Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
Design short peptides that bind mutant SOD1.
Then decide which ones are worth advancing toward therapy.
You will use three models developed in our lab:
• PepMLM: target sequence-conditioned peptide generation via masked language modeling
• PeptiVerse: therapeutic property prediction
• moPPIt: motif-specific multi-objective peptide design using Multi-Objective Guided Discrete Flow Matching (MOG-DFM)
Part 1: Generate Binders with PepMLM
*I initially did this part wrong as I did not introduce the mutation into the sequence, therefore had to do it again. The following is the latest attempt
1. Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
From UniProt:
sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
After adding the A4V mutation in position 5, taking in Methione into account:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
2. Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
Using PepMLM, four candidate peptides of length 12 were generated conditioned on the mutant SOD1 (A4V) sequence. The generated peptides were WRYGPYAIELAX (pseudo-perplexity 11.85), WRYYVAALEWWE (28.73), WHNYAAAIRLKX (15.20), and WHSYAAAAELKX (9.48). For comparison, the known SOD1-binding peptide FLYRWLPSRRGG was scored against the same target, yielding a pseudo-perplexity of 20.64. Lower pseudo-perplexity values indicate higher model confidence in the predicted binder. Three of the four generated peptides outperformed the known binder, with WHSYAAAAELKX achieving the lowest score of 9.48. Notably, two of the four generated peptides, WRYGPYAIELAX and WHNYAAAIRLKX, contained a terminal X residue, representing an unknown or masked amino acid. This suggests a mismatch between the specified peptide length and the model’s generation process, and these sequences should be treated with caution or re-generated with corrected parameters before advancing to downstream evaluation.
3. Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
| Binder | Pseudo Perplexity |
|---|---|
| WRYGPYAIELAX | 11.85063412 |
| WRYYVAALEWWE | 28.7286821 |
| WHNYAAAIRLKX | 15.20319465 |
| WHSYAAAAELKX | 9.482601001 |
4. To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
To find the perplexity score for FLYRWLPSRRGG, I added this code (with help from LLM) to generate perplexity score on the collab notebook:
known_peptide = “FLYRWLPSRRGG”
ppl_score = compute_pseudo_perplexity(model, tokenizer, protein_seq, known_peptide)
print(f"Peptide: {known_peptide}")
print(f"Pseudo Perplexity: {ppl_score}")
This resulted in:
| Binder | Pseudo Perplexity |
|---|---|
| WRYGPYAIELAX | 11.85063412 |
| WRYYVAALEWWE | 28.7286821 |
| WHNYAAAIRLKX | 15.20319465 |
| WHSYAAAAELKX | 9.482601001 |
| FLYRWLPSRRGG | 20.63523127 |
5. Record the perplexity scores that indicate PepMLM’s confidence in the binders.
After generating the 12 amino acid peptides with PepMLM on the mutant SOD1 sequence, I recorded the pseudo-perplexity scores for each (lower scores indicate higher model confidence). I then added the known SOD1-binding peptide FLYRWLPSRRGG as a reference for comparison, yielding a pseudo-perplexity of 20.64. Of the four generated peptides, three outperformed the known binder: WHSYAAAAELKX (9.48), WRYGPYAIELAX (11.85), and WHNYAAAIRLKX (15.20), while WRYYVAALEWWE (28.73) scored worse. The best performing generated peptide, WHSYAAAAELKX, achieved nearly half the perplexity of the known binder, suggesting a strong model confidence in its predicted binding to the A4V mutant SOD1 target.
Part 2: Evaluate Binders with AlphaFold3
1. Navigate to the AlphaFold Server: alphafoldserver.com
2. For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
3. Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
SOD1_ProtPeptide1 (WRYGPYAIELAX)
ipTM: 0.33
The predicted complex produced an ipTM score of 0.33, indicating low confidence in the interaction between the peptide and mutant SOD1. In the structural model, the peptide appears detached from the protein surface and does not localize near the N-terminal region where the A4V mutation occurs. Instead, it remains largely solvent-exposed and does not form clear contacts with the β-barrel region of SOD1.
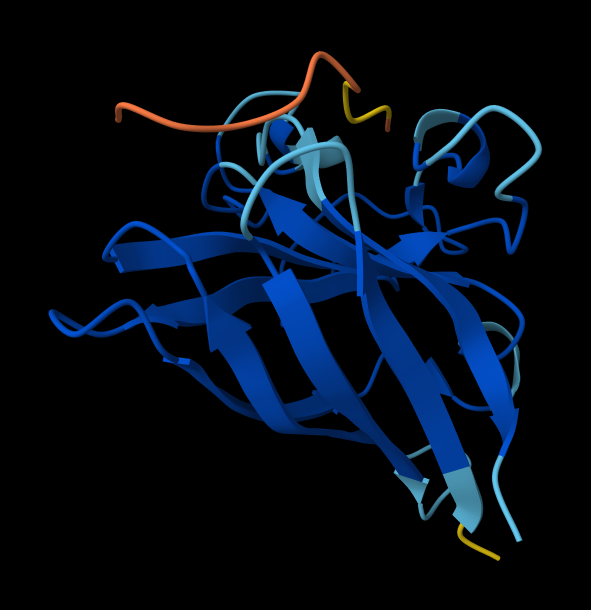
SOD1_ProtPeptide2 (WRYYVAALEWWE)
ipTM: 0.28
The predicted complex produced an ipTM score of 0.28, indicating low confidence in the interaction between the peptide and mutant SOD1. In the structural model, the peptide appears detached from the protein surface, adopting a partially helical conformation in the periphery of the structure but failing to localize near the N-terminal region where the A4V mutation occurs. The peptide does not form clear contacts with the β-barrel core and remains largely solvent-exposed.
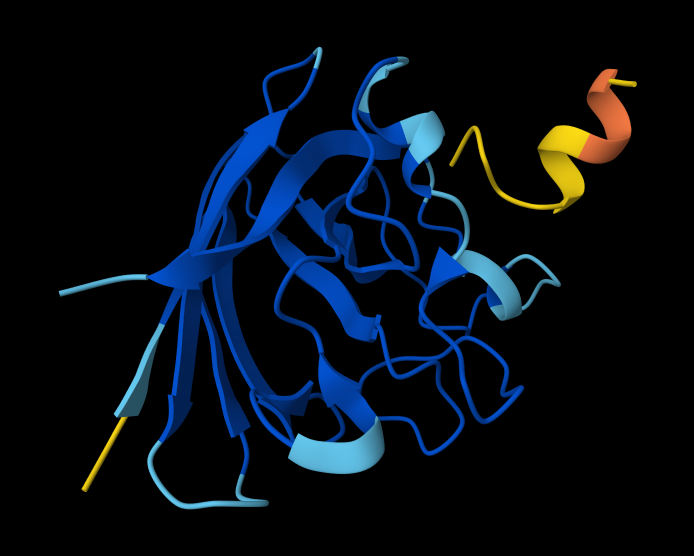
SOD1_ProtPeptide3 (WHNYAAAIRLKX) ipTM: 0.39
While this model has a higher ipTM score, it still has the same problems as a detached peptide, and no clear contacts make it solvent exposed.
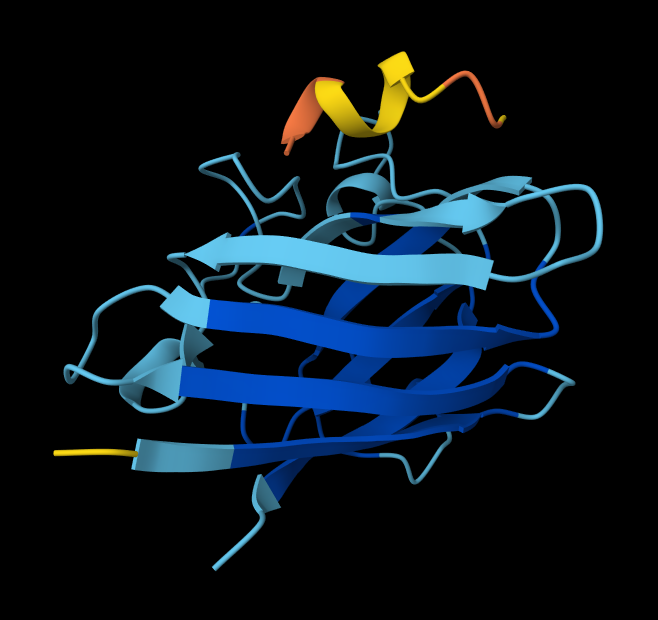
SOD1_ProtPeptide4 (WHSYAAAAELKX)
ipTM: 0.26
Similar trends with this peptide.
4. In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
AlphaFold predictions of SOD-1 peptides produced relatively low ipTM scores ranging from 0.26¬–0.39. This indicates low confidence in stable interactions between the generated peptides and mutant SOD1. In the predicted structures, the peptides generally appear surface-exposed and do not consistently localize near the N-terminal region where the A4V mutation occurs. As a result, they are loosely structured, and do not form clear interfaces with the β-barrel core or the dimer interface.
Part 3: : Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
Paste the peptide sequence.
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes:
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Choose one peptide you would advance and justify your decision briefly.
*Candidate Peptides:
| # | Binder | Pseudo Perplexity |
|---|---|---|
| 0 | WRYGPYAIELAX | 11.85063412 |
| 1 | WRYYVAALEWWE | 28.7286821 |
| 2 | WHNYAAAIRLKX | 15.20319465 |
| 3 | WHSYAAAAELKX | 9.482601001 |
(0)
(1)
(2)
(3)
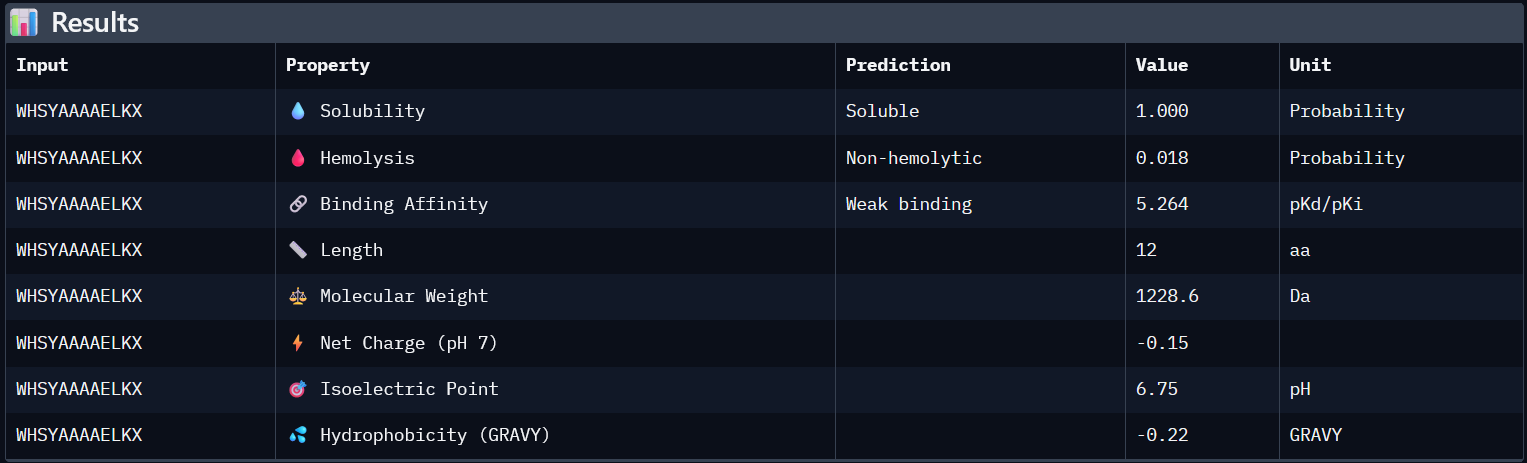
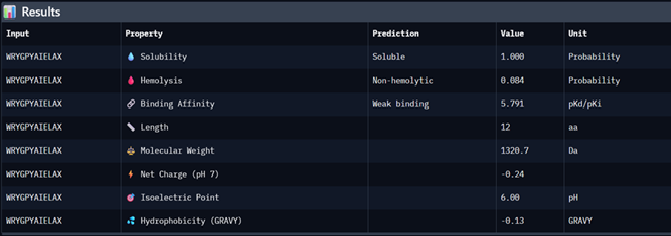
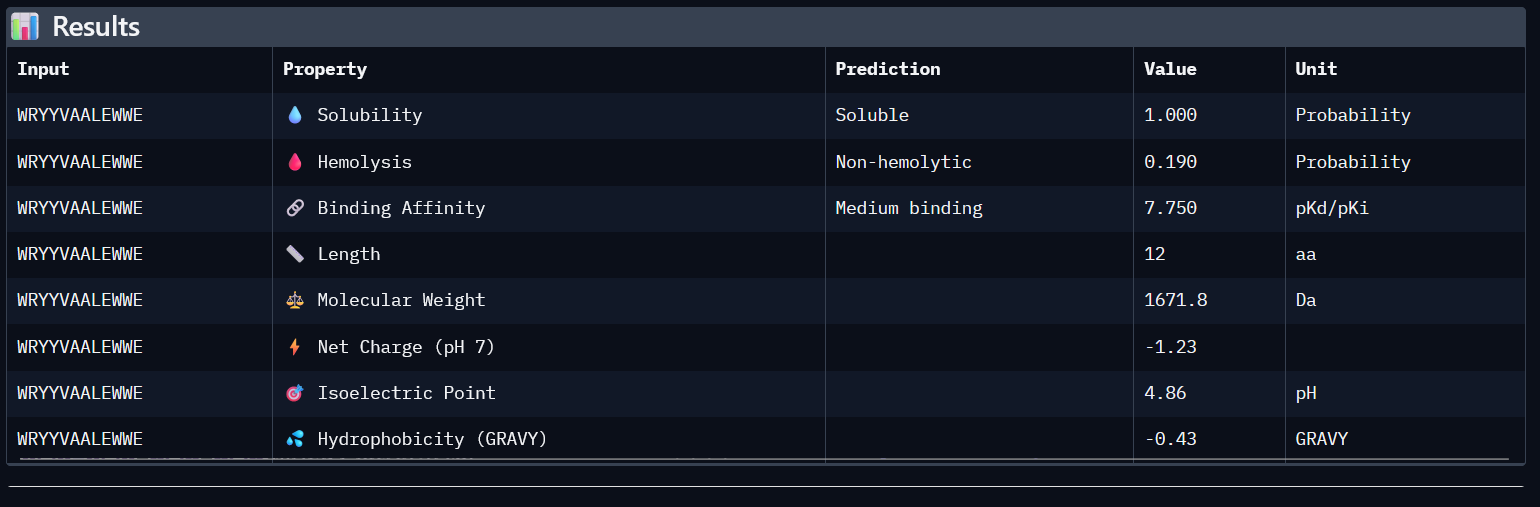
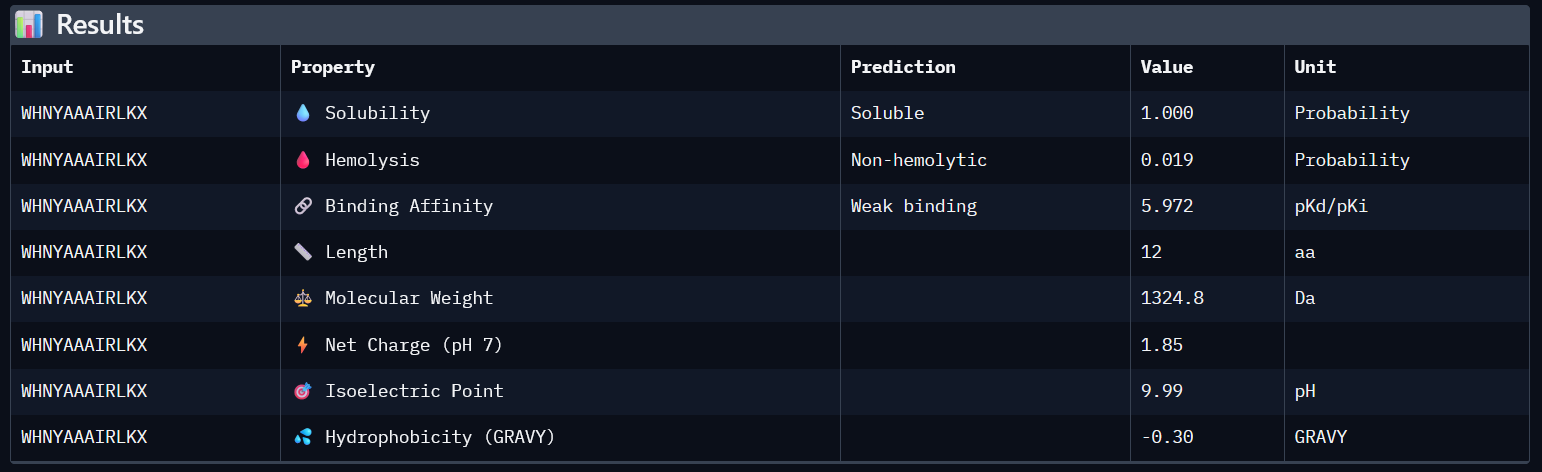
| # | Binder | ipTM | Predicted binding affinity | Solubility | Hemolysis probability | Netcharge (pH7) | Molecular weight |
|---|---|---|---|---|---|---|---|
| 0 | WRYGPYAIELAX | 0.33 | 5.791 | 1 | 0.084 | -0.24 | 1320.7 |
| 1 | WRYYVAALEWWE | 0.28 | 7.750 | 1 | 0.190 | -1.23 | 1671.8 |
| 2 | WHNYAAAIRLKX | 0.39 | 5.972 | 1 | 0.019 | 1.85 | 1324.8 |
| 3 | WHSYAAAAELKX | 0.26 | 5.972 | 1 | 0.019 | 1.85 | 1324.8 |
Higher ipTM scores do not consistently correspond to stronger predicted binding affinity in this dataset. For example, WHNYAAAIRLKX (ipTM 0.39) has a predicted binding affinity of 5.972, while WRYYVAALEWWE (ipTM 0.28) shows a higher affinity of 7.750 despite its lower structural confidence score. This suggests that ipTM and binding affinity capture different aspects of peptide-target interaction and should be considered together rather than in isolation.
All four generated peptides are highly soluble and show low hemolysis probabilities, indicating a favourable therapeutic safety profile. WHNYAAAIRLKX stands out as the most balanced candidate; it achieves the highest ipTM score (0.39), a competitive predicted binding affinity (5.972), perfect solubility, the lowest hemolysis probability in the dataset (0.019), and a positive net charge (1.85) which may favour interaction with the negatively charged surface regions of SOD1. However, it also has an unknown terminal amino acid, which is a problem for synthesis. Alternatively, WRYYVAALEWWE could also be a candidate due to its higher binding affinity and absence of X residue. Given its higher structural confidence (approx. 0.4) compared to the others, WHNYAAAIRLKX would be the most promising candidate to advance for further investigation.
Part 4: Generate Optimized Peptides with moPPIt
To edit the code, given the sliders were static, I used:
########################################################################################################################################################### *# For meet new selections
SELECTED_OBJECTIVES = [“Hemolysis”, “Solubility”, “Affinity”, “Motif”] OBJECTIVE_WEIGHTS_DICT = { “Hemolysis”: 1.0, “Solubility”: 1.0, “Affinity”: 1.5, “Motif”: 1.0 } OBJECTIVE_WEIGHTS_LIST = [1.0, 1.0, 1.5, 1.0] OBJECTIVES_CFG = { “selected_objectives”: SELECTED_OBJECTIVES, “weights_dict”: OBJECTIVE_WEIGHTS_DICT, “weights_list”: OBJECTIVE_WEIGHTS_LIST, “motif_positions”: “1-10” }
print(“Saved:”) print(“SELECTED_OBJECTIVES =”, SELECTED_OBJECTIVES) print(“OBJECTIVE_WEIGHTS_DICT =”, OBJECTIVE_WEIGHTS_DICT) print(“OBJECTIVE_WEIGHTS_LIST =”, OBJECTIVE_WEIGHTS_LIST) print(“motif_positions =”, OBJECTIVES_CFG[“motif_positions”])
###########################################################################################################################################################
| Binder | Hemolysis | Solubility | Binding Affinity | Motif |
|---|---|---|---|---|
| KKKKYITECLVM | 0.9794966895133257 | 0.6666666269302368 | 7.177585601806641 | 0.6455004811286926 |
| ECYYVWTEQGTT | 0.9729829281568527 | 0.8333333134651184 | 6.359397888183594 | 0.5219646692276001 |
| KLKQKKFTEKVC | 0.9676016941666603 | 0.7500000 | 6.8997617 | 0.7254035472869873 |
| SFQKINEKVKNA | 0.9103980 | 0.6666666269302368 | 6.861388206481934 | 0.6815867 |
Peptides generated with moPPIt differ from those generated by PepMLM through controlled, residue-specific generation targeting positions 1-10 of the A4V mutant SOD1 sequence, with simultaneous optimisation of hemolysis, solubility, affinity, and motif objectives.
The four generated peptides show different balances across the optimised properties. KKKKYITECLVM achieves the highest affinity score (7.178) and a strong hemolysis score (0.979), though its solubility is moderate (0.667). KLKQKKFTEKVC shows the highest motif score (0.725) alongside competitive affinity (6.900), suggesting strong localisation near the targeted N-terminal residues. ECYYVWTEQGTT offers the best solubility (0.833) but the lowest affinity and motif scores of the four. SFQKINEKVKNA presents a balanced profile across all objectives with the lowest hemolysis score (0.910).
Compared to the PepMLM-generated peptides, the moPPIt peptides benefit from explicit multi-objective optimisation, producing sequences with higher predicted affinities and targeted motif engagement rather than purely sequence-conditioned sampling.
Before advancing toward therapeutic development, these peptides would require further evaluation through in vitro binding assays to confirm SOD1 interaction, proteolytic stability testing to assess degradation resistance, and cytotoxicity screening to verify safety before progressing to in vivo studies. Special emphasis should be placed on the haemolysis, given the high scores generated by this model; this may or may not indicate high toxicity.
Part C: Final Project: L-Protein Mutants
After running the code for analysis between predicted mutations and the experimental dataset, there is little to no overlap.
Process
After running the code for analysis between predicted mutations and the experimental dataset, there is little to no overlap. Process:
The model evaluated mutations using a log-likelihood ratio (LLR) derived from the probability distribution predicted by the ESM-2 protein language model. Mutations were then ranked by their LLR score, and predicted mutations were compared with experimental mutations using dataset merging.
The mutation C29R is present in both datasets. Experimental data shows no lysis activity, highlighting the difficulty in modelling predictions, as they do not always correspond to functional outcomes.
The model evaluated mutations using a log-likelihood ratio (LLR) derived from the probability distribution predicted by the ESM-2 protein language model. Mutations were then ranked by their LLR score, and predicted mutations were compared with experimental mutations using dataset merging.
Intiailly I tried to geenrate the full length sequences via Excel through updating mutations at specific positions:
This was very tedious, therefore I switched to Python on the desktop. Python was used instead of manual editing in Excel. A script was written to apply selected point mutations to the wild-type sequence by modifying specific residue positions. The code I used is below:
###########################################################################################################################################################
*#### HTGAA W5_HW_Part C: Multimer Assembly ####
*## Base sequence
base_seq = “METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT”
*## Mutations based on experimental dataset and model
*# K50L was done manually on MS EXcel
mutations = {
"Variant1": {"50":"L"},
"Variant2": {"39":"L"},
"Variant3": {"29":"R"},
"Variant4": {"13":"L"},
"Variant5": {"15":"A"}
}
*## Generate list
def apply_mutation(seq, mutation_dict):
seq_list = list(seq)
for pos, aa in mutation_dict.items():
seq_list[int(pos) - 1] = aa # -1 because Python is 0-indexed
return “".join(seq_list)
*# Store sequences
variant_sequences = {}
for name, mut in mutations.items():
variant_sequences[name] = apply_mutation(base_seq, mut)
*## Save variants in text file
with open(“Af2_variants.txt”, “w”) as f:
for name, seq in variant_sequences.items():
f.write(f"{name}: {seq}\n")
###########################################################################################################################################################
| Position of the mutation in L | Base Pair Changed | Amino Acid Position | Amino Acid Change | Lysis | Protein Levels |
|---|---|---|---|---|---|
| 38 | C->T | 13 | P->L | 1 | 1 |
| 38 | C->T | 13 | P->L | 1 | 1 |
| 43 | T->G | 15 | S->A | 1 | 1 |
| 52 | A->G | 18 | R->G | 1 | 1 |
| 53 | G->T | 18 | R->I | 1 | 1 |
From the experimental dataset, I chose the following:
| Position of the mutation in L | Base Pair Changed | Amino Acid Position | Amino Acid Change | Lysis | Protein Levels |
|---|---|---|---|---|---|
| 38 | C->T | 13 | P->L | 1 | 1 |
| 43 | T->G | 15 | S->A | 1 | 1 |
From the model, I then selected mutations with the highest LLR scores as they are the most strongly predicted from the model.
| Position | Wild_Type_AA | Mutation_AA | LLR_Score |
|---|---|---|---|
| 50 | K | L | 2.561468 |
| 29 | C | R | 2.395427 |
| 39 | Y | L | 2.24178 |
K50L and Y39L introduce hydrophobic residues that can help stabilize packed or core regions of the protein, consistent with the tendency for hydrophobic side chains to support structural integrity [1]. C29R adds a charged residue in a position the model favours, which may create new stabilizing interactions without disrupting folding [2]. Together these selections balance predicted stability, polarity, and structural compatibility, supporting the goal of designing functional L protein variants [3].
References
Pace CN, Fu H, Fryar KL, Landua J, Trevino SR, Shirley BA, et al. Contribution of hydrophobic interactions to protein stability. J Mol Biol. 2011;408(3):514-28.
Doig AJ, Williams DH. Is the hydrophobic effect stabilizing or destabilizing in proteins? The contribution of disulphide bonds to protein stability. J Mol Biol. 1991;217(2):389-98.
Hendsch ZS, Tidor B. Do salt bridges stabilize proteins? A continuum electrostatic analysis. Proteins. 1994;20(1):1-10.