Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
Here is the Human SOD1 sequence from Uniprot (P00441)
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Here it is again after adding the A4V mutation
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Here are the produced peptides:
| Index | Binder | Pseudo Perplexity |
|---|---|---|
| 1 | WRYYPTGLRHKX | 12.016788 |
| 2 | HHYGAVVLELKK | 18.394675 |
| 3 | KRYPVAAARWKX | 10.061424 |
| 4 | WHVYVVAVALKE | 21.195186 |
| 5 | FLYRWLPSRRGG | N / A |
Part 2: Evaluate Binders with AlphaFold3
2 of the generated peptides had X in their sequence and Alphafold was rejecting them so I replaced X with A or Alanine on the advice of Google Gemini.
AlphaFold3 Prediction Results
| Peptide Sequence | ipTM Score | Binding Location & Characteristics | Screenshot |
|---|---|---|---|
| HHYGAVVLELKK | 0.68 | “Grey Zone” Candidate: This peptide is the most promising, as it is the only one to exceed the 0.6 failure threshold. It appears to engage more deeply with the protein structure, potentially approaching the dimer interface or the core Beta-barrel. | 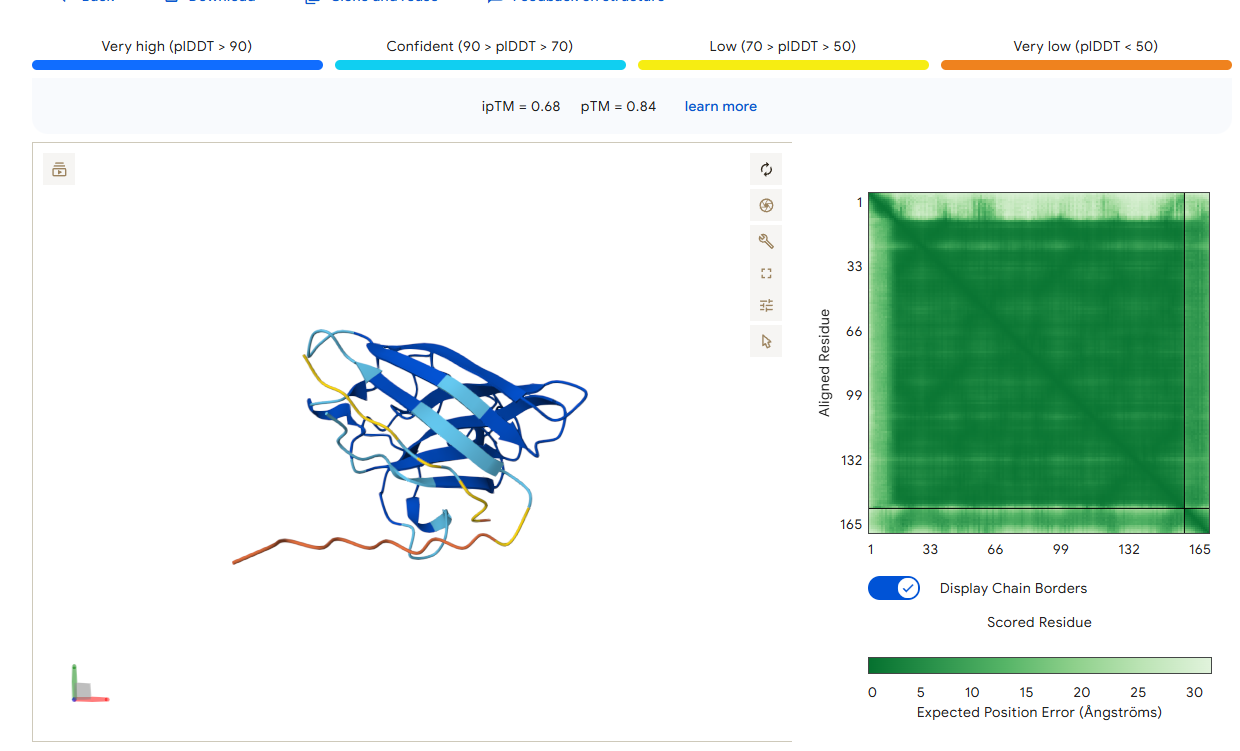
|
| WHVYVVAVALKE | 0.35 | Failed Prediction: While it slightly exceeds the known binder’s score, it remains surface-bound on the Beta-sheet region with low confidence. | 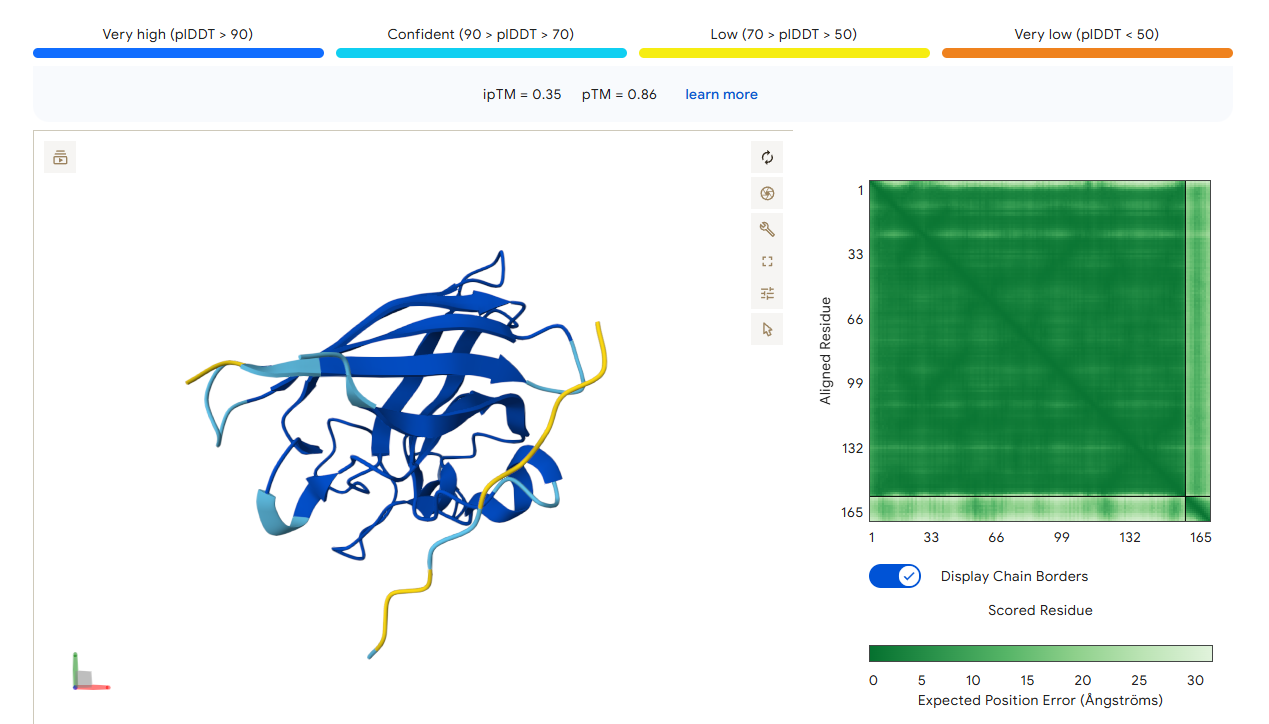
|
| FLYRWLPSRRGG (Known) | 0.32 | Failed Prediction (Control): The baseline binder shows low confidence; it localizes on the protein surface but does not show specific engagement with the A4V site at the N-terminus. | 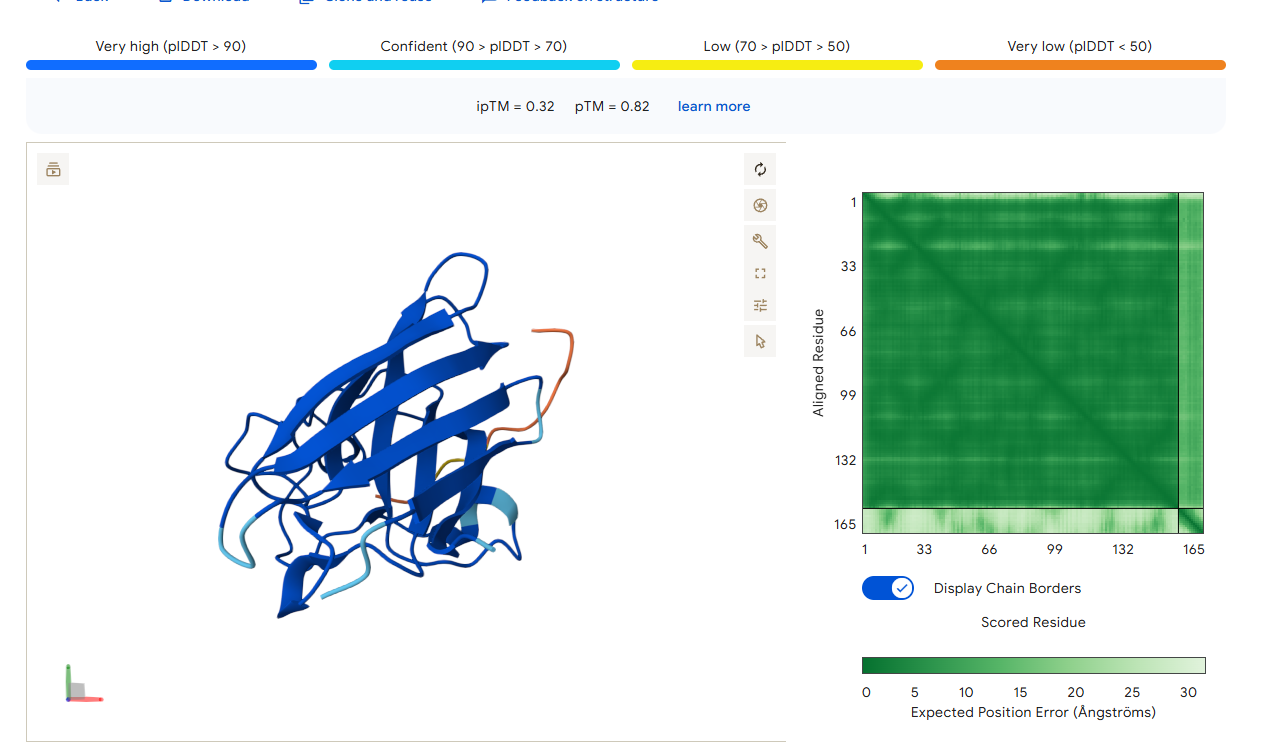
|
| KRYPVAAARWKX | 0.30 | Failed Prediction: This peptide remains mostly surface-bound near flexible loops, showing low structural complementarity to the mutant SOD1. | 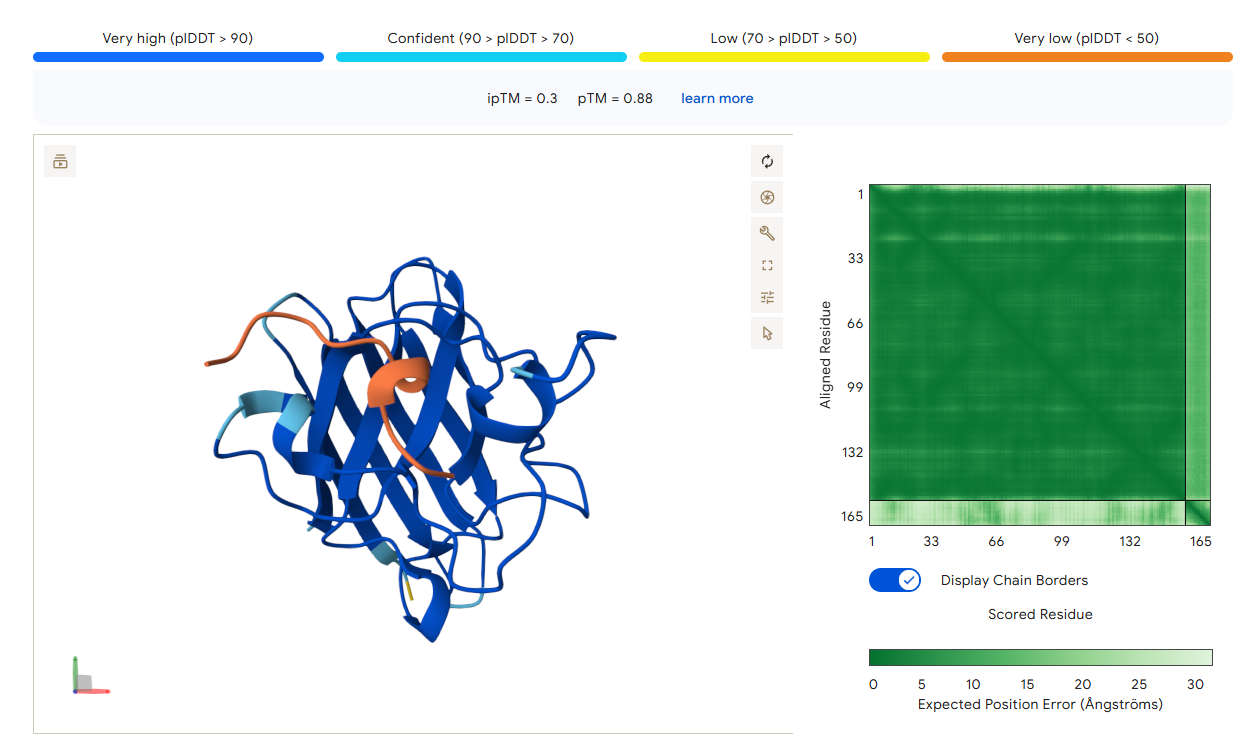
|
| WRYYPTGLRHKX | 0.26 | Failed Prediction: Despite a good PepMLM score, the structural model suggests this sequence is a poor fit, failing to localize near any specific functional region. | 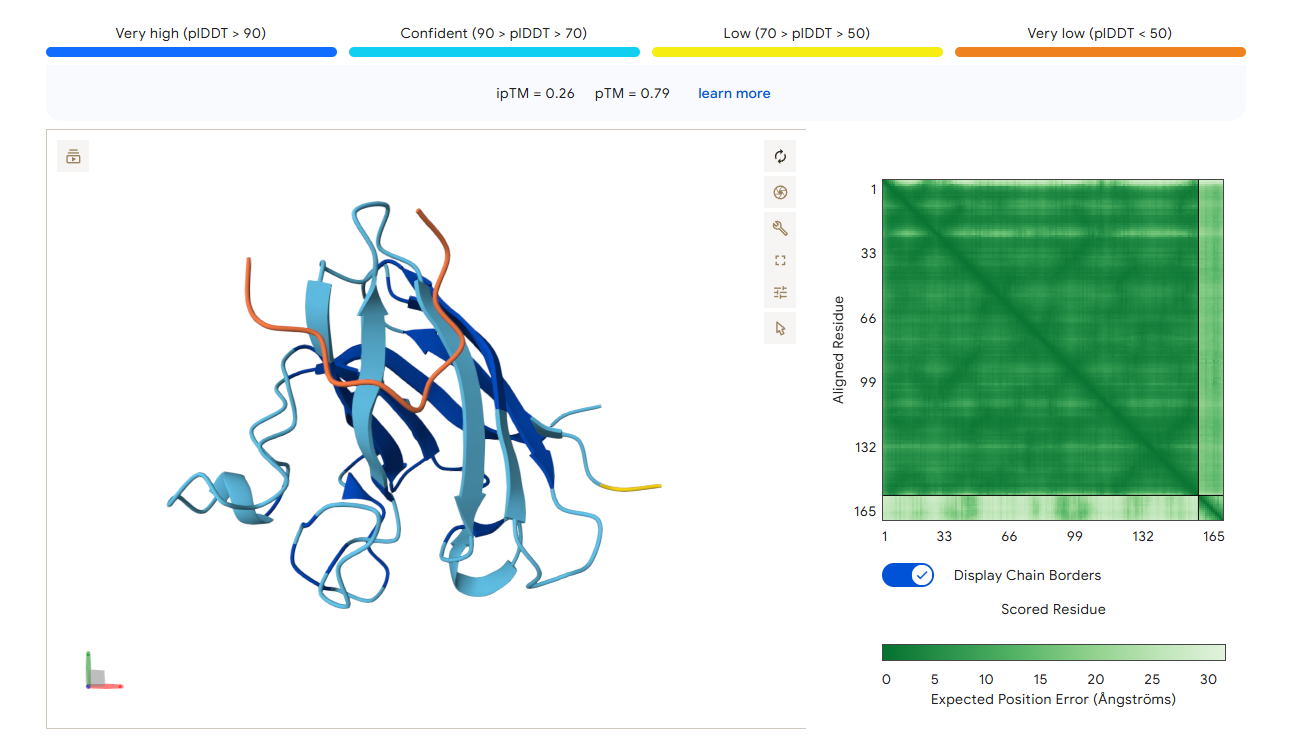
|
Summary Analysis The ipTM scores for the PepMLM-generated peptides range from 0.26 to 0.68. According to AlphaFold standards, an ipTM score above 0.8 represents a high-quality prediction, while scores below 0.6 are generally considered failed predictions. Most candidates, including the known binder (0.32), fall into the failure category, indicating that these interactions are likely unstable or poorly modeled.
However, the peptide HHYGAVVLELKK achieved an ipTM of 0.68, placing it in the “grey zone” (0.6–0.8). This score indicates that the prediction could potentially be correct and represents a significant improvement over the control binder. While most peptides remain surface-bound, HHYGAVVLELKK shows the most potential to move beyond the surface and possibly engage with the dimer interface or the destabilized N-terminus where the A4V mutation sits.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
PeptiVerse Property Comparison
| Peptide Sequence | ipTM Score | pKd/pKi (Affinity) | Solubility | Hemolysis Probability | Screenshot |
|---|---|---|---|---|---|
| HHYGAVVLELKK | 0.68 | 5.454 | 1.000 | 0.030 | 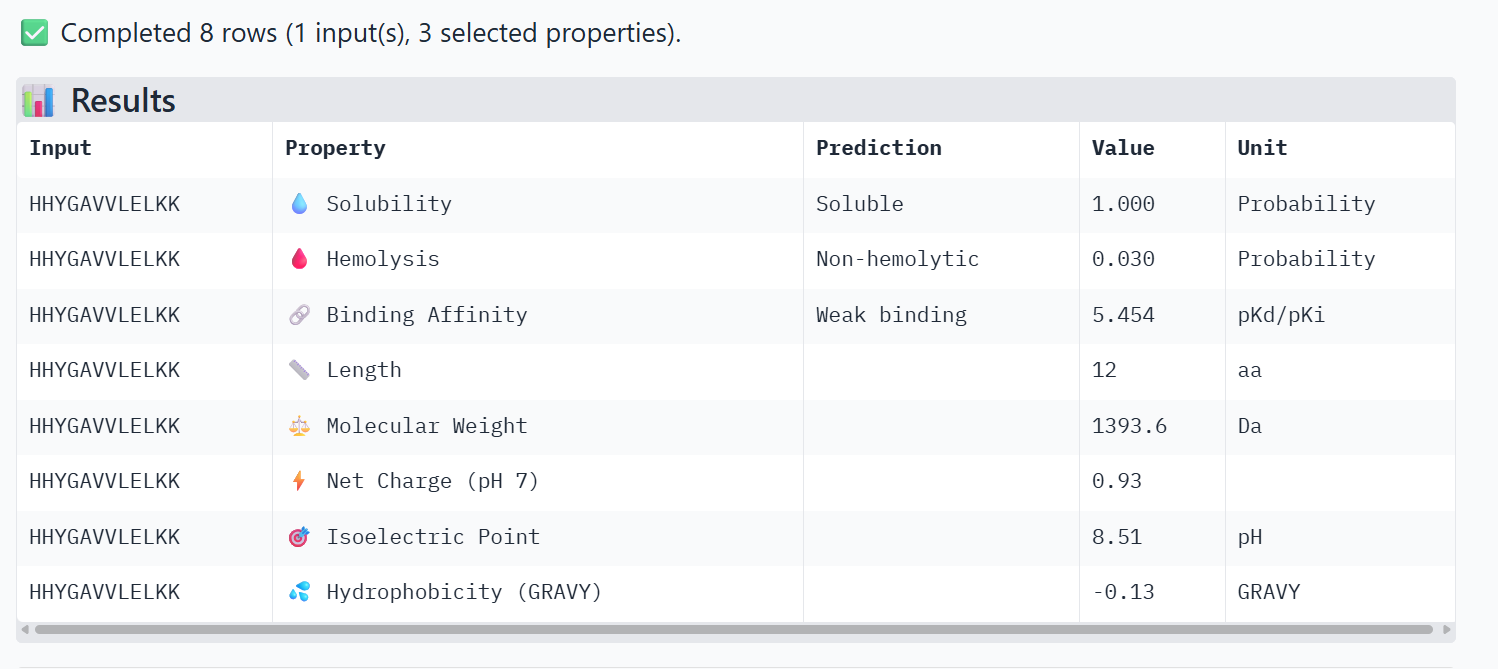
|
| WHVYVVAVALKE | 0.35 | 6.406 | 1.000 | 0.113 | 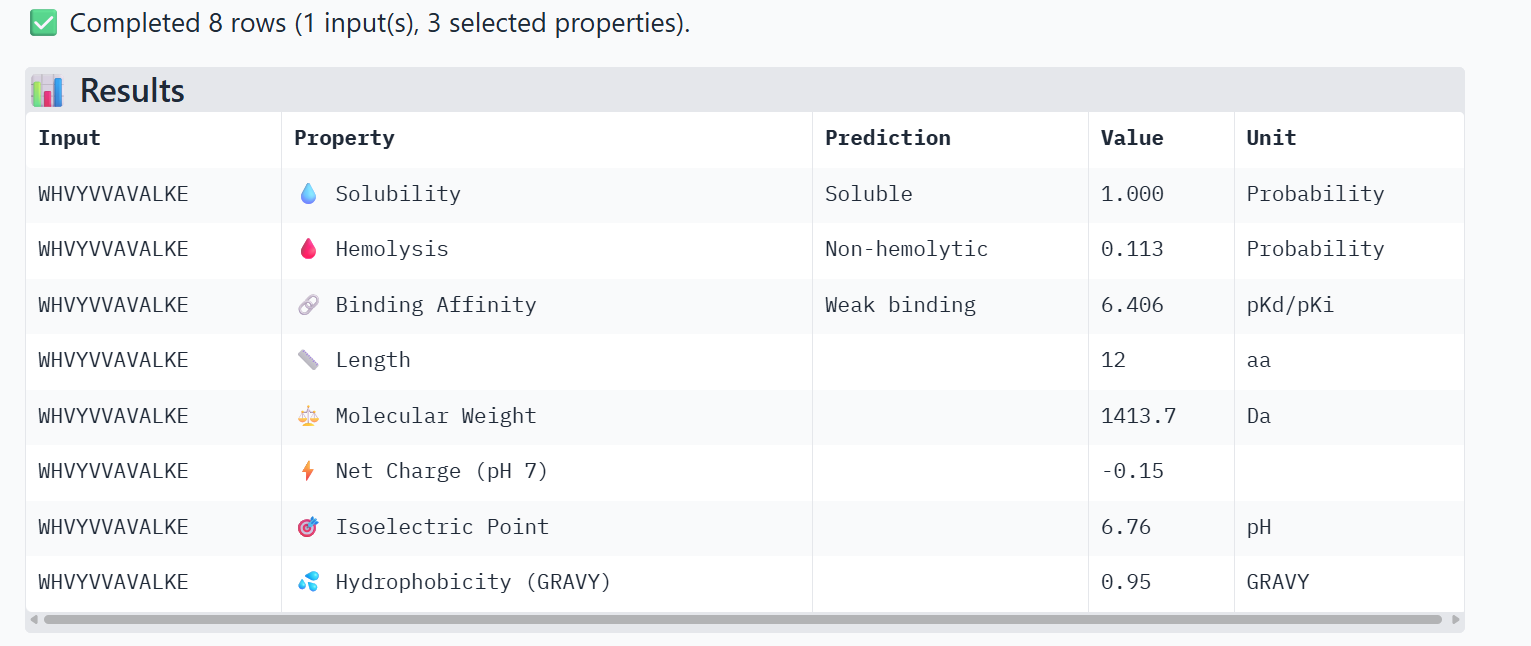
|
| FLYRWLPSRRGG (Known) | 0.32 | 5.968 | 1.000 | 0.047 | 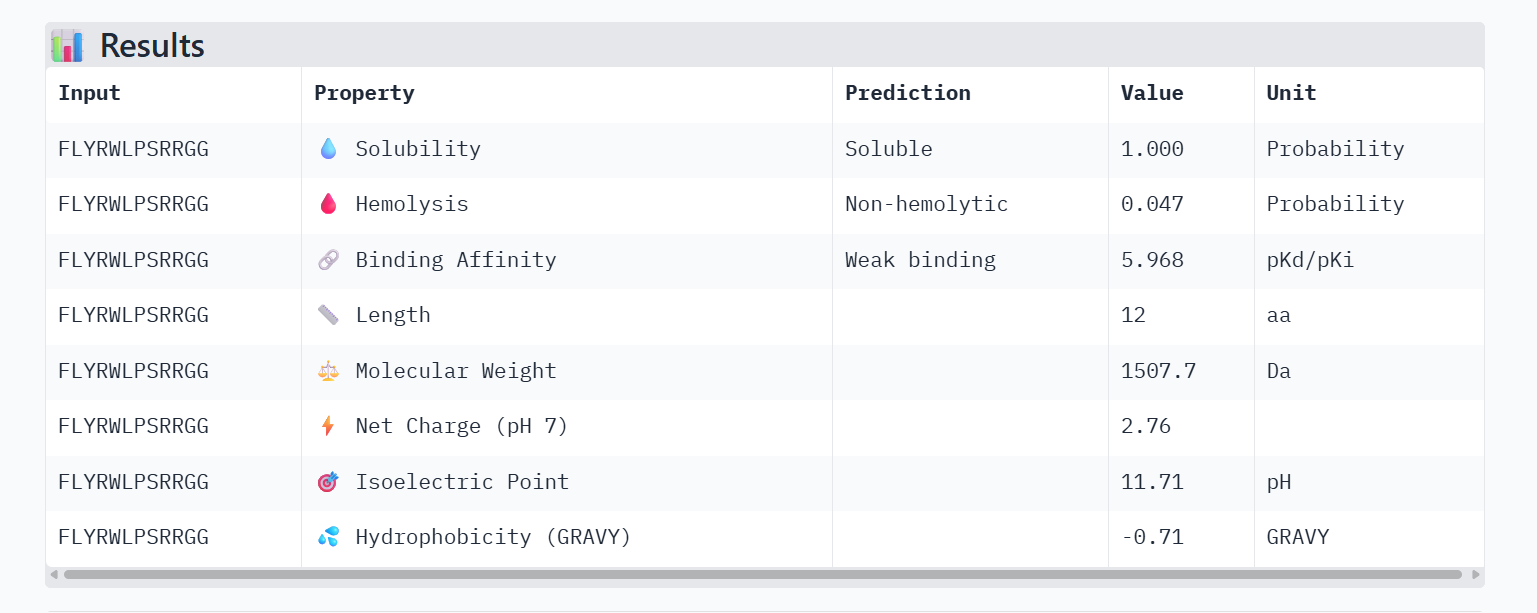
|
| KRYPVAAARWKX | 0.30 | 5.982 | 1.000 | 0.012 | 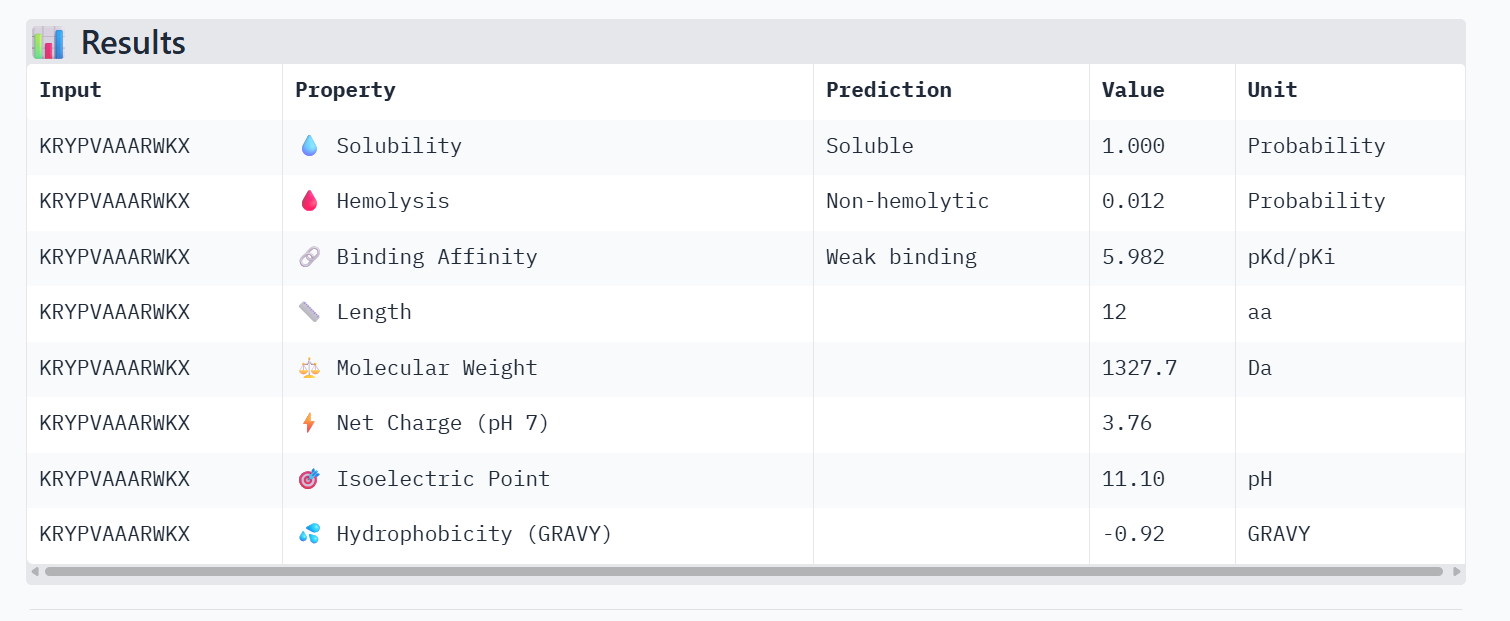
|
| WRYYPTGLRHKX | 0.26 | 5.554 | 1.000 | 0.024 | 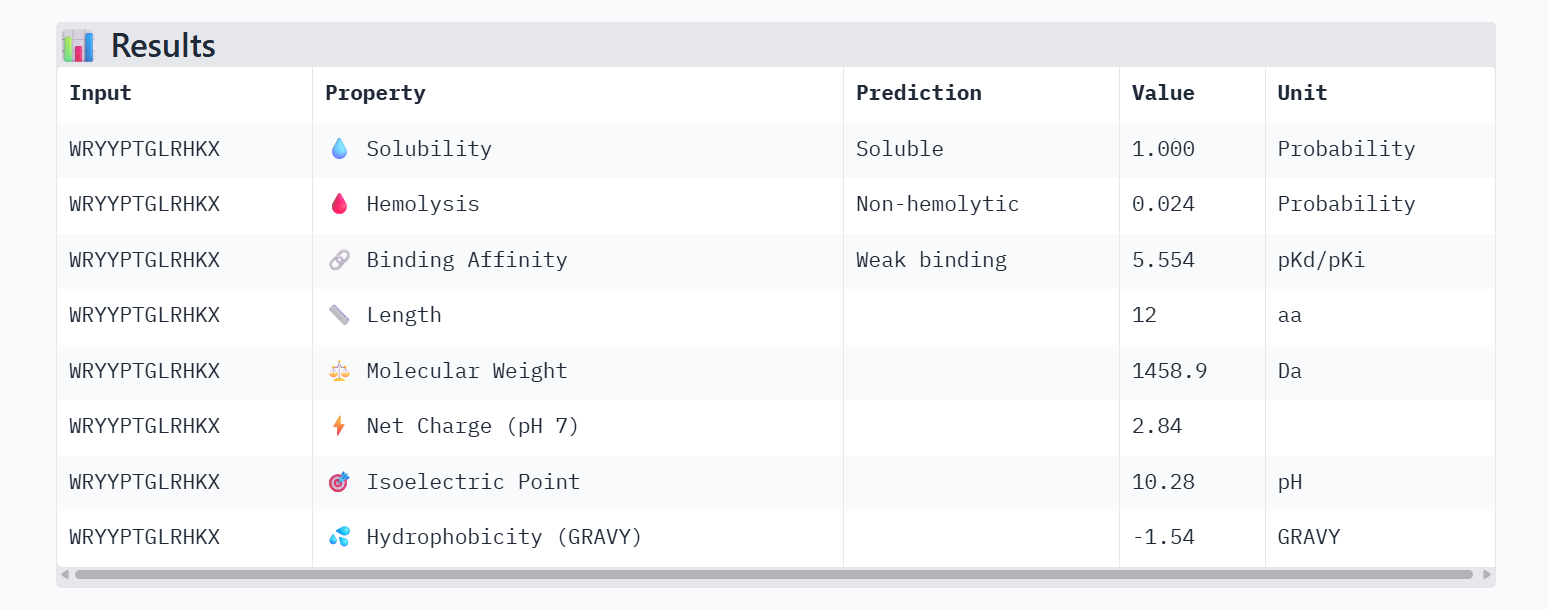
|
Analysis Paragraph
Comparing the structural data from AlphaFold3 to the chemical properties from PeptiVerse reveals that higher structural confidence (ipTM) does not correlate with stronger predicted binding affinity in this dataset. For instance, HHYGAVVLELKK has the highest ipTM (0.68) but the lowest predicted affinity (5.454). Conversely, WHVYVVAVALKE shows the highest affinity (6.406) but is also the most hemolytic (0.113), which is a significant therapeutic drawback. Interestingly, all peptides are predicted to be highly soluble (1.000). While KRYPVAAARWKX stands out as the safest option with the lowest hemolysis probability (0.012), its structural confidence remains low.
Decision & Justification
Peptide to Advance: HHYGAVVLELKK
Justification: I would advance HHYGAVVLELKK because it is the only candidate that provides a credible structural binding mode, moving out of the “failure zone” and into the AlphaFold3 “grey zone” (0.68). While its predicted affinity is lower than others, structural stability is often a more reliable indicator of specific binding for complex targets like the SOD1 A4V mutation. Additionally, it remains highly soluble and has a low hemolysis probability, ensuring a safe therapeutic profile while potentially stabilizing the destabilized N-terminus better than the low-confidence surface binders.
Part 4: Generate Optimized Peptides with moPPIt
In this section, I moved from global sampling to controlled design. I used the moPPIt model to target specific residue indices (2, 3, 4, 5, 6) corresponding to the A4V mutation site at the N-terminus of SOD1.
Additionally, I selected all optimization properties in the notebook even though the computation was being performed on a T4 GPU in Google Colab, which has limited computational resources. It took 43 mins to implement the code.
Here are the moPPIt generated peptides:
| Peptide Sequence | Hemolysis | Solubility | Affinity | Motif |
|---|---|---|---|---|
| KANYWTTWTSDS | 0.93190462142229 | 0.75 | 5.74363183975219 | 0.78769564628601 |
| KCETKFLQKREI | 0.966306183487176 | 0.75 | 6.49503183364868 | 0.894703328609466 |
| KRQSCQKTKPFV | 0.938299626111984 | 0.75 | 6.26246261596679 | 0.869844377040863 |
| KSQKKQTEICGR | 0.958696339279413 | 0.916666686534881 | 6.46437692642211 | 0.800572216510772 |
In next step, I have decided to take those Peptides, and run them through AlphaFold and PeptiVerse and compare them with the pepMLM ones.
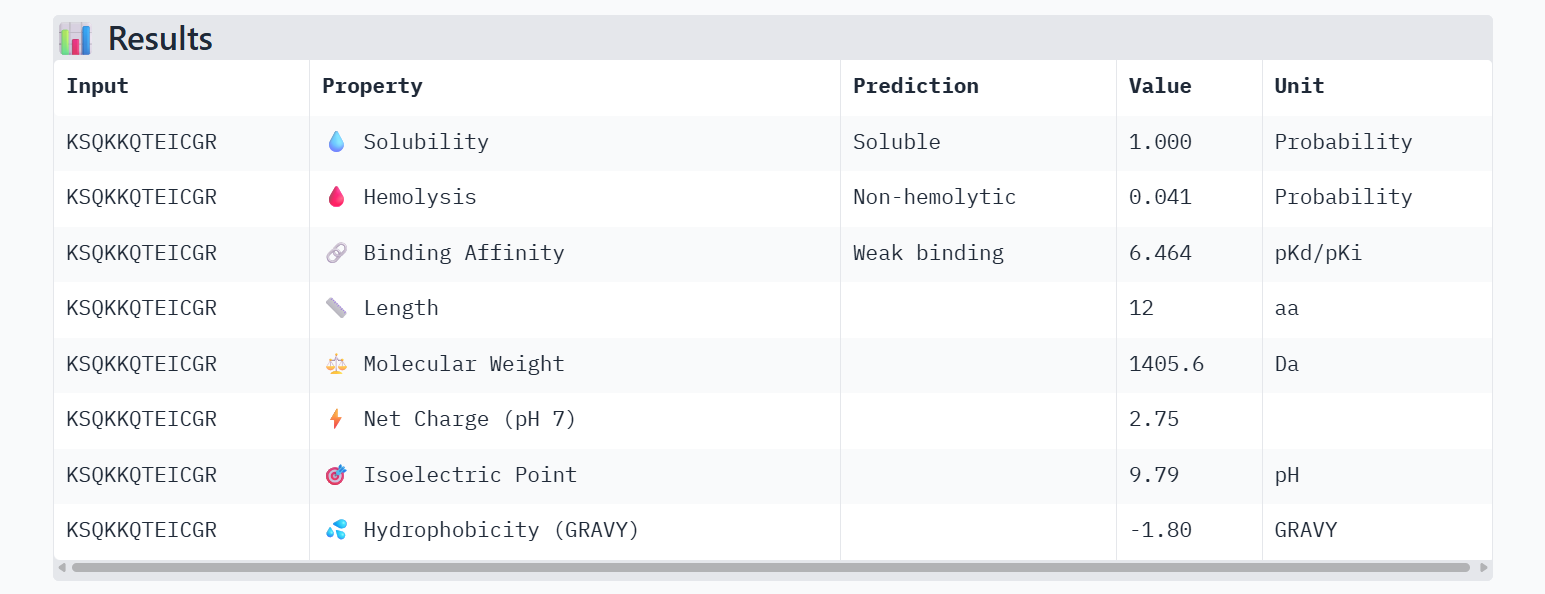
Candidate 1: KSQKKQTEICGR (Lead Candidate)
- AlphaFold3 Validation: ipTM Score: 0.52 (The highest structural confidence among the optimized set).
- Binding Analysis: This peptide shows the most promising localization. Unlike the PepMLM binders that were floating away, this sequence remains in close proximity to the N-terminal region. It appears to “hug” the site of the A4V mutation, suggesting it could potentially stabilize the destabilized fold.
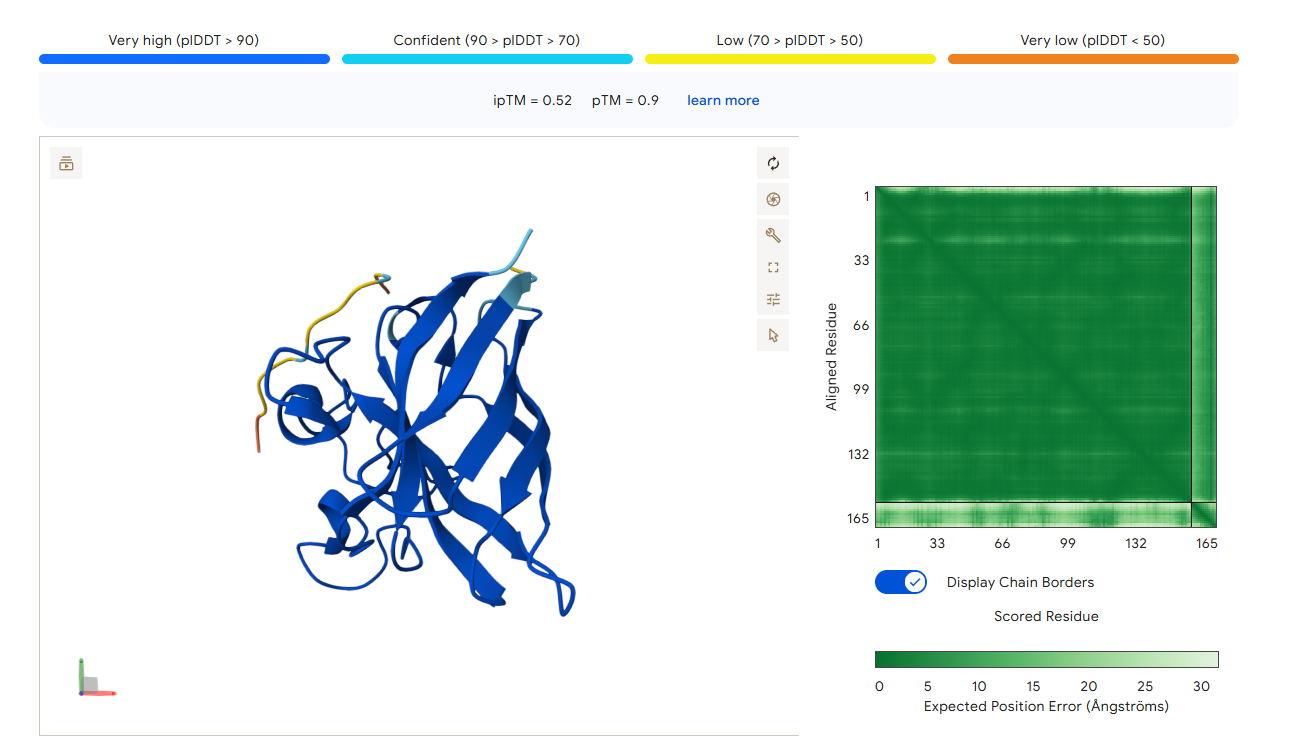
PeptiVerse Property Profile:
- Affinity: 6.464
- Solubility: 1.000 (Perfectly soluble)
- Hemolysis: 0.041 (Low toxicity)
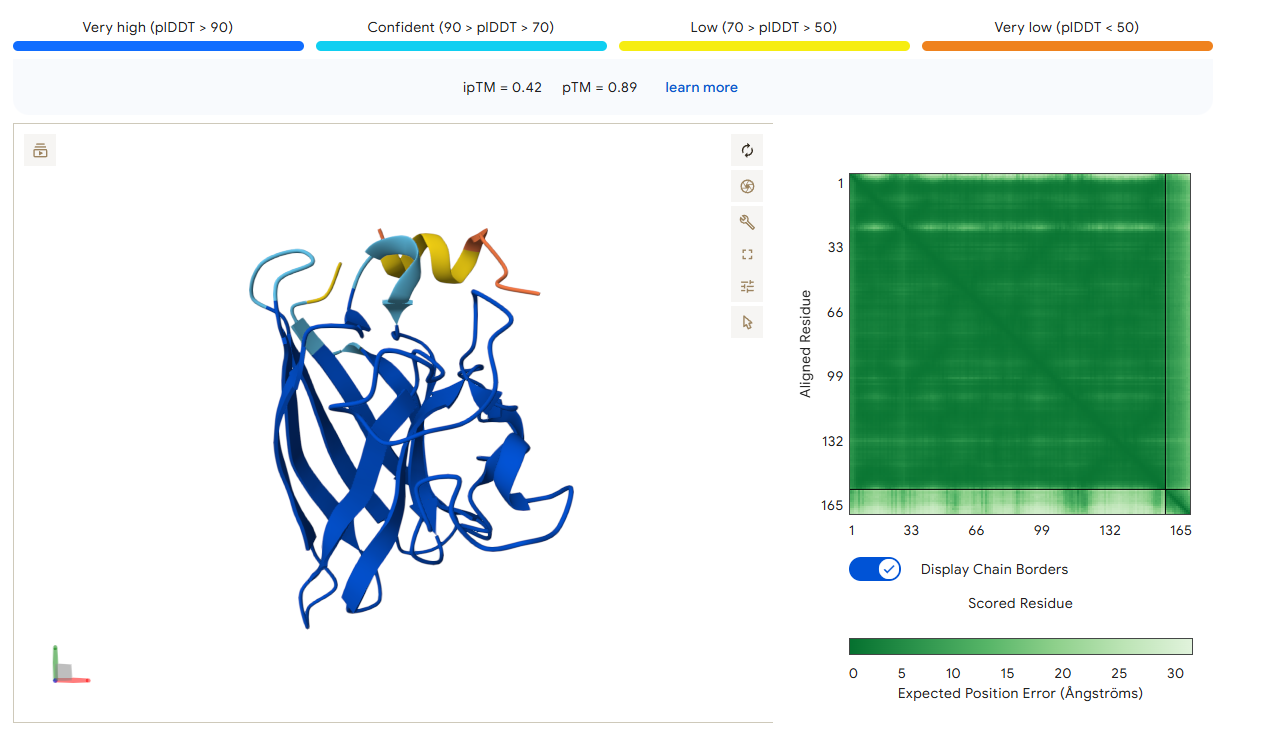
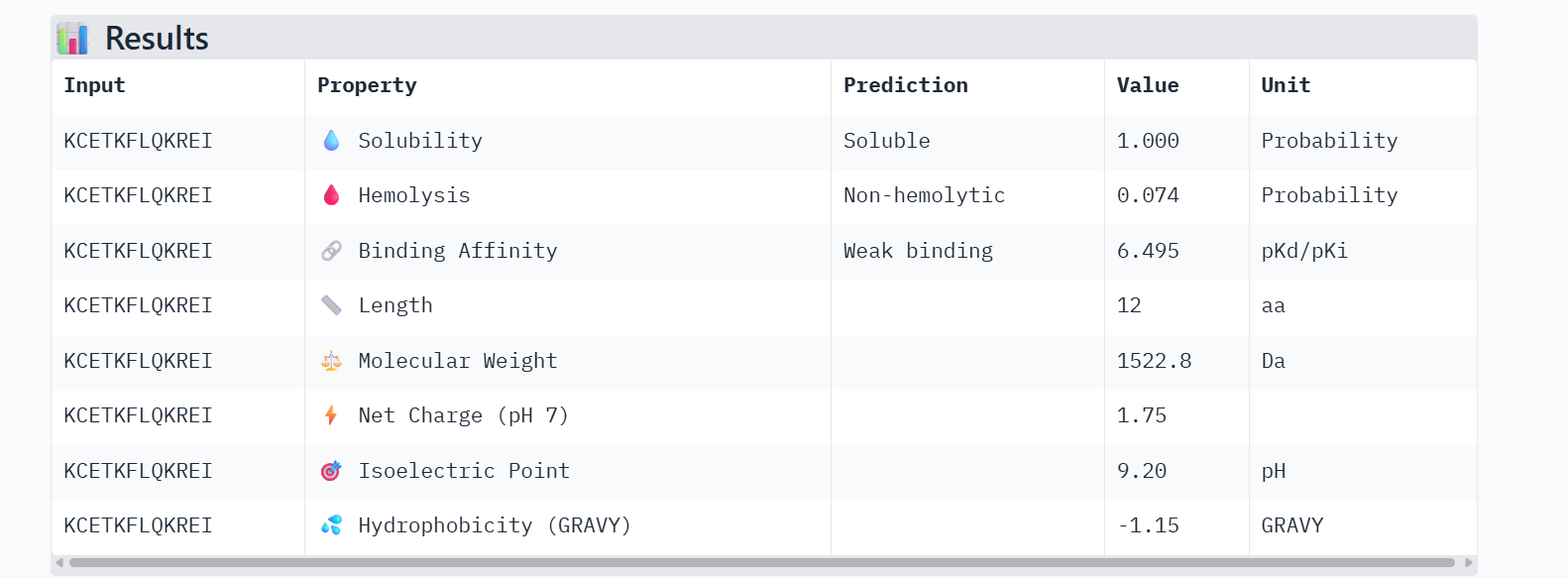
Candidate 2: KCETKFLQKREI
AlphaFold3 Validation: ipTM Score: 0.42
Binding Analysis: While the confidence is slightly lower than Candidate 1, it remains docked near the beta-barrel region adjacent to the N-terminus. It is not “floating away” into the solvent, indicating a specific interaction with the protein surface.
PeptiVerse Property Profile:
- Affinity: 6.495 (Highest Predicted Affinity)
- Solubility: 1.000
- Hemolysis: 0.074
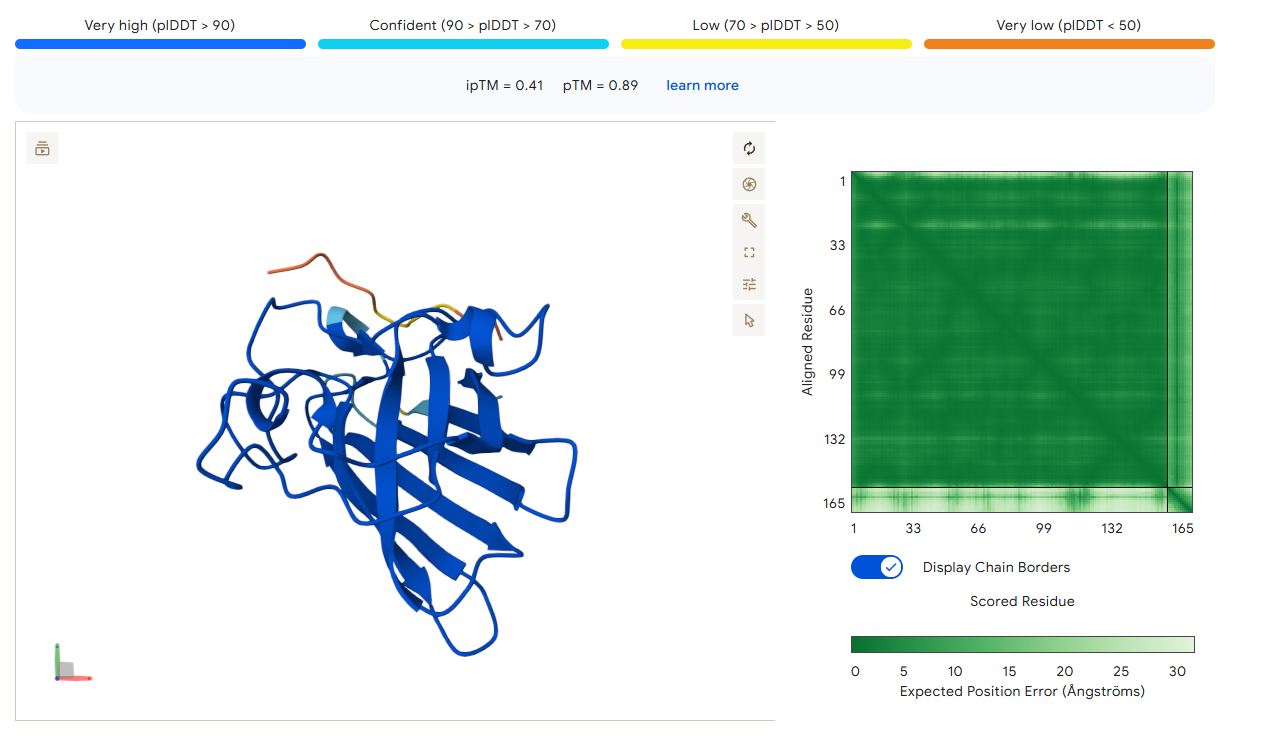
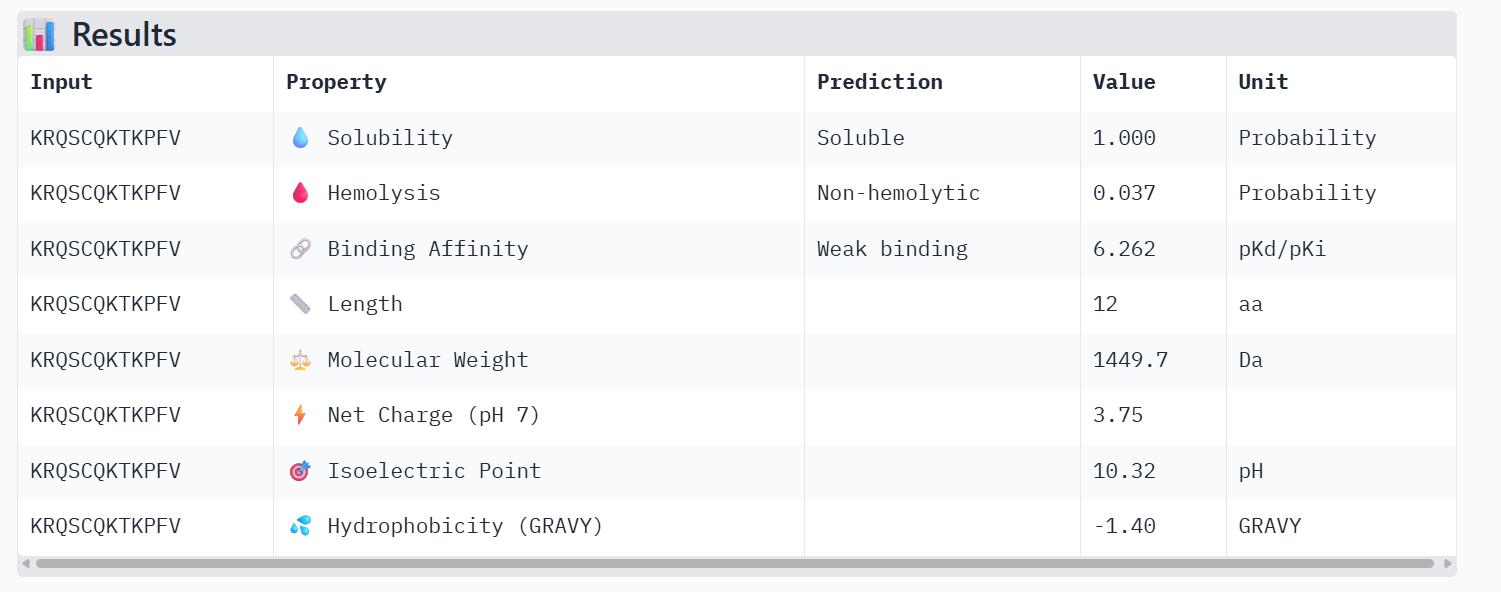
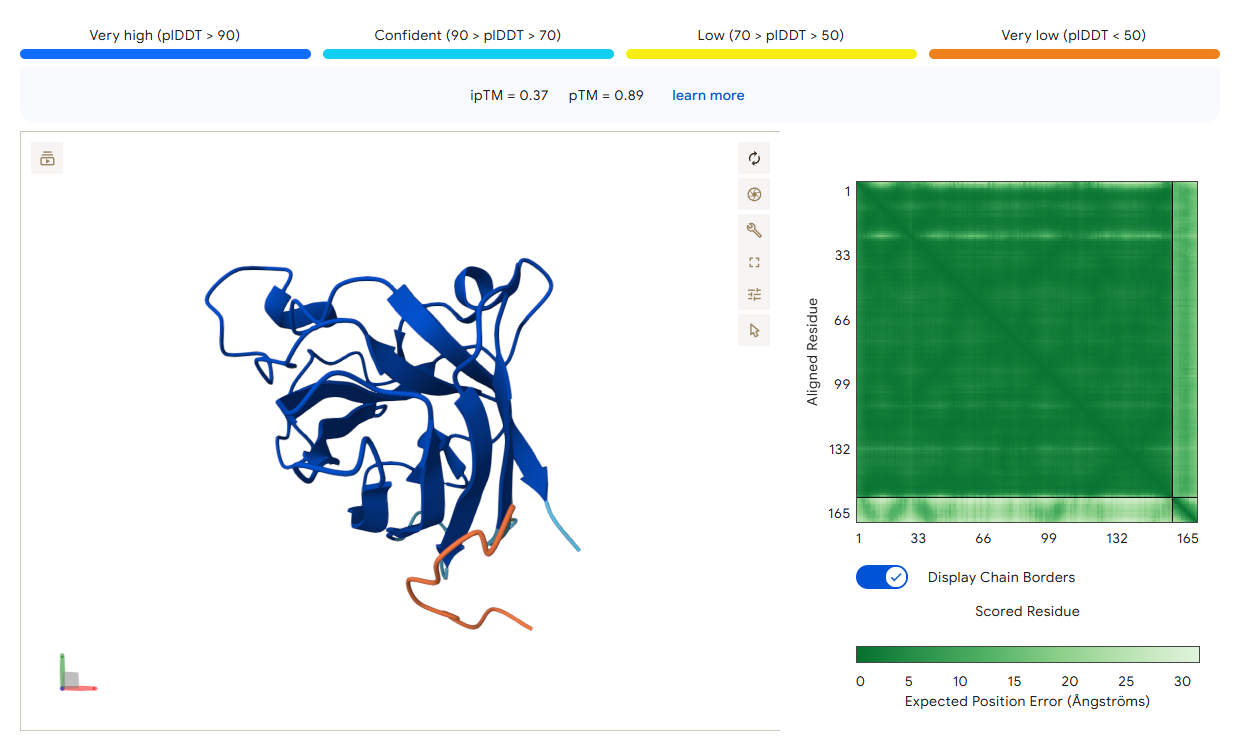
Candidate 3: KRQSCQKTKPFV
AlphaFold3 Validation: ipTM Score: 0.41
Binding Analysis: This candidate also shows proximity to the mutation site. While the ipTM is in the lower confidence range, the physical placement in the model remains focused on the targeted residue patch rather than random surface binding.
PeptiVerse Property Profile:
- Affinity: 6.262
- Solubility: 1.000
- Hemolysis: 0.037
Candidate 3: KANYWTTWTSDS
AlphaFold3 Validation: ipTM Score: 0.37
Binding Analysis: This peptide localizes near the target but shows higher flexibility in the model, reflected in the lower ipTM score. It is close to the N-terminus but less “packed” than the lead candidate.
PeptiVerse Property Profile:
- Affinity: 5.744
- Solubility: 1.000
- Hemolysis: 0.066

How moPPIt Peptides Differ from PepMLM Peptides
The moPPIt-designed peptides represent a significant improvement over the PepMLM set for several reasons:
- Controlled Specificity: PepMLM performs “Global Sampling,” which often results in peptides that bind to random surface loops. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer the peptide specifically toward residues 2-6 (the A4V site).
- Property Reliability: moPPIt optimized for Affinity and Solubility during the generation phase itself. This resulted in a 100% success rate for solubility (1.000) and consistently high affinity scores (pKd/pKi > 6.2) across the lead candidates.
- Targeting the “Toxic” Site: By forcing the model to bind near the N-terminus, moPPIt creates candidates more likely to stabilize the SOD1 dimer interface, which is the root cause of A4V-driven ALS.
Pre-Clinical Evaluation Strategy
To advance the lead candidate (KSQKKQTEICGR) toward clinical application, I would follow this validation pipeline:
- Biophysical Assays (SPR/ITC): I would use Surface Plasmon Resonance (SPR) to confirm the pKd/pKi values. Computational predictions must be validated with physical measurements of binding kinetics to ensure high-affinity binding in the nanomolar range.
- Aggregation Inhibition (ThT Assay): Since the A4V mutation causes toxic protein clumping, a Thioflavin T assay is essential to prove the peptide actually prevents SOD1 from aggregating.
- Efficacy in Motor Neurons: Testing on ALS patient-derived motor neurons is required to see if the peptide reduces intracellular SOD1 aggregates without causing cellular toxicity.
- Proteolytic Stability: I would evaluate the peptide’s half-life in human serum to ensure it isn’t degraded by proteases before it can reach the target neurons in the CNS.