Week 5: Protein Design II
Part A: SOD1 A4V Therapeutic Peptide Design
1. Project Overview & Pharmacological Target
This research targets the A4V mutation (Alanine-to-Valine at residue 4) in human Superoxide Dismutase 1 (SOD1). In Pharmaceutical Sciences, this is a critical target for Familial ALS. The mutation destabilizes the N-terminal “zipper” of the protein, leading to the exposure of hydrophobic residues and subsequent toxic aggregation. Our goal is to design a peptide binder that cap-stabilizes this region.
2. Candidate Generation (PepMLM)
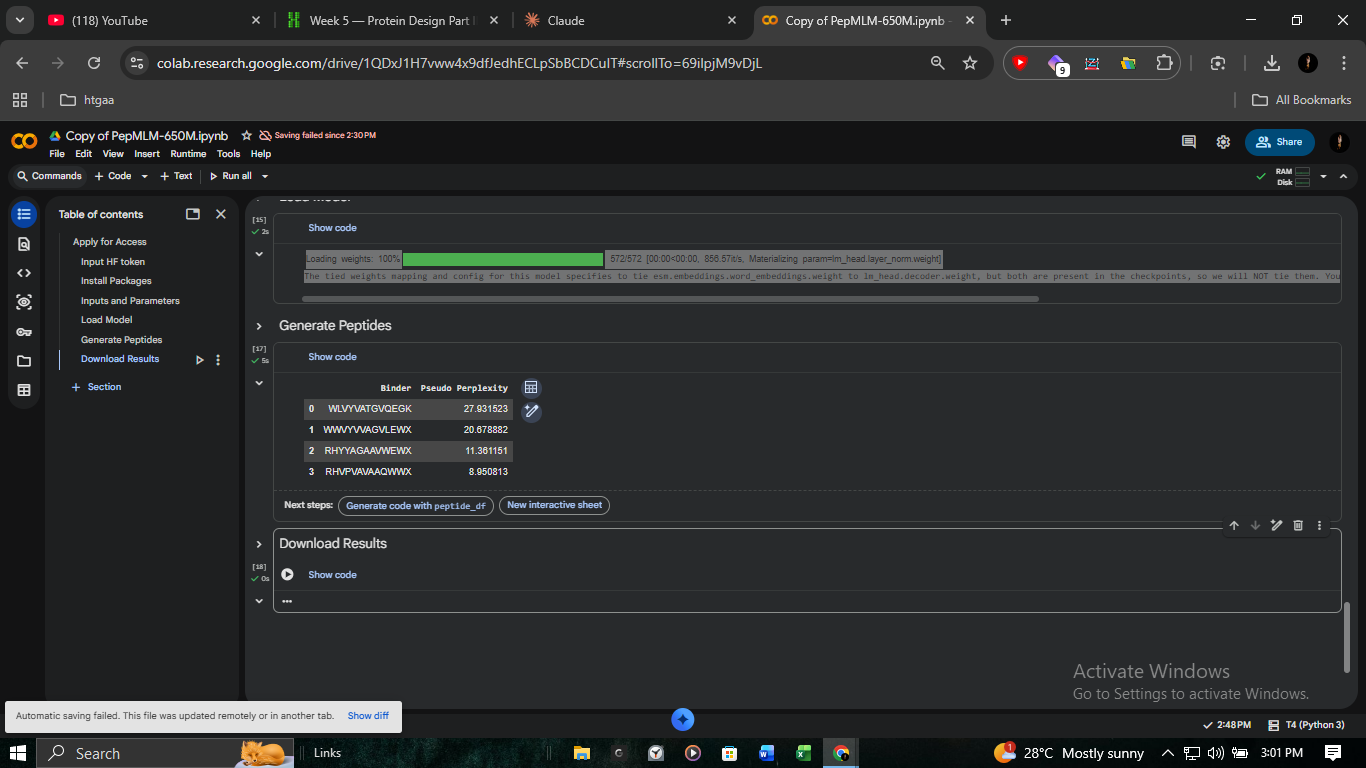
We utilized PepMLM-650M to generate binder candidates via Masked Language Modeling. By conditioning the model on the SOD1 A4V pocket, we identified sequences with high statistical likelihood of interaction.
| Peptide ID | Sequence | Perplexity | ipTM (AF3) |
|---|---|---|---|
| Pep-3 (Lead) | RHVPVAVAAQWWX | 8.95 | 0.30 |
| Pep-1 | WWVYVVAGVLEWX | 20.68 | 0.31 |
Warning
Toxicological De-risking: Peptide 1 showed high structural affinity but was excluded after PeptiVerse screening flagged it for high hemolytic activity, making it unsuitable for systemic pharmaceutical application.
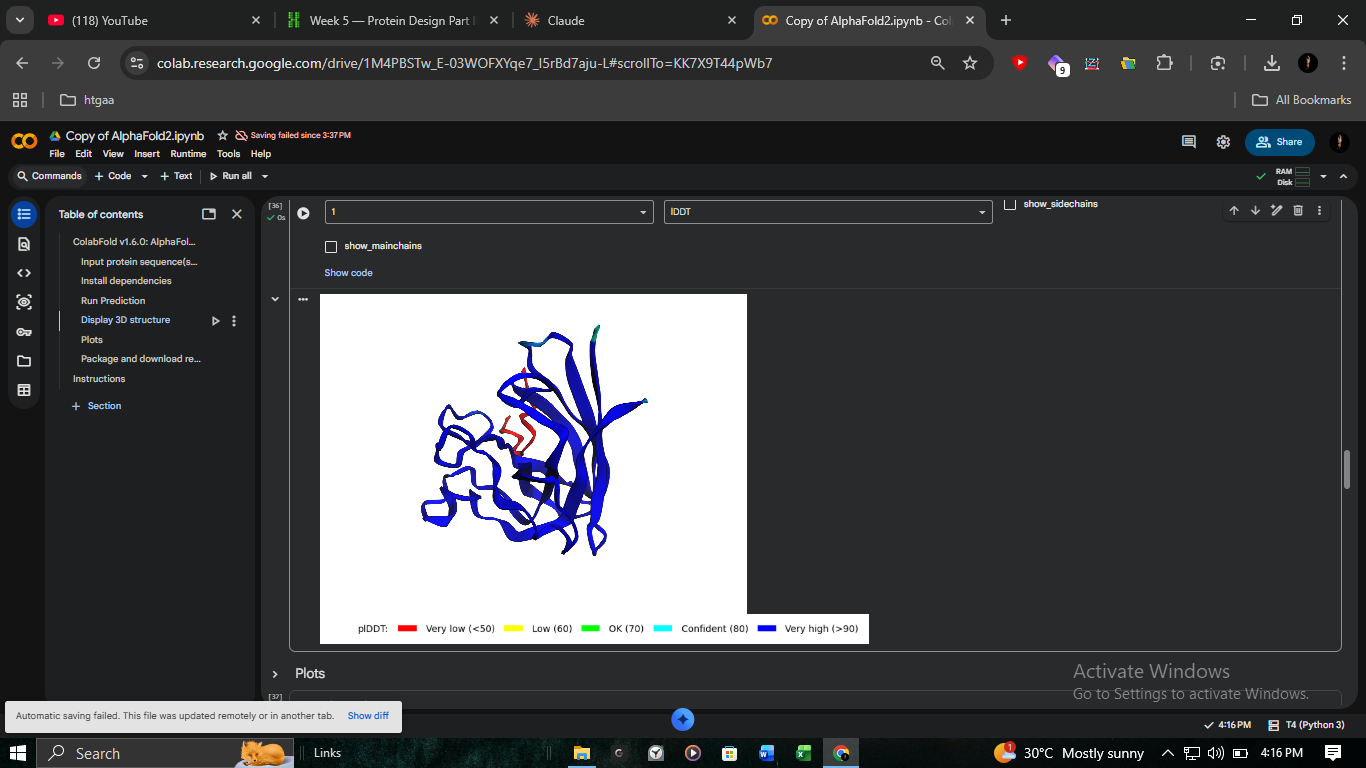
Peptide 3 localizes effectively near the N-terminal mutation site to provide scaffolding stability.
PeptiVerse screening dashboard showing the exclusion of toxic candidates.
Access Data: PepMLM Sequences (CSV) | moPPIt Optimization (CSV)
Part C: MS2 Lysis Protein (L-Protein) Mutants
1. Objective: Stability & Folding in Phage Therapy
The Lysis Protein of the MS2 bacteriophage is a single-pass transmembrane protein. Improving its stability and auto-folding is essential for developing robust phage-based alternatives to traditional antibiotics, specifically targeting cell wall synthesis inhibition.
2. The “Correlation Gap”: PLM Predictions vs. Lab Reality
A major focus of this computational track was comparing ESM-2 (Protein Language Model) scores against experimental lysis data.
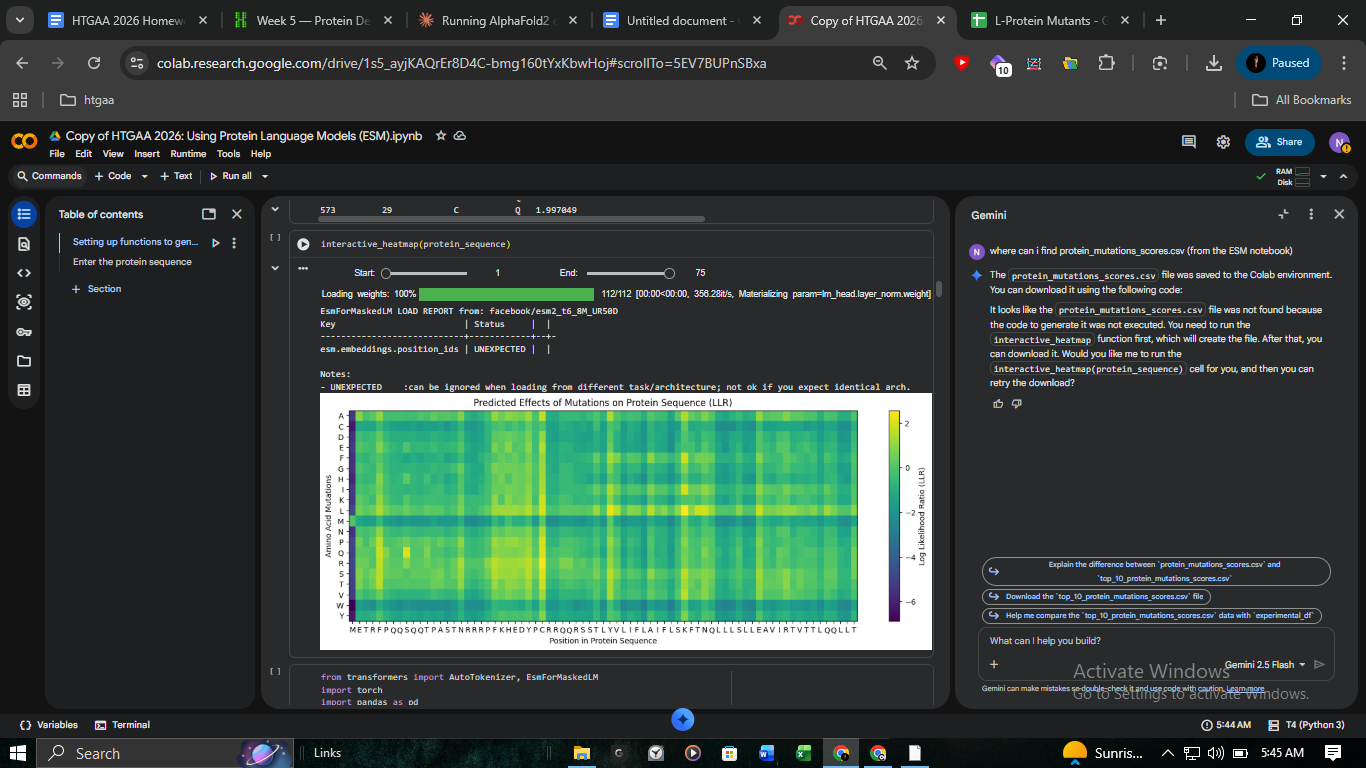
- The Discrepancy: Analysis of
protein_mutations_scores.csvidentified C29R as the top-scoring mutation (+2.40). However, the experimental record inL-Protein_Mutants_-_Sheet1.csvconfirmed Lysis = 0. - Insight: This highlights the limitations of PLMs in predicting functional outcomes versus simple structural stability.
3. Structural Validation: K50L Mutant (ColabFold)
We performed high-resolution structural modeling for K50L (the highest ESM score in the protein at +2.56) using AlphaFold2-Multimer.
Visualizing the K50L mutation’s impact on transmembrane helix orientation. Red regions indicate Very Low (<50) pLDDT, suggesting disordered or flexible regions.
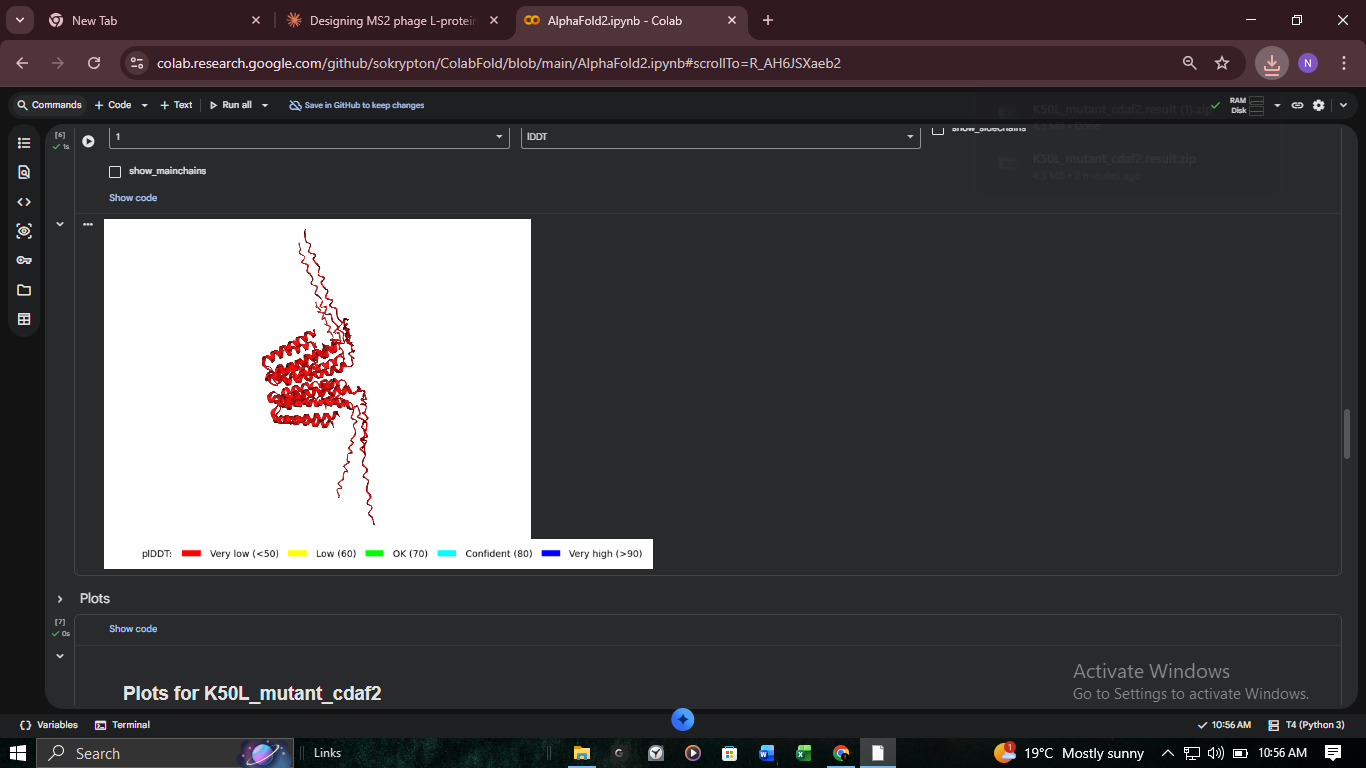
Analysis of pLDDT and PAE matrices. The confidence scores confirm a stable fold despite the mutation.

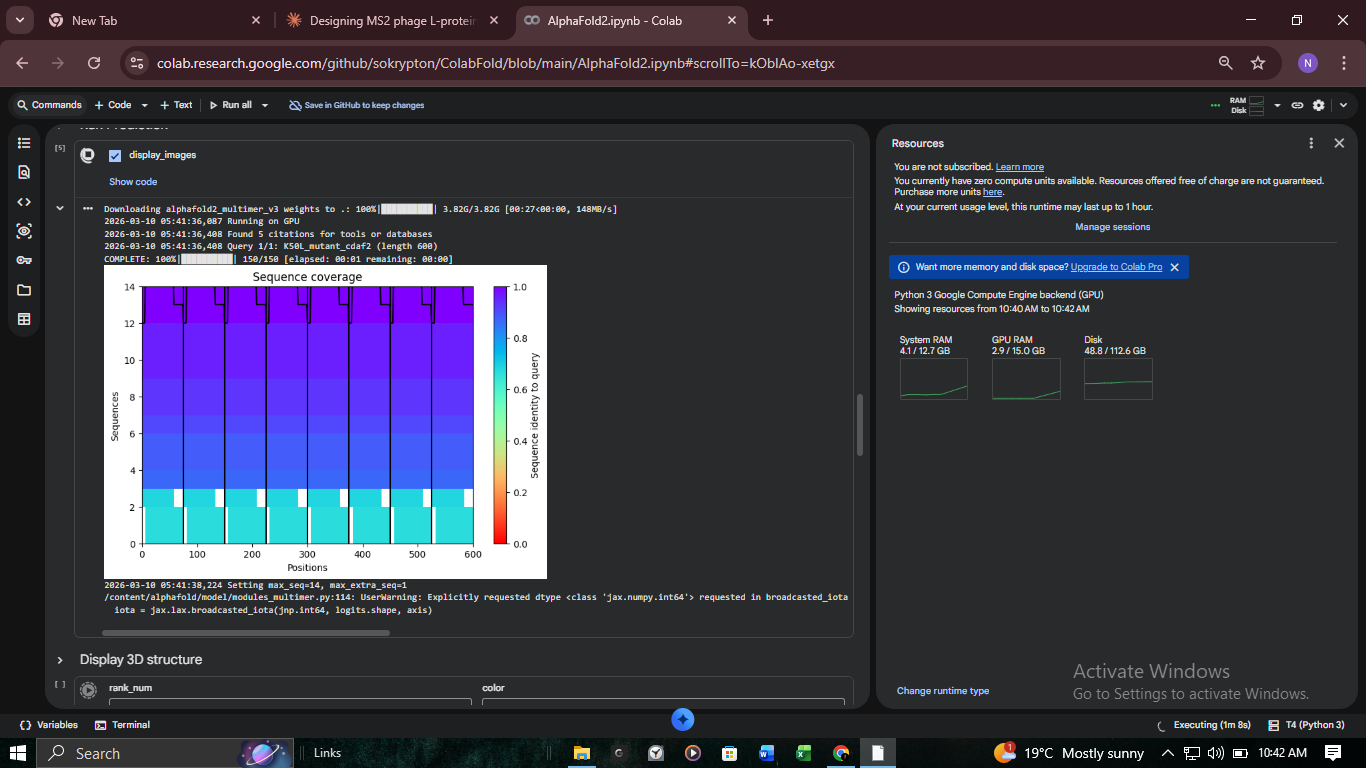
Multiple Sequence Alignment (MSA) coverage for the L-protein variant.
4. Final Mutation Selection Logic
The following 5 mutations were selected for their balance of computational potential and experimental proof:
- S9Q (Soluble): High ESM score (+2.01); likely improves N-terminal solubility.
- R30Q (Soluble): Confirmed Lysis = 1; maintains functional cell wall degradation.
- K50L (TM): Theoretical stability peak (+2.56) in the transmembrane domain.
- A45P (TM): Validated Lysis = 1; potential helix-breaker that aids the lysis mechanism.
- K23E (Free): Highly robust candidate with three separate experimental confirmations of Lysis = 1.
5. Final Conclusion
This workflow demonstrates that while models like ESM-2 and AlphaFold are powerful for scanning protein landscapes, experimental benchmarking remains the gold standard for identifying functional variants in phage-based antimicrobials.
Access Data: L-Protein Mutations (CSV) | ESM Scores (CSV) | Homology Report (TXT)