Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
1. Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Original:
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS
AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV
HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Mutated:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS
AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV
HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
2. Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence, add the known SOD1-binding peptide FLYRWLPRSRRGG and record the perplexity scores of your generated binders.
Binder | Pseudo Perplexity |
WRYPVTVLRHKX | 11.329218109003818 |
WRYYAVVLEHWX | 14.262868870651067 |
WRYPVAALAHGX | 7.885580275805914 |
WLYYATGAAWKK | 15.833767982778536 |
FLYRWLPSRRGG |
The perplexity scores of my polypeptides are relatively low, as all except one are under 15. This indicates a high confidence in the generated samples.
3. Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
Binder | ipTM scores | Binding description |
WRYPVTVLRHKA | 0.46 | The polypeptide appears to bind to the length of the beta barrel, at approximately a third of the diameter of the beta barrel away from the N-terminus and dimer interface. It appears to be surface bound. |
WRYYAVVLEHWA | 0.19 | It seems to superficially bind to the protein at the furthest distance away from the beta-barrel, with low proximity to the N-terminus. Perhaps one end could be described as close to the dimer interface. |
WRYPVAALAHGA | 0.36 | The polypeptide seems to bind far away from the N-terminus and the dimer interface, it approaches the beta barrel region somewhat, but still keeps a relatively large distance from it as well. It appears to be surface bound. |
WLYYATGAAWKK | 0.26 | The polypeptide appears to superficially bind at the edge of the beta-barrel, with very low proximity to both the N-terminus and the dimer interface. |
FLYRWLPSRRGG | 0.31 | The polypeptide appears to be partially buried at the c-terminus, this terminus appears to be the closest part to the beta-barrel even though overall there seems to be very little interaction with that specific structure. There seems to be very low proximity to both the N-terminus and the dimer interface. |
4. In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
All of my ipTM are below the threshold of acceptable probability, the lowest being 0.19. However, two of my PepMLM-generated peptides do exceed the known binder’s score with a score of 0.36 and 0.46.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
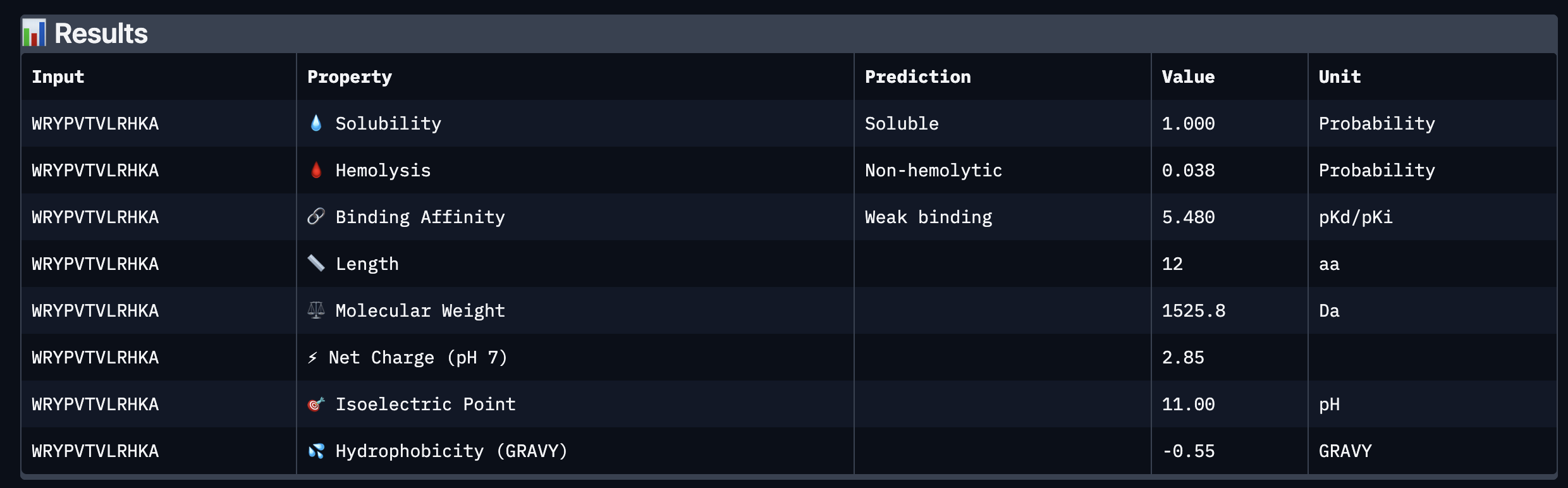
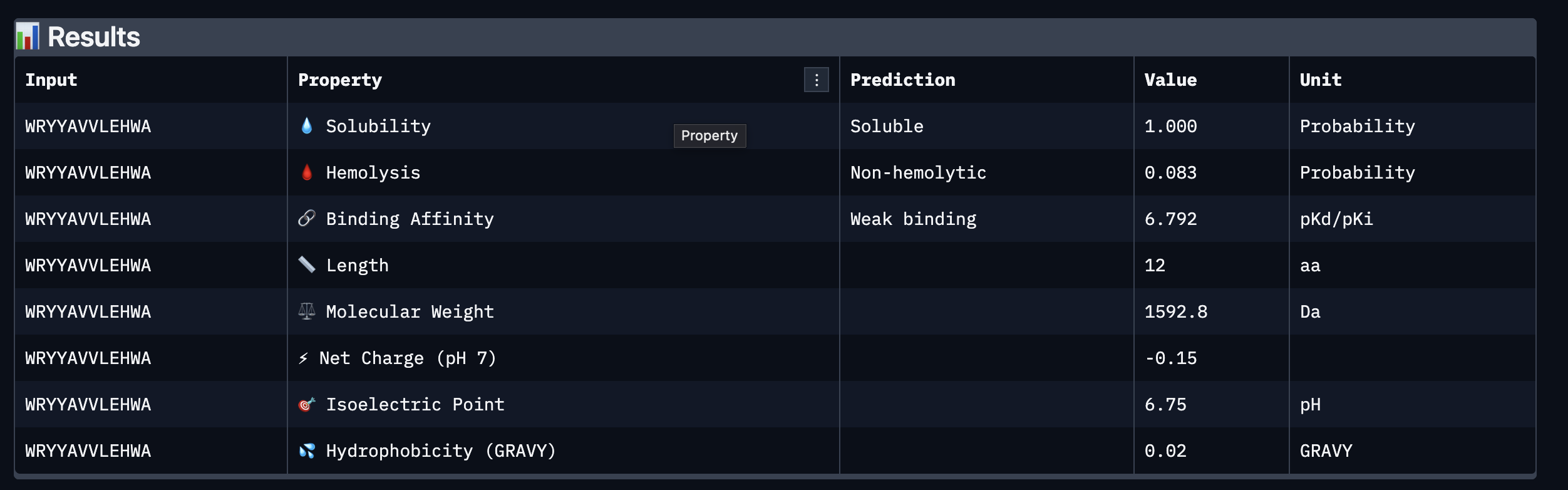
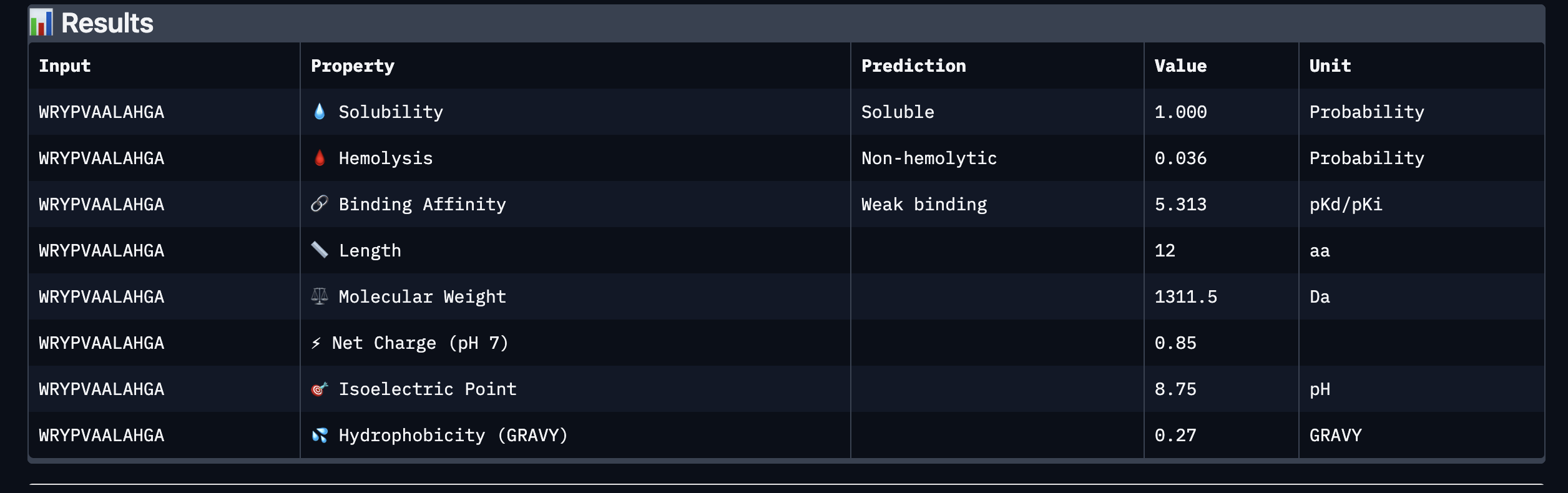
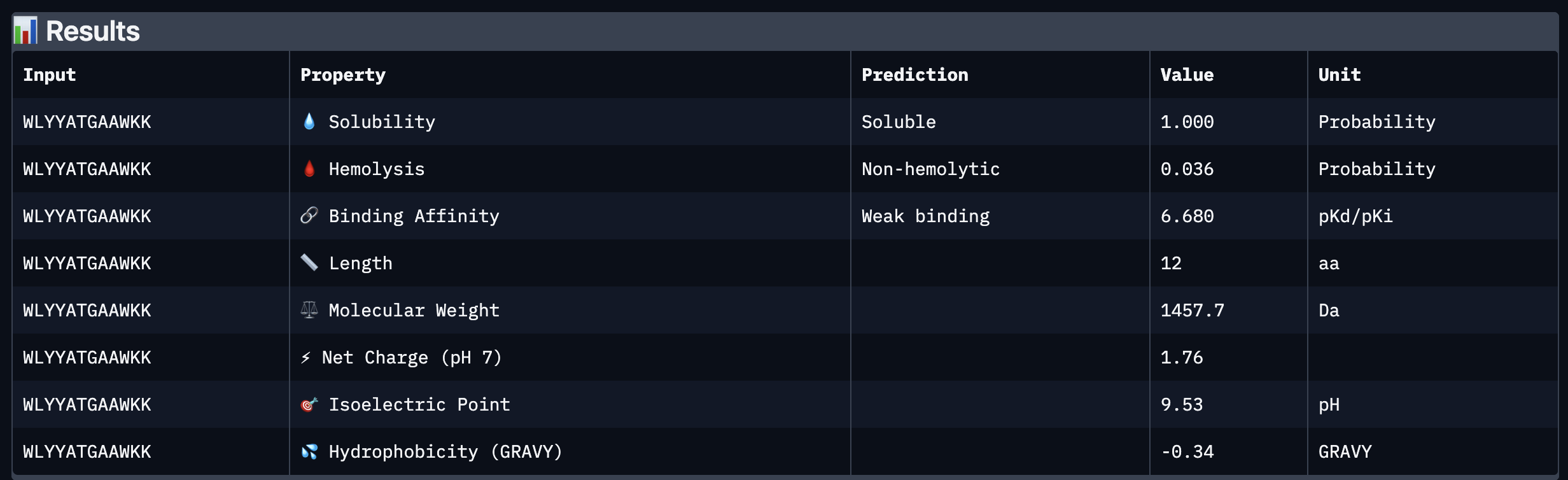
Based on the property prediction by PeptiVerse, it can be stated that all of the polypeptides are water soluble and non-hemolytic. These are highly necessary factors for potential application as they could prove to be detrimental if otherwise.
All polypeptides are predicted to be weak binding, which matches the ipTM scores of the AlphaFold models. This weak binding was also clearly seen in the visualisation of the polypeptides’ binding to the target protein within the AlphaFold models.
The molecular weight of all pp’s ranges between 1311 and 1592 Da, which is within a low range. This could indicate a lower aggregation rate, which would be good.
The SOD-1 protein is negatively charged (-6 per monomer) at physiological pH (7.4) (Shi, 2014), the binders have a small positive charge with the exclusion of WRYVAVVLEHWA having a minute negative charge of -0.15. A strongly positively charged binder would be more prone to non-specific binding to the protein and would result in a higher toxicity, therefore it is good that they are only slightly positively charged.
WRYPVTVLRHKA is the peptide that I would advance as it has the highest ipTM score, it also binds to the protein over the length of the beta-barrel. It has a very low hemolytic score of 0.038. It does however have a relatively weak binding score, however, so do all the other options.
Reference:
Shi Y, Abdolvahabi A, Shaw BF. Protein charge ladders reveal that the net charge of ALS-linked superoxide dismutase can be different in sign and magnitude from predicted values. Protein Sci. 2014 Oct;23(10):1417-33. doi: 10.1002/pro.2526. Epub 2014 Aug 7. PMID: 25052939; PMCID: PMC4287002.
Part C: Final Project: L-Protein Mutants
Information about Lysis protein in Bacteriophage MS2
Amino acid sequence of Lysis
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
DNA Sequence of Lysis
atggaaacccgctttccgcagcagagccagcagaccccggcgagcaccaaccgccgccgcccgtttaaacatgaagattatccgtgccgccgccagcagcgcagcagcaccctgtatgtgctgatttttctggcgatttttctgagcaaatttaccaaccagctgctgctgagcctgctggaagcggtgattcgcaccgtgaccaccctgcagcagctgctgacc
DNAj Sequence
tcgacgctga atttgaagaa gtcaaagaca aaaaataatc gccctataaa cgggtaatta 60
tactgacacg ggcgaagggg aatttcctct ccgcccgtgc attcatctag gggcaattta 120
aaaaagatgg ctaagcaaga ttattacgag attttaggcg tttccaaaac agcggaagag 180
cgtgaaatca gaaaggccta caaacgcctg gccatgaaat accacccgga ccgtaaccag 240
ggtgacaaag aggccgaggc gaaatttaaa gagatcaagg aagcttatga agttctgacc 300
gactcgcaaa aacgtgcggc atacgatcag tatggtcatg ctgcgtttga gcaaggtggc 360
atgggcggcg gcggttttgg cggcggcgca gacttcagcg atatttttgg tgacgttttc 420
ggcgatattt ttggcggcgg acgtggtcgt caacgtgcgg cgcgcggtgc tgatttacgc 480
tataacatgg agctcaccct cgaagaagct gtacgtggcg tgaccaaaga gatccgcatt 540
ccgactctgg aagagtgtga cgtttgccac ggtagcggtg caaaaccagg tacacagccg 600
cagacttgtc cgacctgtca tggttctggt caggtgcaga tgcgccaggg attcttcgct 660
gtacagcaga cctgtccaca ctgtcagggc cgcggtacgc tgatcaaaga tccgtgcaac 720
aaatgtcatg gtcatggtcg tgttgagcgc agcaaaacgc tgtccgttaa aatcccggca 780
ggggtggaca ctggagaccg catccgtctt gcgggcgaag gtgaagcggg cgagcatggc 840
gcaccggcag gcgatctgta cgttcaggtt caggttaaac agcacccgat tttcgagcgt 900
gaaggcaaca acctgtattg cgaagtcccg atcaacttcg ctatggcggc gctgggtggc 960
gaaatcgaag taccgaccct tgatggtcgc gtcaaactga aagtgcctgg cgaaacccag 1020
accggtaagc tattccgtat gcgcggtaaa ggcgtcaagt ctgtccgcgg tggcgcacag 1080
ggtgatttgc tgtgccgcgt tgtcgtcgaa acaccggtag gcctgaacga aaggcagaaa 1140
cagctgctgc aagagctgca agaaagcttc ggtggcccaa ccggcgagca caacagcccg 1200
cgctcaaaga gcttctttga tggtgtgaag aagttttttg acgacctgac ccgctaacct 1260
ccccaaaagc ctgcccgtgg gcaggcctgg gtaaaaatag ggtgcgttga agatatgcga 1320
gcacctgtaa agtggcgggg atcactccca taagcgct 1358
Conserved sites
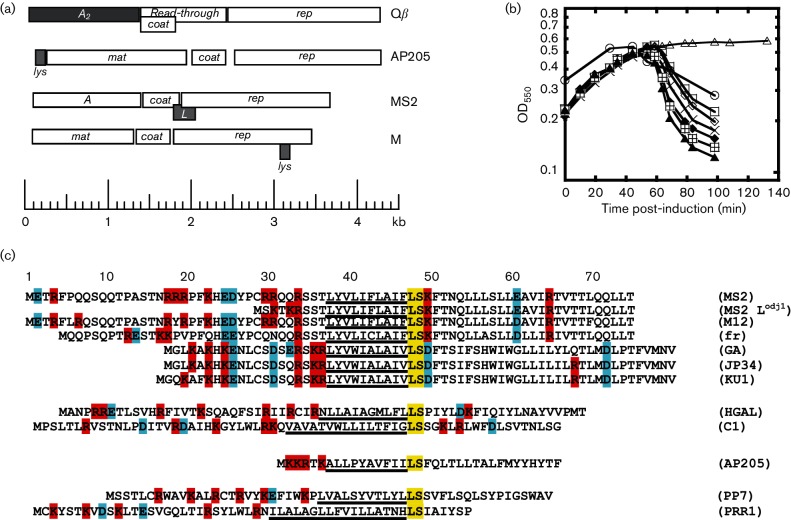
The provided article “Mutational analysis of the MS2 lysis protein L. Microbiology” describes how throughout homologs, position 48 and 49 are consistently conserved sites (LEU en SER). In the central domain there is a high conservation level across most homologs in position 38 39 40, (Leu, Tyr and Val) (Chamakura, 2017)
Known mutational effects from research.
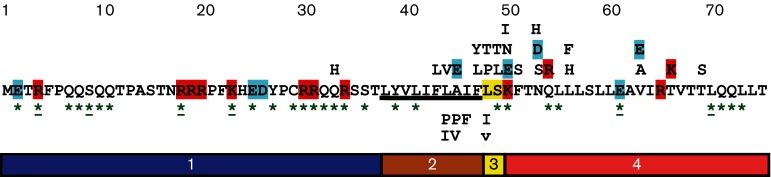
The above figure obtained from the aforementioned paper shows an overview of a mutational analysis of MS2 L. The analysis shows that certain missense mutations in the second domain do not affect lytic function (indicated below the L sequence). In the second, third and fourth domain, numerous mutations did negatively affect lysis function but did not result in protein accumulation (indicated above the L sequence). Green asterisks represent positions where a single nucleotide mutation would result in a nonsense mutation.
References:
Chamakura, K. R., Edwards, G. B., & Young, R. (2017). Mutational analysis of the MS2 lysis protein L. Microbiology (Reading, England), 163(7), 961–969. https://doi.org/10.1099/mic.0.000485
My selected approach to determining sequence variations is cross referencing the provided experimental data on L-protein mutants with generated point mutations and accompanying success scores from the provided Protein Language Models.
As the assignment dictates, 2 mutations should be located in the soluble N-terminal domain (position 1 - 39) and 2 in the transmembrane domain ( position 40 - 75)
Top 10 of colab
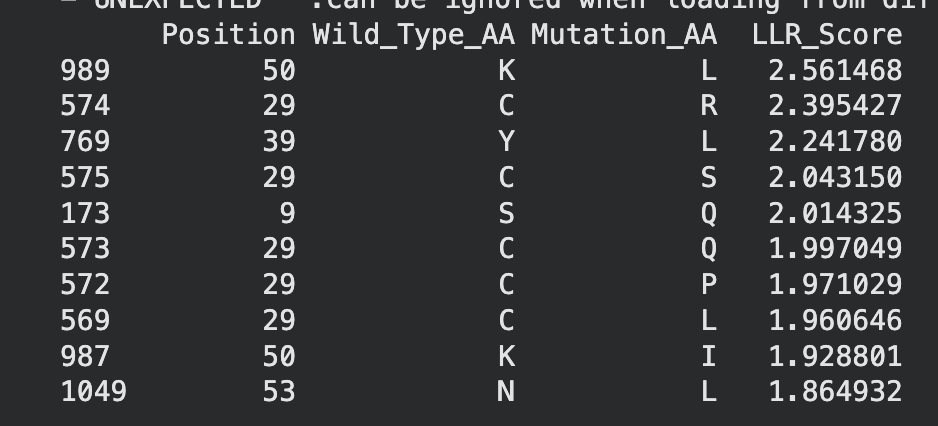
When crossreferencing this list with the original experimental mutations, two mutations, namely C29R and K50I appeared to result in neither lysis or protein formation. Therefore they will be excluded from potential mutation candidates.
This leaves the following mutations:
K50L - While this mutation has the highest LLR score, further research indicates that Lysine (K) and Leucine (L) are structurally very different residues. This could indicate the potential for problematic protein formation. However, due to its high score it should still be considered.
Y39L - Tyrosine and Leucine
S9Q - Serine and glutamine
C29Q - Cystine and glutamine
C29P - Cystine and proline
C29L - cystine and leucine
N53L - Asparagine and leucine
I asked Perplexity to compare the structural properties of the wild-type amino acids to their mutated amino acids.
Mutation | Amino Acids | Structural Similarity? | Reason |
Y39L | Tyrosine → Leucine | No | Aromatic ring vs. branched alkyl chain; different bulk and polarity. www2.chemistry.msu |
S9Q | Serine → Glutamine | Low | Short hydroxyl vs. longer amide; size/volume mismatch despite some polarity. benchchem |
C29Q | Cysteine → Glutamine | No | Thiol (-SH) vs. amide; polarity similar but chain length and reactivity differ. wikipedia |
C29P | Cysteine → Proline | No | Linear thiol vs. rigid cyclic; proline disrupts helices unlike cysteine. youtubeweb02.gonzaga |
C29L | Cysteine → Leucine | No | Small polar thiol vs. large hydrophobic branch; major size/chemistry shift. www2.chemistry.msu |
N53L | Asparagine → Leucine | No | Polar amide vs. hydrophobic branch; polarity and H-bonding lost. www2.chemistry.msu |
However, I will continue the assignment with the following five mutations:
K50L
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSLFTNQLLLSLLEAVIRTVTTLQQLLT
N53L
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTLQLLLSLLEAVIRTVTTLQQLLT
S9Q
METRFPQQQQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Y39L
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLLVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
C29L
METRFPQQSQQTPASTNRRRPFKHEDYPLRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
