Week 6 Lab: PCR & Gibson Assembly
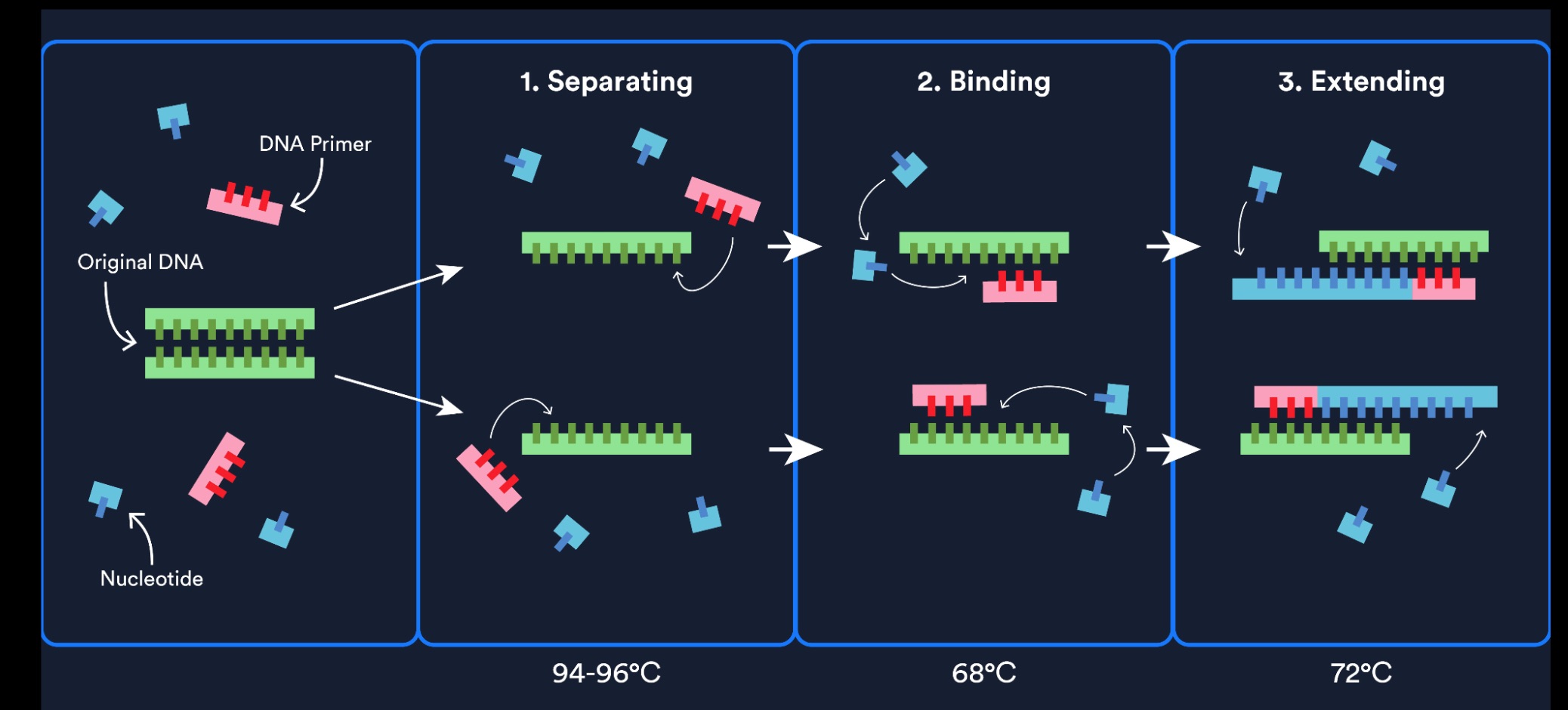
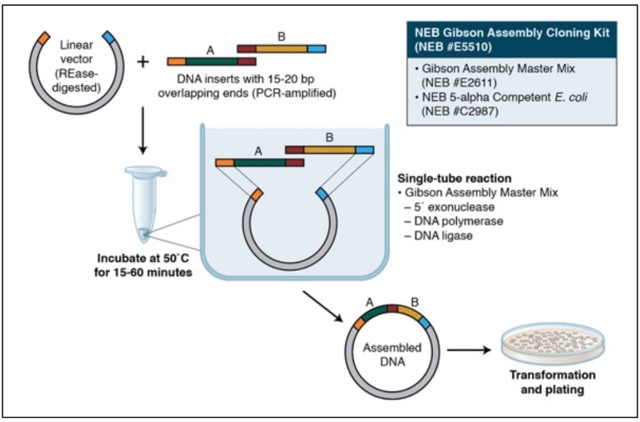
In this two-day lab, we used PCR and Gibson Assembly to engineer mutations in the chromophore region of the purple Acropora millepora chromoprotein (amilCP) in order to generate a range of orange, pink, and blue colour variants. Two separate PCR reactions were performed to generate the DNA fragments required for Gibson Assembly. The insert PCR region extended from 24 base pairs upstream of the chromophore to just beyond the transcription terminator of the gene. The forward primer was specifically designed with an intentional mismatch to introduce a site-directed mutation into the mUAV plasmid DNA. After assembly, the mutated plasmids were transformed into chemically competent E. coli cells for expression and analysis of the resulting colour phenotypes.
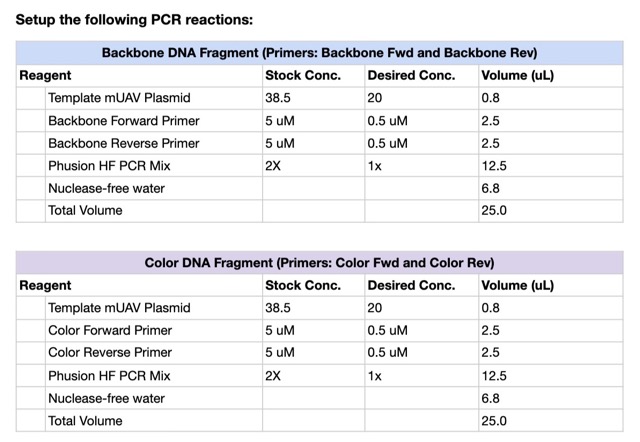
We prepared a backbone reaction alongside four color-specific reactions: Blue, Light Pink, Magenta, and Orange.
Setup the following PCR reactions:

After the reaction mixtures were prepared, the tubes were placed into thermocyclers. The plasmid backbone PCR was run using a specialized program, while the colour mutation PCR reactions were run using a separate optimized cycling program.
We purified the PCR products using the Zymo DNA Clean & Concentrator Kit following the Zymo Research protocol based on silica adsorption.
We used Zymo-Spin purification columns to clean up the PCR products, performing two wash steps before eluting the purified DNA for storage. Since the provided protocol was designed for 50 μL PCR reactions, but our PCR reactions had a total volume of 25 μL, we used 20 μL of PCR product for purification while saving 5 μL separately, and scaled the remaining reagent volumes proportionally. Gel electrophoresis was then performed to verify successful DNA amplification.

As you can see from gel electrophoresis, the furthest left lane contained the DNA ladder, while Lane 1 contained the native plasmid control. Lanes 2–5 showed the expected PCR-amplified fragments for the Gibson Assembly, each appearing at approximately 650 bp. The gel results were very convincing, with strong bands at the expected fragment size and little to no evidence of primer dimers or nonspecific bands, indicating good PCR efficiency and high polymerase fidelity. The purified samples were then stored in the fridge until the following lab session.
On day 2, we took our PCR-generated DNA fragments and assembled them using Gibson Assembly. We diverged slightly from the standard protocol by using unpurified PCR products directly for the assembly reaction. This decision was made for a few reasons. First, we realized that we had very limited sample volumes for each colour mutant in both the purified and unpurified conditions, meaning we effectively had to choose between measuring concentration via NanoDrop or using the maximum possible amount of DNA for the assembly itself. In addition, several other groups reported extremely low, almost negligible, DNA concentrations after purification. Given our strong gel electrophoresis results, we felt it was reasonable to assume that the majority of DNA present in the reactions corresponded to the desired amplicon. We therefore proceeded with the unpurified PCR products while following the remainder of the Gibson Assembly protocol as written.
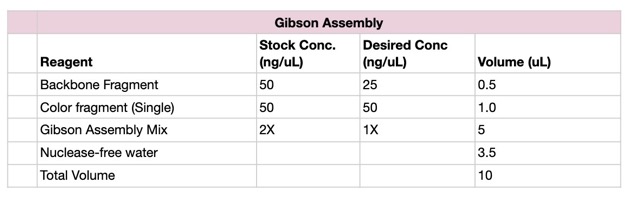

The reaction was incubated at 50°C in the thermocycler for 30 minutes.
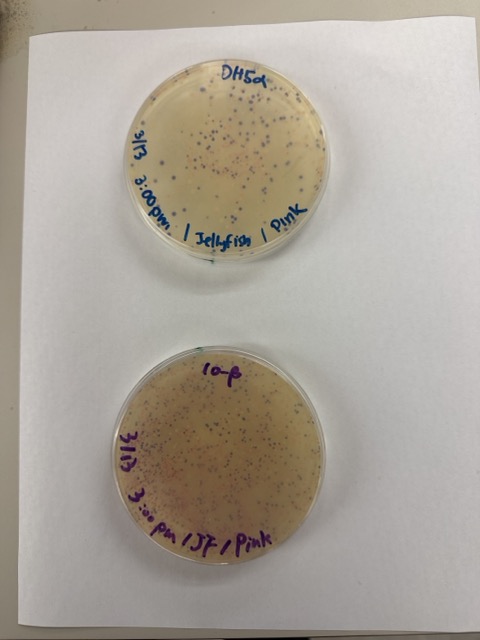
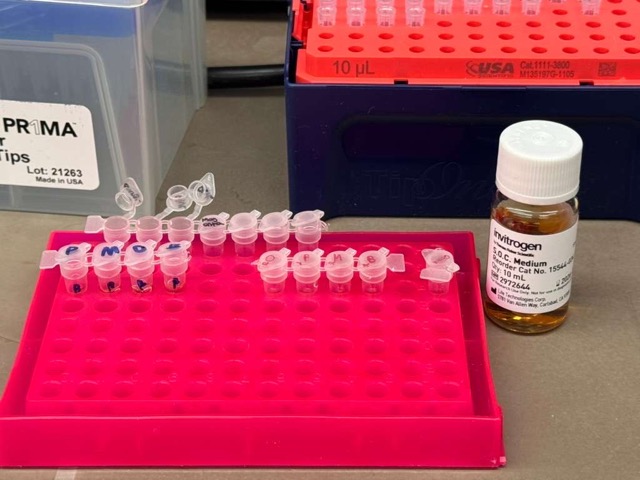
We compared two chemically competent E. coli strains: DH5α and 10-beta. After thawing the competent cells on ice, we mixed 20 μL of cells with 4 μL from each Gibson Assembly reaction and incubated the mixtures on ice for 30 minutes to allow the plasmid DNA to associate with the bacterial membranes.
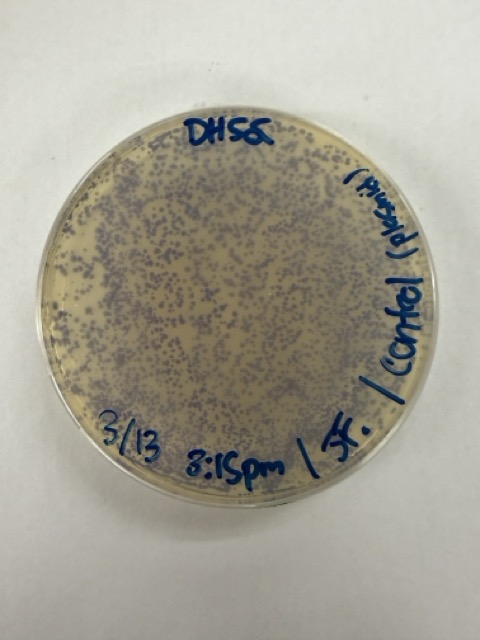
We also prepared an additional transformation reaction using only the native mUAV plasmid in DH5α cells as a positive control for successful transformation. For this control, I used 1 μL of plasmid DNA in an attempt to roughly match the DNA concentration of the Gibson Assembly samples.
Next, we again diverged slightly from the standard protocol. Instead of performing heat shock directly in the original transformation mixture, we transferred each transformation reaction into PCR tubes containing 100 μL of SOC medium and carried out the heat shock step in the thermocycler. The heat shock itself lasted only 45 seconds, after which the cells were immediately returned to ice to stabilize the bacterial membranes.
Following this, the cells were incubated in SOC medium for outgrowth to allow recovery and expression of the antibiotic resistance gene carried by the plasmid. Although the protocol recommended a 60-minute recovery period, our samples were incubated for closer to 45 minutes. Since we did not have access to a standard shaking incubator, we improvised a makeshift shaker using a pipette tip box to keep the cultures gently agitated during incubation.
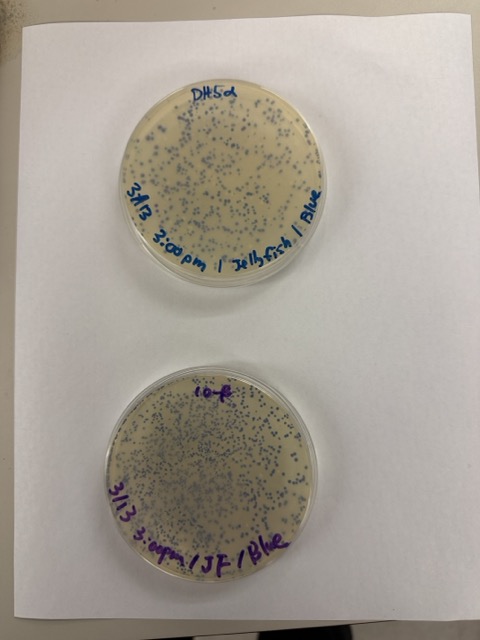
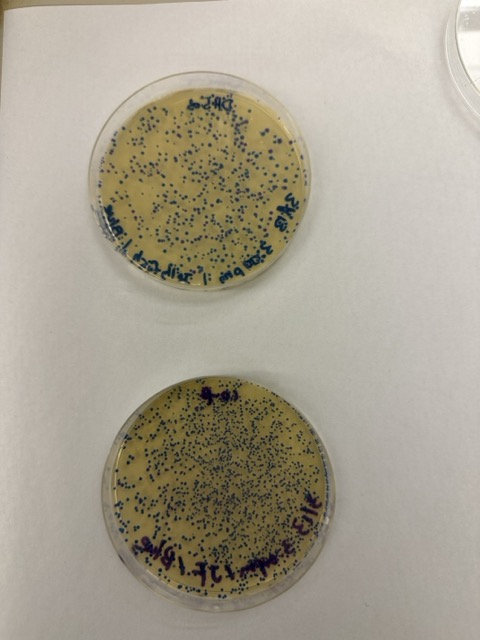
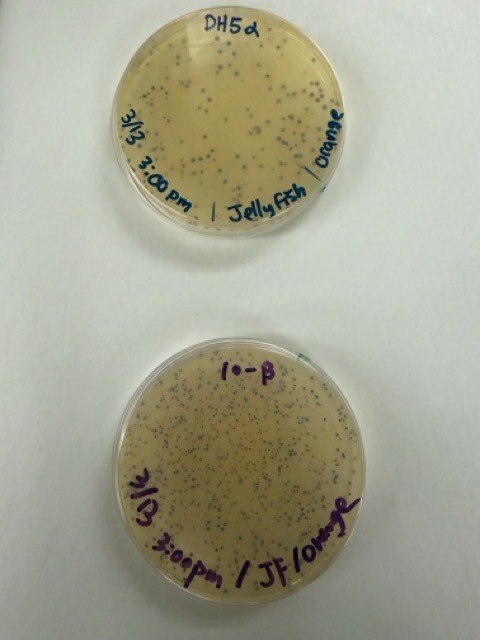
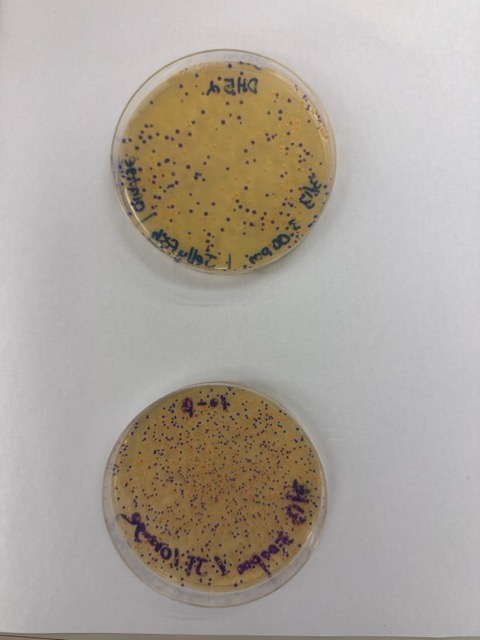
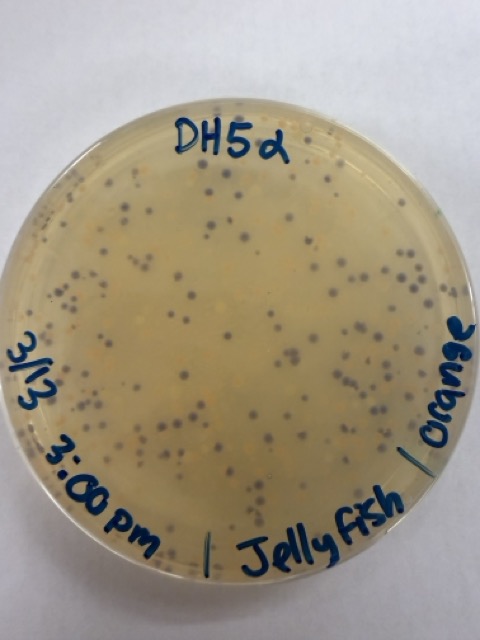
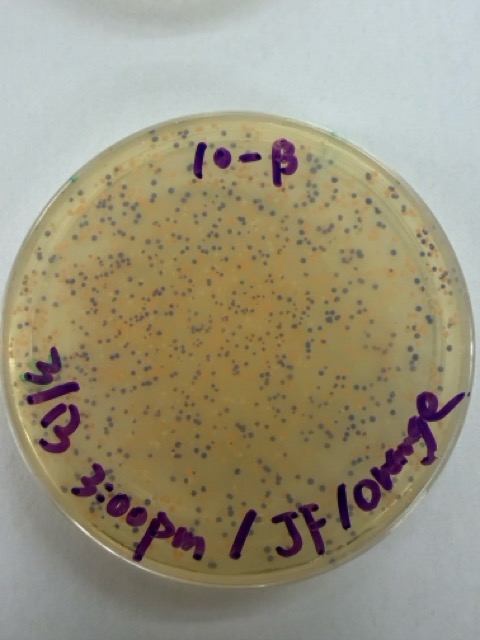
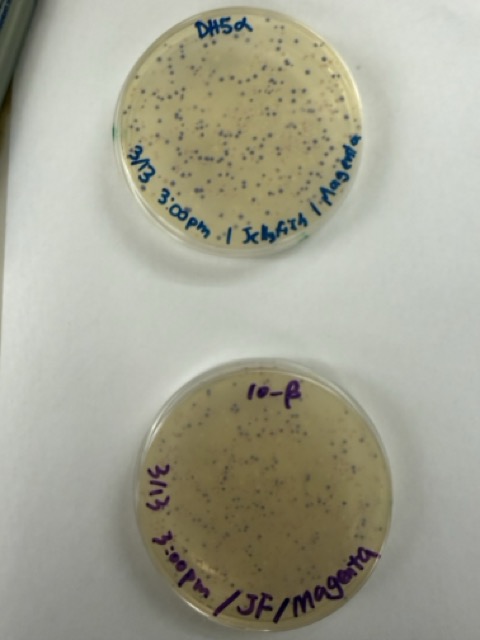
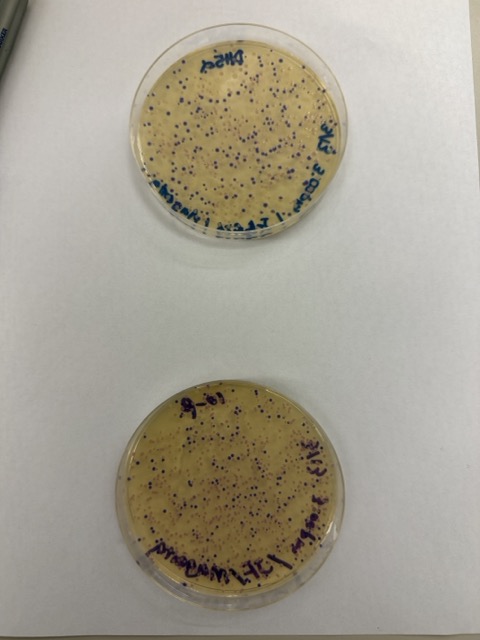
We plated the entire incubation volume (~124 μL) from each transformation reaction onto LB-agar plates containing chloramphenicol. Glass beads were then used to evenly spread the bacterial suspension across the surface of the plates to promote uniform colony growth.

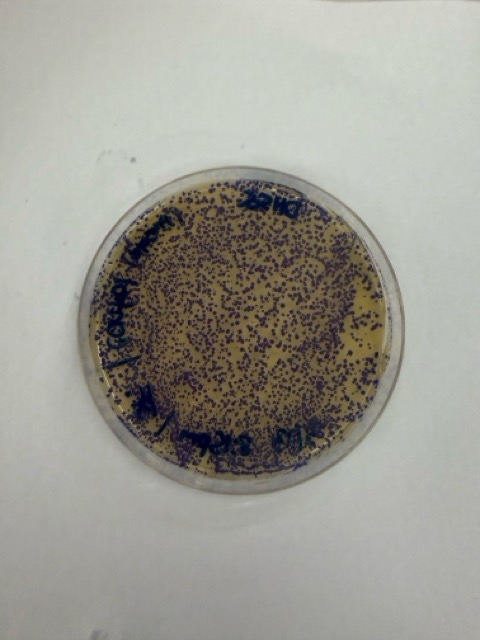
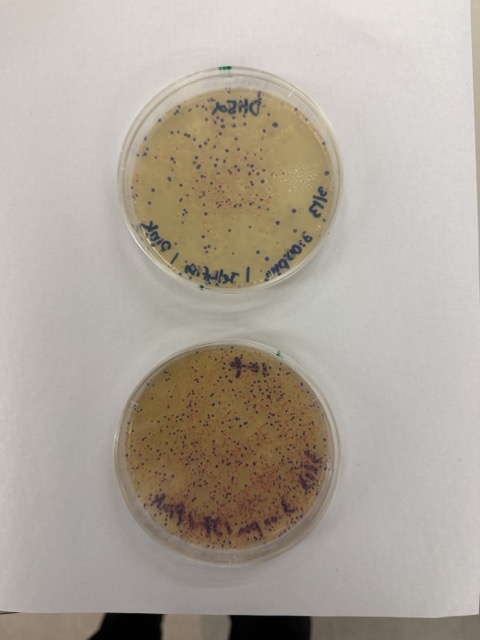
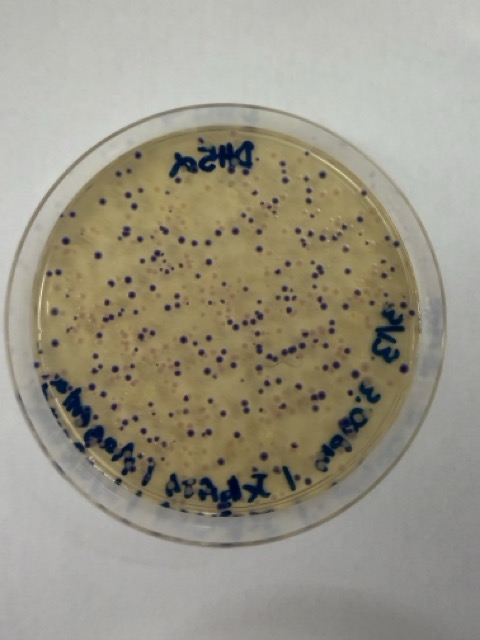
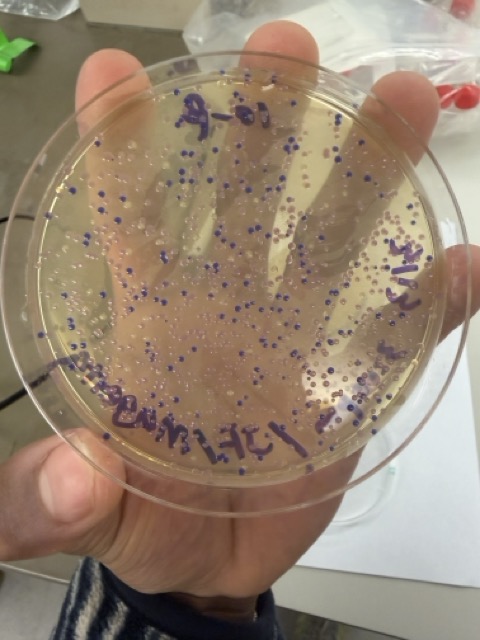
After 72 hours of incubation, the results were highly successful. We observed the targeted chromophore mutations across both E. coli strains.
The positive control confirmed that the transformation process had worked effectively. While some purple colonies corresponding to the native plasmid were present on all plates, each plate also displayed distinct coloured colonies including orange, pink, blue, and magenta. This indicated successful Gibson Assembly, transformation, and expression of the mutated chromoprotein variants.