do so due to its instability (Mylon, 2010).
Objectives
To use computational protein design tools to engineer possible stable variants of the MS2 L-protein. To identify variants where structure is preserved but higher stability is shown Analyse if the variants can fold like the original protein and if they interact correctly with DnaJ, its chaperone Methods
Obtain an initial protein backbone for MS2 L-protein using ESMFold.
Obtain alternative sequences to the protein using ProteinMPNN.
Mutate the alternative sequences to the protein using ESM-2.
Model the variants using ESMFold and analyse if the folding is maintained in comparison to the initial protein (MS2 L-protein). Create a ranking based on the confidence metric.s
Assess the top 3 variants’ interactions with DnaJ using AlphaFold-Multimer to predict 3D structures of protein complexes (co-folding multiple chains)
Expected Outcomes
Hopefully, these methods are able to identify some stabilized MS2 L-protein variants with correct folding and interaction with its chaperone, DnaJ. If successful, these designs could serve as templates for further experimental testing in E. coli and provide a methodology adaptable to other phage‑derived membrane proteins that also show decreased stability.
Potential Challenges
The main concern would be generating variants with correct folding, but incorrect interaction with DnaJ, as computational models sometimes can not predict the dynamics of those interactions (Chamakura et al., 2017 & Mondal et al., 2024), thus disrupting the lysis mechanism.
References
Mezhyrova, J., Martin, J., Börnsen, C., Dötsch, V., Frangakis, A. S., Morgner, N., & Bernhard, F. (2023). In vitro characterization of the phage lysis protein MS2-L. Microbiome Research Reports, 2(4), 28.
Mylon, S. E., Rinciog, C. I., Schmidt, N., Gutierrez, L., Wong, G. C., & Nguyen, T. H. (2010). Influence of salts and natural organic matter on the stability of bacteriophage MS2. Langmuir, 26(2), 1035-1042.
Chamakura, K. R., Tran, J. S., & Young, R. (2017). MS2 lysis of Escherichia coli depends on host chaperone DnaJ. Journal of Bacteriology, 199(12), 10-1128.
Mondal, A., Singh, B., Felkner, R. H., De Falco, A., Swapna, G. V. T., Montelione, G. T., … & Perez, A. (2024). A Computational Pipeline for Accurate Prioritization of Protein‐Protein Binding Candidates in High‐Throughput Protein Libraries.Angewandte Chemie International Edition, 63(24), e202405767.