from opentrons import types
import math
metadata = { # see https://docs.opentrons.com/v2/tutorial.html#tutorial-metadata
'author': 'Saba Saeed',
'protocolName': 'Opentrons Bacteriophage Structural Artwork',
'description': '',
'source': 'HTGAA 2026 Opentrons Lab',
'apiLevel': '2.20'
}
##############################################################################
### Robot deck setup constants - don't change these
##############################################################################
TIP_RACK_DECK_SLOT = 9
COLORS_DECK_SLOT = 6
AGAR_DECK_SLOT = 5
PIPETTE_STARTING_TIP_WELL = 'A1'
well_colors = {
'A1' : 'Red',
'B1' : 'Yellow',
'C1' : 'Green',
'D1' : 'Cyan',
'E1' : 'Blue'
}
def run(protocol):
##############################################################################
### Load labware, modules and pipettes
##############################################################################
tips_20ul = protocol.load_labware(
'opentrons_96_tiprack_20ul',
TIP_RACK_DECK_SLOT,
'Opentrons 20uL Tips'
)
pipette_20ul = protocol.load_instrument(
"p20_single_gen2",
"right",
[tips_20ul]
)
temperature_module = protocol.load_module(
'temperature module gen2',
COLORS_DECK_SLOT
)
temperature_plate = temperature_module.load_labware(
'opentrons_96_aluminumblock_generic_pcr_strip_200ul',
'Cold Plate'
)
color_plate = temperature_plate
agar_plate = protocol.load_labware(
'htgaa_agar_plate',
AGAR_DECK_SLOT,
'Agar Plate'
)
center_location = agar_plate['A1'].top()
pipette_20ul.starting_tip = tips_20ul.well(PIPETTE_STARTING_TIP_WELL)
##############################################################################
### Helper functions for this lab
##############################################################################
def location_of_color(color_string):
for well, color in well_colors.items():
if color.lower() == color_string.lower():
return color_plate[well]
raise ValueError(f"No well found with color {color_string}")
def dispense_and_detach(pipette, volume, location):
above_location = location.move(types.Point(z=location.point.z + 5))
pipette.move_to(above_location)
pipette.dispense(volume, location)
pipette.move_to(above_location)
##############################################################################
### Patterning & Auto-Scaling
##############################################################################
# Raw GUI coordinate arrays
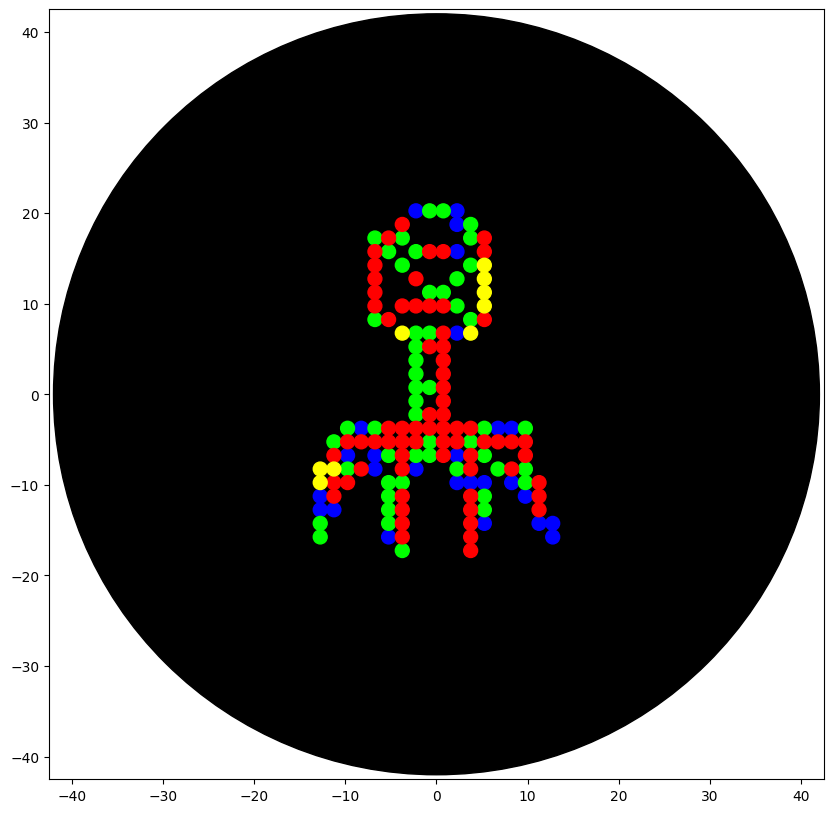
azurite_points = [
(-2.25,20.25),(2.25,20.25),(2.25,18.75),(2.25,15.75),(2.25,6.75),
(-8.25,-3.75),(6.75,-3.75),(8.25,-3.75),(-9.75,-6.75),(-6.75,-6.75),
(2.25,-6.75),(-6.75,-8.25),(-2.25,-8.25),(2.25,-9.75),(3.75,-9.75),
(5.25,-9.75),(8.25,-9.75),(-12.75,-11.25),(9.75,-11.25),
(-12.75,-12.75),(-11.25,-12.75),(5.25,-14.25),(11.25,-14.25),
(12.75,-14.25),(-5.25,-15.75),(12.75,-15.75)
]
mclover3_points = [
(-0.75,20.25),(0.75,20.25),(3.75,18.75),(-6.75,17.25),(-3.75,17.25),
(3.75,17.25),(-5.25,15.75),(-2.25,15.75),(-3.75,14.25),(3.75,14.25),
(2.25,12.75),(-0.75,11.25),(0.75,11.25),(2.25,9.75),(-6.75,8.25),
(3.75,8.25),(-2.25,6.75),(-0.75,6.75),(-2.25,5.25),(-2.25,3.75),
(-2.25,2.25),(-2.25,0.75),(-0.75,0.75),(-2.25,-0.75),(-2.25,-2.25),
(-9.75,-3.75),(-6.75,-3.75),(5.25,-3.75),(9.75,-3.75),
(-11.25,-5.25),(-0.75,-5.25),(3.75,-5.25),(-5.25,-6.75),
(-2.25,-6.75),(-0.75,-6.75),(5.25,-6.75),(-9.75,-8.25),
(2.25,-8.25),(6.75,-8.25),(9.75,-8.25),(-5.25,-9.75),
(-3.75,-9.75),(9.75,-9.75),(-5.25,-11.25),(5.25,-11.25),
(-5.25,-12.75),(5.25,-12.75),(-12.75,-14.25),(-5.25,-14.25),
(-12.75,-15.75),(-3.75,-17.25)
]
mrfp1_points = [
(-3.75,18.75),(-5.25,17.25),(5.25,17.25),(-6.75,15.75),
(-0.75,15.75),(0.75,15.75),(5.25,15.75),(-6.75,14.25),
(-6.75,12.75),(-2.25,12.75),(-6.75,11.25),(-6.75,9.75),
(-3.75,9.75),(-2.25,9.75),(-0.75,9.75),(0.75,9.75),
(-5.25,8.25),(5.25,8.25),(0.75,6.75),(-0.75,5.25),
(0.75,5.25),(0.75,3.75),(0.75,2.25),(0.75,0.75),
(0.75,-0.75),(-0.75,-2.25),(0.75,-2.25),(-5.25,-3.75),
(-3.75,-3.75),(-2.25,-3.75),(-0.75,-3.75),(0.75,-3.75),
(2.25,-3.75),(3.75,-3.75),(-9.75,-5.25),(-8.25,-5.25),
(-6.75,-5.25),(-5.25,-5.25),(-3.75,-5.25),(-2.25,-5.25),
(0.75,-5.25),(2.25,-5.25),(5.25,-5.25),(6.75,-5.25),
(8.25,-5.25),(9.75,-5.25),(-11.25,-6.75),(-3.75,-6.75),
(0.75,-6.75),(3.75,-6.75),(9.75,-6.75),(-8.25,-8.25),
(-3.75,-8.25),(3.75,-8.25),(8.25,-8.25),(-11.25,-9.75),
(-9.75,-9.75),(11.25,-9.75),(-11.25,-11.25),(-3.75,-11.25),
(3.75,-11.25),(11.25,-11.25),(-3.75,-12.75),(3.75,-12.75),
(11.25,-12.75),(-3.75,-14.25),(3.75,-14.25),
(-3.75,-15.75),(3.75,-15.75),(3.75,-17.25)
]
sfgfp_points = [
(5.25,14.25),(5.25,12.75),(5.25,11.25),(5.25,9.75),
(-3.75,6.75),(3.75,6.75),(-12.75,-8.25),
(-11.25,-8.25),(-12.75,-9.75)
]
# Color grouping
color_groups = [
('Blue', azurite_points),
('Green', mclover3_points),
('Red', mrfp1_points),
('Yellow', sfgfp_points)
]
# Auto-scaling logic
all_points = azurite_points + mclover3_points + mrfp1_points + sfgfp_points
max_r = 0
for x, y in all_points:
r = math.sqrt(x**2 + y**2)
if r > max_r:
max_r = r
scale_factor = 38.0 / max_r if max_r > 38.0 else 1.0
# Physical compilation loop
for color_name, points in color_groups:
if not points:
continue
pipette_20ul.pick_up_tip()
for i, (x, y) in enumerate(points):
if i % 20 == 0:
drops_remaining = len(points) - i
volume_to_aspirate = min(20, drops_remaining)
pipette_20ul.aspirate(
volume_to_aspirate,
location_of_color(color_name)
)
scaled_x = x * scale_factor
scaled_y = y * scale_factor
adjusted_location = center_location.move(
types.Point(x=scaled_x, y=scaled_y)
)
dispense_and_detach(
pipette_20ul,
1,
adjusted_location
)
pipette_20ul.drop_tip()