Week 2 HW: Read, write & edit
Homework Week 2
Part 1: Benchling & In-silico Gel Art
Import the Lambda DNA. Simulate Restriction Enzyme Digestion with the following Enzymes:
EcoRI
HindIII
BamHI
KpnI
EcoRV
SacI
SalI
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
You might find Ronan’s website a helpful tool for quickly iterating on designs!
Playing around with the digest enzymes

.png)
Getting an “S”, well…sort of:

Part 3: DNA Design Challenge
3.1. Choose your protein.: In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose. [Example from our group homework, you may notice the particular format — The example below came from UniProt]
I have chosen Tumor Necrosis Factor- Alpha (TNF-α).
Why:
Reasons for choosing this protein include my interest in dermatology and chronic diseases. It is a key inflammatory cytokine in many skin and insulin resistant conditions. I am interested in psoriasis, particularly plaque psoriasis and its relation to insulin resistance and diabetes [1]. This is because this is something my Mum has suffered from the last couple of years, recently developing some pre-diabetes.
Protein Sequence:
NP_000585.2 tumor necrosis factor [Homo sapiens] MSTESMIRDVELAEEALPKKTGGPQGSRRCLFLSLFSFLIVAGATTLFCLLHFGVIGPQREEFPRDLSLI SPLAQAVRSSSRTPSDKPVAHVVANPQAEGQLQWLNRRANALLANGVELRDNQLVVPSEGLYLIYSQVLF KGQGCPSTHVLLTHTISRIAVSYQTKVNLLSAIKSPCQRETPEGAEAKPWYEPIYLGGVFQLEKGDRLSA EINRPDYLDFAESGQVYFGIIAL
References
- Moller DE. Potential role of TNF-α in the pathogenesis of insulin resistance and type 2 diabetes. Trends Endocrinol Metab. 2000 Aug;11(6):212-217. doi:10.1016/S1043-2760(00)00272-1.
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
To obtain the nucleotide sequence encoding TNF-α, I retrieved the validated human mRNA record (NCBI RefSeq: NM_000594.4) from NCBI. From this record, I extracted the coding sequence (CDS), which corresponds to the protein sequence NP_000585.2. Only the CDS was used for downstream codon optimization. See below:
ATGAGCACTGAAAGCATGATCCGGGACGTGGAGCTGGCCGAGGAGGCGCTCCCCAAGAAGACAGGGGGGCCCCAGGGCTCCAGGCGGTGCTTGTTCCTCAGCCTCTTCTCCTTCCTGATCGTGGCAGGCGCCACCACGCTCTTCTGCCTGCTGCACTTTGGAGTGATCGGCCCCAGAGGGAAGAGTTCCCCAGGGACCTCTCTCTAATCAGCCCTCTGGCCCAGGCAGTCAGATCATCTTCTCGAACCCCGAGTGACAAGCCTGTAGCCCATGTTGTAGCAAACCCTCAAGCTGAGGGGCAGCTCCAGTGGCTGAACCGCCGGGCCAATGCCCTCCTGGCCAATGGCGTGGAGCTGAGAGATAACCAGCTGGTGGTGCCATCAGAGGGCCTGTACCTCATCTACTCCCAGGTCCTCTTCAAGGGCCAAGGCTGCCCCTCCACCCATGTGCTCCTCACCCACACCATCAGCCGCATCGCCGTCTCCTACCAGACCAAGGTCAACCTCCTCTCTGCCATCAAGAGCCCCTGCCAGAGGGAGACCCCAGAGGGGGCTGAGGCCAAGCCCTGGTATGAGCCCATCTATCTGGGAGGGGTCTTCCAGCTGGAGAAGGGTGACCGACTCAGCGCTGAGATCAATCGGCCCGACTATCTCGACTTTGCCGAGTCTGGGCAGGTCTACTTTGGGATCATTGCCCTGTGA
3.3. Codon optimization. Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
For codon optimization, I chose the online codon optimizing tool:
https://en.vectorbuilder.com/tool/codon-optimization.html
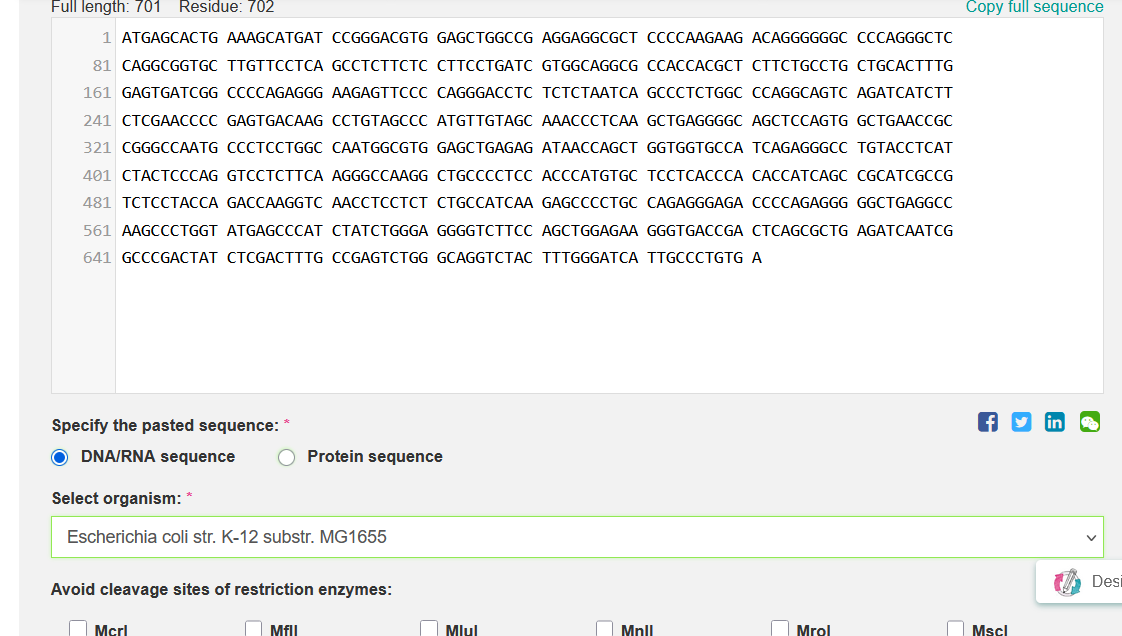
From my input:
I got: Pasted Sequence: GC=59.84%, CAI=0.49
From my output:
Improved DNA[1]: GC=59.97%, CAI=0.92
For CAI (Codon Adaptation Index), this indicates strong expected expression.
For GC content, after optimization it remained near 60%, within a suitable range for Escherichia coli, supporting stable and efficient gene synthesis.
I selected Escherichia coli strain K-12 MG1655 as the target organism for codon optimization because it is a well-studied laboratory strain with a completely sequenced and annotated genome [1–2].
Codon Optimized TNF-Alpha for improved expression of Escherichia coli
CTGAGCCCGTTCAACAACCCGCTGCTGCGCCCGTTTCTGATTCTGTATGAACATTAAAAACATGATCCGGGCCGTGGCGCAGGTCGCGGCGGCGCGCCGCAGGAAGATCGTGGCGCACCGGGCTTACAGGCCGTGCTGGTTCCGCAGCCGCTGCTGCTGCCGGATCGCGGCCGTCGTCACCATGCCCTGCTGCCGGCGGCCCTGTGGTCGGATCGTCCGCAGCGTGAAGAATTTCCGCGCGATCTGAGCCTGATTAGCCCGCTGGCGCAGGCCGTGCGTAGCAGCAGCCGCACCCCGTCAGATAAACCGGTGGCGCACGTGGTGGCAAATCCGCAGGCCGAAGGTCAGCTGCAGTGGCTGAATCGTCGCGCGAATGCCCTGTTAGCCAATGGTGTGGAACTGCGCGATAATCAGCTGGTGGTGCCGTCAGAAGGTCTGTACCTGATCTATTCGCAGGTGCTGTTTAAAGGCCAGGGCTGTCCGAGCACCCATGTGCTGCTGACCCACACCATTAGCCGCATTGCGGTGAGCTACCAGACCAAAGTGAACCTGCTTTCTGCGATTAAAAGCCCGTGCCAGCGTGAAACCCCGGAAGGCGCGGAAGCGAAACCGTGGTACGAACCGATTTATCTGGGCGGCGTGTTCCAGCTGGAAAAAGGCGATCGTCTGAGCGCGGAAATTAATCGCCCGGATTATCTGGATTTTGCGGAAAGCGGTCAGGTGTATTTCGGCATTATTGCCTTGTAA
References
Lukjancenko O, Wassenaar TM, Ussery DW. Comparison of 61 sequenced Escherichia coli genomes. Microb Ecol. 2010 Nov;60(4):708-20. doi:10.1007/s00248-010-9717-3. PMID:20623278; PMCID:PMC2974192.
Yannai A, Katz S, Hershberg R. The codon usage of lowly expressed genes is subject to natural selection. Genome Biol Evol. 2018 May;10(5):1237–46. doi:10.1093/gbe/evy084.
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
After codon optimizing the TNF- α DNA sequence, it can be used to produce protein either through cell-dependent or cell-free systems.
For cell-dependent systems, the DNA will first need to be cloned using and inserted into an expression vector, this is then introduced into live host cells such as E. coli or eukaryotic cells, where cellular machinery transcribes the DNA into mRNA and then translates the mRNA into TNF‑α protein during growth and metabolism; this is seen in standard biotechnology production processes [1–2].
For cell-free systems, crude cell extracts provide all the machinery for transcription, translation, protein folding, and energy metabolism [3]. Therefore, when the codon optimized DNA is added, the TNF‑α protein will be produced in-vitro and under controlled conditions.
Both these methods rely on the flow of information from DNA to mRNA to protein; the Central Dogma of Molecular Biology.
References
Lukjancenko O, Wassenaar TM, Ussery DW. Comparison of 61 sequenced Escherichia coli genomes. Microb Ecol. 2010 Nov;60(4):708-20. doi: 10.1007/s00248-010-9717-3. Epub 2010 Jul 11. PMID: 20623278; PMCID: PMC2974192.
Swartz JR. Advances in Escherichia coli production of therapeutic proteins. Curr Opin Biotechnol. 2001 Oct;12(5):195–201. doi:10.1016/s0958-1669(00)00198-5. PMID:11513436.
Carlson ED, Gan R, Hodgman CE, Jewett MC. Cell-free protein synthesis: Applications come of age. Biotechnol Adv. 2012 Sep-Oct;30(5):1185-94. doi:10.1016/j.biotechadv.2011.09.016. PMID:22001003; PMCID:PMC3359644.
3.5. [Optional] How does it work in nature/biological systems?
1. Describe how a single gene codes for multiple proteins at the transcriptional level. 2. Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!! See example below. [Example shows the biomolecular flow in central dogma from DNA to RNA to Protein] Special note that all “T” were transcribed into “U” and that the 3-nt codon represents 1-AA.
Part 4: Prepare a Twist DNA Synthesis Order
4.2. Build Your DNA Insert Sequence
Link to the sequence (first attempt):
https://benchling.com/s/seq-92QKTmxOZ4NOBZloFYXH?m=slm-ih8RIVqVkxJpGYbdm50f
Link to corrected sequence:
https://benchling.com/s/seq-AKpYnuHnRmdf5XnJxSv8?m=slm-sqc6y4bFyGTTvcXYx3Q9
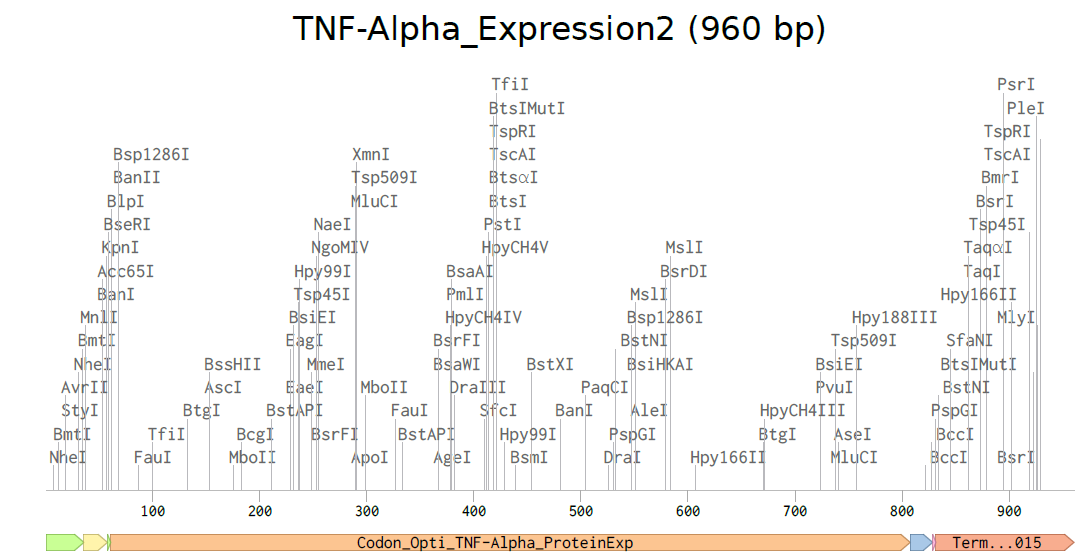
4.3-4.5. Building Expression Cassette and Plasmid
Plasmid with Expression Cassette
https://benchling.com/s/seq-dx10o3kwSJPyNLgmJDGo?m=slm-V5wHDO0G8ZxGTwWVp2A7
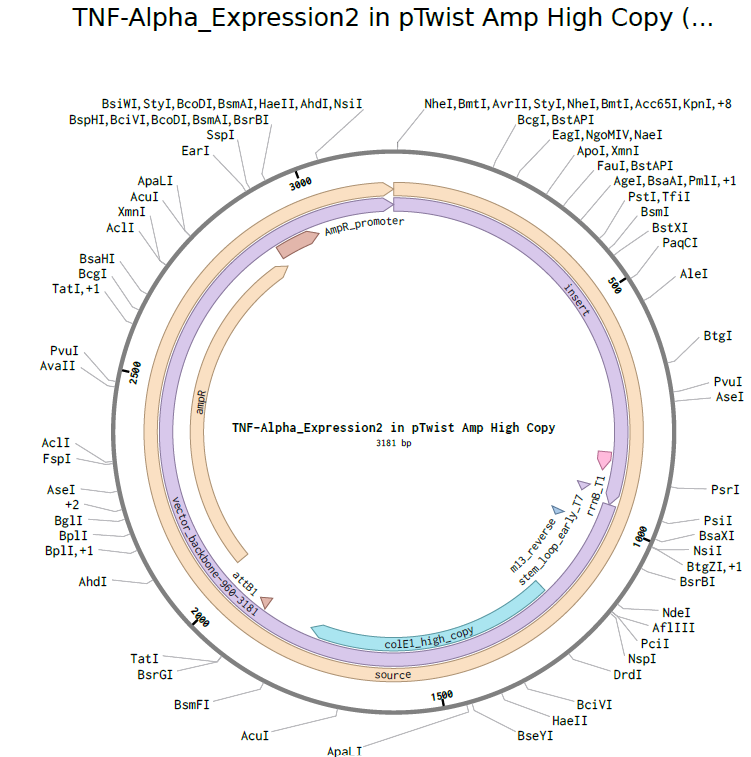
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I would want to sequence Mycobacterium Tuberculosis DNA. I would like to focus on virulence‑associated loci such as phoR, mymA and the mce1, and lineage defining SNPs, such as rpoB, katG, inhA promoter, gyrA, embB.
To integrate with surveillance, I would potentially try to store drug resistance and mutation outputs from my detection bio-tool into a DNA-based archive. This could help build a long-term genomic repository.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why? Also answer the following questions: 1. Is your method first-, second- or third-generation or other? How so? 2. What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps. 3. What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)? 4. What is the output of your chosen sequencing technology?
To perform sequencing for the drug-resistant DNA, short-read sequencing is ideal for identifying the key resistance driving genes for profiling and analysis. In contrast, long-read sequencing (e.g. Oxford Nanopore) would make rapid detection, which is useful in high-burden regions, but has slightly lower accuracy. Therefore, short-read sequencing is ideal for identifying key resistance-driving genes for profiling and analysis (for e.g. using Illumina) [1]. It involves DNA extraction, fragmentation, adapter ligation, cluster amplification, and sequencing by synthesis, with base-calling software decoding the sequence from fluorescent signals. The output includes high-quality short reads, aligned sequences, and variant calls for resistance and lineage analysis. In contrast, long-read sequencing enables rapid detection in high-burden regions but has slightly lower accuracy and may require deeper coverage.
References
- The CRyPTIC Consortium and the 100,000 Genomes Project. Prediction of Susceptibility to First-Line Tuberculosis Drugs by DNA Sequencing. N Engl J Med. 2018;379:1403–1415. doi:10.1056/NEJMoa1800474.
5.2 DNA Read
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
I would like to design a genetic circuit that could be integrated into a microbial chassis or a cell-free system, which would enable it to detect molecular signatures for key multi-drug /extra-drug-resistant tuberculosis and activates a fluorescent reporter when present in a sample. Examples of this have been seen in research that looks at how biosensors are used to detect heavy metal in water through recombinase-based logic gates [1]. Such CRISPR‑based detection systems can be programmed with guides targeting lineage‑specific SNPs (e.g., Beijing/East Asian, Indo-American) [2] alongside resistance mutations so that the circuit only activates a fluorescent reporter when both types of signatures are present. Potentially, CRISPR‑Cas12/13 coupled with allele‑specific amplification can discriminate single‑base changes for lineage and resistance detection with high specificity. There is also a possibility of integrating all of this into a microfluidic biosensor, enabling automated, low-volume, rapid, and multiplexed detection suitable for environmental and point-of-care surveillance [3].
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why? Also answer the following questions:
- Is your method first-, second- or third-generation or other? How so?
- What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
- What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
- What is the output of your chosen sequencing technology?
For this synthesis, I would use synthetic DNA platforms and include CRISPR guide sequences, promoters, and fluorescent reporter proteins. These technologies would allow for quick prototyping, flexibility with design and would allow for automated printers to synthesize sequences up to multiple kilobases accurately.
Essential steps would include: full sequence of nucleotides and CRISPR guides, promoters and reporter proteins; setting the oligonucleotide assembly, this includes making assemblies of short oligos through PCR or ligation. These would need to be further tested and validated to ensure proper functioning of the circuit.
Limitations include, time, fixing errors, and scaling the device. These large constructs and may take time due to the complexity associated with multiple variants.
References
Mathur S, Singh D, Ranjan R. Genetic circuits in microbial biosensors for heavy metal detection in soil and water. Biochem Biophys Res Commun. 2023 Apr 16;652:131–137. doi:10.1016/j.bbrc.2023.02.031.
Napier, G., Campino, S., Merid, Y. et al. Robust barcoding and identification of Mycobacterium tuberculosis lineages for epidemiological and clinical studies. BMC Genome Med 12, 114 (2020). https://doi.org/10.1186/s13073-020-00817-3
Didarian R, Azar MT. Microfluidic biosensors: revolutionizing detection in DNA analysis, cellular analysis, and pathogen detection. Biomed Microdevices. 2025;27:10. doi:10.1007/s10544-025-00741-6.
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
For editing, I would use CRISPR-Cas systems to introduce lineage specific SNPs and resistant mutations into safe mycobacterial strains or cell-free systems [1]. This allow me to test the genetic circuit, validate the CRISPR guides, and generate controls for MDR-TB detection.
(ii) What technology or technologies would you use to perform these DNA edits and why? Also answer the following questions:
- How does your technology of choice edit DNA? What are the essential steps?
- What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
- What are the limitations of your editing methods (if any) in terms of efficiency or precision?
Essential steps would include: designing guide RNAs to target SNPs/loci related to drug resistance; integrating of editing components into cells or a cell-free platform; functional testing to ensure sequences properly activate the fluorescent reporter protein within the circuit.
Preparation would require designing the guide RNAs and providing either a cell-free system or microbial framework as the host.
Limitations include: possible off-target edits; increased complexity when introducing multiple edits or larger constructs, which can affect throughput and precision.
References
- Molla KA, Yang Y. CRISPR/Cas mediated base editing: technical considerations and practical applications. Trends Biotechnol. 2019 Oct;37(10):1121–1142. doi:10.1016/j.tibtech.2019.03.008. Review of CRISPR base editing systems and how they introduce precise nucleotide changes without double strand breaks.