Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
I have answered the following questions based on the concepts provided by Shuguang Zhang regarding protein structure and design.
1. How many molecules of amino acids do you take with a piece of 500 grams of meat?
To calculate the total molecules, we must first determine the actual protein content and molar mass:
- Protein Content: 500g of meat is typically 20-25% protein, which is approximately 125g of protein. The remaining mass consists of water, fat, and minerals.
- Molar Mass: The average mass of an amino acid is 100 Daltons, which is equivalent to 100 g/mol.
- Moles Calculation: Using the formula n = m/M, we find 125g / (100 g/mol) = 1.25 moles of amino acids.
- Total Molecules: Multiplying the moles by Avogadro’s constant (6.022 × 10²³): 1.25 × 6.022 × 10²³ = 7.53 × 10²³.
- Result: Consuming a 500g piece of meat means taking in approximately 753 sextillion molecules of amino acids.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
The biological reason lies in the process of digestion and genetic reassembly:
- Digestion: When we consume beef, our body does not recognize it as “cow protein"Instead, it sees a long chain of chemicals that must be broken down.
- Enzymatic Breakdown: Our digestive system uses enzymes such as pepsin and trypsin to break the peptide bonds of bovine proteins. This reduces the meat to individual amino acids, which serve as “LEGO bricks”.
- Identical Building Blocks: An amino acid like Leucine from a cow is chemically identical to one from a fish or a human.
- Genetic Instructions: Our DNA contains the specific instructions for building human proteins. Ribosomes take these cow-derived “bricks” and reassemble them according to our own unique genetic code.
- Analogy: If you take apart a LEGO castle (the cow) and have the instructions for a LEGO spaceship (the human), it doesn’t matter that the bricks were previously a castle; they will become a spaceship.
3. Why are there only 20 natural amino acids?
The selection of only 20 standard amino acids is a result of evolutionary optimization:
- Frozen Accident: These 20 amino acids represent a “frozen accident” of early evolution, optimized nearly 4 billion years ago.
- Chemical Diversity: They provide a balanced set of hydrophobic, polar, and charged side chains, which is sufficient for creating stable, soluble protein cores and specific binding pockets.
- Metabolic Efficiency: Using 20 amino acids strikes an optimal balance between the metabolic cost of synthesis and the structural complexity required for life.
- Folding Precision: This specific set allows for the precise and complex folding necessary for protein functionality.
- Rarity of Others: While over 500 amino acids exist in nature, and others like selenocysteine are used rarely, the genetic code primarily selected these 20 to be incorporated directly by ribosomes.
4. Can you make other non-natural amino acids? Design some new amino acids.
It is entirely possible to design and synthesize non-natural (non-canonical) amino acids by manipulating the amino group, the carboxyl group, or the R-group (side chain). Below are three designed examples:
- Boron-Phenylalanine (B-Phe): This amino acid is designed for Boron Neutron Capture Therapy (BNCT). By swapping a hydrogen atom on a Phenylalanine benzene ring for a boronic acid group, we create a tool for precision cancer treatment. When incorporated into a cancer-targeting antibody, it captures thermal neutrons and “explodes” on a microscopic scale, killing the cancer cell from the inside while sparing healthy tissue.
- The “Magneto-Protein”: Ferro-Alanine (Fe-Ala): Inspired by magnetotactic bacteria, this design replaces the methyl group of Alanine with an organometallic complex containing an Iron (Fe) atom. This gives the protein paramagnetic properties. It could be used to create “magnetic enzymes” that can be easily pulled out of industrial bioreactors using magnets, significantly lowering production costs.
- The “Light-Switch”: Azobenzene-Tyrosine (Azo-Tyr): This is a photo-switchable amino acid that allows for the control of biology with light. It uses an azobenzene group that “kinks” (cis-configuration) under UV light and “straightens” (trans-configuration) under visible light. This can be used to “cage” a drug, keeping it inactive until a specific wavelength of light is applied directly to a tumor site to activate it.
5. Where did amino acids come from before enzymes that make them, and before life started?
Amino acids likely originated through a process called abiotic synthesis:
- Laboratory Evidence: Experiments such as the Miller-Urey experiment demonstrated that simple organic compounds can form from inorganic precursors like methane, ammonia, and water when exposed to energy sources like lightning or UV radiation.
- Extraterrestrial Origins: Amino acids have also been discovered in meteorites, which suggests they may have arrived on Earth via extraterrestrial impacts before life began.
6. If you make an alpha-helix using D-amino acids, what handedness (right or left) would you expect?
An alpha-helix composed of D-amino acids will form a left-handed helix:
- Mirror Images: Because D-amino acids are the mirror images of the L-amino acids used in natural proteins, they produce a mirror image of the standard right-handed helix.
- Chirality: D-amino acids do not fit properly into a right-handed configuration due to chiral interactions; therefore, they naturally adopt the left-handed form.
7. Can you discover additional helices in proteins?
Yes, researchers have identified several helices beyond the common alpha-helix:
- Identification: While alpha-helices are the most abundant, other types such as 3-10-helices, pi-helices, and polyproline helices exist.
- Discovery Tools: These structures can be identified by searching database entries like the PDB or using visualization tools like PyMOL.
- pi-helices: These occur in about 15% of known proteins and are often associated with functional sites, such as active sites.
- 3-10-helices: These are shorter and tighter than alpha-helices and are frequently found at the ends of standard alpha-helical structures.
- Methodology: Discovering these involves analyzing the specific backbone hydrogen-bonding patterns.
8. Why are most molecular helices right-handed?
Right-handedness is the preferred orientation due to thermodynamic stability:
- Energetic Stability: Most molecular helices, including DNA and protein alpha-helices, are right-handed because this configuration is more energetically stable.
- Steric Hindrance: This arrangement reduces collisions (steric hindrance) between atoms compared to left-handed forms.
- Optimal Bonding: In proteins, right-handed alpha-helices allow for the most favorable hydrogen bonding between amino acids.
9. Why do beta-sheets tend to aggregate?
beta-sheets have a natural tendency to clump together due to their structural chemistry:
- Exposed Groups: beta-sheets possess exposed hydrogen-bonding groups (NH and C=O) along their edges.
- Sticky Edges: Unlike alpha-helices, where hydrogen bonds are satisfied internally, beta-sheets have “sticky” edges.
- Complex Formation: These edges can easily form new hydrogen bonds with the edges of other beta-sheets.
10. What is the driving force for beta-sheet aggregation?
The aggregation is driven by two primary physical forces:
- Hydrogen Bonding: The main driving force is the formation of hydrogen bonds between the backbones of adjacent sheets.
- Hydrophobic Effect: In many beta-sheets, hydrophobic side chains are exposed on the surface.
- Energy Minimization: When these sheets aggregate, they bury these hydrophobic surfaces away from water.
Part B. Protein Analysis and Visualization
1. Protein Selection and Description
- Protein Name: Green Fluorescent Protein (GFP)
- Organism: Aequorea victoria
- Why Selected: GFP is extensively used in cancer research, primarily as a molecular marker to track, visualize, and study tumor cells in real-time. It allows researchers to label cancer cells, enabling the study of tumor growth, metastasis, angiogenesis, and the effect of drugs on cancer in vivo.
- Structure: It consists of a “β-can” motif, which is an 11-stranded β-sheet barrel with an α-helix running through the center.
2. Sequence Analysis
- Amino Acid Sequence:
MSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTLVTTFSYGVQCFSRYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVNFKIRHNIEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITHGMDELYK - Length: 238 amino acids.
- Most Frequent Amino Acid: Glycine (G), which appears 22 times.
- UniProt ID: P42212
- Homologs: There are 205 homologs identified for this protein using UniProt’s BLAST tool.
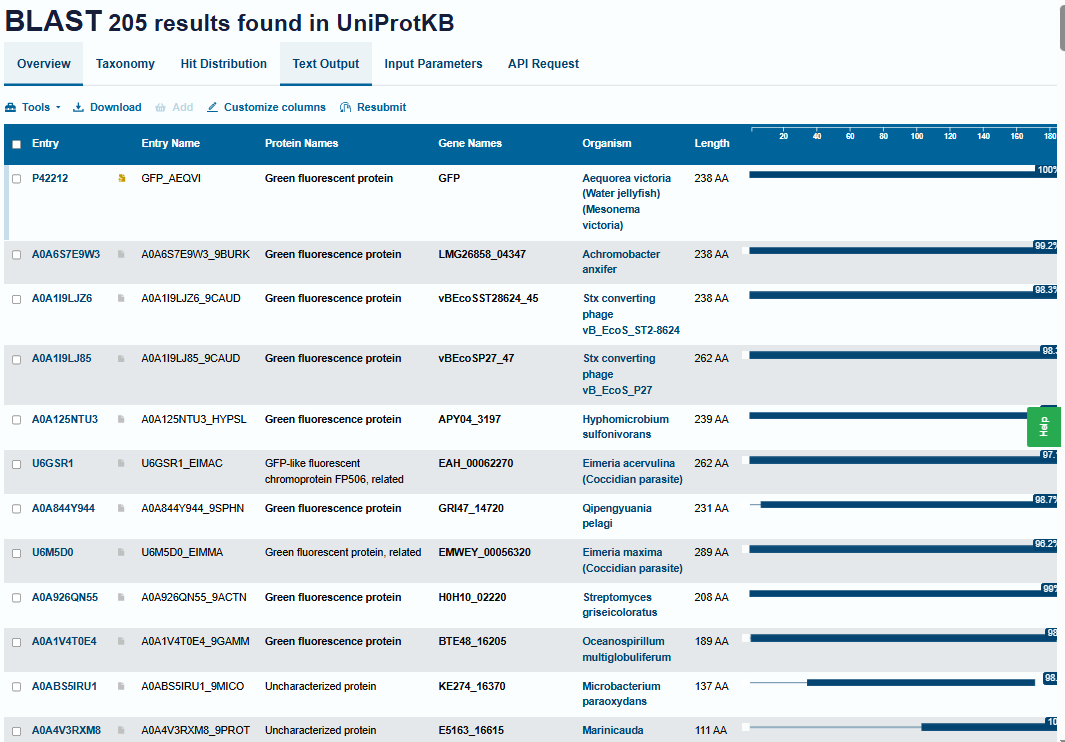
- Protein Family: It belongs to the superfamily of GFP-like proteins, including chromoproteins and photoproteins.
3. Structural Analysis (RCSB PDB)
- RCSB PDB ID: 1GFL
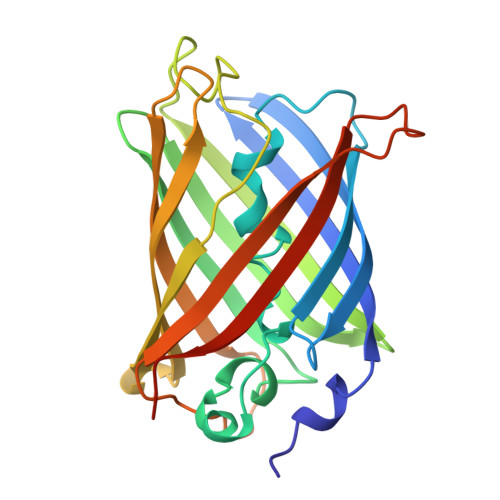
- Structure Solved: The structure was first solved and deposited on August 23, 1996.
- Quality/Resolution: It has a resolution of 1.90 Å. Since this is significantly smaller than the 2.70 Å threshold, it is considered a high-quality structure.
- Other Molecules: Apart from the protein chains, the structure contains water (HOH) molecules. The internal chromophore (CRO) is part of the protein sequence itself, formed by the cyclization of residues Ser65, Tyr66, and Gly67.
- Structure Classification: According to CATH and SCOP, this protein belongs to the Beta Barrel architecture and the GFP-like fold family.
4. 3D Visualization and Structural Analysis (PyMOL)
I used PyMOL software to perform a detailed 3D visualization and analysis of the 1GFL (Green Fluorescent Protein) structure. Below are the steps and findings from this session:
A. Initial Loading
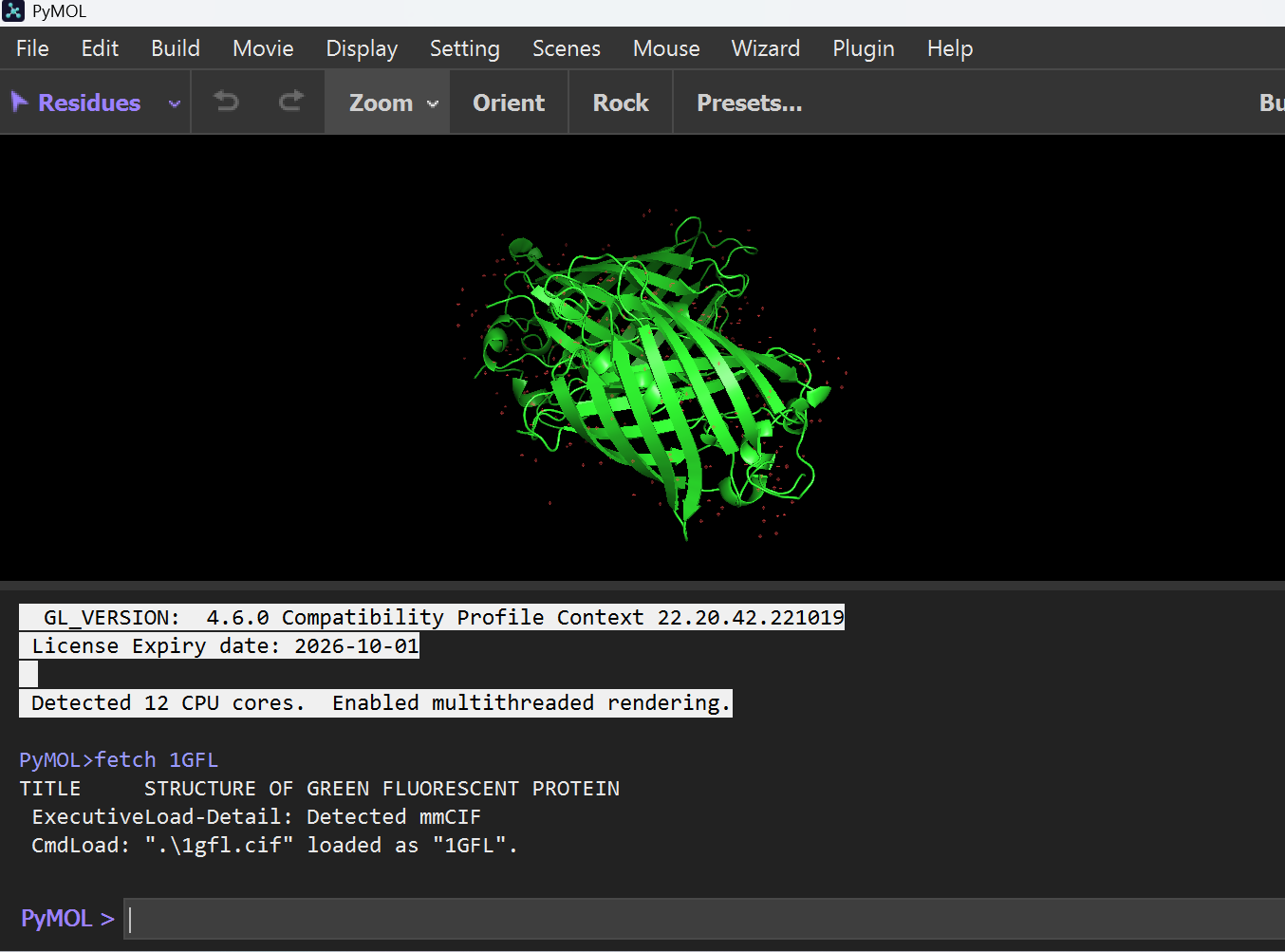
I started by fetching the 1GFL protein structure directly from the PDB database.
- Command:
fetch 1GFL
B. Visualization Formats
I explored different structural representations to understand the “Beta-can” architecture:
Cartoon Representation: Highlights the 11-stranded $\beta$-sheet barrel and the central $\alpha$-helix.
- Command:
hide everything; show cartoon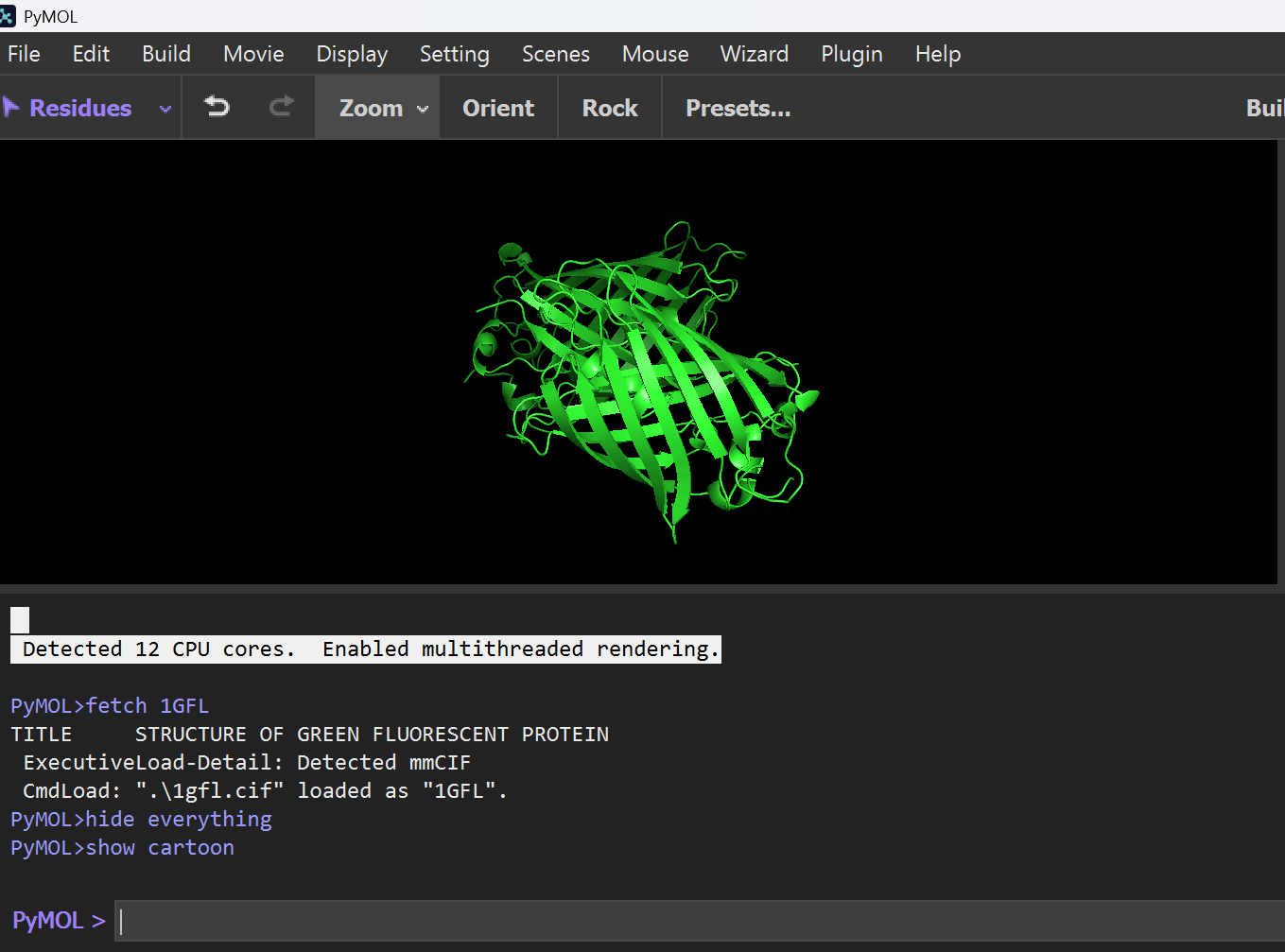
- Command:
Ribbon Representation: Shows the smooth path of the polypeptide backbone.
- Command:
hide everything; show ribbon
- Command:
Ball and Stick Representation: Used to visualize specific atomic interactions and side chains.
- Command:
hide everything; show sticks
- Command:
C. Secondary Structure Analysis
To determine the dominant structural elements, I assigned and colored the secondary structures.
- Command:
hide everything show cartoon dss color red, ss h color yellow, ss s color green, ss l
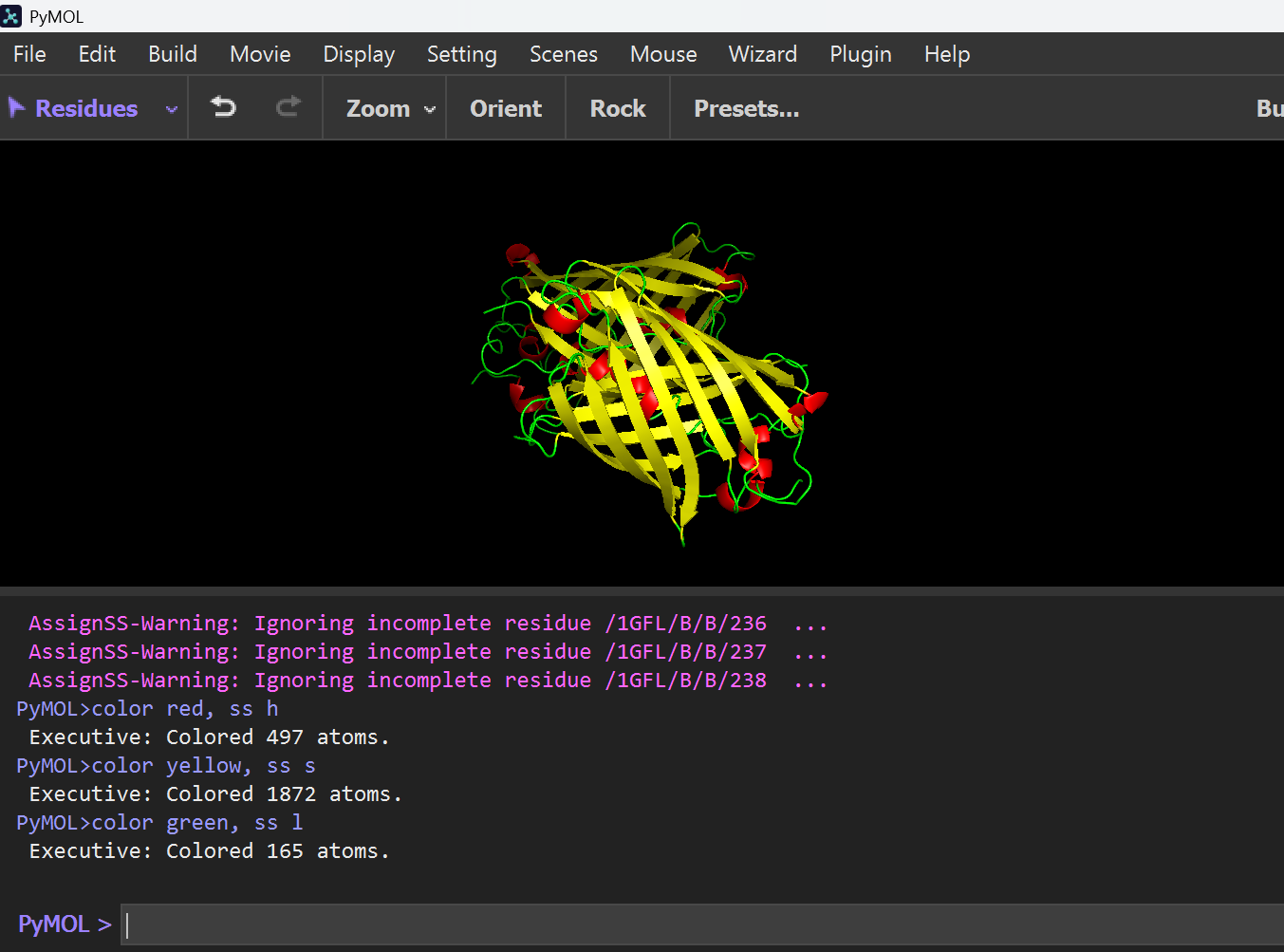
The image shows that the protein is predominantly composed of Beta-sheets (yellow) forming a cylindrical Beta-barrel structure, with very few Alpha-helices (red). Therefore, the structure is sheet-dominated.
D. Color by Residue Type
Then I typed this command to color by residue type (hydrophobic vs hydrophilic):
hide everything; show cartoon; # Hydrophobic residues; select hydrophobic, resn ala+val+leu+ile+met+phe+trp+pro; color orange, hydrophobic; # Hydrophilic residues; select hydrophilic, resn arg+lys+asp+glu+asn+gln+his+ser+thr+tyr; color blue, hydrophilic
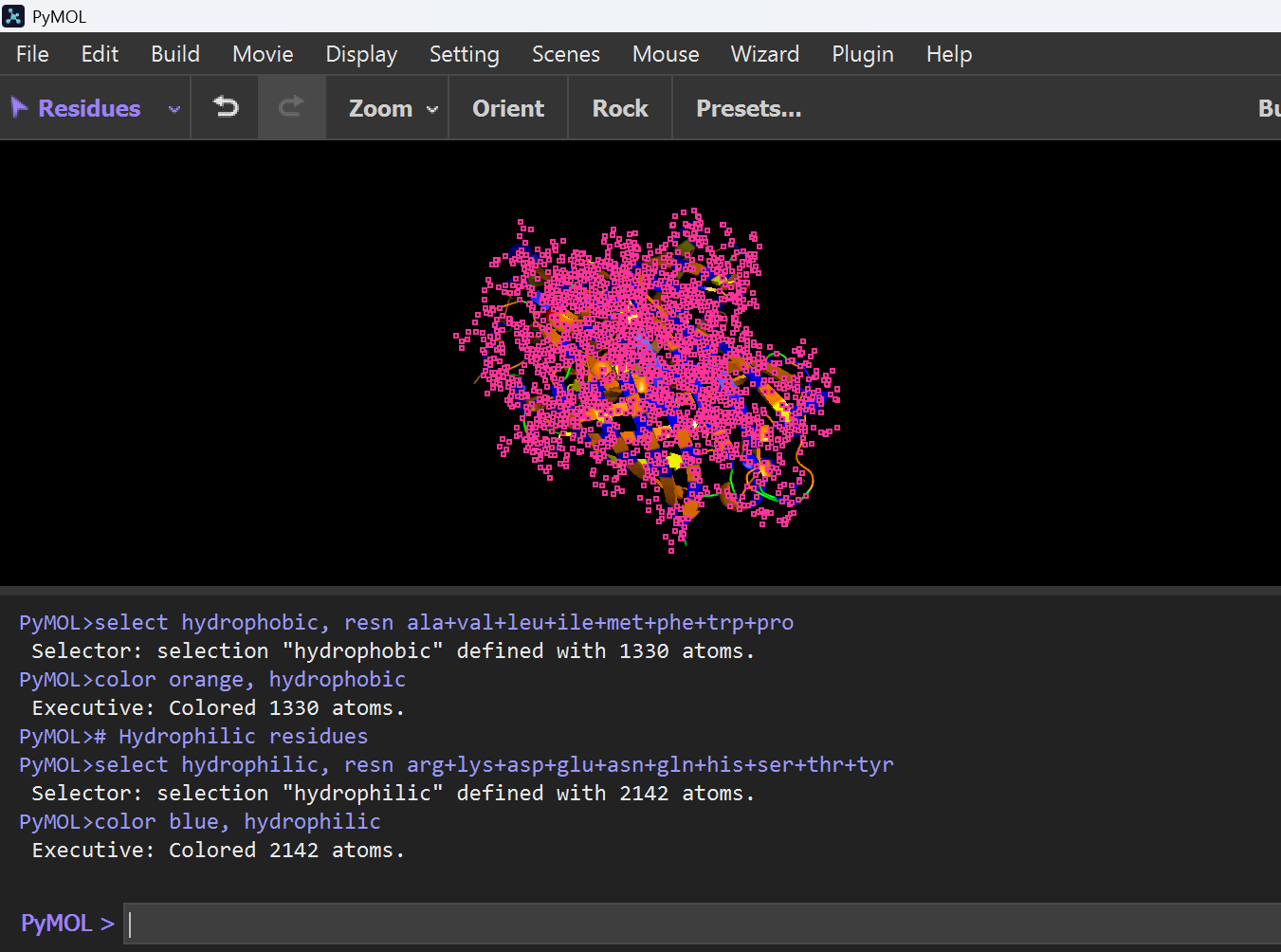
Image shows that Hydrophobic residues (Orange) are primarily located in the interior of the protein, stabilizing the structure, while hydrophilic residues (blue) are exposed on the surface, allowing interaction with the aqueous environment.
E. Binding Pocket
Then I typed this command to show surface visualization and binding pockets:
hide everything show surface color white set transparency, 0.2
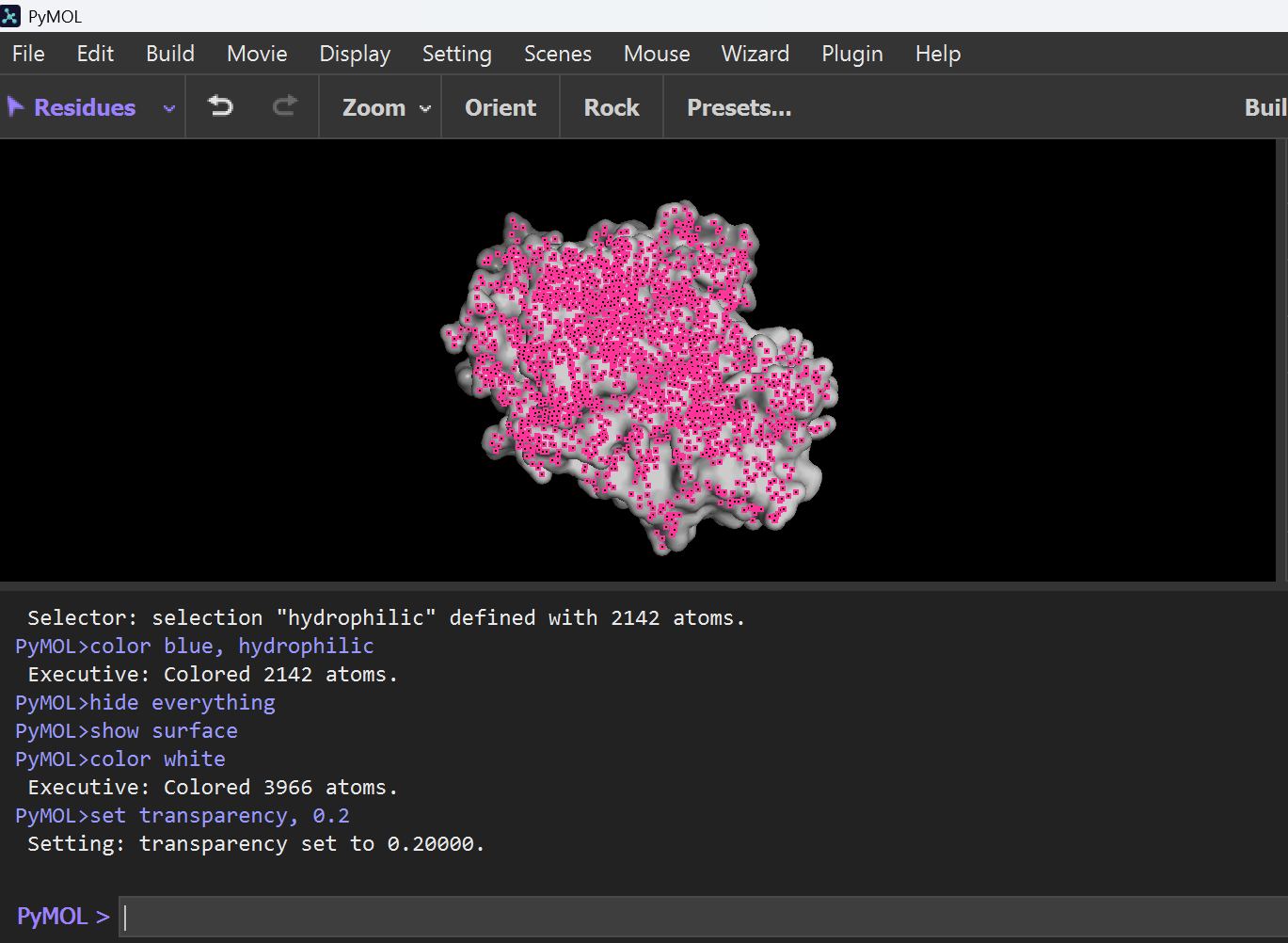
To highlight pockets, I typed this this command:
set surface_quality, 1
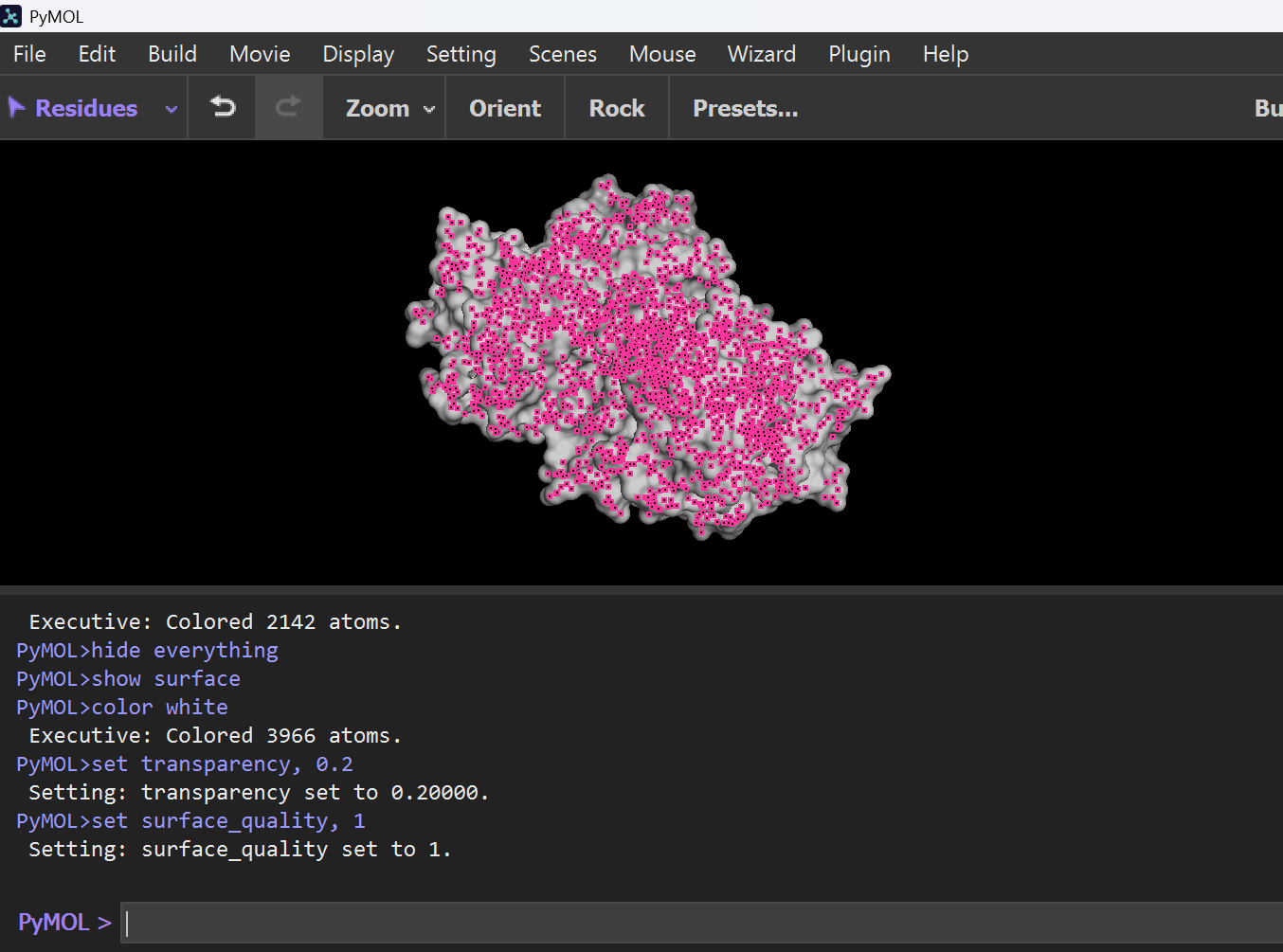
Note: I got help from chatgpt for PyMol Portion
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling: Deep Mutational Scan
I used the ESM-2 (650M parameters) model in relative mode to generate an unsupervised deep mutational scan for the GFP (1GFL) sequence. The heatmap represents the log-likelihood of every possible single-point mutation across the 238 amino acids.
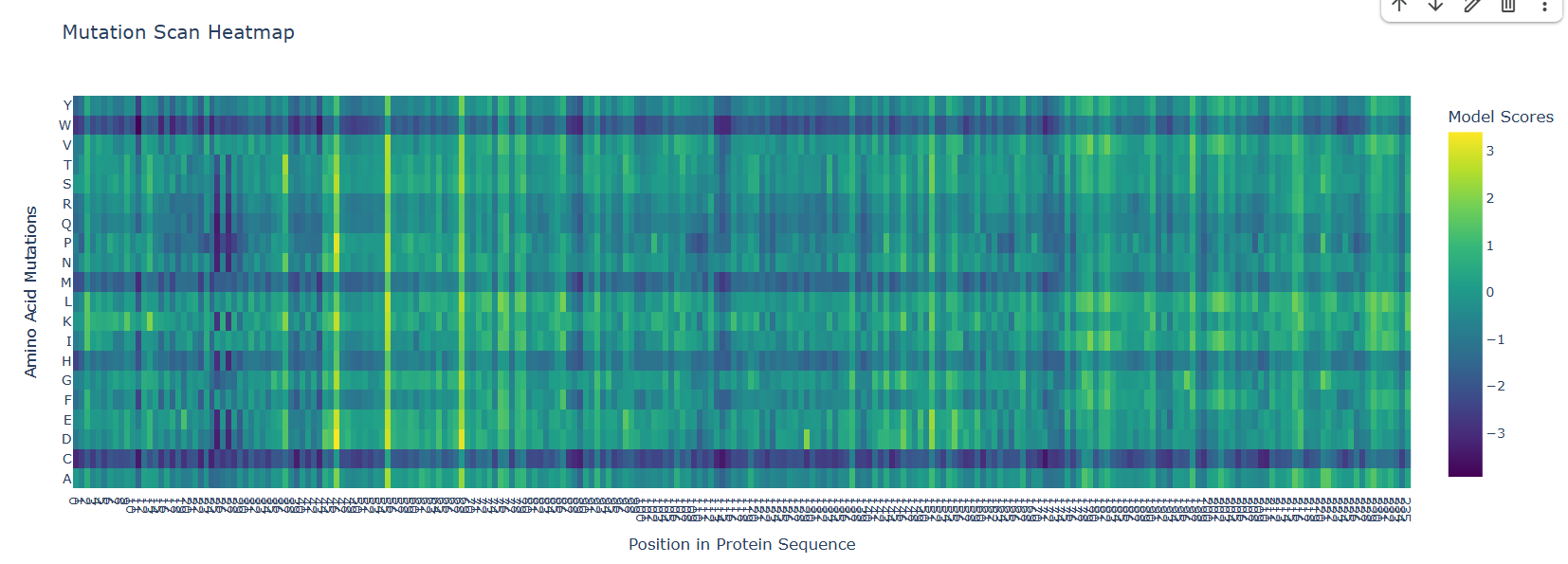
Pattern Analysis & Biological Interpretation:
Based on the heatmap results, I identified several distinct biological patterns that align with the structural functional requirements of GFP:
The “Kill Zone” (Chromophore: Positions 65–67):
- Observation: A dense, dark vertical stripe is visible at positions Ser65, Tyr66, and Gly67.
- Interpretation: These residues are essential for the spontaneous chemical reaction that forms the glowing fluorophore. The model correctly predicts that almost any mutation here is deleterious, as it would disrupt the electronic conjugation required for light emission.
The “Zebra Stripe” Pattern (Beta-Barrel: Residues 80–200):
- Observation: There is a repeating pattern of low tolerance (dark) and high tolerance (light) spots.
- Interpretation: This reflects the alternating nature of the β-barrel. Inward-facing residues are hydrophobic and hold the core together; the model punishes polar mutations here. Outward-facing residues interact with water and show high tolerance for mutations as they do not destabilize the primary fold.
Flexible Termini (N and C Tails):
- Observation: Positions 1–10 and 230–238 show high tolerance (lighter colors).
- Interpretation: These “floppy” ends do not contribute significantly to the structural stability of the barrel, allowing for mutations or the addition of affinity tags without breaking the protein’s function.
Specific Residue Highlight:
- Residue: Glycine 67 (G67).
- Mutation: G67A (Alanine).
- Pattern: Even a small change to Alanine shows a high negative score (approx -3.0).
- Explanation: G67 is structurally required because it is the only amino acid small enough to allow the protein backbone to “kink” into the specific shape needed for chromophore formation. Replacing it with Alanine introduces steric hindrance that prevents the cyclization reaction entirely.
Note: Google Cheat Sheet of GFP Protein for interpretation

C1. Latent Space Analysis
I used the ESM-2 model to generate 320-dimensional embeddings for a dataset of protein sequences, to which I manually added my target protein GFP (1GFL). To visualize this high-dimensional data, I reduced it to 3 dimensions using t-SNE.
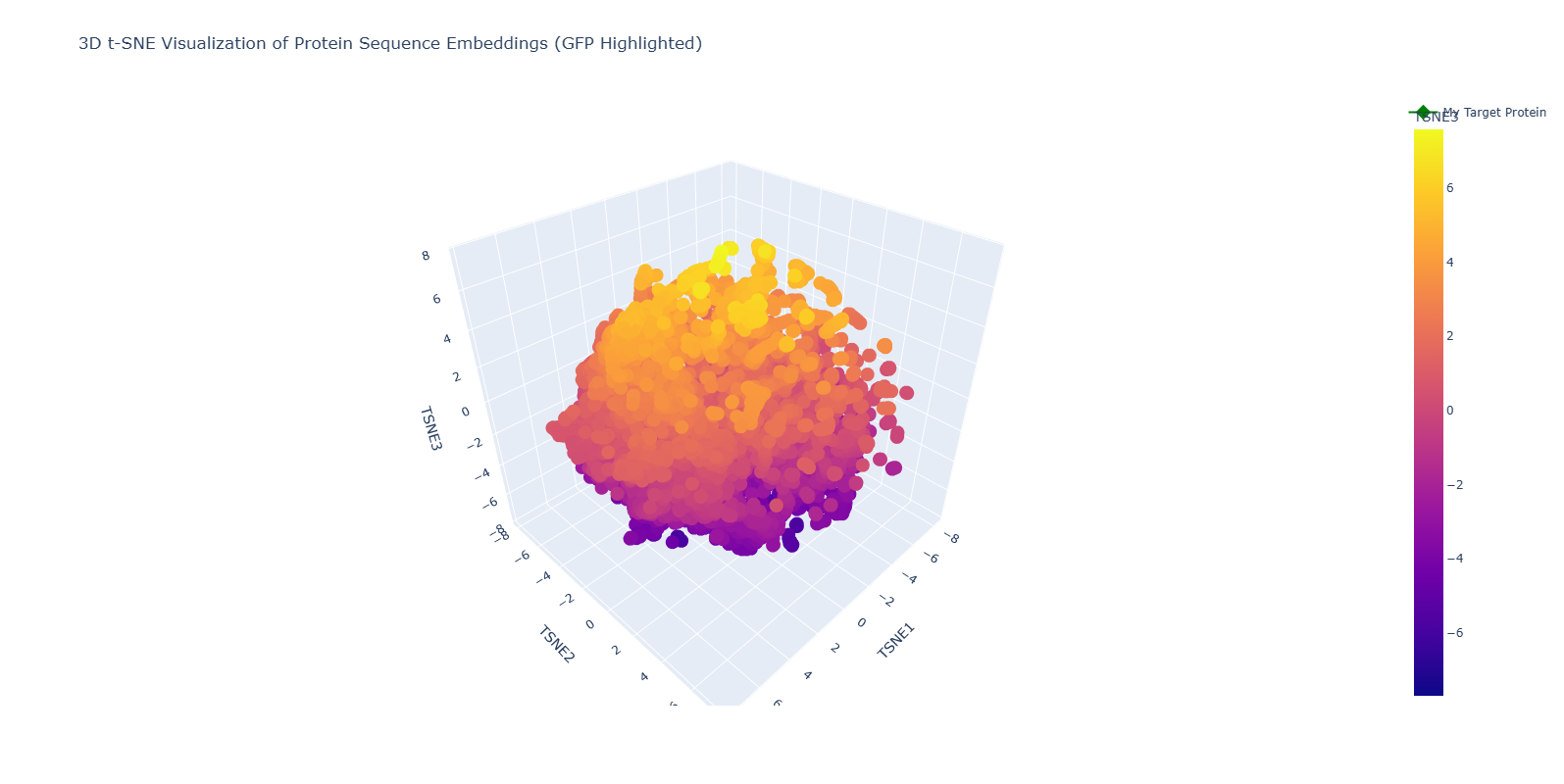
1. Neighborhood Analysis: Structural Approximation
- Observations: The resulting 3D map shows that the model effectively approximates similar proteins into “neighborhoods.” While exploring the interactive plot, I found clusters of specific families, such as interleukins, ribonuclear proteins, and globins from various species (Norway rat, mouse, etc.).
- 3D Perspective: Because this is a 3D visualization, the proximity of proteins changes depending on the orbital angle. This complexity reflects how the model evaluates multiple biochemical features (charge, hydrophobicity, length) simultaneously.
2. Positioning and Similarity of My Protein (GFP) To easily identify my protein, I highlighted it as a Green Diamond in the plot.
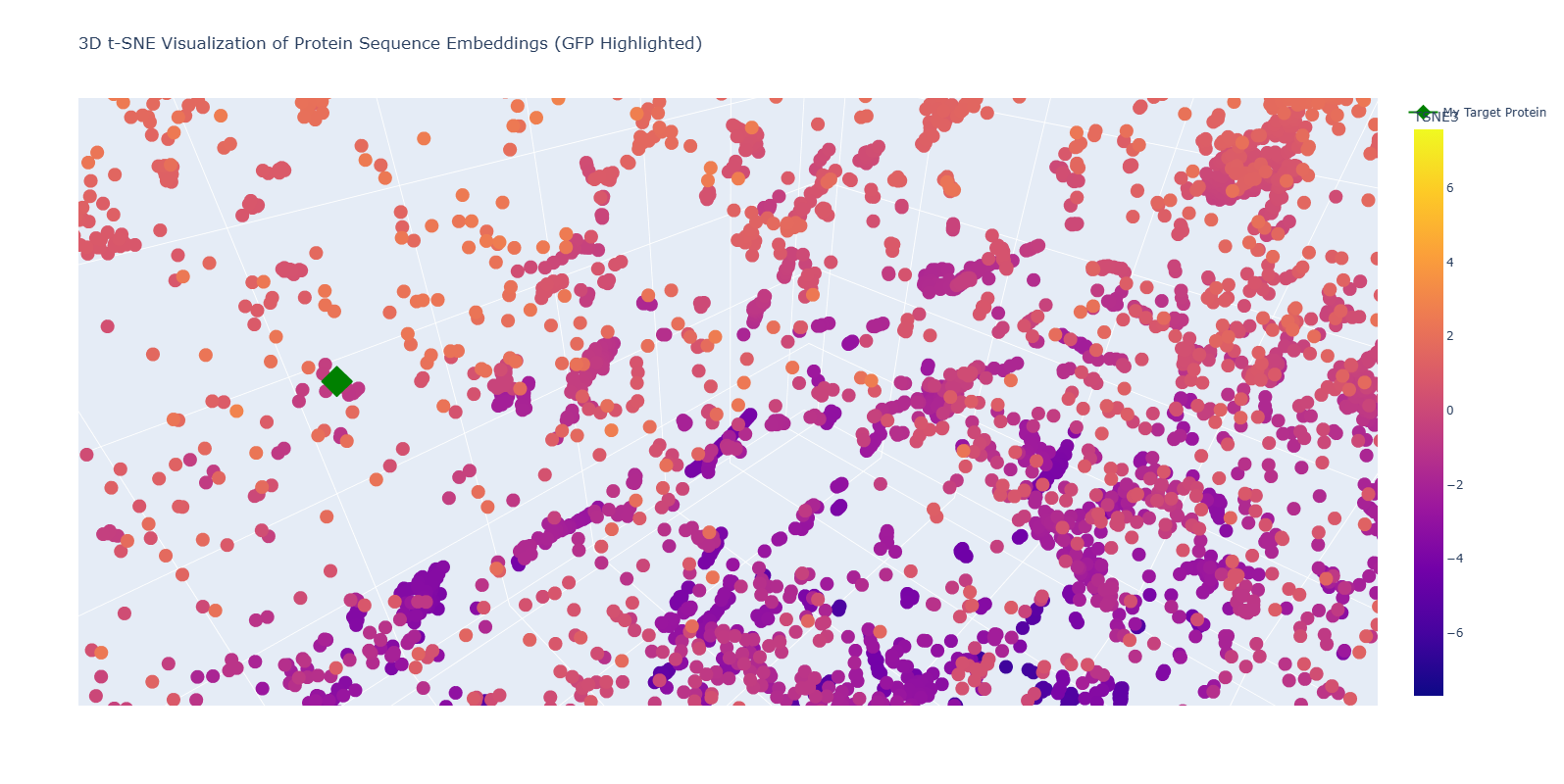
- The “Twin” Discovery: Upon zooming into the neighborhood of my highlighted point (1GFL), I discovered another protein entry: “d6l27a_ Green fluorescent protein, GFP {Jellyfish (Aequorea victoria)}”.
- Biological Significance: This is a powerful validation of the ESM-2 model. Without being provided any structural data, the model recognized the unique amino acid patterns of the 11-stranded beta-barrel and placed my sequence directly next to its biological homolog (Jellyfish GFP).

- Conclusion: The positioning confirms that the latent space of the language model successfully captures the “grammar” of protein folds. My protein is surrounded by other Beta-sheet dominated proteins, proving that the model can distinguish between different structural motifs (alpha vs. beta) based purely on the primary sequence.
How to visualize your protein in latent space


C2. Protein Folding (ESMFold Analysis)
I used ESMFold to predict the structure of GFP and tested its resilience by introducing specific mutations.
| Version | Visual Description | Structural Analysis |
|---|---|---|
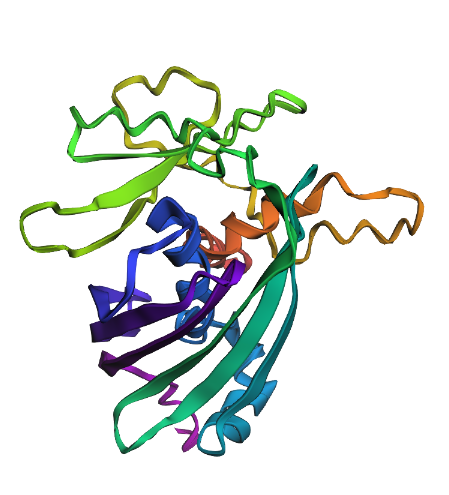
| Normal | Perfect 11-stranded $\beta$-barrel. | The model accurately predicts the native fold of 1GFL. |
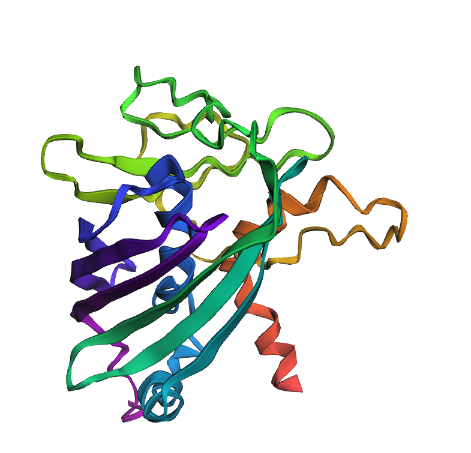
| Mutation 1 | Structure remains intact and stable. | Resilient: Changing Y66 to A66 does not disrupt the overall “beta-can” architecture. |
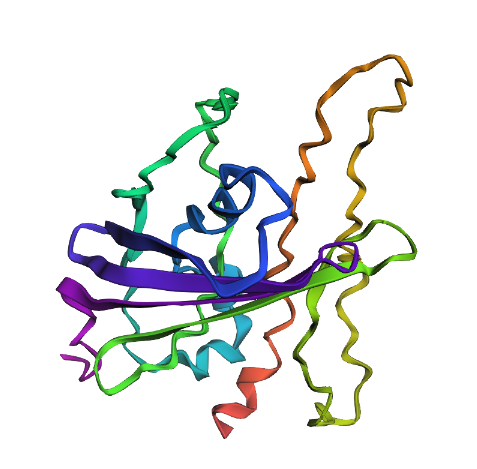
| Mutation 2 | Significant structural collapse and loss of fold. | Not Resilient: Deleting a 20-residue segment from 100 to 120 amino acid breaks the hydrogen bond network, causing the barrel to unfold. |
Normal GFP:
Mutation 1 (Point Mutation):
Mutation 2 (Large Deletion):
C3. Protein Generation (Inverse Folding)
In this final section, I explored Inverse Folding. While traditional protein folding (Forward) predicts a structure from a sequence, Inverse Folding uses a fixed 3D backbone to propose entirely new amino acid sequences that could stabilize that specific shape.
1. Sequence Proposal with ProteinMPNN
I used the 3D backbone of GFP (1GFL) as the input for ProteinMPNN. The model analyzed the geometric constraints of the 11-stranded $\beta$-barrel to suggest novel sequence candidates.
- Probability Heatmap Analysis: The generated heatmap illustrates the likelihood of each amino acid at every position across the 238-residue sequence. Bright yellow regions indicate “fixed” residues that are structurally essential for the fold, while darker regions show areas where the model allows for more sequence diversity.
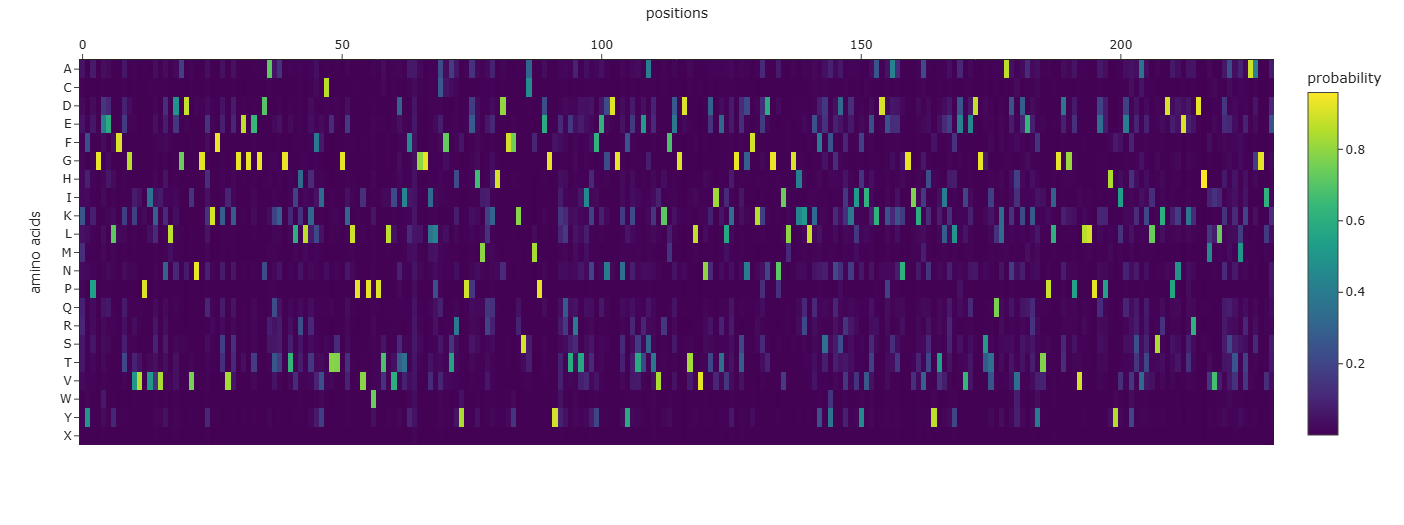
- Sequence Recovery: My best-designed sequence achieved a Sequence Recovery of 66.38%. This indicates that while the AI-generated sequence is ~34% different from the natural one, it preserved the core chemical logic required to maintain the “beta-can” architecture.
2. Round-trip Validation with ESMFold
To verify if the AI-designed sequence (SYPGDELFEGVVPIKVNLKGDVNGEKFSVEGEGEGDAKKGEITLKFVCTTGKLPVPWPTLVDIFSGGIPCFTKYPEHMKHHDFFKSCMPEGYKQERTIYFEGDGKFETRATVKFEGDTLVNEIELKGSGFKKDGNILGHKLKFSYQSYKRYITADKAKNGIKATYTLEYPVEDGSVQKAKVEETYTPLGDGPVLLPEPHYLEVEVELSKDPNEKRDHVVLKAKMVAAGIE) actually folds into the intended shape, I performed a “round-trip” validation by folding the new sequence back into 3D space using ESMFold.
- Observations: As shown in the predicted structure below, the new sequence successfully folded into a near-perfect $\beta$-barrel.
- Structural Fidelity: The alignment of the $\beta$-strands and the overall topology remained consistent with the original 1GFL structure, despite the significant sequence divergence.
Conclusion
This experiment demonstrates the power of ProteinMPNN in de novo protein design. It proves that the model can “invent” new sequences that nature has not yet explored, which still satisfy the physical and geometric requirements of the GFP fold. The structural integrity remained robust, confirming that the “language” of proteins allows for multiple sequence solutions for a single structural fold.
For Part C: Extensive help was taken from Google Gemini to understand google collab notebook, methodology and results.
Part D. Group Brainstorm on Bacteriophage Engineering
By: Sami Ur Rehman (2026a-sami-ur-rehman) and Edna Wanjiru Macharia (2026a-ednah-wanjiru)
Goals and Strategy
- Primary Goal: Increase the toxicity of the MS2 bacteriophage lysis protein (L) to achieve faster and more complete bacterial lysis, leading to higher phage titers for therapeutic applications.
- Secondary Goal: Modulate the protein’s interaction with the host chaperone DnaJ by redesigning the N-terminal interface rather than simply deleting it. This allows for tunable control of lysis timing, not just complete bypass.
- Design Focus: The strategy involves targeting the N-terminal regulatory domain (residues 1-36) while protecting essential functional elements: the C-terminal transmembrane domain (residues 46-75), the critical L48-S49 motif (which Chamakura, Edwards, et al. (2017) shows is essential for function), and the protein’s ability to form high-order oligomeric pores (>10 subunits) as demonstrated in Mezhyrova et al. (2023).
Computational Pipeline
The project utilizes a multi-step computational protein engineering pipeline to rationally design mutations: Homolog Discovery (BLAST): Identifying related lysis proteins from related leviviruses to find evolutionarily conserved residues and natural sequence variations that inform design choices
Multiple Sequence Alignment (Clustal Omega): Mapping essential structural regions and differentiating between highly conserved zones (LS motif, pore-forming helix) that must be protected versus mutable sites (N-terminal domain) that can be engineered
In Silico Mutagenesis (Evo 2): Using phage-trained protein language models from King et al. (2025) to generate mutation heatmaps across the N-terminal domain and rationally select amino acid substitutions that improve protein fitness while maintaining “phage-like” sequence characteristics
Structure Prediction (ESMFold): Modeling the 3D structures of promising mutants to ensure the essential transmembrane helix is not distorted and the LS motif remains intact (pLDDT >70)
Aggregation Screening (TANGO/AGGRESCAN): Filtering out mutants with high aggregation propensity that could impair membrane insertion Complex Prediction (AlphaFold Multimer): Evaluating whether mutated proteins can successfully form the required oligomeric pore complex (>10 subunits) and assessing if N-terminal mutations successfully reduce interactions with DnaJ while preserving the core lytic domain
Overlapping Gene Check: Verifying that codon changes do not disrupt the overlapping Coat and Replicase reading frames essential for phage viability.
Expected Outcomes and Applications
The pipeline is expected to yield MS2 L variants with faster lysis kinetics, reduced DnaJ dependency, proper transmembrane insertion, and preserved pore-forming capability.
These optimized proteins have potential downstream applications in synthetic phage engineering for therapeutic cocktails, antimicrobial protein development, bacterial ghost cell production for vaccines, and tools to study lysis timing regulation in phage infection cycles.
Challenges and Future Validation
- Key computational challenges include limited structural data for the L-DnaJ complex (no experimental structure exists), model bias toward globular proteins rather than small transmembrane toxins, poor database annotation of single-gene lysis proteins (amurins), and the risk of over-stabilization leading to rigid structures that fail to transition into the active pore-forming state.
- Biological risks include mutations might inadvertently affect L’s interaction with its unknown lethal target, modified proteins may become vulnerable to host proteases, disruption of overlapping genes could break essential phage functions, and context-dependent interactions in the membrane environment are difficult to model computationally.
- Future validation steps will involve experimentally expressing the computationally identified mutants in E. coli, measuring lysis kinetics (OD600), assessing protein stability via Western blot, testing DnaJ independence through pulldown assays in wild-type and DnaJ mutant backgrounds, and visualizing pore formation using cryo-EM in lipid nanodiscs (following Mezhyrova et al. (2023) protocols).
You can check out the fully detailed Project Proposal here
Note: I acknowledge help from DeepSeek in understanding the complex research papers and computational tools. I also thank my batchmate 2026a-nourelden-rihan (BioClub Tokyo node) for sharing his approach, which helped clarify the project expectations. *