Week 6 HW: Genetic Circuits Part I
Assignment: DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion High-Fidelity PCR Master Mix contains Phusion DNA Polymerase, nucleotides, and optimized reaction buffer including MgCl2.
The DNA Polymerase synthesizes new strands of DNA by assembling nucleotides based on a template strand, the nucleotides are necessary for assembly and the optimized reaction buffers assure variable factors are optimised for PCR.
What are some factors that determine primer annealing temperature during PCR?
The main factor that determines primer annealing during PCR is the melting temperature of the primer.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Polymerase Chain Reaction (PCR) is a widely used, highly sensitive laboratory technique invented by Kary Mullis in 1983 to amplify specific DNA sequences, producing millions to billions of copies in just a few hours. UPCR is a nucleic acid amplification technique involving denaturation, annealing, extension, and amplification of short DNA or RNA segments. The reaction employs DNA polymerase derived from Thermus aquaticus, known as Taq polymerase. Using a thermal cycler, DNA polymerase (like Taq polymerase), and primers, it enables rapid diagnosis of diseases, forensic analysis, and genetic research.
The thermostability of Taq polymerase preserves the physical and chemical integrity of nucleic acids throughout repeated high-temperature cycles, making it suitable for the PCR-based detection of bacterial and viral pathogens and for screening genetic disorders.
During denaturation, prepared samples are heated to 94 to 96 degrees Celsius for 30 seconds, this separates the double stranded DNA in single strands. Next, during annealing the sample temperature is lowered to 50 to 65 degrees Celsius for 30 seconds. This allows for the primers (short, single-stranded nucleic acid sequences (oligonucleotide) that serves as a necessary starting point for DNA synthesis and replication) to bind to the target sites on the ssDNA. Then the samples are heated to 72 degrees for at least another 30 seconds. This allows for the Taq polymerase to bind to the primers and extend a new DNA strand onto the ssDNA.
The cycle is then restarted.
Restriction enzyme digests use bacterial enzymes to cut DNA at specific sites. Restriction enzymes recognize short, palindromic DNA sequences (typically 4-8 base pairs) and cleave the phosphodiester backbone. Type II enzymes, most common in labs, cut within or near the recognition site, producing either blunt ends (straight cuts) or sticky ends (overhanging single strands that facilitate ligation).
PCR is a simple method to replicate DNA sequences exponentially in a short amount of time with a large variety of applications from diagnostics to genetics research. Restriction enzyme digests are used to cut (out) specific strands of DNA that would for example be used for genetic engineering. Relationally, RED could be done to then perform PCR cloning, this would allow one to isolate a sequence and then amplify it for further
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
An important step to ensure the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning is purifying your samples. This would remove any contaminants, such as reagents from prior steps, that interfere with further processing.
Moreover, one could perform gel electrophoresis to establish that the target sequence length is present in the sample.
How does the plasmid DNA enter the E. coli cells during transformation?
There are two methods through which plasmid DNA enters E.coli cells during transformation:
Chemical competence method - E. coli cells are grown to log phase, chilled on ice, and treated with cold calcium chloride (CaCl₂), which binds to the cell membrane's phospholipids and DNA phosphates. This neutralises charges, making the negatively charged plasmid DNA less repelled by the membrane. The DNA-cell mix is then heat-shocked (42°C for 30-90 seconds), temporarily fluidising the membrane to form pores that let the plasmid slip inside; cells are quickly iced again to reseal the membrane.
Reference:
Froger, A., & Hall, J. E. (2007). Transformation of plasmid DNA into E. coli using the heat shock method. Journal of visualized experiments : JoVE, (6), 253. https://doi.org/10.3791/253
Electroporation
Cells are mixed with DNA and zapped with a high-voltage pulse (e.g., 2.5 kV), creating transient membrane holes via dielectric breakdown. The electric field drives charged DNA into the cell before pores close.
Biology LibreTexts (2021) Transforming E. coli. Available at: https://bio.libretexts.org/Bookshelves/Biotechnology/Lab_Manual:_Introduction_to_Biotechnology/01:_Techniques/1.13:_Transformation
Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Model this assembly method with Benchling or Asimov Kernel!
Golden Gate Assembly is a modular DNA cloning method for joining multiple fragments in a precise, scarless manner. It employs Type IIS restriction enzymes like BsaI, which cut outside their recognition sites to produce custom 4-base sticky overhangs. These overhangs are designed via PCR primers or pre-made parts to ensure fragments only ligate in the intended order. A one-pot reaction cycles between enzyme digestion at 37°C and T4 ligase joining at 16°C, self-correcting mismatches. Domesticated sequences—lacking internal cut sites—prevent unwanted breaks, resulting in clean final constructs without residual enzyme motifs. This approach outperforms traditional cloning by enabling efficient, hierarchical assembly of complex genetic circuits.
Bird, J.E. et al. (2022) 'A User's Guide to Golden Gate Cloning Methods and Standards', ACS Synthetic Biology, 11(12), pp. 3895–3909. Available at: https://pubs.acs.org/doi/10.1021/acssynbio.2c00355
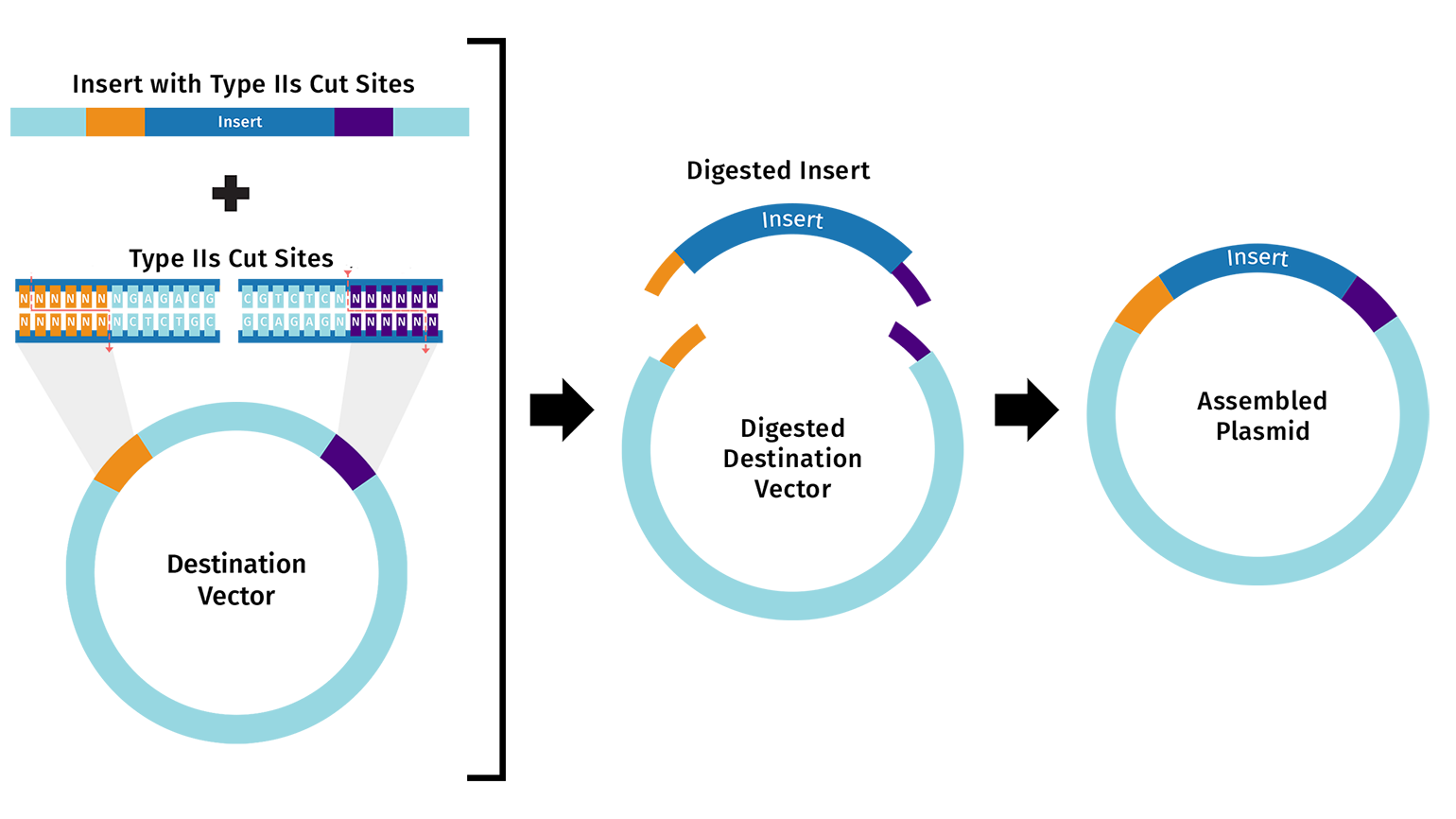
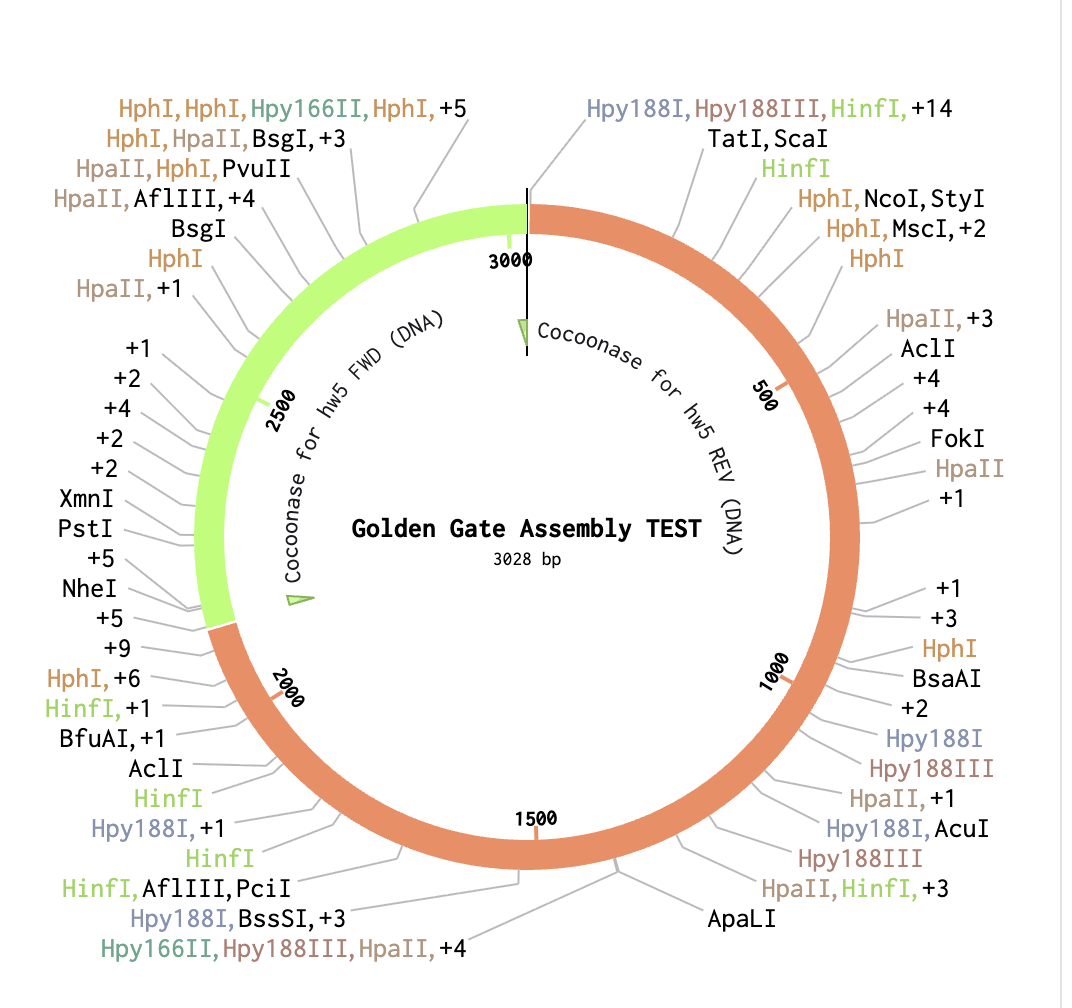
I chose to model Golden Gate Assembly using Benchling.
First, I looked up a suitable backbone vector. Through quick research I found that for golden gate assembly has domesticated vectors like pGGA which I ended up selecting for this exercise. I then opened the sequence in Benchling.
From a previous homework assignment I selected the sequence for cocoonase as my insert sequence.
With both tabs open, I started the assembly wizard for Golden Gate Assembly.
As a backbone I selected pGGA, with BsaI cut sites. For the Insert selection, I selected the entire cocoonase sequence and pressed insert. This resulted in the following circular sequence: