Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Assuming the meat is a red meat like beef, there would be approximately 20-25g of protein per 100g of meat [1, 2].
So, taking the upper end of that range for 500g:
500g x 0.25 = 125g
Given on average 1 amino acid ≈ 100 Daltons, then 1g/mol ≈ 1 Dalton
Therefore,
125 Daltons ≈ 125g/mol
Converting grams to moles:
Moles = mass/molar mass = 125g/125g/mol = 1 mole of amino acids
Converting moles to molecules using Avogadro’s constant [3]:
1 mole ≈ 6.02214076 x 1023 ≈ 6.02 x 1023
University Hospitals Sussex NHS Foundation Trust. Protein fact sheet [Internet]. West Sussex (UK): University Hospitals Sussex NHS Foundation Trust; [cited 2026 Mar 1]. Available from: https://www.uhsussex.nhs.uk/resources/protein-fact-sheet/
Nuffield Health. Best high protein foods [Internet]. Epsom (UK): Nuffield Health; [cited 2026 Mar 1]. Available from: https://www.nuffieldhealth.com/article/best-high-protein-foods
Metric System. Avogadro constant [Internet]. 2024 [cited 2026 Mar 1]. Available from: https://metricsystem.net/si/defining-constants/avogadro-constant/
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
When we digest meat or fish, we are breaking them down into their basic constituents, which include amino acids. These amino acids are further used by ribosomes (through DNA instruction) to build human proteins, not that of a cow or fish.
3. Why are there only 20 natural amino acids?
The standard 20 amino acids were formed through evolutionary pressures which selected the acids based on folding capabilities, catalysis, and molecular recognition. These were most likely adopted in pre-biotic conditions through early metabolism/pre-biotic chemistry [1]. Once incorporated into the genetic code, it got fixed given other additions may have created disruptions towards survival.
Exceptions include Pyrrolysine and Selenocysteine, which are naturally occurring amino acids incorporated into proteins via specialized mechanisms; with pyrrolysine encoded by UAG stop codon in certain areas using dedicated tRNA and biosynthetic enzymes, and selenocysteine inserted at UGA codons with a Selenocysteine Insertion Sequence (SECIS) in the mRNA.
- Doig AJ. Frozen, but no accident – why the 20 standard amino acids were selected. FEBS Lett. 2016 Dec 7;590(21):3977–3985. doi:10.1111/febs.13982. Available from: https://doi.org/10.1111/febs.13982
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes, it is possible to make non-natural amino acids as well as incorporate them into proteins using engineered tRNA synthase pairs with reassigned codons [1]. Initially will choose a base amino acid modify side chain to add new function Synthesize and by introducing protein with engineered tRNA, so that the amino acid can be recognized insert in specific codon.
To design a new amino acid, I would modify non‑natural amino acid is para‑azido‑L‑phenylalanine (pAzF), which contains an azide (‑N₃) group. When pAzF is genetically incorporated into a protein at a chosen site, the azide can act as a chemical handle attaching a fluorescent dye or imaging agent to that protein. This can help label or track proteins in cells and animals [2].
Bag SS, Saraogi I, Guo J. Editorial: Expansion of the Genetic Code: Unnatural Amino Acids and their Applications. Front Chem. 2022;10:958433. doi:10.3389/fchem.2022.958433.
Lightle HE, Kafley P, Lewis TR, Wang R. Site‑specific protein conjugates incorporating para‑azido‑L‑phenylalanine for cellular and in vivo imaging. Methods. 2023;219:95–101. doi:10.1016/j.ymeth.2023.10.001
5. Where did amino acids come from before enzymes that make them, and before life started?
The origins of amino acids are hypothesized to have emerged from primordial earth [1], and have undergone abiotic synthesis under early environmental conditions (such as electrical discharges and impact‑driven reactions during the Hadean Eon) before life existed; over time, as organisms evolved in the Archean and Proterozoic Eons, they developed enzyme‑mediated biosynthetic pathways to produce amino acids internally, eventually supporting the diversity of life seen in the three domains of Archaea, Bacteria, and Eukarya.
- Nature Education. **An evolutionary perspective on amino acids. Nature Scitable. 2014. Available from: https://www.nature.com/scitable/topicpage/an-evolutionary-perspective-on-amino-acids-14568445
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
If an α-helix is made from D-amino acids instead of L-amino acids, it would form a left-handed helix [1]. In natural proteins, L-amino acids are used, and they form right-handed α-helices. Therefore, the helix built from D-amino acids reverse that twist to make it left-handed.
Perlego. Alpha helix. Perlego Chemistry Index. Available from: https://www.perlego.com/index/chemistry/alpha-helix OpenAI. ChatGPT (version 5.2)
7. Can you discover additional helices in proteins?
Yes, it is possible to have additional helices in proteins, both natural and artificially designed. This includes the 310 helix which is a secondary structure found in proteins and polypeptides.
Another is the pi/π helix, which is a secondary structure found in proteins.
8. Why are most molecular helices right-handed?
9. Why do β-sheets tend to aggregate?
β-sheets tend to aggregate due to their structure as they have exposed edges with available hydrogen bonding groups [1]. This leaves it susceptible to interactions with other β-sheets.
o What is the driving force for β-sheet aggregation?
The intermolecular backbone formed from β-sheet aggregation from hydrogen bonds forming between the backbone groups. Once aligned, hydrophobic side-chain interactions and van der Waals forces between tightly packed residues further stabilize the β-sheet aggregates [1].
- Richardson JS, Richardson DC. Natural β-sheet proteins use negative design to avoid edge-to-edge aggregationProc Natl Acad Sci U S A. 2002 Mar 5;99(5):2754–9. doi:10.1073/pnas.052706099.
10. Why do many amyloid diseases form β-sheets?
Give β-sheets allow for extensive intermolecular backbones, it enables multiple proteins to stick together. For example, in Alzheimer’s disease, amyloid-β peptides misfold and aggregate into fibrils that are rich in β-sheet structure. These facilitate plaque formation in the brain [1].
o Can you use amyloid β-sheets as materials?
Amyloid β-sheets can be used as materials because their cross-β structure forms highly stable, self-assembling nanofibers. These properties allow them to be developed into biomaterials such as hydrogels and nanofibers.
- Ow SY, Dunstan DE. A brief overview of amyloids and Alzheimer’s disease. Protein Sci. 2014 Oct;23(10):1315–31. doi:10.1002/pro.2524.
Part B. Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
1. Briefly describe the protein you selected and why you selected it.
I selected the Sonic Hedgehog protein. One, because when I was a massive Sonic the Hedgehog fan. Later as I started studying and working, I became interested in biology, neuroscience, and mental health. I found out that the protein has important functions in information exchange at fetal stage, the central nervous system development, tooth enamel growth, and it has also been that it may have potential regenerative functions for hair growth. Whereas, dysregulation can lead to aging-related neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis
2. Identify the amino acid sequence of your protein.
From UniProt:
sp|Q15465|SHH_HUMAN Sonic hedgehog protein OS=Homo sapiens OX=9606 GN=SHH PE=1 SV=1
MLLLARCLLLVLVSSLLVCSGLACGPGRGFGKRRHPKKLTPLAYKQFIPNVAEKTLGASG RYEGKISRNSERFKELTPNYNPDIIFKDEENTGADRLMTQRCKDKLNALAISVMNQWPGV KLRVTEGWDEDGHHSEESLHYEGRAVDITTSDRDRSKYGMLARLAVEAGFDWVYYESKAH IHCSVKAENSVAAKSGGCFPGSATVHLEQGGTKLVKDLSPGDRVLAADDQGRLLYSDFLT FLDRDDGAKKVFYVIETREPRERLLLTAAHLLFVAPHNDSATGEPEASSGSGPPSGGALG PRALFASRVRPGQRVYVVAERDGDRRLLPAAVHSVTLSEEAAGAYAPLTAQGTILINRVL ASCYAVIEEHSWAHRAFAPFRLAHALLAALAPARTDRGGDSGGGDRGGGGGRVALTAPGA ADAPGAGATAGIHWYSQLLYQIGTWLLDSEALHPLGMAVKSS
Results from the collab notebook:
Length: 462 amino acids
Most frequent: A (57 times, 12.3%)
This matches the number provided on UniProt
o How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
After inputting the sequence into UniProt’s BLAST tool, there are 244 homologs identified.
o Does your protein belong to any protein family?
It belongs to the hedgehog family.
3. Identify the structure page of your protein in RCSB o When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
After the search, I selected:
8Z2V | pdb_00008z2v
Crystal structure of Sonic hedgehog in complex with antibody 5E1 mutant H-R102A with metals
Resolution: 1.89 Å
This was deposited on April 13, 2024 and released in the PDB on April 16, 2025.
The resolution indicates that it is of good quality due to its resolution of a more detailed structure. This presents a more accurate interpretation of its structure.
o Are there any other molecules in the solved structure apart from protein?
The solved structure 8Z2V includes the heavy and light chains of the antibody 5E1 to which it is bound, as well as several small molecules: glycerol, zinc ions, calcium ions, and a chloride ion.
o Does your protein belong to any structure classification family?
It belongs to the immune system.
4. Open the structure of your protein in any 3D molecule visualization software:
o PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
After loading the protein, I got:

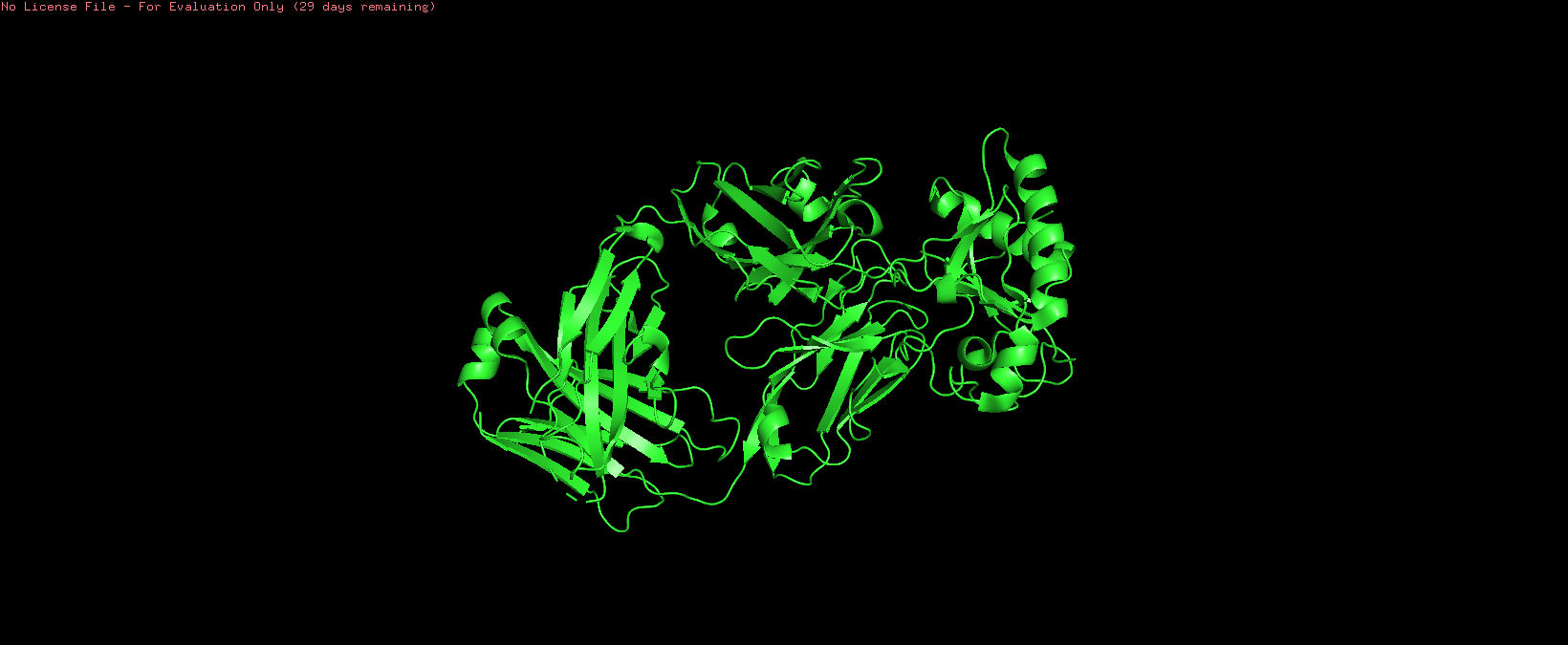
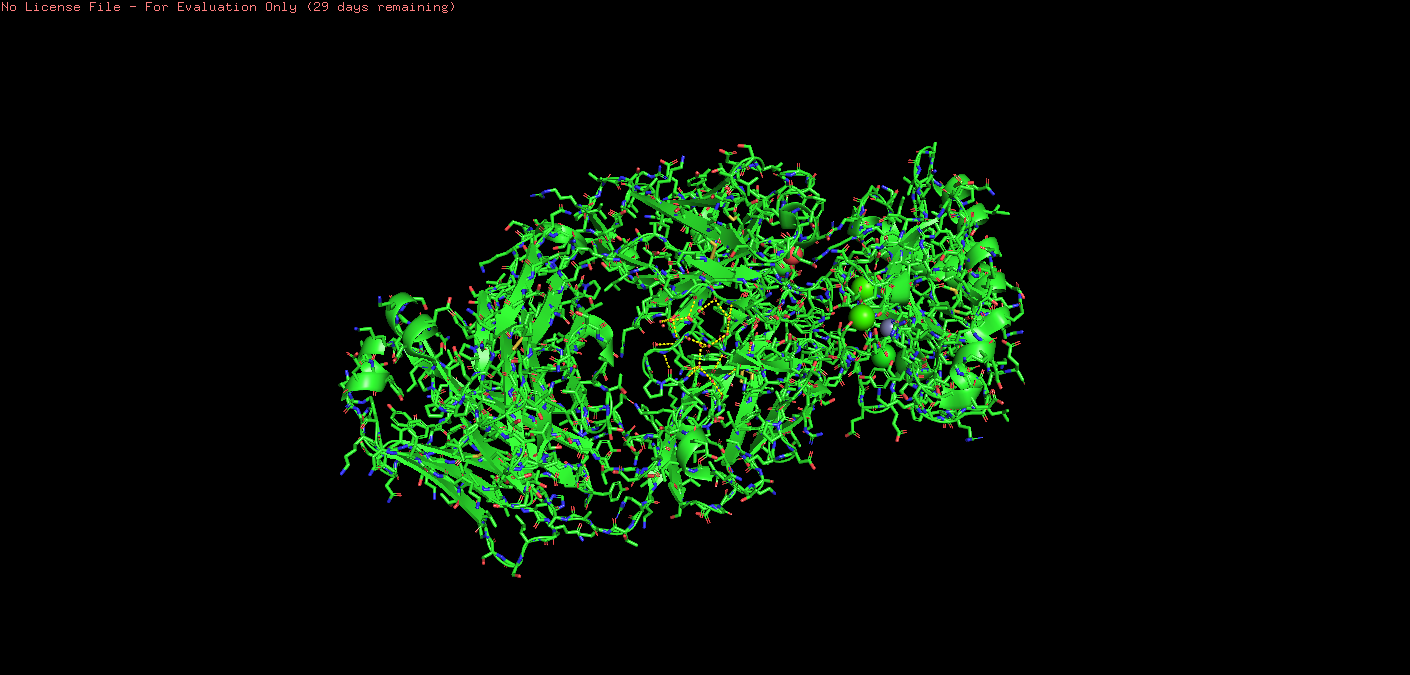
o Visualize the protein as “cartoon”, “ribbon” and “ball and stick”
Cartoon:
Ball & Stick:
Ribbon:

o Color the protein by secondary structure. Does it have more helices or sheets?
On PyMol I used:
*# color by secondary structure color red, ss h # helices color yellow, ss s # sheets color green, ss l # loops/coils
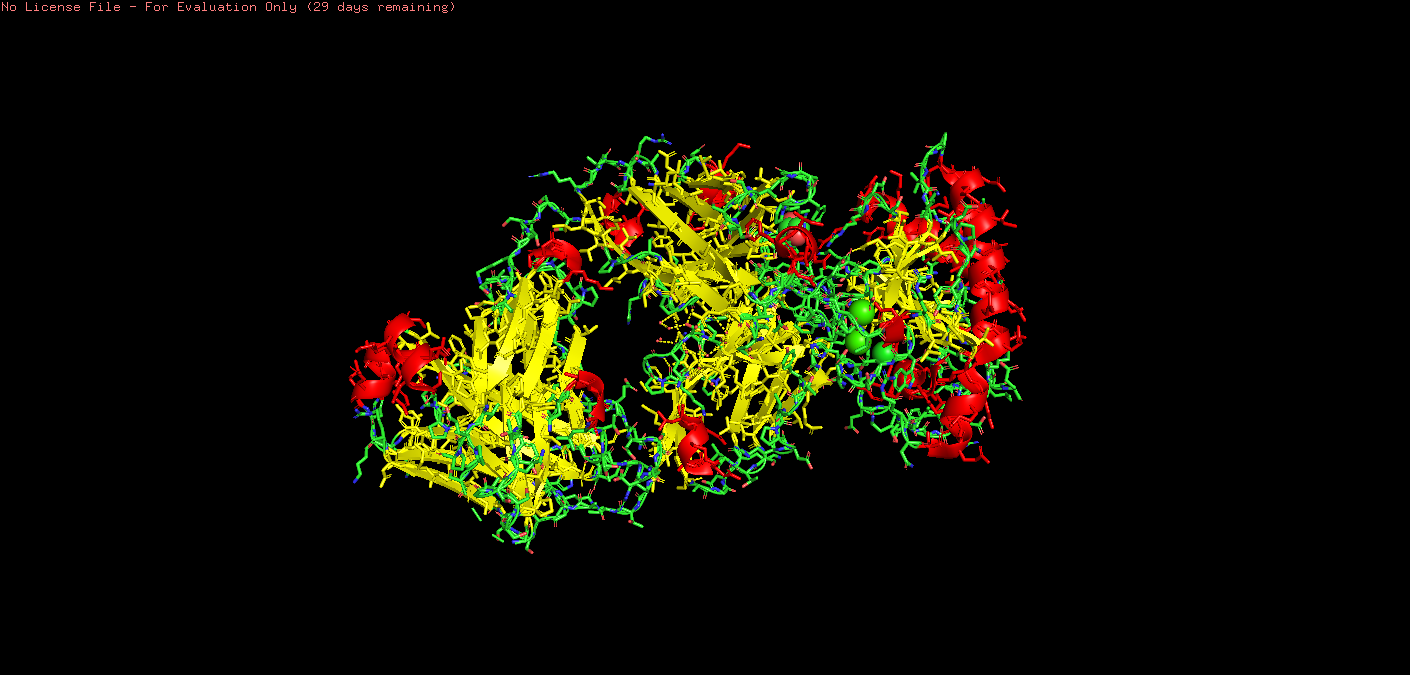
Upon visual inspection, there seems to be more sheets.
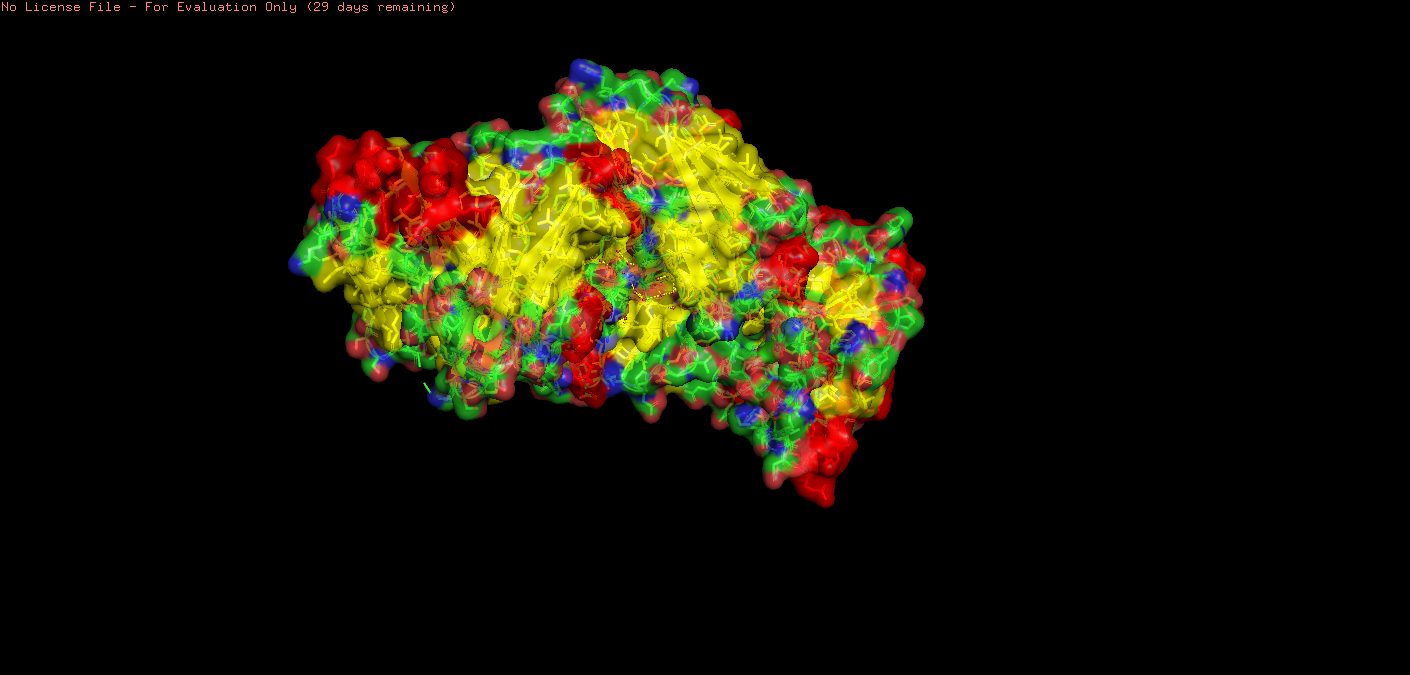
o Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
For inspection of holes/pockets, I reduced transparency.
Setting: transparency set to 0.30000.
scene: scene stored as “004”.
I then restarted with the following code:
fetch 3m1n show surface show spheres, organic set transparency, 0.3
This showed spheres:

This showed some deeply embedded pockets, with one (I think!) more towards the surface.
May need some help with this!!
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
1. Deep Mutational Scans
a. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
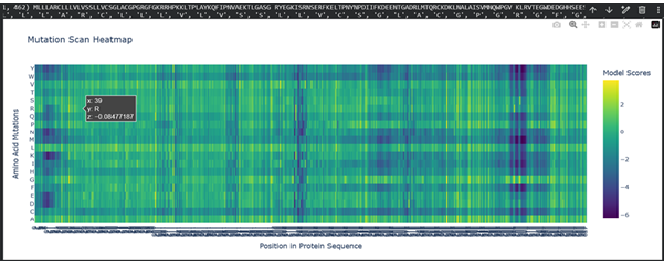
b. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
Dark vertical stripes in the heatmap indicate positions where nearly all mutations score negatively; highly conserved residues critical for SHH function. Position 141 (His), part of the zinc-binding motif, shows strongly negative LLR scores for most substitutions, reflecting its essential role in zinc coordination. Interestingly, our ESMFold experiments confirmed that mutating this site (H141A/H142A) preserved the backbone fold while likely abolishing function, consistent with the language model’s predictions. In contrast, position 39 showed a near-neutral score (-0.08) for arginine substitution, expected given its location in the signal peptide which is cleaved after translation and therefore under weaker evolutionary pressure.
c. (Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
No systematic DMS dataset exists for SHH as far as I searched. Though, with more time I could do deeper searches. However, I would need some help with this question.
2. Latent Space Analysis
a. Use the provided sequence dataset to embed proteins in reduced dimensionality.
After realising that SHH was not included in the code, with the help of ChatGPT, I coded:
#####################################################################################################
*# 1. Embed SHH sequence shh_tokens = tokenizer( [protein_sequence], # your already-defined protein_sequence variable return_tensors=“pt”, padding=True, truncation=True, max_length=tokenizer.model_max_length )
with torch.no_grad(): shh_outputs = esm2( input_ids=shh_tokens[‘input_ids’], attention_mask=shh_tokens[‘attention_mask’], output_hidden_states=True, )
*# Mean pool the embedding shh_embedding = shh_outputs.hidden_states[-1][0] shh_mask = shh_tokens[‘attention_mask’][0] shh_mean_embedding = shh_embedding[shh_mask == 1].mean(dim=0).cpu().numpy()
*# 2. Stack with existing embeddings and re-run t-SNE all_embeddings = np.vstack([embeddings_array, shh_mean_embedding])
tsne_3d_new = TSNE(n_components=3, perplexity=30, n_iter=300, random_state=42) embeddings_3d_new = tsne_3d_new.fit_transform(all_embeddings)
*# 3. Build dataframe tsne_df_new = pd.DataFrame(embeddings_3d_new, columns=[‘TSNE1’, ‘TSNE2’, ‘TSNE3’])
*# Add labels — SCOP proteins + SHH labels = protein_sequence_annotations[:len(embeddings_array)] + [‘SHH (Sonic Hedgehog)’] tsne_df_new[’label’] = labels tsne_df_new[‘is_SHH’] = [‘SHH’ if i == len(embeddings_array) else ‘Other’ for i in range(len(tsne_df_new))]
*# Create a numerical column for marker size tsne_df_new[‘marker_size’] = tsne_df_new[‘is_SHH’].apply(lambda x: 10 if x == ‘SHH’ else 3)
*# 4. Plot with SHH highlighted fig_shh = px.scatter_3d( tsne_df_new, x=‘TSNE1’, y=‘TSNE2’, z=‘TSNE3’, color=‘is_SHH’, color_discrete_map={‘SHH’: ‘red’, ‘Other’: ’lightblue’}, hover_name=‘label’, title=‘3D t-SNE with SHH Highlighted’, size=‘marker_size’ # Use the new numerical size column )
fig_shh.update_layout(height=800) fig_shh.show() #####################################################################################################
This produced:
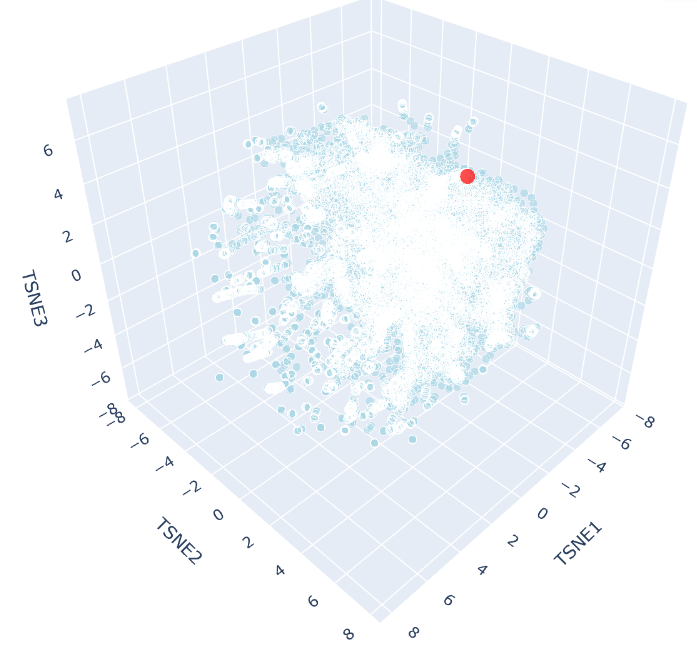
b. Analyze the different formed neighborhoods: do they approximate similar proteins?
The 3D t-SNE plot shows a single continuous distribution of SCOP protein embeddings with no sharply defined clusters, suggesting protein sequence space varies gradually across structural families. Outlier points at the periphery represent the most divergent sequences, consistent with the known continuity of protein fold space.
c. Place your protein in the resulting map and explain its position and similarity to its neighbors
SHH appears as a distinct red point near the periphery of the t-SNE cloud, reflecting its unusual biochemical features, including autocatalytic processing and lipid modification, that makes it distinct from most SCOP representatives. Despite this, it remains within the main cloud boundary, indicating shared broad sequence features with neighbouring proteins. Its nearest neighbours would be expected to include other hedgehog family members (IHH, DHH), consistent with ESM2 capturing evolutionary relationships through sequence alone.
C2. Protein Folding
Folding a protein
- Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Total sequence length: 462
Running ESMFold inference for sequence with length 462…
Prediction complete. ptm: 0.603 plddt: 78.225
Results saved to SHH_Fold_V1_3a3ca/
CPU times: user 1min 26s, sys: 8.6 s, total: 1min 35s
Wall time: 2min 8s
ESMFold predicted the SHH structure with a pTM of 0.603 and mean pLDDT of 78.2. The pTM score above 0.5 suggests the overall fold topology is likely correct, while the pLDDT of 78.2 indicates confident but not perfect local coordinate prediction. Regions of lower confidence likely correspond to flexible loops and the signal peptide. A full structural comparison via RMSD alignment to the crystal structure 1VHH would further quantify coordinate accuracy.
- Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
Mutation:
MLLLARCLLLVLVSSLLVCSGLACGPGRGFGKRRHPKKLTPLAYKQFIPNVAEKTLGASG RYEGKISRNSERFKELTPNYNPDIIFKDEENTGADRLMTQRCKDKLNALAISVMNQWPGV KLRVTEGWDEDGAHSEESLHYEGRAVDITTSDRDRSKYGMLARLAVEAGFDWVYYESKAH IHCSVKAENSVAAKSGGCFPGSATVHLEQGGTKLVKDLSPGDRVLAADDQGRLLYSDFLT FLDRDDGAKKVFYVIETREPRERLLLTAAHLLFVAPHNDSATGEPEASSGSGPPSGGALG PRALFASRVRPGQRVYVVAERDGDRRLLPAAVHSVTLSEEAAGAYAPLTAQGTILINRVL ASCYAVIEEHSWAHRAFAPFRLAHALLAALAPARTDRGGDSGGGDRGGGGGRVALTAPGA ADAPGAGATAGIHWYSQLLYQIGTWLLDSEALHPLGMAVKSS
Changed HH→AA at zinc-binding site (positions 141-142)
Total sequence length: 462
Running ESMFold inference for sequence with length 462…
Prediction complete. ptm: 0.602 plddt: 78.128
Results saved to test_2cd60/
CPU times: user 1min 25s, sys: 8.45 s, total: 1min 33s
Wall time: 2min 3s
A double point mutation at the zinc-binding site (H141A/H142A) had negligible effect on predicted structure (pTM 0.603 vs. 0.602, pLDDT 78.2 vs. 78.13), suggesting SHH’s fold is resilient to point mutations even at functionally critical residues.
Mutation:
Alanine substitution was chosen as it removes side chain functionality while preserving backbone geometry, representing a conservative but informative structural perturbation.
MLLLARCLLLVLVSSLLVCSGLACGPGRGFGKRRHPKKLTPLAYKQFIPNVAEKTLGASG RYEGKISRNSERFKELTPNYNPDIIFKDEENTGADRLMTQRCKDKLNALAISVMNQWPGV KLRVTEGWDEDGHHSEESLHYEGRAVDITTSDRDRSKYGMLARLAVEAGFDWVYYESKAH IHCSVKAENSVAAKSGGCFPGSATVHLEQGGTKLVKDLSPGDRVLAADDQGRLLYSAAAAAAAAAAAAAAAAAAAAAAAAFLDRDDGAKKVFYVIETREPRERLLLTAAHLLFVAPHNDSATGEPEASSGSGPPSGGALG PRALFASRVRPGQRVYVVAERDGDRRLLPAAVHSVTLSEEAAGAYAPLTAQGTILINRVL ASCYAVIEEHSWAHRAFAPFRLAHALLAALAPARTDRGGDSGGGDRGGGGGRVALTAPGA ADAPGAGATAGIHWYSQLLYQIGTWLLDSEALHPLGMAVKSS
Replaced 25 residues with Alanines in a surface region
This resulted in:
Total sequence length: 482
Running ESMFold inference for sequence with length 482…
Prediction complete. ptm: 0.554 plddt: 72.026
Results saved to SHH_FinalMut_d0fdf/
CPU times: user 1min 40s, sys: 10.3 s, total: 1min 51s
Wall time: 2min 27s
A large segment mutation was introduced by replacing a surface region with a polyalanine stretch (26 residues), resulting in a slight sequence length increase from 462 to 482 residues due to insertion. This caused a moderate reduction in predicted structural confidence (pTM 0.603 –> 0.554, pLDDT 78.2 –> 72.0), while the fold remained above the 0.5 pTM threshold, indicating overall structural resilience.
C3. Protein Generation
sequence candidates via ProteinMPNN
- Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
After installing necessary packages for ProteinMPNN, I input the latest PDB for SHH protein, 8Z2V.
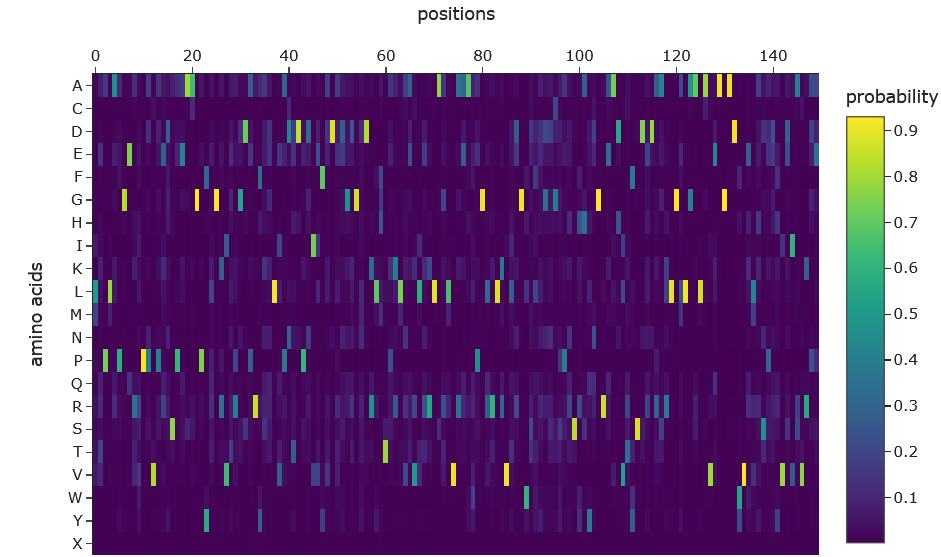
Heat map:
Sequence comparison:
Generating sequences…
8Z2V, score=1.5464, fixed_chains=[], designed_chains=[‘A’], model_name=v_48_020
LTPLAYKQFIPNVAEKTLGASGRYEGKISRNSERFKELTPNYNPDIIFKDEENTGADRLMTQRCKDKLNALAISVMNQWPGVKLRVTEGWDEDGHHSEESLHYEGRAVDITTSDRDRSKYGMLARLAVEAGFDWVYYESKAHIHCSVKAE
T=0.1, sample=0, score=0.8104, seq_recovery=0.4467
LTPLAPGERVPPVPEDSPEAAGPYLGRVERGDPRFADLVPDTDPDIEFADADGDGNDRLHTPKLVAVLRRLARLVREAWPGLRLRVLRGWSLDGDGSPRSHHYNGREADVTFSDEDAARLGALAALAVEAGADWVELASPDYVEIAVRPE
The ProteinMPNN probability heatmap shows that most positions along the SHH backbone are highly constrained, with single amino acids receiving probabilities exceeding 0.9 (yellow). This reflects strong structural determinism; the backbone geometry dictates specific residue preferences at key positions. A minority of positions, particularly around residues 95–105, show broader probability distributions across multiple amino acids, indicating structurally tolerant surface-exposed regions. The overall sparsity of high-probability assignments is consistent with the 44.67% sequence recovery observed, where roughly half of positions were confidently recovered while the remainder tolerate sequence variation.
- Input this sequence into ESMFold and compare the predicted structure to your original.
Inputting designed sequence back into ESMFold:
Original:
LTPLAYKQFIPNVAEKTLGASGRYEGKISRNSERFKELTPNYNPDIIFKDEENTGADRLMTQRCKDKLNALAISVMNQWPGVKLRVTEGWDEDGHHSEESLHYEGRAVDITTSDRDRSKYGMLARLAVEAGFDWVYYESKAHIHCSVKAE
Designed:
LTPLAPGERVPPVPEDSPEAAGPYLGRVERGDPRFADLVPDTDPDIEFADADGDGNDRLHTPKLVAVLRRLARLVREAWPGLRLRVLRGWSLDGDGSPRSHHYNGREADVTFSDEDAARLGALAALAVEAGADWVELASPDYVEIAVRPE
Total sequence length: 150
Running ESMFold inference for sequence with length 150…
Prediction complete. ptm: 0.910 plddt: 90.664
Results saved to SHH_Inverse_FinalMut_3f7ad/
CPU times: user 10.2 s, sys: 8.37 s, total: 18.6 s
Wall time: 47.2 s
The ProteinMPNN designed sequence, when folded by ESMFold, achieved a pTM of 0.910 and pLDDT of 90.664; substantially higher than the native SHH sequence (pTM 0.603, pLDDT 78.2). This improvement reflects two factors: first, the designed sequence covers only the structured core of SHH (150 residues vs 462), excluding disordered regions such as the signal peptide that reduce confidence scores; second, ProteinMPNN explicitly optimises sequences for backbone compatibility, producing a sequence more thermodynamically suited to the given fold than the evolutionarily derived native sequence.
Part D. Group Brainstorm on Bacteriophage Engineering
Proposal by: Sameen Nasar, Robert C Beck
Group Project Goal: Engineering a chaperone-independent efficient MS2 lysis protein
Project Rationale:
The efficacy of bacteriophage MS2 as an antibacterial agent is currently limited by the host’s ability to evolve resistance. Specifically, E. coli can mutate the molecular chaperone DnaJ (e.g., at position P330), disrupting the essential interaction required for the MS2 lysis (L) protein to fold and function [1.] This interaction is required for proper function of the lysis protein, as DnaJ binds to the N-terminal domain of MS2 lysis protein and alleviates its inhibitory effect on lytic activity.
We propose engineering a self-activating L protein by replacing its inhibitory, chaperone-dependent N-terminal region with a computationally designed, thermodynamically stable scaffold. As this original domain is dispensable for actual lysis but creates the DnaJ dependency [2], our redesign conceptually eliminates the need for the molecular “handshake” between host and phage, allowing MS2 to fold independently and bypass bacterial control mechanisms entirely.
Schematic
MS2 Protein & DnaJ Sequences
↓
AlphaFold-Multimer
Map the DnaJ binding interface
↓
RFDiffusion
Design a stable, independent N-terminal scaffold
↓
ProteinMPNN
Generate amino acid sequences for the new scaffold
↓
ESMFold
Confirm the new single-chain mutant folds correctly
↓
AlphaFold-Multimer
Confirm the mutant no longer binds to DnaJ
↓
Final L Protein Mutant for Synthesis
References
Chamakura KR, Tran JS, Young R. MS2 lysis of Escherichia coli depends on host chaperone DnaJ. J Bacteriol. 2017;199(9):e00058-17. doi:10.1128/JB.00058-17.
Chamakura KR, Edwards GB, Young R. Mutational analysis of the MS2 lysis protein L. Microbiology (Reading). 2017;163(7):961–969. doi:10.1099/mic.0.000485.