Final Project
Project Aims
Experimental Aim
Development Aim
Visionary Aim
Background and Literature Context
How does SNIPR work?
Engineering Material-to-Cell Signalling with synNotch Systems
Neuromorphic Circuits
Novelty and Innovation
Why This Project Matters and Potential Impact
Ethical Implications
Experimental Design
1. Produce and purify GFP
2. Conjugate GFP to magnetic nanoparticles
3. Build the magnetic actuation device
4. Design the synthetic SNIPR and neuromorphic circuit plasmids
5. Transfect mammalian cells across different circuit configurations
6. Measure activation and compare outcomes
Results & Quantitative Expectations
Fluorescent Microscopy Results
Flow Cytometry Results
Flow Cytometry Distribution Plot
Additional Analysis
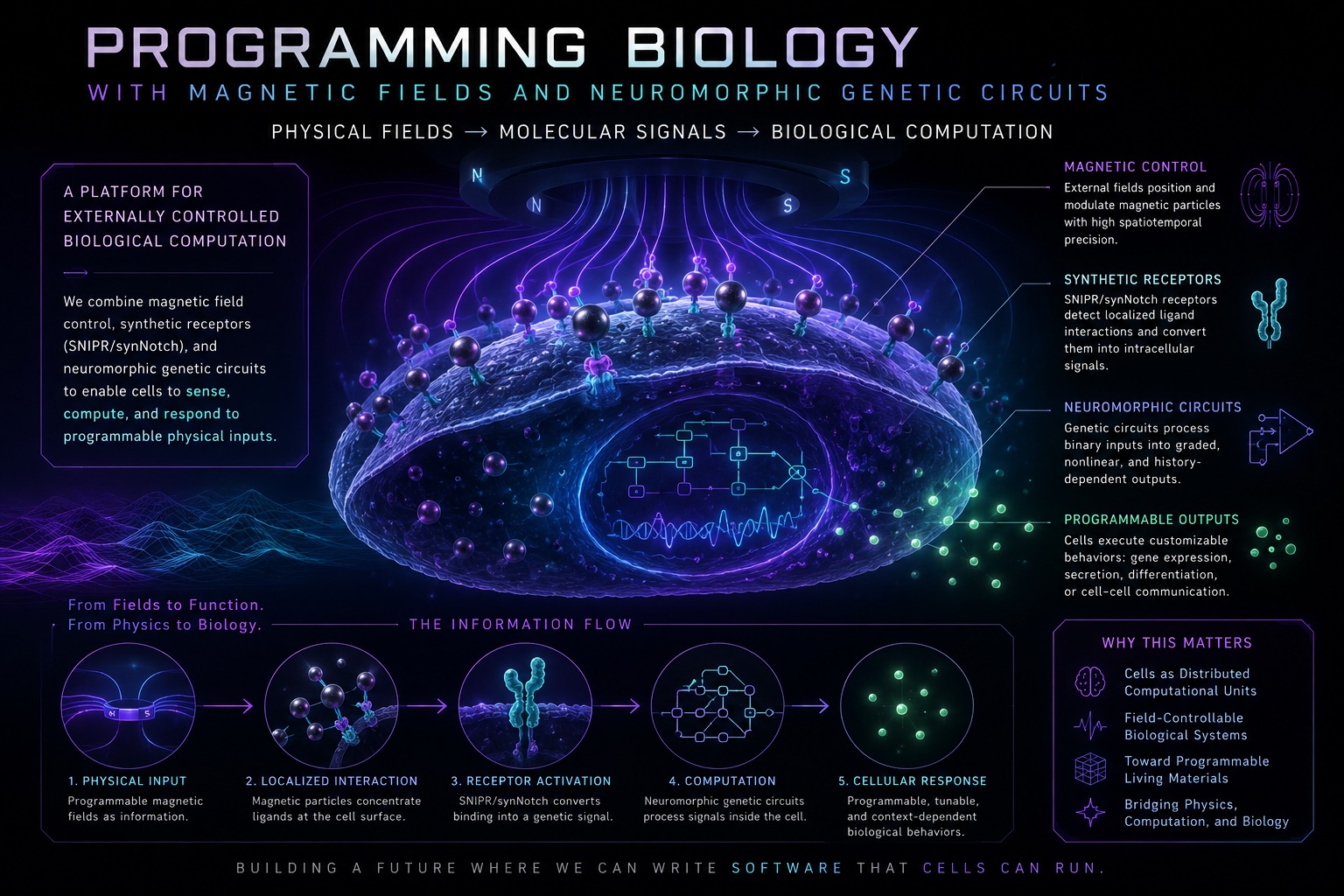
This project explores whether magnetic fields can be used to spatially localize, focus, and control biological computation inside mammalian cells.
Many signalling and gene expression processes in synthetic biology rely on diffusion, where molecules randomly spread through space before interacting with their targets. As a result, we have very limited control over where receptor activation and downstream cellular computation actually occur. This raises an interesting question: what if we could spatially control signalling interactions with microscale precision inside multicellular systems?
In this project, we explore whether magnetic nanoparticles may provide a solution by actively guiding signalling molecules toward engineered cellular receptors. By using external magnetic fields to localize these interactions, we aim to shape where signalling and cellular computation occur. More broadly, the goal is to explore magnets as a targeted external control layer for programmable biological systems, enabling more precise, localized, and dynamically tuneable cellular behaviour.
If successful, this work could introduce an entirely new way of controlling biology through externally applied physical fields. The ability to localize signalling and computation with magnetic precision could fundamentally change how engineered cells are programmed, coordinated, and controlled within living systems. Rather than cells passively responding to random molecular encounters, magnetic fields could allow biological computation to be dynamically focused in space and time, opening the door to programmable tissues, adaptive cell therapies, remotely tunable biological systems, and future forms of biological computation that blur the boundary between physical control systems and living matter.
To explore this idea, the project combines engineered cellular receptors, magnetic nanoparticles, and neuromorphic genetic circuits to convert external magnetic inputs into biological responses. Signalling proteins attached to magnetic nanoparticles are used to interact with synthetic receptors on mammalian cells, triggering receptor cleavage and downstream gene activation. These signals are then processed through a neuromorphic genetic circuit capable of producing graded, nonlinear responses rather than simple binary outputs.
The central hypothesis is that magnetically localized signalling can alter receptor activation compared to standard diffusion-driven interactions.
To test this, we will use protein conjugation protocols to generate protein-coated magnetic nanoparticles, engineer synthetic receptor and genetic circuit plasmids, transfect mammalian cells, and build magnetic actuation systems to control nanoparticle localization. We will then measure changes in spatial activation patterns and downstream circuit behaviour using fluorescence microscopy and flow cytometry.
click to expand
Mammalian cells (HEK cells)
Cells grown in the lab that come from mammals (often human-derived cell lines). HEK cells are a common “workhorse” cell type because they are easy to grow and easy to genetically modify.
Cell culture
Growing cells in dishes/plates with warm nutrient media so experiments can be run in a controlled environment.
DNA, RNA, and proteins (the “flow of information”)
DNA stores genetic instructions. RNA is the message copied from DNA. Proteins are the functional molecules built from RNA instructions (including receptors, enzymes, and fluorescent reporters).
Gene expression
The process of turning a gene “on” to make RNA (transcription) and then protein (translation). In this project, “more expression” often means “brighter fluorescence.”
Signalling
How cells detect an input (like a ligand binding a receptor) and convert it into an internal response (like gene expression).
Diffusion
Random spreading of molecules in liquid. If GFP is free in solution, diffusion largely determines how often it reaches and binds receptors.
synNotch vs SNIPR (Synthetic Notch receptors)
Engineered receptors based on the Notch pathway that convert an external binding event into gene expression. Ligand binding + mechanical pulling triggers proteolytic cleavage, releasing an intracellular transcription factor that turns on a chosen promoter.
Receptor (and receptor activation)
A receptor is a protein “sensor” on the cell surface. Activation means the receptor has bound its ligand (and, for Notch-like systems, experienced the physical force needed to trigger cleavage).
Mechanosensitive
Force-sensitive. synNotch/SNIPR receptors don’t just need binding — they typically need a small physical pull/tension to trigger the cleavage step.
Ligand
A molecule presented outside the cell that binds the receptor. In this project, GFP is used as a “designer ligand” and is either free in solution or immobilized on magnetic beads to control spatial presentation.
GFP (Green Fluorescent Protein)
A protein that glows green under the right light. Here it plays two roles: (1) a ligand the receptor can recognize, and (2) a fluorescent tag you can directly see/measure.
Magnetic nanoparticles / microparticles
Magnetic beads that can be functionalized with proteins (GFP) and repositioned with external magnets. They act as a physically steerable scaffold for ligand presentation rather than a soluble signal.
Carboxylated beads (–COOH surface)
Magnetic beads whose surfaces have carboxyl groups that make it easier to chemically attach proteins like GFP.
External magnetic field
A magnetic force applied from outside the dish (using permanent magnets). It can pull magnetic beads up, down, or to the side depending on where the magnet is placed.
Spatial localization (magnetic localization)
Controlling where in space ligand–receptor interactions occur (or do not occur). Here, magnets can concentrate particles near cells (activation) or pull/suspend particles away from the cell layer (inhibition), changing effective receptor engagement.
“Inhibition” vs “activation” by magnets (in this experiment)
Activation would mean magnets concentrate ligand-coated particles near cells to increase receptor engagement. Inhibition (the strategy used here) means magnets pull particles away from the cell layer to reduce engagement and toxicity.
Protein conjugation (EDC coupling; “EDC/NHS chemistry”)
Covalent attachment of proteins to carboxylated bead surfaces. EDC activates surface –COOH groups (often stabilized by NHS in similar protocols) so they react with protein amines, producing stable GFP-coated beads.
Covalent bond
A strong chemical bond that permanently links molecules. Covalent attachment helps keep GFP stuck to beads during washes and experiments.
PBS and MES buffer
Common lab buffers that keep pH and salt conditions stable. PBS is cell-friendly; MES is often used for EDC coupling reactions.
Washing / magnetic separation
Using a magnet to pull beads to the side of a tube so liquid can be removed and replaced. This removes unbound GFP and leftover reagents after conjugation.
Mammalian cell transfection (poly-transfection)
Delivery of plasmid DNA into mammalian cells (e.g., HEK cells), typically using lipid reagents. Poly-transfection forms separate DNA–lipid complexes for different plasmids to create a distribution of component ratios across single cells.
Lipid transfection (Lipofectamine)
A method that packages DNA into tiny lipid particles that fuse with cell membranes, delivering DNA into cells.
Plasmid
Circular DNA used to encode the receptor, response program, and computational layer. Including constitutive fluorescent markers helps identify which cells received which components.
Promoter
A DNA “switch” that controls when a gene is expressed. Some promoters are always on (constitutive); others only turn on when a transcription factor binds (inducible/regulated).
Transcription factor (Gal4-VP64) and UAS
Gal4-VP64 is the released activator domain in the receptor design. It binds UAS (Upstream Activating Sequences) to drive a minimal promoter, converting receptor cleavage into transcription of the response cassette.
Proteolytic cleavage
Cutting a protein at a specific site. For synNotch/SNIPR, cleavage releases the transcription factor from the membrane so it can enter the nucleus.
Nucleus
The cell compartment containing DNA. Many transcription factors must enter the nucleus to turn genes on.
Minimal promoter + Kozak sequence
A weak promoter that becomes active mainly when the upstream transcription factor is present. The Kozak sequence improves translation initiation of the downstream protein in mammalian cells.
Reporter fluorescent proteins (mNeonGreen, mKO2, eBFP2, mMaroon)
Fluorescent markers used either as the circuit output (e.g., mNeonGreen) or as constitutive markers to track expression of specific plasmids and enable gating/normalization in flow cytometry.
mNeonGreen (output readout)
A very bright green fluorescent protein used here as the main “how activated is the circuit?” output.
Neuromorphic genetic circuit
A genetic circuit designed to produce graded / nonlinear input–output responses (rather than simple ON/OFF logic), conceptually inspired by neural computation.
Csy4 + Csy4 target site
Csy4 is an RNA-processing endonuclease that recognizes a specific hairpin target sequence embedded in transcripts. By placing a Csy4 site in the response transcript, expression of the output can be tuned post-transcriptionally, shaping the circuit’s transfer function.
Post-transcriptional regulation
Control that happens after RNA is made (but before or during translation into protein). Csy4 is used here to shape how much protein gets produced from the RNA message.
Dose / concentration (e.g., mg/mL)
How much of a substance is present per volume of liquid. Higher concentration generally means more potential binding events (unless limited by toxicity or saturation).
Toxicity / cell viability
Whether the experimental conditions damage or kill cells. Magnetic particles can be toxic if they accumulate on cells or create stressful physical/chemical conditions.
Flow cytometry (FSC/SSC; fluorescence channels)
Single-cell fluorescence measurement used to quantify circuit activation distributions and cell health. FSC/SSC reflect size/complexity (helpful for viability/debris gating); fluorescence channels read out each reporter.
FSC and SSC (scatter signals)
“Shadows” of each cell measured by the instrument. FSC roughly relates to cell size; SSC relates to internal complexity/granularity. They help distinguish healthy cells from debris.
Gating
The process of selecting the subset of events that are real, healthy cells (and excluding debris/dead cells) before comparing fluorescence between conditions.
Fluorescence microscopy
Imaging used to qualitatively inspect spatial patterns (e.g., particle distribution, apparent reporter expression) before or alongside cytometry.
The first aim of my final project is to test whether magnetic fields can act as a control input for receptor activation by comparing standard diffusion-driven signalling against magnetically localized signalling in engineered mammalian cells.
This will be achieved using ligand-coated magnetic nanoparticles, SNIPR receptors, and a Csy4-based neuromorphic genetic circuit capable of producing graded nonlinear responses rather than simple binary outputs. The project will involve protein conjugation protocols, mammalian cell transfection, plasmid engineering, magnetic actuation systems, fluorescence microscopy, and flow cytometry to measure how magnetic localization changes receptor activation and downstream genetic circuit behaviour.
The second aim of this project is to develop a more spatially precise and controllable magnetic signalling system beyond the scope of this course. This includes exploring engineered or biologically derived magnetic particles with improved localization properties, optimizing magnetic field architectures for finer spatial control, and developing methods for rigorously tuning receptor activation strength in space and time. Ultimately, this would enable highly targeted control of signalling interactions and biological computation within complex multicellular environments.
The long-term vision of this project is to create remotely programmable and continuously tuneable living systems that bridge external physical control with biological computation. If fully realized, magnetic fields could become a programmable control layer for biology, enabling cells to compute, communicate, and respond in spatially organized ways directed by external fields rather than passive molecular diffusion alone.
One potential application is adaptive cell therapies, where engineered immune cells could be remotely activated only at specific regions within the body using magnetic localization, reducing off-target effects and enabling dynamically adjustable therapeutic responses in real time. Rather than cells remaining permanently ON or OFF after administration, magnetic control could allow therapies to be spatially focused, modulated, or reprogrammed after delivery depending on the patient’s condition.
Another possibility is programmable tissues, where external magnetic fields organize how groups of cells signal, differentiate, and interact across space. Instead of tissues behaving as static engineered structures, magnetic localization could allow cellular behaviour and computation to be dynamically patterned over time, enabling responsive biomaterials and living systems whose properties can be externally adjusted after formation.
More broadly, this points toward a new paradigm of field-controlled biology, where physical systems such as magnetic fields act as external control architectures for living matter, blurring the boundary between biological systems, computation, and programmable physical control.
One major limitation in cellular engineering is that traditional receptor systems are difficult to precisely program and control. Most receptors rely on endogenous signalling pathways, meaning they feed into the cell’s natural communication networks which evolved for complex biological behaviour rather than clean engineered outputs. For example, immune cells use receptors and antibodies to recognize antigens such as viruses or abnormal proteins, but once activated, these signals propagate through large interconnected networks involving many genes, signalling cascades, and feedback loops simultaneously. As a result, receptor activation often produces broad and difficult-to-predict downstream responses rather than precise programmable behaviour.
A major development addressing this limitation has been the creation of synthetic Notch receptor systems such as SNIPRs (Synthetic Notch Intramembrane Proteolysis Receptors). Unlike traditional receptor systems, SNIPRs are engineered to convert external binding events directly into customized gene expression outputs. When a ligand binds to the receptor, mechanical pulling forces trigger receptor cleavage, releasing an intracellular transcription factor which enters the nucleus and activates specific engineered genes chosen by the researcher. This allows cells to execute highly programmable responses to external signals with much cleaner input-output control. The recent paper Engineering precise cell-therapeutic function via synthetic Notch receptors demonstrates how synthetic Notch systems can be engineered to tightly control cellular therapeutic behaviour and programmable responses.
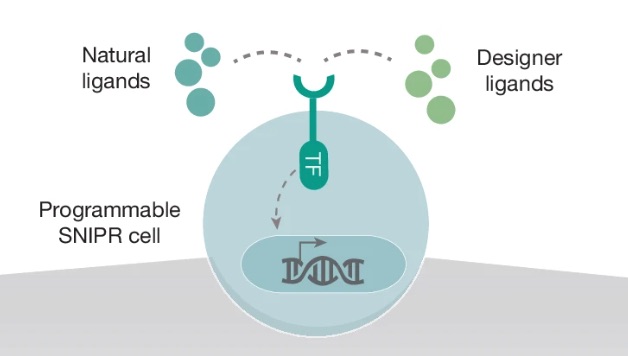
Figure 1: Programmable SNIPR cell
A ligand is simply a molecule that binds to a receptor, similar to how a key fits into a lock. In biology, ligands are often proteins or signalling molecules that tell cells when to activate certain behaviours. Natural ligands are the molecules cells normally encounter inside the body, while designer ligands are engineered molecules created by researchers to intentionally control cellular behaviour.
This diagram shows a programmable SNIPR cell. The receptor sticking out from the surface of the cell is the SNIPR receptor itself. The outside part of the receptor acts like a sensor and is designed to recognize a specific ligand, either natural or engineered. In this project, an example ligand is GFP attached to magnetic nanoparticles.
When the ligand binds to the receptor, it physically activates the SNIPR system. This causes the receptor to be cleaved, meaning part of it is cut and released inside the cell. The released component is the transcription factor (TF) shown in the diagram.
A transcription factor is a protein that controls gene expression by binding to DNA and turning specific genes ON or OFF. You can think of it like a biological switch or control signal for the cell’s genetic program. In SNIPR systems, the transcription factor acts as the messenger connecting an external event outside the cell to a programmed response inside the nucleus.
After being released from the receptor, the transcription factor travels into the nucleus, where the cell stores its DNA. Once inside, it binds specific engineered DNA sequences and activates genes chosen by the researcher. These genes can produce fluorescent proteins, therapeutic molecules, or other programmed cellular behaviours.
The key idea behind SNIPRs is that they decouple sensing from the cell’s natural signalling systems. Instead of triggering large messy biological pathways, ligand binding directly activates a clean programmable output, allowing researchers to engineer cells with much more precise input-output behaviour.
A major inspiration for this project comes from the paper Engineering programmable material-to-cell pathways via synthetic Notch receptors, which explores how engineered materials can directly control cellular behaviour using synNotch receptors.
In the paper, researchers attached signalling ligands directly onto materials such microparticles and extracellular matrix scaffolds (structural protein networks that surround and support cells in tissues) so that cells expressing synNotch receptors would only activate when they physically touched or came very close to regions containing the ligand. Rather than targeting individual cells directly, they controlled where signalling occurred by controlling where the ligands were positioned within the material itself. Cells located near different patterned regions encountered different ligands, activated different synNotch programs, and produced different cellular behaviours depending on their position. This allowed the researchers to spatially organize gene expression and cellular differentiation patterns across the tissue with microscale precision.
A major takeaway from this work is that spatial organization itself can act as a powerful control mechanism in biology. However, in the original paper, signalling interactions still largely depended on passive diffusion and static material positioning.
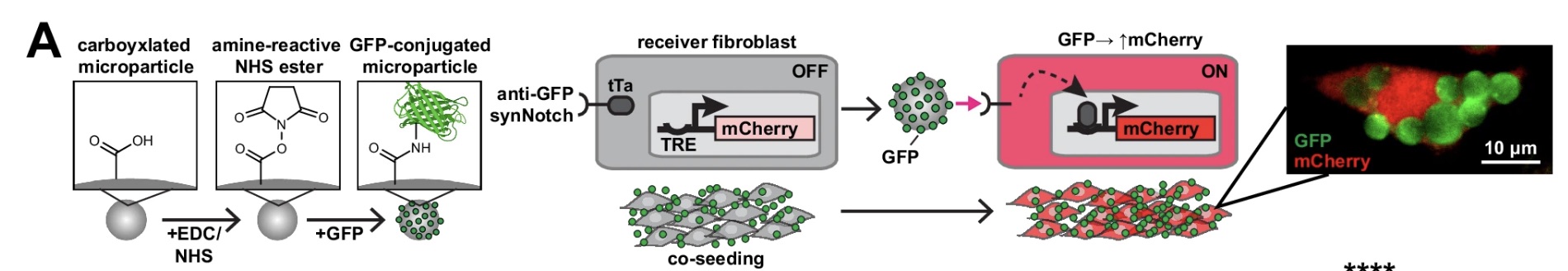
Figure 2: GFP-conjugated microparticles activating anti-GFP synNotch receptors and downstream mCherry expression.
In this work, the researchers demonstrated conjugating GFP proteins onto magnetic microparticles using EDC/NHS coupling chemistry. The microparticles first contained carboxyl (-COOH) groups on their surface. EDC/NHS chemistry converted these carboxyl groups into reactive NHS esters, creating chemically active attachment sites. GFP proteins, which contain primary amine groups (-NH₂), were then added and covalently bound to the particle surface, producing stable GFP-coated microparticles.
These GFP-coated particles then interacted with mammalian cells expressing anti-GFP synNotch receptors. When GFP bound the receptor, mechanical pulling forces triggered receptor cleavage, releasing a tTa transcription factor which activated a TRE promoter and turned on downstream mCherry gene expression. In effect, the physical interaction between the particle and the receptor became directly converted into programmable gene activation inside the cell.
The paper demonstrated that localized ligand presentation could directly shape gene expression patterns and cellular organization with microscale precision. However, although magnetic microparticles were used, their magnetic properties were primarily used for preparation and separation rather than active biological control. Once positioned within the material environment, signalling still relied largely on passive diffusion and cell-generated pulling forces.
Our project extends this concept by investigating whether external magnetic fields can actively manipulate these magnetic nanoparticles in space and time to directly influence signalling interactions and the dynamics of a synNotch system. Rather than using the particles as static signalling materials, we explore whether magnetic control can dynamically localize, focus, and modulate receptor activation itself.
Another important paper for our project is Synthetic neuromorphic computing in living cells. This paper introduced a way to build genetic circuits that behave less like simple ON/OFF switches and more like biological computing units capable of processing graded inputs.
Neuromorphic genetic circuits are engineered gene-regulation systems that behave more like analog computing elements than binary logic gates. A useful way to think about them is as single computing units that can be tuned so the cell produces a wide range of input-to-output relationships. In this sense, they act like biological versions of function-approximating systems, where a cell can process an input signal and convert it into a more complex output response.
In our project, the synNotch/SNIPR receptor provides the input. When the receptor binds to its ligand, it releases a transcription factor that activates gene expression. That transcriptional signal is then passed into a neuromorphic genetic circuit, which shapes the final output.
The neuromorphic unit can be understood as having two opposing sides:
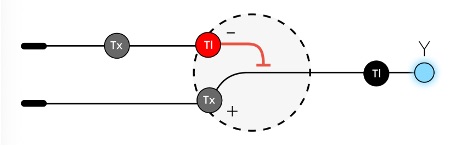
Figure 3: Neuromorphic Unit
Positive terminal:
The expression-driving side of the circuit. This terminal contains the reporter gene, such as mNeonGreen, along with a Csy4 target site built into its RNA transcript. When the positive terminal is transcribed, it produces mRNA that could be translated into fluorescent protein. However, because that mRNA contains the Csy4 target site, it can also be recognized and cleaved by Csy4 from the negative terminal.
Negative terminal:
The inhibitory side of the circuit. The input to this terminal could be, for example, Csy4, an RNA-processing enzyme that directly regulates the output from the positive terminal. Csy4 recognizes a specific target sequence placed on the positive-terminal mRNA and cleaves that transcript. Once the mRNA is cut, it becomes less stable or less efficiently translated, reducing how much mNeonGreen protein is ultimately produced.
By combining these two terminals, the circuit compares an expression-driving signal against an inhibitory RNA-processing signal. The final level of mNeonGreen depends on the balance between how much transcript is produced and how much of that transcript is repressed, cleaved, or destabilized. This allows the system to convert different levels of receptor activation into graded or nonlinear fluorescence outputs, rather than behaving like a simple binary reporter.
This is important because receptor activation in our system is unlikely to be purely ON or OFF. Magnetic particle positioning, ligand density, receptor engagement time, and local receptor clustering can all vary continuously, creating different levels of transcriptional activation. A simple reporter circuit could show whether activation occurred, but a neuromorphic circuit allows the cell to process the magnitude of that activation and convert it into a programmable graded or nonlinear output. In this way, the circuit does not just detect receptor activation; it interprets it. This makes the system better suited for field-controlled biology, where the input signal may vary spatially and temporally rather than behaving like a clean binary switch.
This project is novel because it proposes using magnetic nanoparticles not simply as passive ligand carriers, but as an active and dynamically controllable signalling system. Existing synNotch and SNIPR systems rely heavily on diffusion-driven interactions, meaning researchers have limited control over where signalling and computation occur once ligands are introduced into a biological environment. In contrast, this project explores whether external magnetic fields can spatially direct signalling interactions with microscale precision, potentially allowing biological computation itself to be focused and organized in space.
The project is also innovative because it combines magnetic control systems with neuromorphic genetic circuits. Instead of producing only binary ON/OFF outputs, the system integrates neuromorphic computation capable of generating graded and nonlinear responses. This creates the possibility of continuously tuneable biological systems where external physical fields dynamically shape cellular computation and behaviour.
More broadly, the project challenges the assumption that biological signalling must primarily rely on passive diffusion. By introducing programmable magnetic localization as an external control layer, the work expands synthetic biology toward more physically programmable and spatially organized living systems.
If we are able to fully control biological signalling using magnetic fields, it could introduce an entirely new paradigm for how engineered cells and therapies interact with the body. Instead of cells passively responding to random molecular encounters through diffusion, magnetic fields could allow signalling and cellular computation to be dynamically focused in space and time with much greater precision. This could make engineered biological systems externally programmable in ways that are currently very difficult to achieve.
One potential application is adaptive cell therapy. Imagine a patient receiving engineered therapeutic cells or magnetic nanoparticle-based treatments through an injection, and then later passing through a localized magnetic field system capable of concentrating signalling interactions only at a diseased region of the body. Rather than activating therapies everywhere, magnetic localization could potentially focus cellular activation only near specific tissues, injuries, or tumour sites. Similar concepts could eventually be explored for spatially targeted cancer therapies, where engineered immune or signalling systems activate preferentially near localized cancerous regions while minimizing effects on surrounding healthy tissue.
More broadly, the project contributes to the growing intersection between synthetic biology, computation, and physical control systems. Even at a small experimental scale, demonstrating magnetically localized signalling would expand current technical capabilities for controlling multicellular behaviour and spatial organization inside living systems. In the long term, this could contribute toward programmable tissues, externally tuneable biomaterials, and future biological systems where physical fields directly organize and control cellular computation.
This project raises important ethical questions surrounding the engineering and external control of living systems. One concern involves non-maleficence, particularly whether remotely controllable biological systems could behave unpredictably or produce unintended effects if eventually translated into therapeutic settings. Because the project introduces external magnetic control over cellular signalling, it is important to carefully evaluate how engineered cells respond under different conditions and whether unintended activation or off-target interactions could occur. The project also raises broader questions about how far synthetic biology should move toward programmable living systems and what responsibilities researchers have when designing technologies capable of externally manipulating cellular behaviour.
To ensure the research is conducted ethically, the project remains entirely within controlled in vitro laboratory environments using standard mammalian cell culture systems and established synthetic biology methods. Experimental validation, controls, and reproducibility are important because one major uncertainty is whether magnetic localization will meaningfully influence signalling dynamics at all. It is possible that magnetic fields may not produce sufficient mechanical or spatial control to significantly alter receptor activation compared to normal diffusion-driven interactions. Alternative approaches, such as optogenetic or chemically inducible signalling systems, already exist and may ultimately prove more practical for certain applications. The broader goal of this work is therefore exploratory: to responsibly investigate whether magnetic fields can become a useful new control modality for synthetic biological systems while carefully considering long-term safety, governance, and societal implications.
Our experimental design tests whether magnetic fields can localize GFP-based signalling and change activation of a synthetic SNIPR/neuromorphic circuit in mammalian cells.
1. Produce and purify GFP
Express GFP in bacteria, lyse the cells, and purify the protein through elution so it can be used as the signalling ligand.
2. Conjugate GFP to magnetic nanoparticles
Attach purified GFP onto magnetic nanoparticles using protein conjugation chemistry, creating magnetic signalling particles that can bind anti-GFP receptors.
3. Build the magnetic actuation device
Construct a device capable of dynamically moving magnets around the cell culture environment to control where the magnetic nanoparticles localize.
4. Design the synthetic SNIPR and neuromorphic circuit plasmids
Design plasmids encoding the anti-GFP SNIPR receptor, downstream reporter outputs, and the Csy4-based neuromorphic circuit.
5. Transfect mammalian cells across different circuit configurations
Introduce the SNIPR and circuit plasmids into mammalian cells, then expose them to GFP-coated magnetic nanoparticles under magnetic and non-magnetic conditions. Test different circuit conditions with and without magnetic fields, including diffusion-only controls, magnetic nanoparticle controls, receptor/no-receptor controls, and reporter controls.
6. Measure activation and compare outcomes
Use fluorescence microscopy and flow cytometry to compare spatial activation patterns and circuit outputs across the different experimental conditions.
To generate the signalling ligand for our system, we expressed His-tagged GFP in bacteria and purified it using nickel affinity purification with Ni-NTA resin
1. Prepare the buffers
Prepare binding, wash, and elution buffers using PBS, NaCl, and different imidazole concentrations.
Binding buffer: 10 mM imidazole
Wash buffer: 30 mM imidazole
Elution buffer: 250 mM imidazole

2. Pellet the bacterial cells
Centrifuge the bacterial culture to collect the cells and discard the supernatant.

Figure 6: Centrifuging the bacterial culture
3. Lyse the cells
Resuspend the pellet in B-PER and binding buffer to break open the cells and release GFP into solution.
4. Clarify the lysate
Centrifuge the lysed sample and collect the clear GFP-containing supernatant.
5. Prepare the Ni-NTA resin
Equilibrate the nickel affinity resin using binding buffer so it is ready to capture His-tagged GFP.
6. Bind GFP to the resin
Incubate the cleared lysate with the Ni-NTA resin, allowing the His-tagged GFP to bind to the nickel surface.

Figure 7: Tube rotator used to incubate
7. Wash the resin
Wash the resin multiple times using wash buffer to remove non-specific proteins and contaminants.
8. Elute purified GFP
Add elution buffer containing high imidazole concentration to release purified GFP from the resin and collect the fluorescent green fractions.

Figure 8: Purified GFP fractions visualized using the E-Gel
To implement magnetically controlled ligand presentation, we adapted the conjugation strategy used in the paper.
Engineering programmable material-to-cell pathways via synthetic Notch receptors
The original paper used Magsphere MCA5UM magnetic microparticles to attach GFP ligands onto particle surfaces for synNotch activation experiments. In our project, we substituted these with MagnaBind Carboxyl Derivatized Beads from Thermo Fisher, which provide similar surface carboxyl (–COOH) chemistry for protein attachment while also possessing the properties required for downstream magnetic manipulation experiments.
MagnaBind beads: MagnaBind Carboxyl Derivatized Beads
Documentation: MagnaBind User Guide
Additional reagents included PBS buffer, EDC coupling reagent, and MES conjugation buffer.
EDC: EDC Coupling Reagent
MES Buffer: MES Buffer
The conjugation process began by washing the magnetic beads several times with PBS to remove storage solution and prepare the bead surface. Purified GFP protein was then added to the beads. To chemically attach the GFP, EDC coupling chemistry was used to activate the carboxyl groups on the bead surface, creating reactive intermediates capable of binding the primary amine groups naturally present on GFP proteins. This forms stable covalent bonds between the GFP and the magnetic particle surface.
After incubation, we used neodymium n52 magnets to isolate the GFP-coated beads from the surrounding solution. The beads were washed multiple times with PBS to remove unbound protein and residual reagents, leaving purified GFP-conjugated magnetic nanoparticles ready for downstream signalling experiments with the synNotch/SNIPR system.
1. Wash the magnetic beads
Wash MagnaBind magnetic beads multiple times with PBS to remove storage solution and prepare the bead surface.

Figure 9: MagnaBind beads vs n52 neodymium magnets
2. Prepare the GFP solution
Dissolve purified GFP protein in conjugation buffer at the appropriate concentration.

Figure 10: Creating the MES conjugation buffer
3. Mix GFP with the magnetic beads
Add the GFP solution to the washed magnetic beads and gently agitate to evenly distribute the protein around the particles.

FIgure 11: Adding the GFP + MES buffer solution to beads.
4. Activate the bead surface using EDC chemistry
Freshly prepare EDC solution in MES buffer and add it to the GFP-bead mixture. EDC activates the carboxyl groups on the bead surface, allowing them to react with amine groups on GFP proteins.

Figure 12: Preparing the EDC solution in the fume cupboard
5. Incubate the conjugation reaction
Incubate the mixture at room temperature to allow covalent attachment of GFP onto the magnetic nanoparticles.
6. Magnetically separate the GFP-coated beads
Use n52 neodymium magnets to isolate the GFP-conjugated magnetic nanoparticles from the surrounding solution.
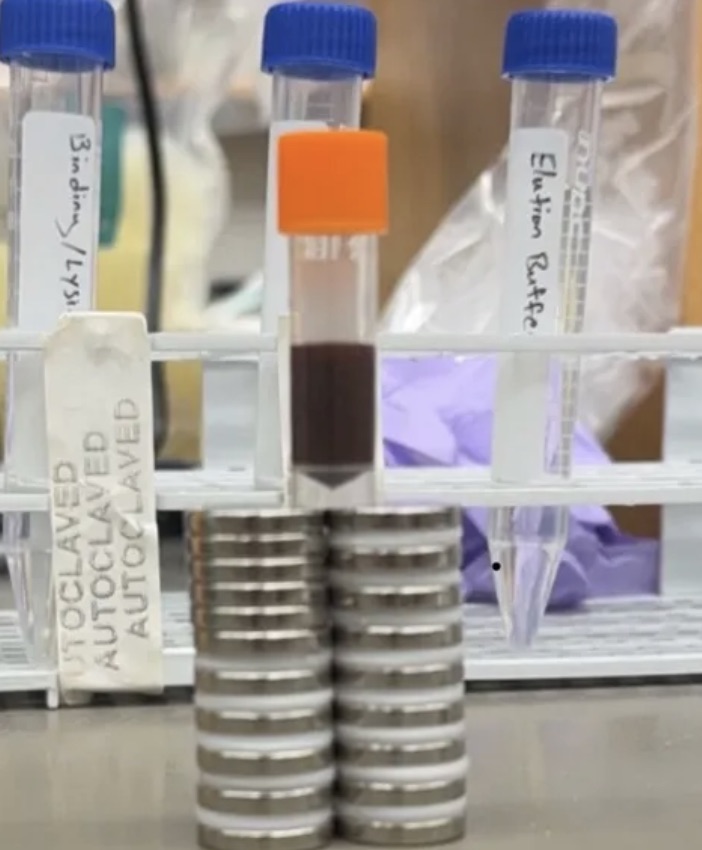
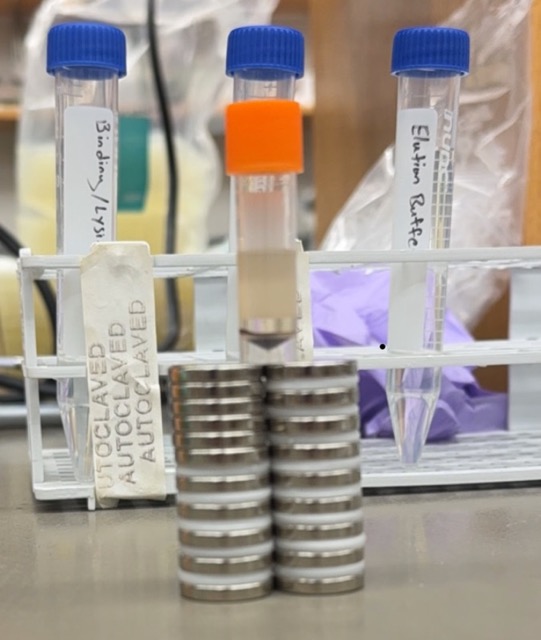
Figure 13: Separating magnetic nanoparticles using N52 neodymium magnets
7. Wash away excess reagents and unbound GFP
Wash the GFP-coated magnetic nanoparticles multiple times with PBS to remove residual chemicals and unconjugated protein.
8. Collect the final GFP-conjugated magnetic nanoparticles
The purified GFP-coated magnetic nanoparticles are then ready for downstream synNotch/SNIPR signalling experiments and magnetic localization studies.
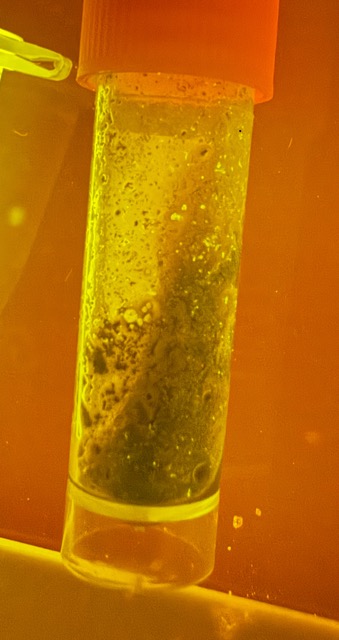
Figure 14: Magnetically Conjugated GFP under E-Gel
A major goal of this project was to use magnetic fields as a dynamic external control layer for synNotch receptor activation. To achieve this, we designed an orbiting magnetic actuation system where strong N52 neodymium magnets rotate around the cell culture dish using a motorized circular track. This setup allows magnetic fields to continuously move around the cells, dynamically repositioning GFP-coated magnetic nanoparticles within the culture environment.
The design was motivated by the mechanosensitive nature of synNotch receptors, where receptor activation depends on physical ligand-receptor interactions and pulling forces. By orbiting the magnets around the dish, we aimed to spatially localize the magnetic nanoparticles near engineered cells and increase the probability of repeated ligand-receptor engagement over time. In principle, this allows the magnetic field to influence where signalling interactions occur and potentially increase downstream synNotch activation and cellular computation.


Figure 15: Soldering the motor Figure 16: Internal Electronics of the device

Figure 17: Fabricated Orbiting Track Design
During development, we also explored an alternative magnetic control strategy based on inhibition rather than activation. Instead of concentrating nanoparticles near the cells, magnets could also be positioned to spatially separate or suspend the GFP-coated magnetic nanoparticles away from the cell surface, reducing ligand-receptor interactions. We ultimately focused on this inhibitory approach because it provided a cleaner and more experimentally controllable way to test whether magnetic localization could directly influence synNotch signalling dynamics.
To implement the system, we designed a set of plasmids that define each layer of sensing, signalling, and computation within the engineered cellular circuit. Together, these plasmids create a single-cell neuromorphic system capable of converting external magnetic signalling inputs into programmable genetic outputs.
1. Sensor Plasmid
The sensor plasmid encodes an anti-GFP synNotch/SNIPR receptor fused to an intracellular transcription factor. This allows engineered mammalian cells to detect GFP-coated magnetic nanoparticles and convert ligand binding into downstream gene activation. The plasmid also constitutively expresses mMaroon as a fluorescent marker, allowing us to identify cells that successfully received the receptor plasmid during transfection. We can simply use the plasmid from the engineering programmable materials https://www.addgene.org/79127/
2. Response Program Plasmid
The response plasmid contains a synNotch-responsive promoter that drives expression of mNeonGreen after receptor activation. This construct also contains a Csy4 target site, allowing downstream post-transcriptional regulation of the output signal by the neuromorphic circuit. A constitutive BFP marker is included to identify cells successfully expressing this plasmid independently of receptor activation. This plasmid is a lot more custom and will require some thoughtful plasmid design.
3. Computation / Neuromorphic Circuit Plasmid
The final plasmid encodes the Csy4 processing enzyme, which acts as the computational layer of the system. Csy4 interacts with the target site on the response plasmid to regulate mNeonGreen expression and generate graded, nonlinear circuit behaviour rather than simple binary ON/OFF outputs.
The fluorescent marker proteins are important because mammalian cell transfection is inherently variable, not every cell receives every plasmid equally. By including constitutive fluorescent markers on each construct, we can identify which cells successfully received specific plasmids and distinguish true circuit behaviour from failed transfection events using flow cytometry. This allows more accurate interpretation of signalling and computation within the engineered cellular system.
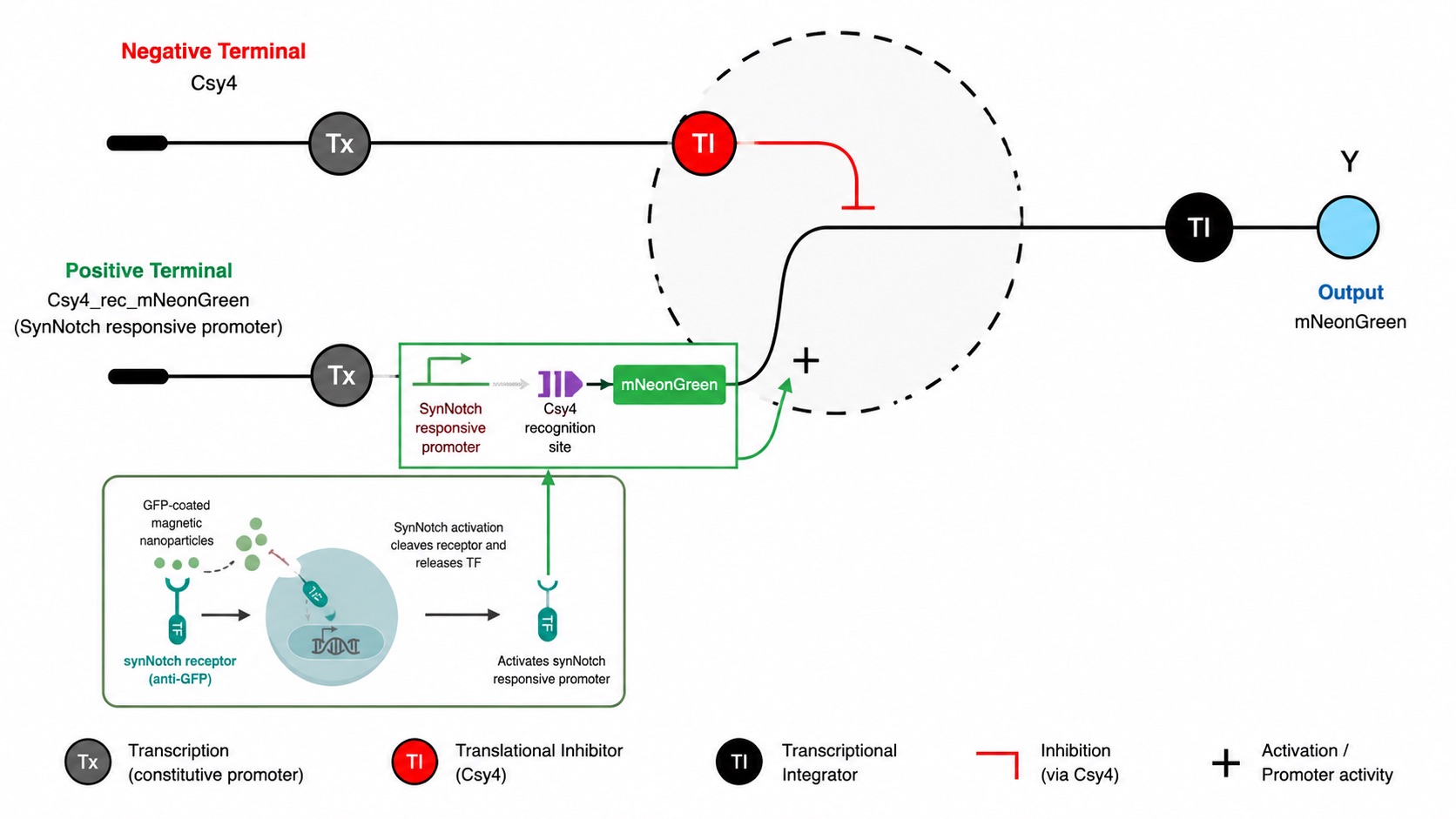
Figure 18: Circuit Architecture of our system
To construct the response layer of our neuromorphic synNotch system, we designed a custom response program plasmid based on the pHR_5x Gal4 UAS backbone from Addgene and modified it using Benchling.
https://www.addgene.org/79119/
The plasmid architecture was designed as:
The 5x Gal4 UAS region acts as the transcription factor binding site for the synNotch-released Gal4 transcription factor. Once synNotch activation occurs, the released transcription factor binds the UAS region and activates the downstream minimal promoter, initiating expression of the response cassette.
1. Identify the synNotch-responsive activation region
We first identified the 5x Gal4 UAS sequence within the template plasmid. This region contains repeated Gal4 transcription factor binding sites and acts as the programmable entry point for synNotch-driven activation. Downstream of this region sits the minimal promoter, which remains largely inactive until Gal4 binding occurs.
During synthesis preparation, we found that Twist Bioscience flagged the highly repetitive UAS regions as difficult to synthesize due to sequence repetition and instability concerns. To address this, we slightly modified the repeated UAS sequences while preserving their overall Gal4 binding functionality. This allowed the plasmid to remain compatible with DNA synthesis constraints while maintaining the intended synNotch-responsive behaviour.
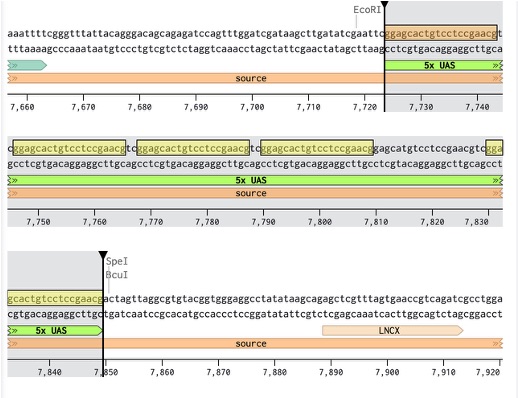
Figure 19: Identified UAS sequence in Benchling
2. Locate the insertion region
Next, we identified the multiple cloning site (MCS), a dense restriction-site region designed for inserting new genetic payloads. This served as the insertion point for our custom response module. Importantly, the insertion region was chosen upstream of the WPRE regulatory sequence so downstream regulatory elements within the plasmid backbone remained intact.
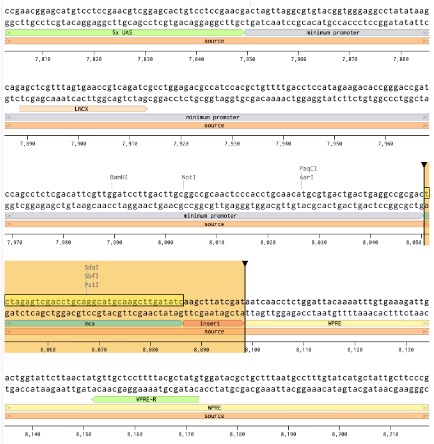
Figure 20: Identification of MCS
3. Insert the response module and output cassette
The original insert region was then replaced with our engineered response module:
The Csy4 target site was positioned directly upstream of the Kozak sequence so that the downstream transcript could later be regulated by the Csy4 neuromorphic circuit layer. The Kozak sequence was included to ensure efficient translation initiation in mammalian cells, while mNeonGreen acts as the fluorescent reporter output for synNotch activation.
Csy4 target sequence used:
Kozak sequence used:
mNeonGreen sequence source: https://www.ncbi.nlm.nih.gov/nuccore/KC295282
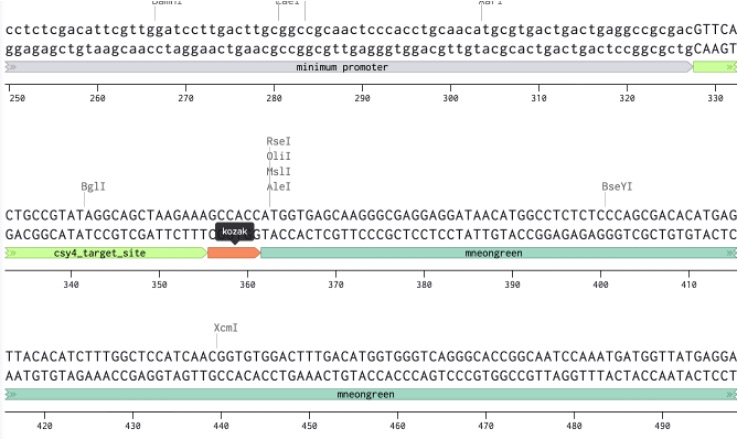
Figure 21: Custom Insert in our Benchling sequence
The final plasmid therefore converts synNotch receptor activation into a fluorescent mNeonGreen output while also enabling downstream post-transcriptional regulation through the Csy4 neuromorphic circuit. The completed plasmid design was then prepared (removed backbone and replaced) and then submitted to Twist Bioscience for synthesis and assembly
https://benchling.com/s/seq-M3U6H8Li4bqkH59ekZzA?m=slm-Be8uJzzgL6c80IIrUaAX
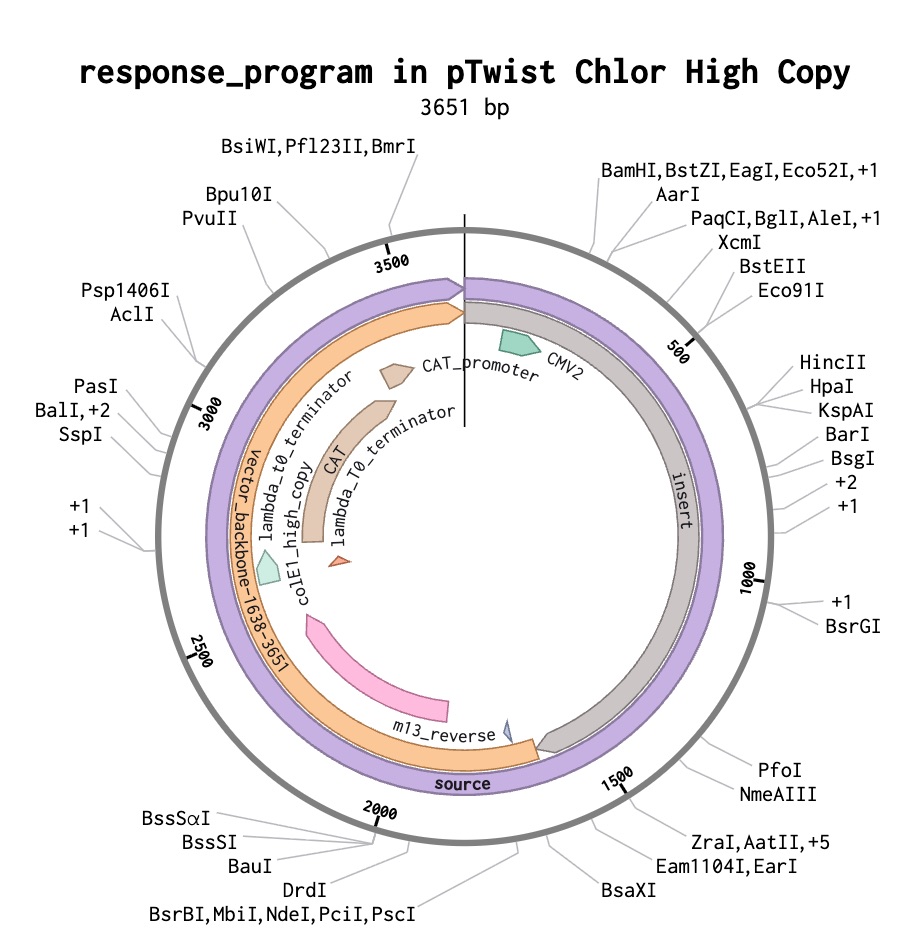
Figure 22: Final Plasmid ordered
To prepare the mammalian cells for transfection, cell culture medium was first warmed using metal bead baths to bring the media to physiological temperature before handling the cells. Maintaining proper temperature is important to reduce stress on the cells and preserve healthy growth conditions during the experiment.

Figure 23: Metal bead bath for mammalian cells media
Mammalian cells were then detached from the bottom of the culture flask using trypsin, an enzyme that breaks down the adhesion proteins holding the cells to the surface. After detachment, the cells were resuspended in fresh culture medium and transferred into tubes for downstream preparation.
Figure 24: Using trypsin to detach mammalian cells
The cells were inspected under a microscope. Cell concentration and viability were then measured using an automated cell counter, which estimates how many cells are alive and suitable for the experiment ~5,280,000.
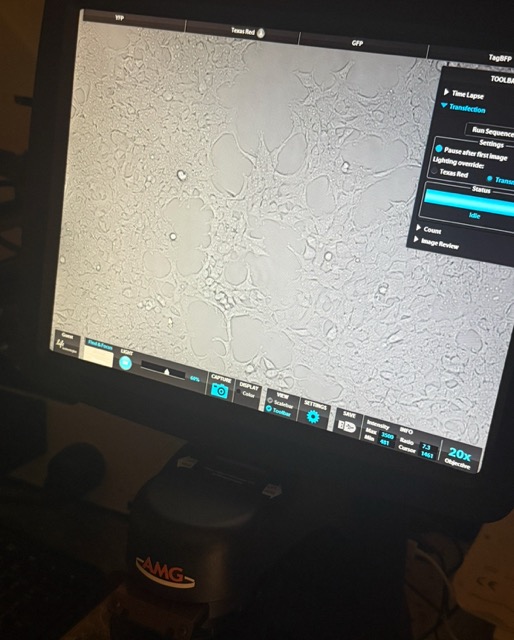

Figure 25: Mammalian cells under microscope and automated cell counter
Following cell counting, the mammalian cells were mixed with fresh medium and plated into multi-well culture plates across different experimental circuit configurations. The plates were then incubated overnight

Figure 26: Mammalian cell medium Figure 27: Mammalian cells incubation

Figure 28: Experiment Notes
We designed the experiment around two plates: one plate without magnets and one plate exposed to magnets. This allowed us to compare normal diffusion-driven signalling against magnetically influenced signalling.
The non-magnetic plate included several control conditions:
- GFP + circuit
Tests whether free GFP alone activates the circuit without magnetic particles.
- Magnetic beads + circuit
Tests whether the magnetic beads themselves affect the cells or circuit without GFP attached.
- Neuromorphic circuit alone
Baseline control to measure circuit behaviour without GFP or magnetic beads.
- MagGFP + circuit
Tests whether GFP-conjugated magnetic particles activate the circuit under normal diffusion-driven conditions without an external magnet.
The magnetic plate included wells containing:
MagGFP + circuit with weaker magnetic exposure
MagGFP + circuit with stronger magnetic exposure
Both wells used GFP-conjugated magnetic nanoparticles with the circuit, but different magnet strengths or positions were applied to test whether magnetic localization changed receptor activation.
We started with GFP at 2 mg/mL and conjugated it to 60 µL of magnetic beads. Assuming complete conjugation, this gave an estimated effective MagGFP concentration of about 8.3 mg/mL on the beads. Based on this concentration, we calculated that each MagGFP condition could receive up to 332 µL per well.
On the second day of the experiment, we transfected the mammalian cells with our synNotch and neuromorphic circuit plasmids using a lipid nanoparticle-based transfection method.
Because the plasmids arrived freeze-dried, they first needed to be resuspended in nuclease-free water to a working concentration of approximately 50 ng/µL. The tubes were briefly vortexed and centrifuged to ensure the DNA fully dissolved and collected at the bottom of the tube before use. Different plasmid combinations were then prepared for the various experimental controls and circuit configurations.

Figure 29: Vortexing the freeze dried DNA with nuclease-free water
To introduce the plasmid DNA into the mammalian cells, we used a lipid nanoparticle-based transfection system. These reagents work by encapsulating DNA inside microscopic lipid particles which can fuse with the cell membrane and deliver the genetic material into the cell interior. To prepare the transfection mixtures, plasmid DNA and lipid reagents were first diluted separately in Opti-MEM reduced-serum media, since full-serum media can interfere with lipid nanoparticle formation and reduce transfection efficiency.
Figure 30: Plasmid DNA and lipid reagents
Reagents such as P3000 and L3000 were then combined with the plasmid mixtures to promote formation of the lipid-DNA complexes and improve cellular uptake efficiency. Different plasmid combinations were prepared across the experimental circuit conditions, including fluorescent marker controls, synNotch receptor plasmids, response program plasmids, and Csy4 neuromorphic circuit components.
Figure 31: Transfection preparation table showing plasmid combinations, fluorescent control markers, and lipid nanoparticle reagent conditions used across the experimental circuit configurations.
We also utilized a poly-transfection strategy developed in the Weiss lab from the paper Poly-transfection enables rapid, quantitative testing of genetic circuits in mammalian cells.
Instead of mixing all plasmids into the same lipid complex, separate lipid-DNA complexes were formed for different plasmids before being added simultaneously to the cells. This creates a wider distribution of plasmid uptake across the cell population, allowing different cells to receive different relative amounts of each circuit component.
Figure 32: Poly-Transfection into Mammalian cells
This approach is particularly useful for synthetic biology circuits because it allows more flexible exploration of how varying receptor, response, and computational plasmid ratios influence downstream circuit behaviour. Once the lipid nanoparticles were formed, the transfection mixtures were added into the mammalian cells across the different experimental well configurations and returned to the incubator for recovery and expression.
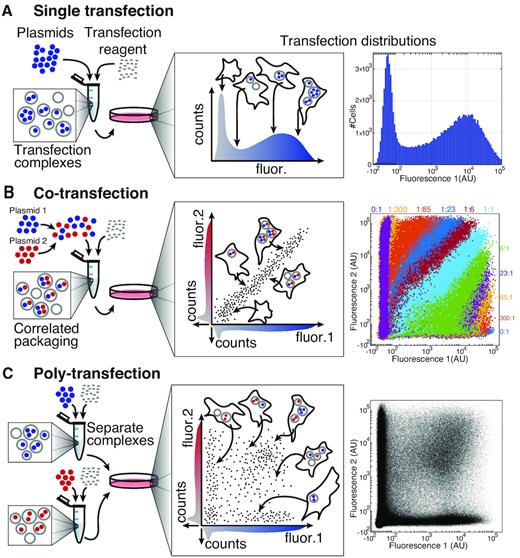
Figure 32: Overview and comparison of plasmid delivery with a single transfection, co-transfection of two plasmids, or poly-transfection of two plasmids.
After incubation, the self-assembled lipid nanoparticles containing the plasmid DNA were added directly into the plated mammalian cells across the different experimental circuit configurations. Depending on the condition, additional signalling components such as free GFP, magnetic beads alone, or GFP-conjugated magnetic beads (MagGFP) were also introduced alongside the circuit plasmids to test how different signalling inputs influenced synNotch activation and downstream circuit behaviour.
Figure 33: Experimental circuit configurations used for the non-magnetic plate. The top row contains fluorescent colour controls used for flow cytometry calibration and gating. The second row contains the main experimental conditions: GFP + circuit, magnetic beads + circuit, circuit-only control, and GFP-conjugated magnetic beads (MagGFP) + circuit.
Originally, our magnetic plate was designed to use the motorized magnetic actuation device to dynamically vary magnetic localization across the cell culture and potentially increase synNotch receptor activation. However, during testing the actuation device short-circuited, so we adapted the experimental design to test an alternative magnetic configuration.
Instead of trying to increase receptor activation through moving magnetic fields, we explored whether magnetic fields could inhibit signalling by spatially pulling GFP-conjugated magnetic nanoparticles (MagGFP) away from the cells. In this setup, neodymium magnets were suspended above selected wells to attract the magnetic nanoparticles upward and reduce ligand-receptor interactions at the cell surface.
To vary the magnetic field strength, we positioned the magnets at different distances from the wells. For the weaker magnetic condition, plastic spacers were placed between the magnet and the plate to increase the distance and reduce the magnetic field experienced by the particles. For the stronger magnetic condition, the magnets were placed directly against the plate without spacing. The hypothesis was that stronger magnetic fields would pull more MagGFP particles away from the cells, resulting in lower synNotch receptor activation compared to the weaker field condition.
Figure 34: Weak magnetic field configuration using spacers on the bottom left well. Strong magnetic field on the top right well Figure 35: Underside view of the magnetic plate showing reduced magnetic nanoparticle accumulation at the bottom of the well under the stronger magnetic field condition, consistent with particles being pulled upward toward the magnet.
The plates were then returned to the incubator to allow the cells to recover and begin expressing the engineered synNotch and neuromorphic circuit plasmids.

Figure 36: Incubation of our magnetic plate
After incubating the transfected mammalian cells overnight, we used fluorescent microscopy to qualitatively analyze circuit activation, GFP localization, and overall cell viability across the different experimental configurations. Fluorescent microscopy allows specific fluorescent proteins to be visualized inside living cells using different excitation and emission wavelengths. In our system, green fluorescence corresponded to GFP or mNeonGreen-related outputs, while red fluorescence acted as a marker for successfully transfected and viable mammalian cells. By comparing fluorescence patterns across the different well conditions, we could begin evaluating how magnetic nanoparticles and magnetic field exposure influenced cellular behaviour.
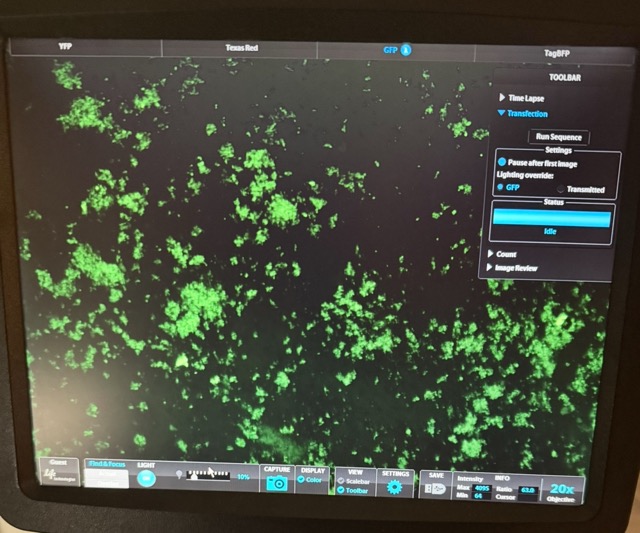
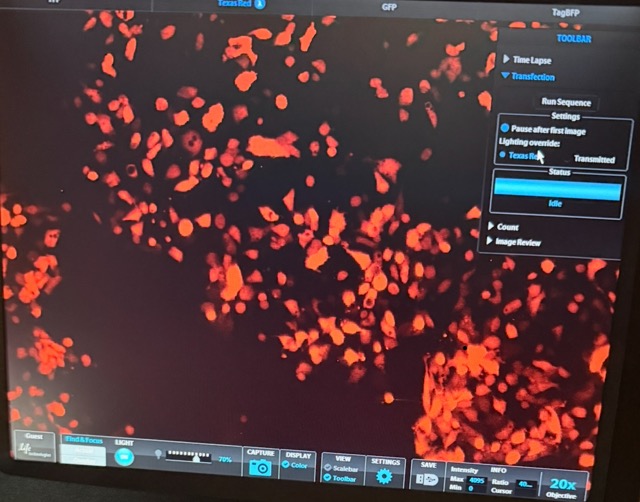
Figure 37 GFP + circuit microscopy results
Under the GFP + circuit well, we observed strong green fluorescence alongside clear red-labelled mammalian cells, indicating successful transfection and healthy cell growth. The green fluorescence confirms that free GFP was there and able to interact with the anti-GFP synNotch system and activate downstream circuit output, while the red fluorescence demonstrates that a large population of mammalian cells remained alive throughout the experiment.
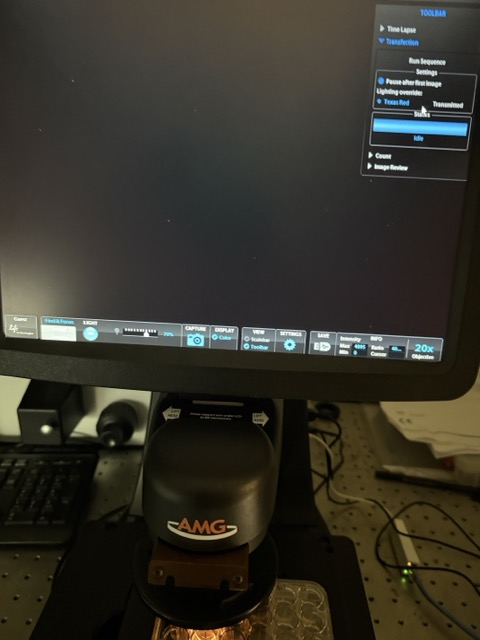
Figure 38: Magnetic beads + circuit - similar MagGFP + circuit microscopy results
In contrast, the magnetic beads + circuit and MagGFP + circuit conditions initially produced very limited visible fluorescence under the microscope. Several explanations may account for this observation. One possibility is that the magnetic nanoparticles interfered with optical imaging by scattering or blocking light, making it difficult to visualize fluorescence clearly. Another possibility is that the GFP conjugation to the magnetic nanoparticles was inefficient, preventing effective receptor activation. Additionally, the magnetic nanoparticles themselves may have negatively affected mammalian cell viability, reducing the number of observable fluorescent cells.
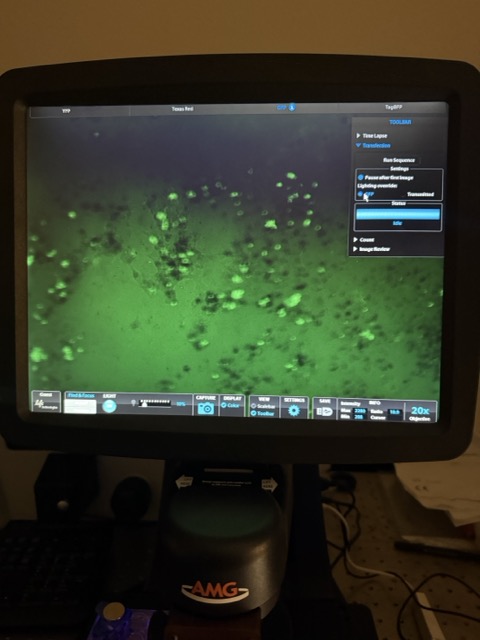
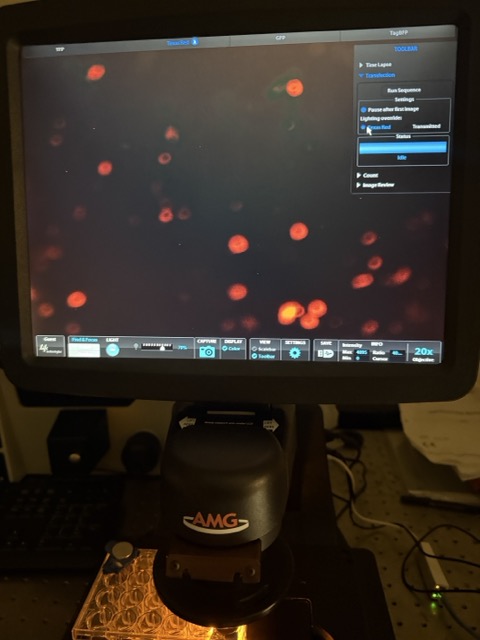
Figure 39: MagGFP + circuit with stronger magnetic exposure microscopy results
Interestingly, under strong magnetic exposure in the MagGFP + circuit condition, green fluorescence became clearly visible again. This strongly suggests that the magnetic GFP conjugation protocol was successful and that GFP remained attached to the magnetic nanoparticles. A likely explanation is that the stronger magnetic field pulled many of the magnetic nanoparticles upward toward the top of the well, reducing optical interference near the microscope focal plane and allowing the GFP signal to become visible. However, despite the visible GFP signal, relatively little red fluorescence was observed, suggesting some of the mammalian cells were killed by the magnetic nanoparticles.
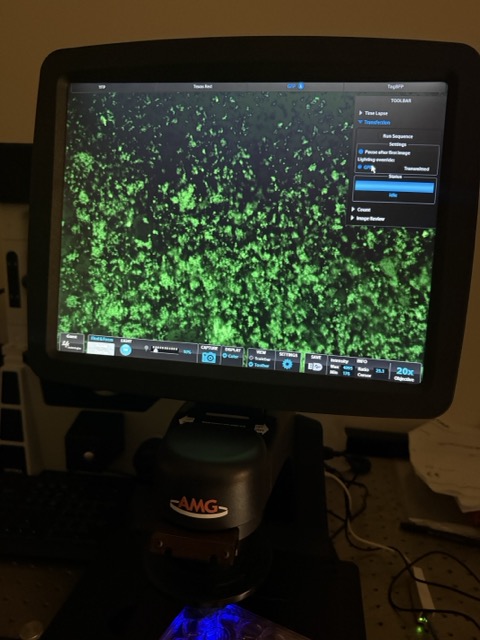
Figure 40: MagGFP + circuit with weaker magnetic exposure
Under weaker magnetic exposure, GFP fluorescence was still detectable. Similar to the strong field condition, very limited red fluorescence was observed, again suggesting either reduced cell survival or continued optical obstruction caused by suspended magnetic nanoparticles. While fluorescence microscopy provided useful qualitative observations, more precise quantitative analysis was required to distinguish between these possibilities. We therefore used flow cytometry to further analyze cell viability and circuit activation across the different experimental conditions.
Flow cytometry is a high-throughput technique that rapidly passes individual cells through lasers to measure fluorescence intensity and light scattering properties on a cell-by-cell basis. Unlike fluorescence microscopy, which provides mainly qualitative visual observations, flow cytometry allows precise quantitative measurement of how strongly each engineered cell is expressing specific fluorescent outputs.
Our experimental design used multiple fluorescent protein markers to track the different plasmids introduced during poly-transfection. The synNotch receptor plasmid was associated with the mMaroon marker, the Csy4 plasmid used the mKO2 fluorescent marker, and the response program plasmid used a BFP marker. The final mNeonGreen output acted as the primary readout of synNotch circuit activation. During flow cytometry analysis, different laser and detector channels were used to isolate each fluorescent signal independently. For example, the FITC-A channel was used to measure mNeonGreen fluorescence, the PE-Texas Red-A channel measured mKO2, the Pacific Blue-A channel measured BFP, and the Alexa Fluor 700-A channel detected mMaroon-associated fluorescence.

Figure 41: Flow Cytometer
Focusing specifically on the FITC-A channel of the flow cytometer, we were able to isolate and measure the mNeonGreen fluorescence output from our engineered synNotch/neuromorphic circuits. Since mNeonGreen acts as the final downstream output of circuit activation, the FITC-A channel provides a direct quantitative readout of how strongly the engineered cells responded under each experimental condition.
For every experimental well, the flow cytometer measured thousands of individual cells one-by-one as they passed through the laser system. Each cell therefore produced its own mNeonGreen fluorescence value depending on how strongly its circuit was activated. Because poly-transfection creates variability in plasmid uptake across the cell population, not all cells expressed the circuit components equally. Some cells received high amounts of plasmid DNA, some received lower amounts, and some received incomplete circuit combinations. Rather than producing a single fluorescence value for an entire well, this generated a broad distribution of fluorescence intensities across the population.
To visualize this, we constructed a density plot of mNeonGreen fluorescence output. In this plot, the x-axis represents mNeonGreen fluorescence intensity (MEF), while the y-axis represents the relative density of cells at each fluorescence level. Importantly, density does not represent fluorescence itself it reflects how many cells fall within a particular fluorescence range. Peaks in the curve therefore correspond to fluorescence values shared by large numbers of cells.
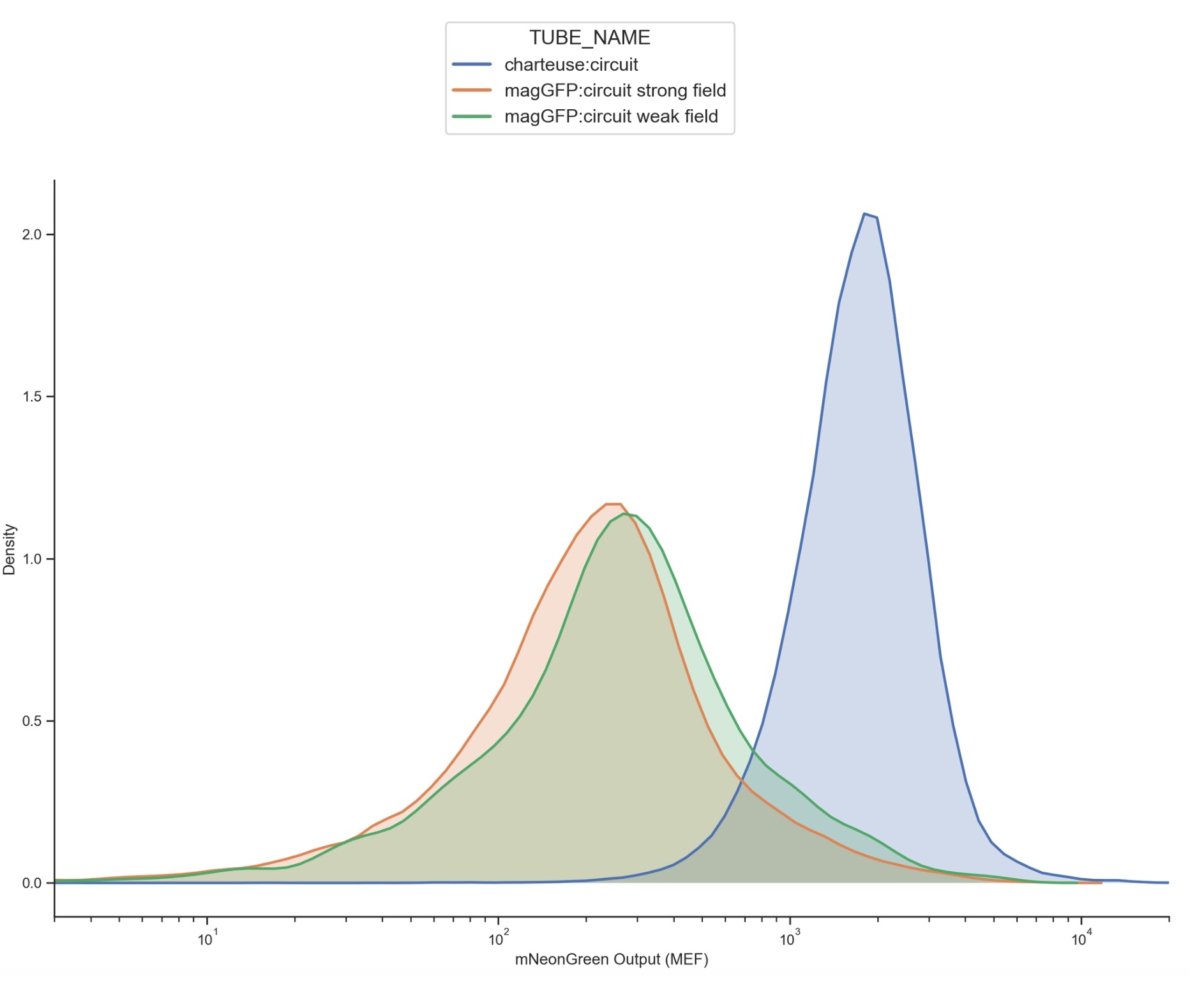
Figure 42: Flow Cytometry distribution plot
The blue distribution represents the baseline circuit-only control without magnetic GFP signalling input. This population is shifted significantly further to the right, indicating substantially higher mNeonGreen fluorescence across the majority of cells. This suggests strong baseline activation of the circuit in the absence of magnetic nanoparticle modulation.
The orange and green distributions represent cells exposed to GFP-conjugated magnetic nanoparticles under strong-field and weak-field magnetic conditions respectively. Both magnetic conditions are shifted leftward relative to the control, indicating substantially reduced mNeonGreen output and therefore lower downstream activation of the synNotch/neuromorphic system when magnetic localization of the nanoparticles was introduced.
Importantly, the weak-field and strong-field conditions also display slightly different fluorescence distributions from one another. The weak-field condition appears modestly shifted relative to the strong-field condition, suggesting that varying magnetic field strength may influence nanoparticle positioning, receptor accessibility, and downstream signalling dynamics. One possible interpretation that we intended is that stronger magnetic fields more effectively pulled GFP-conjugated nanoparticles away from the cell surface, reducing receptor engagement and limiting synNotch activation.
Overall, these results support the central hypothesis that magnetic manipulation of GFP-coated nanoparticles can influence downstream signalling behaviour within the engineered synNotch/neuromorphic system. Although additional controls and replicates would be required for definitive conclusions, the observed shifts in fluorescence distributions suggest that external magnetic fields may provide a mechanism for dynamically modulating cellular computation and receptor activation in space and time.
In addition to measuring fluorescent circuit activation, we also analyzed how the magnetic nanoparticles affected the overall mammalian cell population by examining the SSC-A and FSC-A channels from flow cytometry.
During flow cytometry, cells pass individually through a focused laser beam. As the laser interacts with each cell, light is scattered in different directions depending on the physical structure of the cell. Forward scatter (FSC-A) measures light scattered in the forward direction and is generally correlated with cell size, while side scatter (SSC-A) measures light scattered sideways and is associated with internal cellular complexity or granularity.
Healthy mammalian cells typically cluster within a characteristic FSC-A and SSC-A region because they maintain relatively consistent size and internal structure. In contrast, dead cells, damaged cells, or cellular debris scatter light differently and often appear outside the main population distribution.
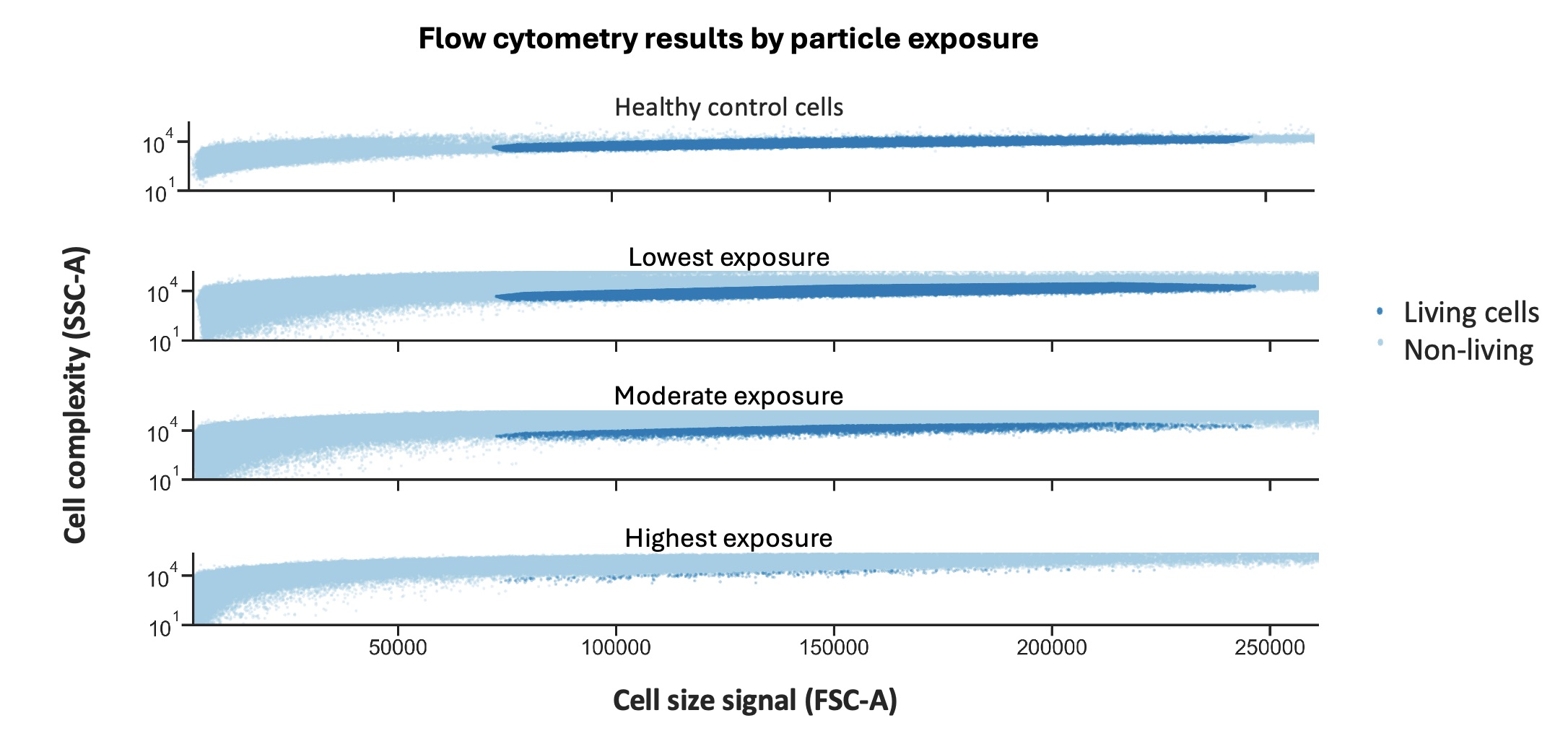
Figure 43: Flow Cytometry Plot of forward scatter vs side scatter with varied magnetic conditions
The flow cytometry graph separates the experiment into four conditions: healthy control cells, lowest nanoparticle exposure, moderate nanoparticle exposure, and highest nanoparticle exposure. Using the healthy control, we can identify the characteristic FSC-A and SSC-A region where viable mammalian cells normally cluster. In the graph, this living-cell population is highlighted in dark blue, while the lighter blue points represent non-living material, dead cells, debris, or background noise.
The highest exposure condition corresponds to the MagGFP + circuit condition without a magnetic field. We call this “highest exposure” because there is no magnetic field pulling the nanoparticles away from the cells. The particles are therefore free to settle directly onto the cell layer at the bottom of the well and interact continuously with the cellular environment. In this condition, very little material remains within the viable-cell region. Instead, most events shift outside the normal mammalian cell cluster. This suggests that the magnetic GFP nanoparticles may have caused substantial cellular stress or toxicity, resulting in widespread loss of viable mammalian cells.
In the moderate exposure condition, which corresponds to the weak magnetic field, a partial viable-cell population begins to reappear within the expected FSC-A and SSC-A range. This suggests that applying even a weak magnetic field may reduce some of the harmful nanoparticle-cell interactions, allowing a subset of mammalian cells to remain viable.
Most interestingly, the lowest exposure condition, which corresponds to the strong magnetic field, shows a much larger population overlapping with the viable-cell region identified by the healthy control. This suggests substantially improved cell survival compared to the no-field condition. One possible explanation is that the stronger magnetic field more effectively pulled or suspended the magnetic nanoparticles away from the cell layer at the bottom of the well, reducing nanoparticle-cell interaction and limiting toxicity.
Together, these results suggest that increased exposure to the magnetic nanoparticles is likely toxic to mammalian cells. In the no-field condition, where the MagGFP nanoparticles were free to settle directly onto the cells, very few viable mammalian cells remained within the normal FSC-A and SSC-A region. Applying magnetic fields appeared to partially reduce this toxicity by physically redistributing the nanoparticles away from the cell layer. The weak-field condition showed partial recovery of viable cells, while the strong-field condition preserved a substantially larger healthy cell population.
This has important implications for future therapeutic applications. If systems like this were ever to be used in engineered cell therapies or programmable biological implants, significantly more biocompatible and non-toxic magnetic nanoparticles would likely need to be developed. Otherwise, prolonged nanoparticle exposure could damage or destroy the engineered cells themselves.
Importantly, despite the strong-field condition showing a larger surviving mammalian cell population, the mNeonGreen activation output in the FITC-A density plot was still lower than the weak-field condition. This is a particularly important observation because it suggests the reduced circuit activation was not simply caused by having fewer living cells. Instead, it supports our central hypothesis that magnetic manipulation of the GFP-conjugated nanoparticles was directly altering receptor activation dynamics. In the stronger magnetic field, more nanoparticles were likely pulled away from the cell surface, reducing synNotch receptor engagement and therefore decreasing downstream mNeonGreen activation, even though more cells remained viable overall.
Working on this project has been one of the most exciting and intellectually inspiring experiences I have had so far. The ability to directly interface physical fields with engineered biological systems feels like an entirely new way of thinking about computation and control in living matter. Throughout this work, I became increasingly fascinated by the broader idea of field-controlled biology using external physical forces such as magnetism to dynamically organize, regulate, and program cellular behaviour in real time. I am extremely excited to continue expanding these ideas with the Weiss Lab and further explore the concept of magnetically controlled ligand presentation within synNotch and SNIPR systems.
What makes this direction particularly exciting is that it feels conceptually analogous to how modern electronics operate. In traditional computers, electric fields are used to precisely control the movement of electrons through circuits to perform computation. In biology, cells already process information through receptor interactions, signalling cascades, and gene regulation networks. Magnetically controlling ligand localization and receptor activation therefore feels like a natural bridge between physical field control and cellular computation. Rather than statically engineering cells once, this approach opens the possibility of dynamically programming living systems in space and time using externally controllable physical signals.
In the short term, an important next step will be to repeat the experiments and expand the number of biological replicates to confirm that the observed effects are reproducible. We also plan to revisit the original magnetic actuation device design, which aimed to dynamically move magnetic fields around the culture environment in real time. This would allow us to test whether actively changing magnetic localization patterns can produce more precise spatial and temporal control over synNotch receptor activation.
Medium-term work will focus on improving the biocompatibility of the system itself. Our results strongly suggest that the current magnetic nanoparticles introduce substantial toxicity to mammalian cells, particularly when particles accumulate directly on the cell layer. To make systems like this viable for real biological applications, we will likely need to develop alternative magnetic materials, coatings, or conjugation strategies that are naturally synthesizable, biologically stable, and significantly less toxic to mammalian cells while still maintaining strong magnetic responsiveness.
Longer term, I believe systems like this could eventually evolve into highly precise platforms for programmable cellular computation. If magnetic fields can be used to dynamically localize ligands, organize signalling patterns, and control receptor activation with fine spatial precision, it may become possible to build biological systems that can be externally programmed in ways somewhat analogous to electronic circuits. Such technologies could eventually enable entirely new approaches to tissue engineering, adaptive therapeutics, synthetic developmental systems, and programmable multicellular behaviour.
Engineering programmable material-to-cell pathways via synthetic Notch receptors
Engineering precise cell-therapeutic function via synthetic Notch receptors
Synthetic neuromorphic computing in living cells
Poly-transfection enables rapid, quantitative testing of genetic circuits in mammalian cells
🧬 Plasmids & Genetic Components
synNotch / SNIPR receptor plasmid (anti-GFP receptor architecture)
Response program plasmid containing modified 5x Gal4 UAS response element
Csy4
mKO2 fluorescent marker
mMaroon fluorescent marker
eBFP2 fluorescent marker
🧫 Mammalian Cell Culture
HEK mammalian cells
Cell culture plates (24-well / multiwell plates)
Complete mammalian growth media
Opti-MEM reduced-serum media
PBS (phosphate-buffered saline)
Trypsin-EDTA
Nuclease-free water
🧪 Transfection Reagents
Lipofectamine L3000
P3000 reagent
Poly-transfection preparation tubes
Pipettes and sterile pipette tips
🧲 Magnetic Nanoparticle Conjugation
MagnaBind carboxylated magnetic beads
GFP / fluorescent protein solution
N52 Neodymium magnets
Special shout out to Evan Holbrook, Ronan Donovan, and David S. Kong, who really made HTGAA such a great experience. David did an incredible job leading the course and creating an environment that genuinely captured the MIT Media Lab spirit. Ronan constantly went above and beyond for students. I still fondly remember staying late debugging DNA plasmids with him before placing our orders. Coming from a background of late-night production code debugging at Goldman Sachs, it honestly felt surreal and incredibly exciting to suddenly be staying up late debugging plasmid designs and engineering DNA instead almost sci-fi like. It was one of those moments where I genuinely felt how much the world is changing and how accessible these powerful technologies are becoming!
Evan was also amazing to work with throughout the semester, especially during my final project. I probably learnt the most from him overall, and his patience, openness, and willingness to help made a huge impact on my experience in the course.

