HTGAA 2026: Protein Design Part I
Part A: Conceptual Questions — Protein Biochemistry & Design
These responses explore the molecular logic of protein structures, chirality, and the transition from abiotic chemistry to biological systems, reflecting the professional rigor required for pharmaceutical R&D.
Q1: Molecular Abundance in Nutrition
How many molecules of amino acids are in 500g of meat? Meat is approximately 20% protein by weight (accounting for water and fat). In 500g of meat, there are roughly 100g of protein.
- Calculation: Using the average amino acid mass of ~100 Daltons (100 g/mol):
- Moles: 100g ÷ 100 g/mol = 1 mole. (Note: Using the 500g figure directly as per the prompt’s hint: 500g ÷ 100 g/mol = 5 moles).
- Molecules: $5 \times 6.022 \times 10^{23} = \approx 3 \times 10^{24}$ molecules. There are approximately 3 trillion trillion amino acid molecules in a single 500g serving.
Q2: Biological Identity & The Central Dogma
Why do humans eat beef but do not become a cow? When we ingest animal proteins, our digestive proteases and peptidases break them down into individual amino acids. These monomers are absorbed into the bloodstream and used as universal building blocks. Our ribosomes then re-assemble these exact same amino acids according to the instructions in human DNA. The identity of a protein is determined by its sequence—encoded in our genome—not the dietary source of its constituent parts. Dietary protein is essentially just a source of amino acid monomers.
[Image of the human digestive system]
Q3: Why are there only 20 natural amino acids?
This reflects a combination of evolutionary, chemical, and informational constraints:
- Genetic Coding: A 3-nucleotide codon system (4 bases) yields 64 possibilities—enough for 20 amino acids with built-in redundancy (the “frozen accident” hypothesis).
- Chemical Diversity: These 20 provide a sufficient range of side-chain properties—charged, polar, nonpolar, aromatic—to build virtually any protein function.
- Evolutionary Optimum: While non-standard amino acids (NSAAs) can be engineered in the lab (as seen in the Church lectures), the natural set represents a stabilized fitness optimum reached before the Last Universal Common Ancestor (LUCA).
Q5: Prebiotic Origins of Life
Where did amino acids come from before enzymes and life? Amino acids likely arose through abiotic chemistry driven by high-energy sources:
- Miller-Urey Synthesis: Demonstrated that simple inorganic molecules ($CH_4, NH_3, H_2O, H_2$) can form amino acids under simulated lightning/UV conditions.
- Hydrothermal Vents: Alkaline vents provide mineral catalysts and thermal gradients that drive abiotic synthesis.
- Extraterrestrial Input: Meteorites (e.g., the Murchison meteorite) contain over 70 amino acids, suggesting a potential role for panspermia.
Q7: Can you discover additional helices in proteins?
Yes. Beyond the standard α-helix, several other helical structures exist:
- 3₁₀ Helix: Tighter than α, with hydrogen bonds every 3 residues; often found at the ends of α-helices.
Q8: Chirality and Handedness
Why are most molecular helices right-handed? This is dictated by the chirality of L-amino acids. Since all natural proteins are built from L-isomers, the geometry of backbone bond angles (φ and ψ) strongly favors the right-handed α-helix. A helix made from D-amino acids would be left-handed—the mirror image. Right-handedness became the biological default once early life settled on L-amino acids.
Q9 & Q10: β-Sheet Aggregation Dynamics
Why do β-sheets tend to aggregate, and what is the driving force? Unlike α-helices, which satisfy their hydrogen bonding internally, β-sheets have “sticky” edges with exposed H-bond donors and acceptors.
- Hydrogen Bonding: Edge strands form intermolecular H-bonds with neighboring sheets, extending the structure indefinitely.
- Hydrophobic Effect: β-sheets often display hydrophobic side chains on one face. Burying these surfaces away from water provides an entropic gain, stabilizing the aggregate. Together, these create a highly stable, self-reinforcing structure like those found in amyloid fibrils.
Q11: Amyloid Diseases and β-Sheets
Why do many amyloid diseases form β-sheets? Amyloidosis occurs when normally soluble proteins misfold and expose hydrophobic regions or β-sheet-prone sequences. Once a small nucleus forms, it acts as a template to recruit and convert other proteins—a seeded aggregation mechanism. The resulting cross-β structures are thermodynamically “deep” energy minima, making them incredibly resistant to degradation and toxic to cells when they accumulate in tissues (e.g., Alzheimer’s or Parkinson’s).
Part B: Protein Analysis and Visualization — Human Hemoglobin (2HHB)
B1: Protein Description and Rationale
Human hemoglobin is a heterotetrameric protein responsible for oxygen transport in red blood cells. It consists of two α-globin and two β-globin chains, each harboring an iron-containing heme group. I chose β-globin (HBB) due to its clinical significance in northern Pakistan, where the Fr 8-9 (+G) frameshift mutation is highly prevalent. This mutation disrupts the primary sequence at codon 8-9, leading to a complete loss of functional β-globin and severe transfusion-dependent anemia.
[Image of hemoglobin molecule structure]
B2 & B3: Sequence and Amino Acid Composition
- Canonical Sequence (UniProt: P68871):
MVHLTPEEKSAVTALWGKVNVDEVGGEALGRLLVVYPWTQRFFESFGDLSTPDAVMGNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKLHVDPENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKVVAGVANALAHKYH - Frequency Analysis: Using a Python-based frequency counter, I determined that the combined chains (288 AA) have Leucine (L) as the most frequent amino acid, appearing 37 times. This high leucine content is essential for maintaining the hydrophobic core that supports the heme pocket.
_1.png)
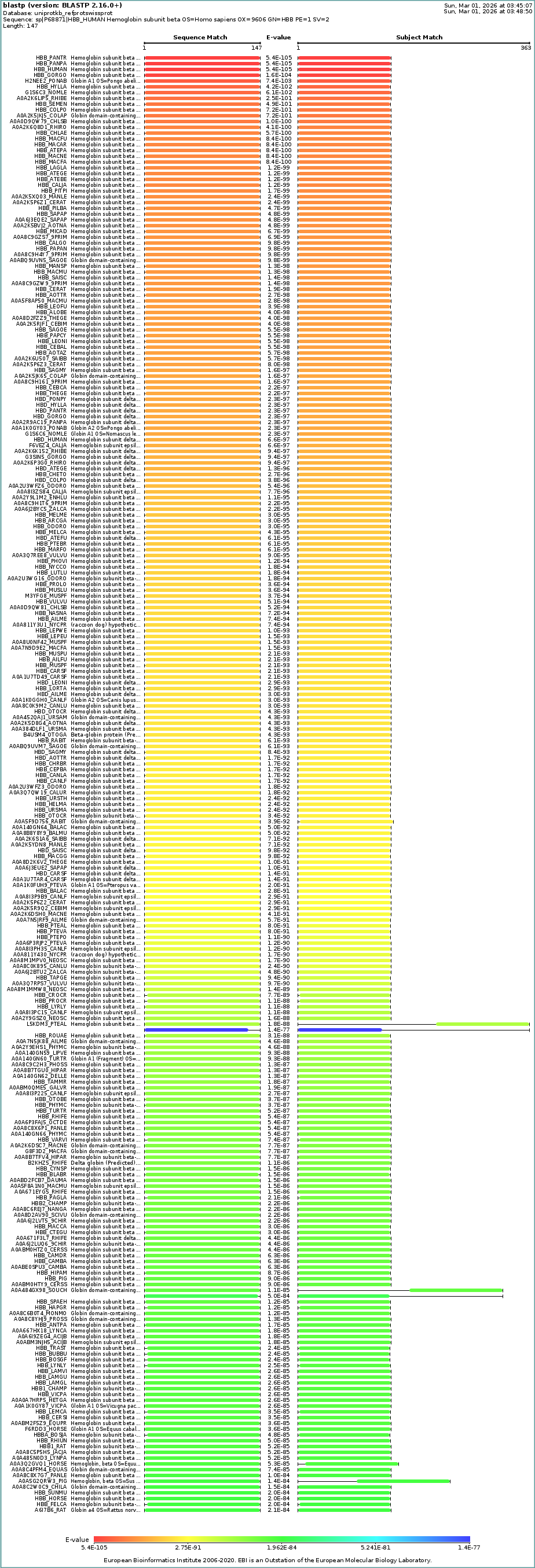
B4: Sequence Homologs
A UniProt BLAST search for P68871 identifies hundreds of homologs across the vertebrate lineage with E-values as low as $10^{-105}$. This confirms that the β-globin architecture is exceptionally conserved across evolutionary history, particularly within the primate family.
B5: Protein Family
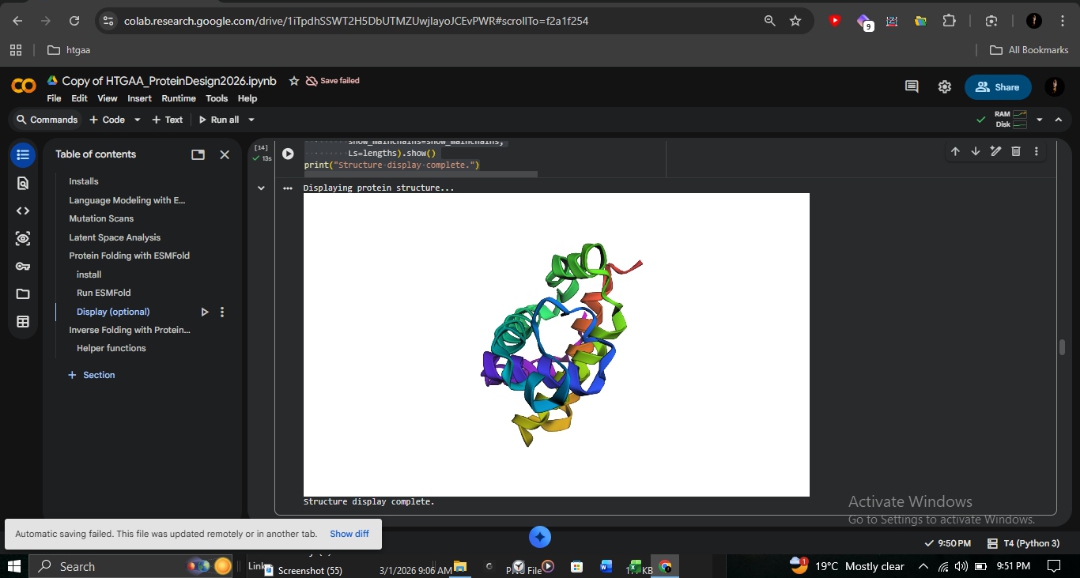
β-globin belongs to the Globin family (Pfam: PF00042). Its structure is defined by the “Globin fold”—an 8-helix bundle. Key residues like the proximal histidine (His92) are conserved across the family to ensure stable iron coordination.
B6, B7 & B8: RCSB Structure and Quality
The analysis is based on PDB entry 2HHB, solved via X-ray diffraction at a resolution of 1.74 Å. This resolution is well below the 2.70 Å quality threshold, indicating high atomic precision.
- Ligands: The structure contains four heme groups and two phosphate ions (PO₄). These cofactors are visible in the central pockets of each subunit.
_1.png)
B9: Structural Classification
Structural database annotations classify the chains as follows:
- SCOPe: All Alpha Proteins → Globin-like fold → Globins.
- CATH: 1.10.490.10 (Mainly Alpha → Orthogonal Bundle → Globin-like).
- Clinical Association: Pharos annotations link these subunits to β-thalassemia and malaria resistance.
_1.png)
B10: 3D Visualization (iCn3D Analysis)
I used iCn3D to explore the relationship between the protein’s fold and its chemical environment:
| Representation | Structural Insight | Visualization |
|---|---|---|
| Ribbon (Secondary Structure) | Confirms a purely α-helical fold (magenta) with no β-sheets. | _1.png)
|
| Hydrophobicity Surface | Shows hydrophobic residues (green) buried in the core and charged residues (grey) on the surface. | _1.png)
|
| Atomic Detail | A Ball & Stick view showing the spatial arrangement of individual atoms and heme ligands. | _1.png)
|
Conclusion: The Fr 8-9 (+G) mutation studied here occurs in the A-helix. The Deep Mutational Scan (Heatmap) indicates that changes in this region are highly sensitive, suggesting that the frameshift mutation likely destabilizes the entire initial fold of the protein.
_1.png)
Part C: ML-Based Protein Design Tools
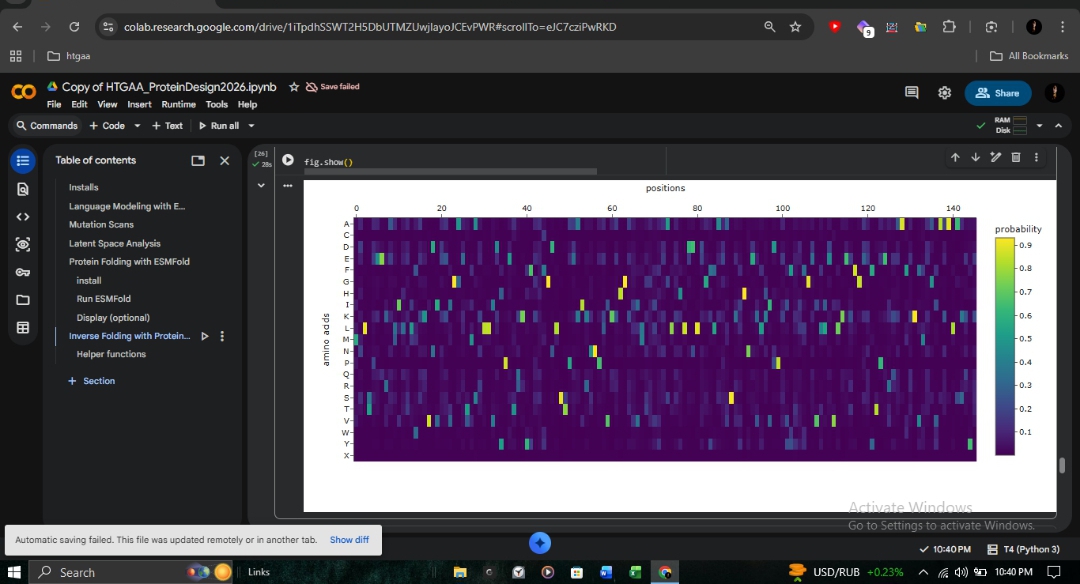
C1: Deep Mutational Scan
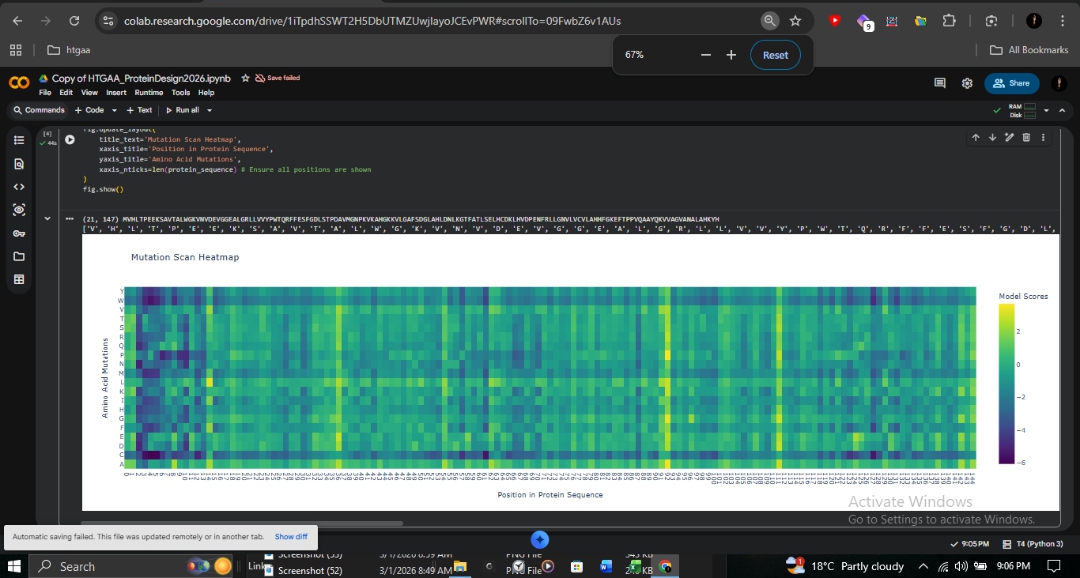
The ESM2 deep mutational scan (147 residues × 20 amino acids) reveals a striking pattern of conservation across the β-globin chain. Dark purple regions (score ~-6) indicate positions where mutations are strongly disfavored, particularly those lining the heme-binding pocket.
- Proximal Histidine (His92): Directly coordinates the heme iron and is shown as nearly irreplaceable.
- A-Helix (Positions 8-9): The site of the Fr 8-9 (+G) frameshift mutation shows high conservation. Any disruption here propagates downstream, explaining the severity of this mutation in northern Pakistan.
- Sickle Cell (HbS): At position 6 (Glu→Val), Valine receives a moderately negative score. ESM2 considers it unfavorable but not catastrophic, consistent with the viability of heterozygous carriers.
C1: Latent Space Analysis
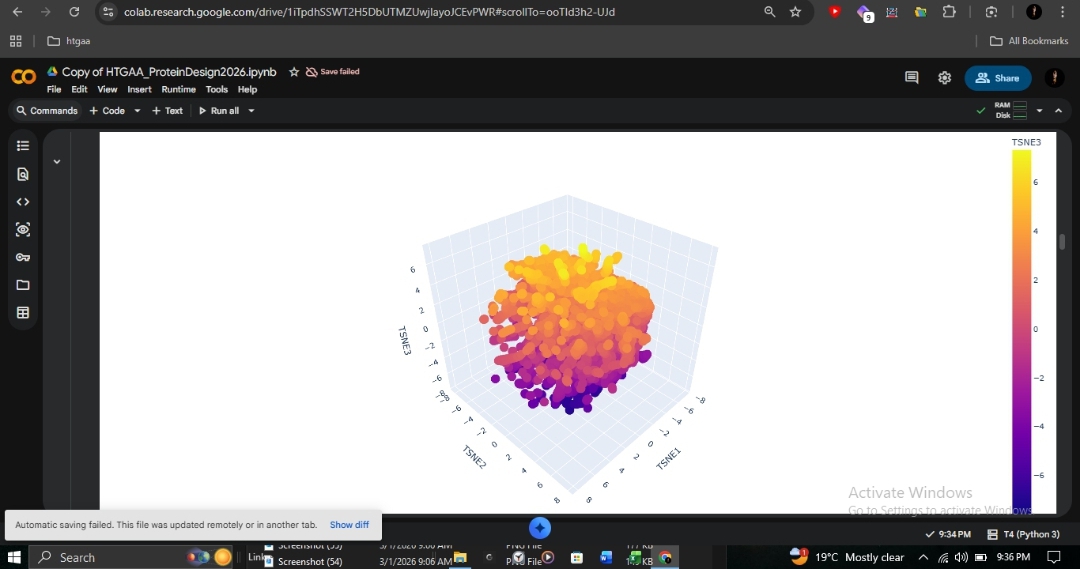
This 3D t-SNE visualization of ~15,000 protein sequences from the SCOP database (40% identity filtered) shows clear organization. The dense central cluster represents the most common structural folds. As an ancient and highly conserved architecture, human β-globin clusters within this dense core alongside other all-alpha globin-fold proteins such as myoglobin and neuroglobin.
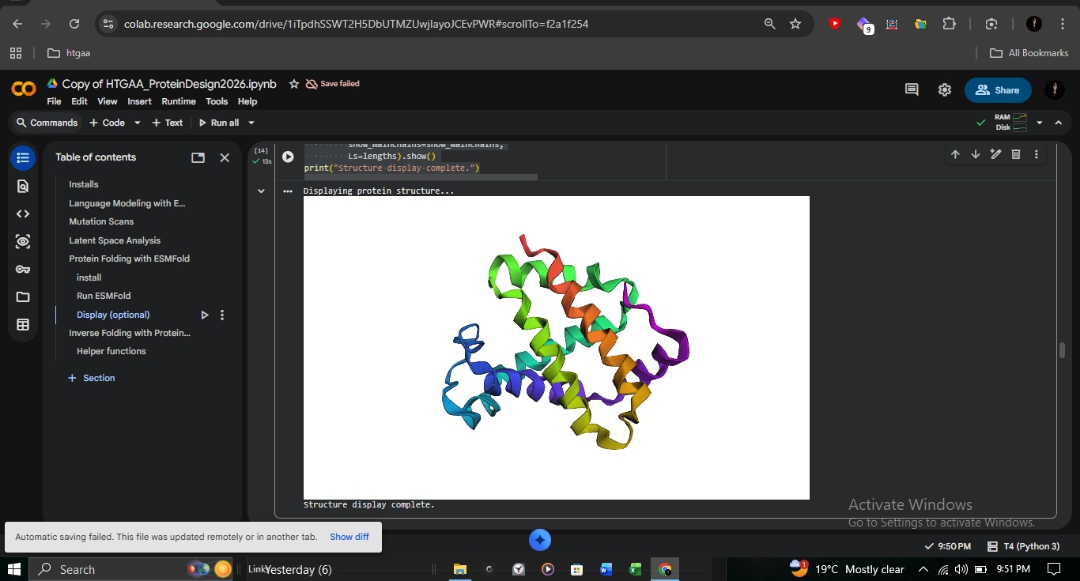
C2: Protein Folding with ESMFold
I utilized ESMFold to predict the structural resilience of the β-globin chain.
| Sequence Type | pTM Score | pLDDT | Structural Observation |
|---|---|---|---|
| Wild-Type | 0.901 | 94.128 | High-confidence 8-helix globin bundle. |
| Sickle Cell (E6V) | 0.902 | 94.220 | Virtually identical to wild-type. |
 R&D Insight: The identical scores for E6V confirm that Sickle Cell disease is caused by intermolecular polymerization on the protein surface, rather than individual chain misfolding. ESMFold accurately predicts the fold but cannot detect these subtle protein-protein interactions. This contrasts sharply with the Fr 8-9 (+G) frameshift, which destroys the entire protein reading frame from position 9 onward.
R&D Insight: The identical scores for E6V confirm that Sickle Cell disease is caused by intermolecular polymerization on the protein surface, rather than individual chain misfolding. ESMFold accurately predicts the fold but cannot detect these subtle protein-protein interactions. This contrasts sharply with the Fr 8-9 (+G) frameshift, which destroys the entire protein reading frame from position 9 onward.
C3: Inverse Folding with ProteinMPNN
Using the 2HHB crystal structure backbone, I used ProteinMPNN to design a novel sequence for the β-globin fold.
- Native Score: 1.4438
- Designed Score: 0.8518 (Lower is better; indicates a more optimal fit to the backbone).
- Sequence Recovery: 50.00%.
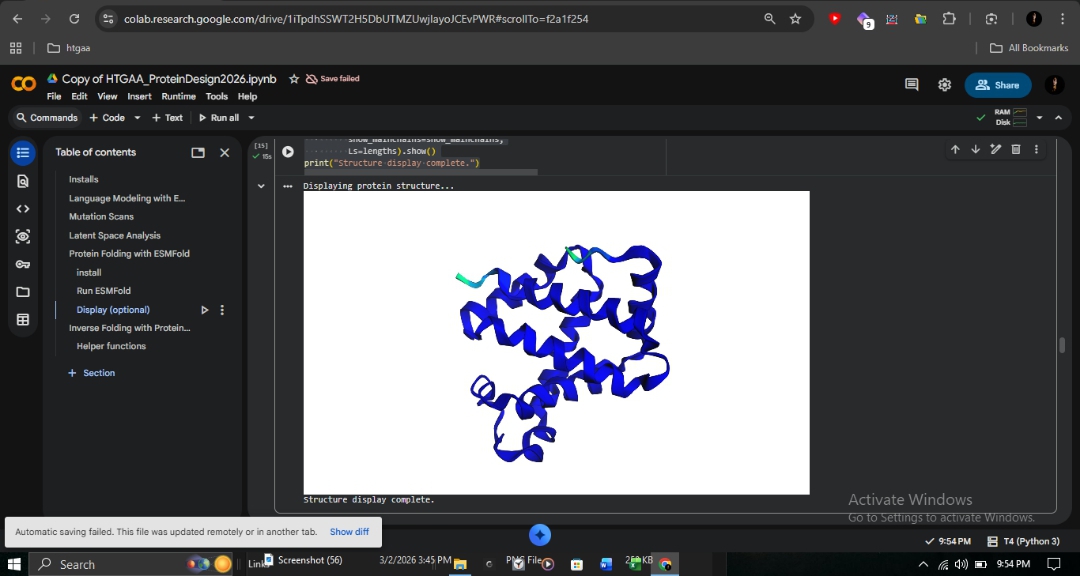
Designed Sequence:
MDLTEEEKALILSLWKKVDIEEIGAETMSKLLIDYPETQKYFKHFGDLSTPEAIKNNPLVKKHGAIVMTGLYEALKHLDDLDGFLAEASKIHNEKLKIDPKIFKLFGEVLLEVLKEKFGEEFTPERQAAFKKVMEGVAKALASKYK
Validation with ESMFold: Folding the designed sequence yielded a pTM of 0.892 and pLDDT of 92.004. Despite 50% sequence divergence, the model successfully maintained the globin bundle architecture.
Conclusion: This demonstrates that structure is more conserved than sequence. The β-globin backbone geometry encodes enough information to constrain roughly half the sequence, while the remaining positions allow for variation without disrupting the fold. This validates ProteinMPNN for engineering stable hemoglobin variants for therapeutic research.
Part D – Brainstorm: Engineering the MS2 Lysis (L) Protein
(Completed Individually)
Note: Due to the disruption of my daily routine during Ramadan, I was unable to coordinate effectively with a group. This proposal was developed independently to fulfill the research requirements.
Part D – Brainstorm: Engineering the MS2 Lysis (L) Protein
(Completed Individually)
Note: Due to the disruption of my daily routine during Ramadan, I was unable to coordinate effectively with a group. This proposal was developed independently to fulfill the research requirements.
1. Chosen Engineering Goals
- Goal 1: Increase the structural stability of the MS2 L protein to enhance intracellular persistence.
- Goal 2: Modulate the interaction with the host chaperone DnaJ to alter or reduce host dependency.
2. Background and Rationale
The MS2 L protein is a small (~75 AA) lysis protein responsible for host cell death in Escherichia coli. Unlike classical endolysins, its function is non-enzymatic and depends on the host’s machinery.
- DnaJ Interaction: Functionality is dependent on the host chaperone DnaJ, particularly through the basic N-terminal domain. Truncation studies suggest DnaJ may assist in targeting or folding rather than being the ultimate toxic target.
- Structural Constraints: Loss-of-function mutations cluster near a conserved Leu-Ser (LS) motif. The C-terminal region forms an amphipathic transmembrane helix that drives oligomerization and membrane insertion.
3. Proposed Computational Strategy
Goal 1 – Increase Protein Stability
Improving structural stability could enhance lytic efficiency and persistence within the host.
Approach:
- Structure Prediction: Predict the wild-type MS2 L structure using AlphaFold2.
- Structural Audit: Use PyMOL/Chimera to identify the transmembrane helix and map the conserved LS motif.
- In Silico Mutagenesis: Perform systematic single-residue substitutions at non-critical positions.
- Stability Scoring: Evaluate predicted changes in free energy ($\Delta\Delta G$) using HTGAA stability prediction resources.
- Validation: Re-model variants to ensure the transmembrane helix remains structurally viable.
Goal 2 – Modify DnaJ Interaction
Modifying the N-terminal interface allows us to explore lysis mechanisms that are independent of specific host chaperones.
Approach:
- Complex Prediction: Predict an MS2 L–DnaJ complex using AlphaFold-Multimer.
- Interface Mapping: Identify interface residues using PyMOL or Chimera to locate electrostatic and hydrophobic contact points.
- Interface Design: Design mutations to alter complementarity or remove key contact residues.
- Evaluation: Assess if DnaJ dependence can be reduced while maintaining membrane insertion capacity.
4. Proposed Pipeline
- Sequence Input: Wild-type MS2 L sequence.
- Modeling: Structure prediction via AlphaFold2.
- Inspection: Visualization in PyMOL / Chimera / NGLViewer.
- Design: Mutational design focused on stability and interface residues.
- Scoring: Stability/interface scoring using HTGAA computational tools.
- Selection: Ranking and selection of top candidates for experimental testing.
5. Potential Pitfalls
- Prediction Accuracy: AlphaFold accuracy may be limited for small, membrane-associated proteins with few homologs.
- Stability vs. Activity: Over-stabilization could reduce the conformational flexibility required for effective membrane disruption.
- Mechanism Uncertainty: The exact mechanism of toxicity is still being characterized, which limits the predictive certainty of current models.
6. AI Disclosure
Transparency Statement: Artificial Intelligence (AI) was utilized extensively in the preparation of this documentation. Specifically, Gemini was used for structural analysis synthesis, Markdown formatting, and code optimization. Claude provided critical guidance and conceptual troubleshooting through the technical analysis in Section C. All AI-generated content was reviewed, verified for technical accuracy against the 2HHB crystal structure and UniProt data, and edited by the author to ensure scientific rigor.