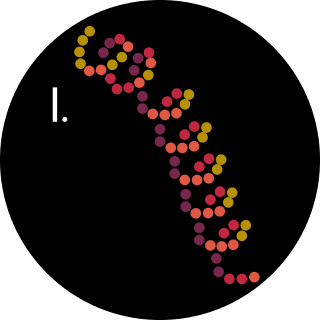My name is Samriddh Sadhukhan. I am a high school student, residing in Maharashtra, India. I don’t have a lot of prior knowledge in the field, except a course on BioArts (by Professor Georg Tremmel), one in Synthetic Biology (by Professor Darius V. Koester), one going deeper into the combination of Media Arts with Biology (by Professor Georg Tremmel), and one on Agent-Based Modelling (NetLogo Coding) (by Professor Aditya Asopa and Abhishek Singh). I am fascinated by what this course and community offers and desire to make the most of this opportunity to learn from all my peers. I know that the information and materials I will receive from this course will surely further my knowledge and ethusiasm towards synthetic biology.
I. Biological Engineering Application or Tool - MechaVita My idea is to create a pseudo living robot in a sense. Living cells (in an organism) will be programmed by humans. The living cells will be provided mechanical exoskeletons to survive in unfavourable conditions. These cells will be able to perform their assigned functions (a specific purpose with which they are created). They will function like living organisms with metabolic processes but utilise mechanical enhancements to aid their purpose.
Part 1: Firstly, I made the enzyme list on Benchling, containing the mentioned enzymes:
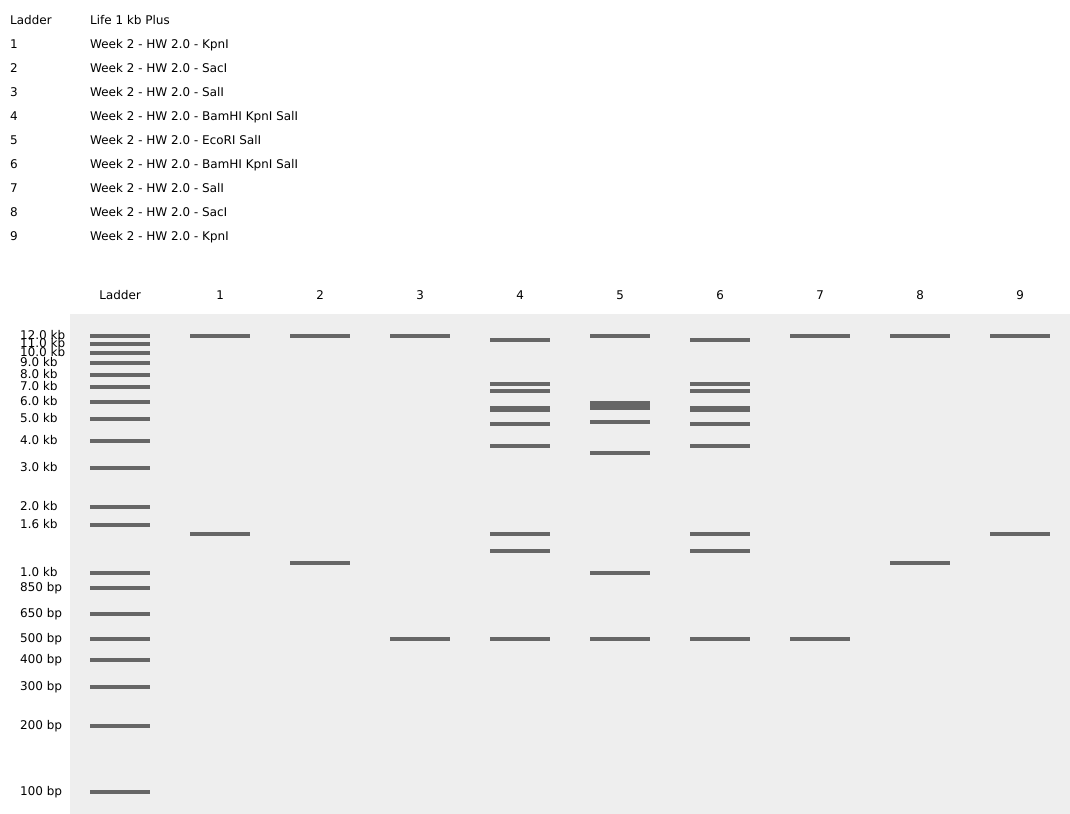
Secondly, I tried playing around with the restriction enzymes to make some kind of resemblance to artwork. I finally made something satisfactory, and had to make the other symmetrical half.
Homework Questions I. From Professor Jacobson Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy? The Error Rate of polymerase is 1:106 when the throughput is 10 mS per base addition. The length of the human genome is ~3.2 giga base pairs. This means that polymerase will make errors in ~3.2 kilo base pairs for a human genome. Biological DNA Synthesis has a process called Proofreading post the copying of the DNA, where most of these discrepancies are taken care of. How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest? An average human protein would have 1036 base pairs. This means there are ~345 amino acids. If every amino acid could be coded with 1 codon, there would be exactly 1 way (not usually possible) but if every amino acid had 6 codons, there would be a maximum of 6345 possible ways to code the protein. In my opinion, the reasons why many of these ways fail is the energy requirement. Only the most efficient route is preferred. Secondly, the error rates must also play a role here. Additionally, many of the methods would simply not have been tried since evolution followed a specific path. II. From Dr. LeProust What’s the most commonly used method for oligo synthesis currently? The solid phase synthesis of oligos on inorganic support (CPG) by Caruthers is the most commonly used method for oligo synthesis. Why is it difficult to make oligos longer than 200nt via direct synthesis? I think it is because of yield losses at each step, which results in the final product having a large efficiency loss, as the errors add up. Why can’t you make a 2000bp gene via direct oligo synthesis? It would be the same reason, as the errors would pile up and the impurities will increase. The end product will not be as intended, especially with large number of base pairs (like 2000), where the number of errors at the end would also be a larger number. Hence, synthesizing smaller oligos and later assembling them is a much more viable option. III. From Professor George Church [Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”? The 10 essential amino acids are:
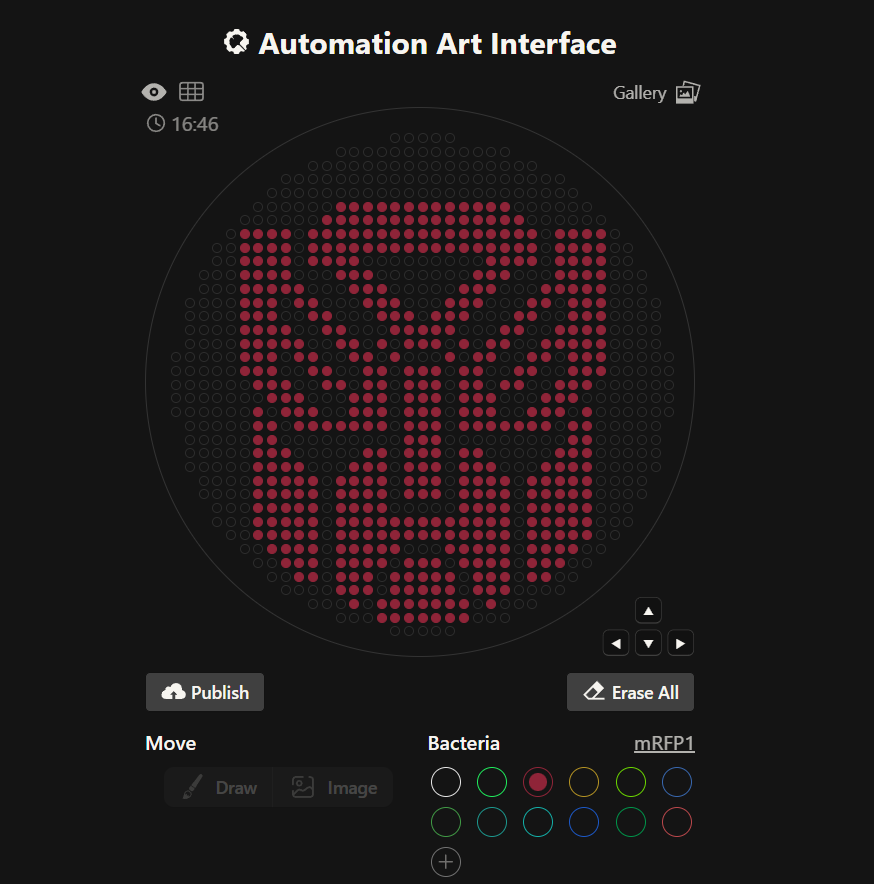
Python Script Having no prior experience in python, I decided to use TA Donovan’s website. I chose the mRFP1 Bacteria to design the Transformers Autobots logo on the agar plate. I manually drew the desired shape and manipulated using only the cursor. I ended up with the following:
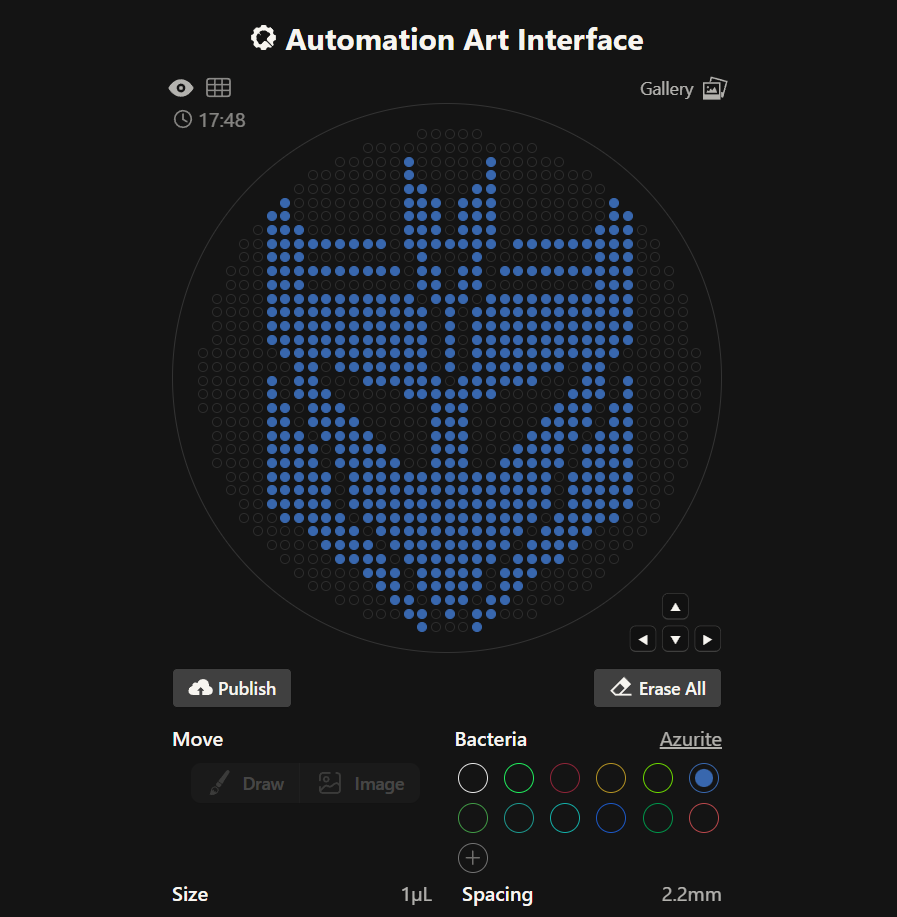
After making the logo of one faction, I decided it would be fun to attempt again with the Transformers Decepticons logo, as well. I chose the Azurite bacteria for this one. Again after careful trial and error (especially to make diagonal lines), I finished with the following:
Part A 1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Meat contains ~20% protein (Kenneth Carpenter et al. Britannica, 2026). In 500 grams of meat, ~100 grams is protein. That is equivalent to 100 grams of amino acids. By the mol formula, the number of mols of amino acids = Mass/RMM = 100 grams/100 Daltons = 100 grams/100 gram mol-1 = 1 mol Therefore, number of molecules = Number of mols x Avogadro’s Number = 1 mol x 6.02214076×1023 mol−1 = 6.02214076×1023 Hence, 6.02214076×10^23 molecules of amino acid are present in 500 grams of meat. 2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
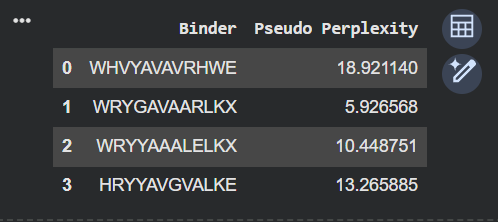
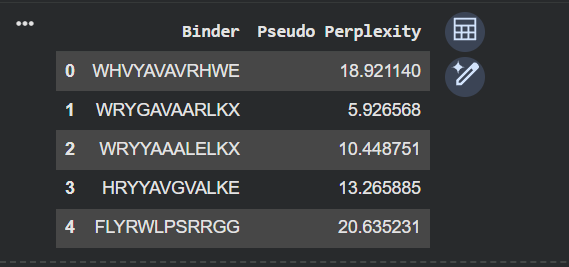
Part A Part 1 I added the protein sequence from UniProt after mutating it, and changed the parameters, as needed. The Sequence: MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ I ended up with these 4 results. I added the given peptide sequence and calculated the Pseudo Perplexity for the same. Part 2
Subsections of Homework
Week 1 HW: Principles and Practices
I. Biological Engineering Application or Tool - MechaVita
My idea is to create a pseudo living robot in a sense. Living cells (in an organism) will be programmed by humans. The living cells will be provided mechanical exoskeletons to survive in unfavourable conditions. These cells will be able to perform their assigned functions (a specific purpose with which they are created). They will function like living organisms with metabolic processes but utilise mechanical enhancements to aid their purpose.
One example could be advanced prosthetics. In the case of limb replacemnts, these cells would be programmed to input neural signals from nerves and nutrients from blood vessels (as detailed or as superficially as necessary) and function how the original limb would. The mechnical parts provide the rigidity and the strength the cells will need to actually fulfil their role. Another use could be tasks with threat to human life. In fields like space exploration, mining, marine exploration, etc. where there is a huge and unyielding risk of losing human life, these living robots could fill in. They would also be able to establish safe routes, set new heights (or depths) to our knowledge of the unknowns, and help decide whether specific locations would be safe enough for humans.
With enough advancements in LLMs, these could evolve into cyborgs, though that is for the future. At the moment, I think the cell-machine interface would be highly beneficial, where the cell thinks (as it is programmed to) and the machine parts help it perform tasks, which humans would normally require lots of resources to achieve.
II. Contribution to An Ethical Future
The more massive the problem, the more we will be willing to sacrifice our standards of “do no harm” in favour of the “greater good”. - From What Ethics for Bioart? by Nora S Vaage
WHO statistics from 2017 state that approximately 35-40 million people worldwide are in need of prosthetics or othotic services. It is safe to assume the need has only increased from then. A subset of these people would be largely benefitted from the fully functioning pseudo living limbs. The living robotic limbs would allow the users to actively participate in society as healthy individuals.
Usually more than hundreds of deaths are reported annually worldwide, not taking into account the deaths caused dude to illegal mining. These activities endangering human lives would become completely safe, as the living robots could perform these dangerous tasks in their stead. As for job security, the robots will possibly require supervision yet no human will have to stake their lives.
In essence, the idea aims to improve quality of life for the differently abled, reduce fatality in fields deemed too dangerous for humans, or where guaranteed loss of life takes place year after year. They perform a crucial role in risk management.
Biosecurity Concerns 🔒
There is concern over biosecurity with this project as there is always potential malfunction that can occur in these living robots. It is important to create technology that will be able to detect anomalies and effectively deal with it too. Hence, the research in the field of detecting and tackling malfunctions will also have to be increased.
Equity Concerns 📊
A critical part of this project’s purpose is related to advanced prosthetics. Therefore, it helps improve equity and accessibility among the differently-abled. It additionally helps improve the quality of life.
Environmental Concerns 🌏
Utilizing metabolic systems like living organisms, the living robots help mitigate the need for conventional sources of energy. Yes, they need other matters for sustenance but do not negatively impact the environment in any way.
III. Governance Actions
1. Purpose
I believe this is a solution to a problem that is not often discussed enough. With this approach, many huamn lives can be saved and/or improved. The radical development will have its hurdles but when achieved, it has immense benefit to humanity and can be developed for more advanced purposes too.
2. Design
The project requires valuable input from computer engineers, biomedical engineers, neuroscientists, and extensive research in the field and with regards to feasibilty and scalabity. Additionaly, funding from the governtment would be the optimal option. The project aims to improve human lives (sharing a vision with governmnet seeking public welfare). Contributions from other NGOs working towards solving problems of accessibility with better prosthetics (or those with similar motives) will be invaluable to the project.
3. Assumptions
There are numerous assumptions made by me:
The concept of living robots can actually be carried out.
The project will be properly funded and developed in the way that I am imagining it to be.
The security concerns can be mitigated and solved.
The prosthetic version will not put an additional toll on the individual and will actually work as programmed.
The version used for exploration and mining purposes will not be misused as a guinea pig, since there is no human life risk involved anymore.
The concept of living robots will be accepted by the larger community and concerns over the loss of life of the living robots is overriden by the concerns over loss of human life.
The complex cell systems, organs, organ systems, etc. can actually be accurately engineered and programmed. Hopefully, these can be resolved by CrispR and complex 3-D modelling.
The living cells can actually survive under the mechanical exoskeleton.
4. Risk of Failure & “Success”
From my perspective, there is a low margin of failure in developing the technology required. However, the idea of unyielding funding till the final project can be made is optimistic at best. Other potential failures would only be in the process of research. Post the success, the only foreseeable risk is malfunctions and I believe these can be easily detected and prevented.
IV. Rubric
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
✔️
• By helping respond
✔️
Foster Lab Safety
• By preventing incident
✔️
• By helping respond
✔️
Protect the environment
• By preventing incidents
✔️
• By helping respond
✔️
Other considerations
• Minimizing costs and burdens to stakeholders
✔️
• Feasibility?
✔️
• Not impede research
✔️
• Promote constructive applications
✔️
V. Prioritising Governance
The governance option with greatest priority would be any international or national government body (health organization) since, this project helps a large part of the general public. It affects and positively impact a significant population and hence, I think it deserves funding as part of healthcare and public welfare. Another potential investor could be space agencies and/or mining corporations as the project would aid in exploring new limits in their respective fields as well. I considered the trade-offs over employment concerns, and I sincerely believe, humans should still not risk their lives for their profression. Not to mention, the living robot would only perform primitive tasks and cannot match an intelligent human. Another assumption is that the extensive data and code can be embedded in the programmed cells to achieve the required results. There are uncertainties like this but if completed, the service as a result of research and development of these living robots will exceed the minor inconveniences.
Week 2 HW: DNA Read, Write and Edit
Part 1:
Firstly, I made the enzyme list on Benchling, containing the mentioned enzymes:
Secondly, I tried playing around with the restriction enzymes to make some kind of resemblance to artwork. I finally made something satisfactory, and had to make the other symmetrical half.
I tried representing a smiling face through the different restriction enzymes on benchling.
Part 3:
3.1
My initial choice was Myosin. However, it was too long of a sequence according to Twist, nearing the end of Part 4. Hence, I have chosen the protein Somatotropin. This is because I find it interesting how such a small protein is responsible for growth in complex creatures like humans.
3.2
I used a Reverse Translate Tool to translate the protein sequence to a nucleotide sequence (based on the Central Dogma). The nucleotide sequence was:
Codon optimization is necessary so that instead of the “rare” codons being used, the host organism can use the codons it possesses. This improves the yield of the protein and helps in boosting the production.
I used a Codon Optimization Tool to optimize the codons for Escherichia. I did this because it is an organism that has a symbiotic relationship with humans and can be used as a vector to produce the proteins.
Cell-Dependent:
This would involve using living cells (such as E. coli) as host cells and vectors, and using them as sort of factories to produce the protein sequences. First, once the DNA is obtained and prepared (likely using PCR), it is inserted into the vector where transcription and translation take place. Post translation, the protein is produced.
Cell-Independent:
This would involve using DNA templates and RNA polymerase to transcribe DNA to mRNA. Ribosomes transcribe mRNA to Protein and voila! This method is preferrable because it takes less time, and does not require cell maintenance (toxic proteins can also be synthesized).
Therefore, I think cell-independent systems might be more efficient but both types should work equally well.
Part 4:
After completing all the steps I built the Plasmid with my Expression Casette. The expression cassete was made on Benchling and can be found here.
Part 5:
5.1 Read
I. What DNA would you want to sequence (e.g., read) and why?
I would like to read DNAs from fossils and prehistoric artifacts. I choose this to help evolutionary research. I am also very invested in paleontology and dream to de-extinct the organisms driven to extinction because of human activities like hunting or poaching, or otherwise.
II. In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would use Second Generation Sequencing DNA Sequencing Techniques like Illumina. It would involve small DNA fragments. It has high accuracy. It can do massive parallel sequencing. Additionally, it has already been used for similar purposes in labs (for Ancient DNA).
The input here is the aDNA extracted from the fossils or remnants. It is already highly fragmented and does not require fragmenting further. It might also be damaged and requires certain steps to fix it. The process involves:
DNA Extraction: After powdering the sample, aDNA is carefully extracted without contamination (usually in highly equipped facilities).
Repair: The DNA fragments may have overhangs and damages. The ends are repaired in this process. Some damages are partially removed, as well.
Adapter Ligation: The DNA adapters are ligated to both ends of the fragment. These adapters have neccessary sequences that make the DNA compatible with sequencing platforms, allowing for Amplification.
PCR: Ancient DNA is scarce and limited. PCR amplifies and makes millions of copies of each fragment.
Flow Cell Binding: The DNA Library is sent to the sequencer (Illumina). The fragments are immobilized onto the surface of a sequencing flow cell.
Sequencing By Synthesis: Using base calling (signal from sequencer is converted to corresponding nucleotide, where Illumina uses light intensities), an output is created as a FASTQ file (FASTA file but also contains quality scores for each base).
5.2 Write
I. What DNA would you want to synthesize (e.g., write) and why?
I would like to synthesize materials like polymers through vectors. The polymers would be biodegradeable and utilised for 3-D Printing, Injection Molding and similar Manufacturing techniques. These could replace the petroleum-based polymers in various industries.
II. What technology or technologies would you use to perform this DNA synthesis and why?
I would use Solid Phase Chemical DNA Synthesis, which is a method for creating custom DNA/RNA sequences by anchoring the growing chain to an insoluble support and adding the nucleotide bases. This technique has high yield, utilizes codon optimization and produces precise sequences. Following the oligo synthesis, Gibson Assembly could be used to combine all the DNA fragments in one reaction. It is also highly accurate and efficient.
Then the genes would need to be inserted in the plasmid vectors (most likely E. coli). Plasmids are commonly used because theyproduce many copies and have a fast growth. This is likely the stage, where TWIST comes in.
Some Metabollic Engineering needs to be done to make the vector more efficient, solve conflicting metabollic pathways and make it into a proper “Cell Factory.”
Accumulation of the products, harvesting and purification of the same will be needed for scaling up the production of the polymers.
5.3 Edit
I. What DNA would you want to edit and why?
I would like to edit Human genes to resolve genetic disorders. When I had first come to know about CRISPR, I had wondered if editing genes could cure genetic diseases or even cancer. After CRISPR had been used to provide therapy for KJ Muldoon (first person to receive in vivo peronalized CRISPR therapy), I have only become more intrigued in the field of using Gene Editing to help treat these kinds of diseases.
II. What technology or technologies would you use to perform these DNA edits and why?
CRISPR-cas9 would be the major technology that would be used to perform these DNA edits. It is a precise, efficient, and customizable gene-editing technology adapted from a natural bacterial immune system. It already has great significance in the clinical industry. For example, Casgevy is the first FDA-approved CRISPR-cas9 gene editing therapy used to treat sickle-cell anaemia and beta-thalassemia.
CRISPR uses cas-9 enzyme (the enzyme that breaks down the molecules) and gRNA (Guide RNA that directs the cas-9 enzyme). The Design Steps:
Target Identification: Used to identify the mutation and figure out the exact target site.
gRNA Design: The gRNA is designed based on the information from the previous step.
Donor Template: This is used to correct mutations. It contains the correct sequence.
The input includes the sequence to be edited, gRNA, cas9 enzyme and the Donor Template.
Limitations of CRISPR-cas9:
The enzyme may cut the wrong sites and there is a risk of mutation.
Delivery Challenges: Might be difficult to deliver in certain tissues and vectors.
Immune Response: cas-9 being a bacterial enzyme may trigger immune reactions.
AI Citations
Tool name and version: Chat GPT, Version GPT-5.2
Time and Date: 14th February, ~7 P.M. IST
Prompts:
what kind of dna sequencing tech would i use to sequence dna from fossils in order to revive the organims. what generation would the dna sequencing tech be
how do you prepare your input ( fragmentation adapter ligation PCR) list the essential steps how does it decode the bases of your DNA sample (base calling) what is the output of your chosen sequencing technology
What technology or technologies would you use to perform DNA synthesis where you hypothetically make vectors produce biodegradeable plastics and why?
What technology or technologies would you use to perform DNA edits to the human gene to fix genetic diseases and why?
Responses:
If you’re talking about sequencing DNA from fossils for something like “reviving” an organism (de-extinction), the type of sequencing technology matters a lot because ancient DNA (aDNA) is extremely degraded, fragmented, and chemically damaged. Here’s how it breaks down: What makes fossil DNA difficult? DNA from fossils (like mammoths or Neanderthals) is: Highly fragmented (often 30–100 base pairs long) Chemically modified (cytosine → uracil deamination) Contaminated with microbial and modern human DNA Present in very low amounts Because of this, you need sequencing technology that works well with short, damaged fragments. Best generation of sequencing technology ✅ Second-Generation Sequencing (Next-Generation Sequencing, NGS) This is the most widely used for ancient DNA. Example platform: Illumina Why Illumina-type sequencing? Works extremely well with short DNA fragments High accuracy Massive parallel sequencing (millions of fragments at once) Standard method used in ancient DNA labs Most famous ancient genome reconstructions (like Neanderthal and mammoth genomes) used this approach. So the answer is: Second-generation (NGS) sequencing is the primary technology used for fossil DNA. What about other generations? 🧬 First Generation (Sanger sequencing) Example: Applied Biosystems systems Too slow Requires longer intact DNA Not practical for ancient DNA ❌ Not suitable for fossil DNA. 🧬 Third Generation (Long-read sequencing) Examples: Oxford Nanopore Technologies Pacific Biosciences These produce very long reads (thousands of bases), but: Ancient DNA is already very short Higher error rates (especially problematic with damaged bases) They can be used supplementally, but they are not the primary tool for degraded fossils. Full Workflow for Fossil DNA If the goal is de-extinction, the process would involve: Extract DNA in a clean-room ancient DNA lab Build sequencing libraries from tiny fragments Sequence using Illumina (2nd generation) Bioinformatically reconstruct genome Compare to closest living relative Fill gaps using reference genome Edit living relative genome using CRISPR Clone via somatic cell nuclear transfer Example of proposed de-extinction target: Woolly Mammoth Closest living relative: Asian Elephant Important Reality Check “Reviving” a fossil organism is not: Extract DNA → grow dinosaur Instead, it’s: Reconstruct genome computationally → Edit living species to resemble extinct one. Even then: DNA rarely survives millions of years Dinosaur DNA is effectively impossible Only relatively recent extinct species (tens of thousands of years old) are feasible Final Answer To sequence DNA from fossils for possible revival: You would use Second-Generation (Next-Generation) sequencing technology, especially Illumina short-read platforms. It is currently the gold standard for ancient DNA research. If you want, I can also explain why Jurassic Park–style dinosaur revival is scientifically impossible from a DNA chemistry perspective.
I’ll answer this assuming you’re using second-generation (NGS) short-read sequencing, specifically an Illumina platform — the standard for ancient/fossil DNA. What is the input? Input material: Extracted ancient DNA (aDNA) from fossil bone, tooth, or preserved tissue. DNA is already: Highly fragmented (30–100 bp typical) Chemically damaged Very low concentration So unlike modern DNA sequencing, you usually do NOT fragment it further — it’s already broken. How do you prepare the input? (Library Preparation) These are the essential steps: Step 1: DNA Extraction Powder fossil sample Use EDTA + proteinase digestion Silica-column or magnetic-bead purification Work done in clean-room aDNA facility Goal: isolate short DNA fragments while minimizing contamination. Step 2: End Repair Ancient DNA fragments: Have overhangs May contain damaged bases (like uracil from cytosine deamination) Enzymes are used to: Repair fragment ends Sometimes partially remove damage (optional UDG treatment) Step 3: Adapter Ligation Short synthetic DNA adapters are ligated to both ends. Adapters: Contain sequencing primer binding sites Include index/barcode sequences (to identify sample) Allow fragments to bind to flow cell Now your DNA becomes a sequencing library. Step 4: PCR Amplification Because ancient DNA is scarce PCR enriches fragments that have adapters Produces millions of copies of each fragment. After this step: You have a ready-to-sequence DNA library. Essential Steps of Illumina Sequencing Now the library goes onto the sequencer. Step 5: Flow Cell Binding Library fragments bind to complementary oligos on the flow cell surface. Step 6: Bridge Amplification (Cluster Generation) Each fragment bends and forms a “bridge.” Through repeated amplification: Forms a dense cluster of identical DNA molecule Each cluster = one original fragment This massively increases signal strength. Step 7: Sequencing by Synthesis (SBS) This is how base calling works. Illumina uses: Fluorescently labeled reversible terminator nucleotides (A, T, C, G) Each base has a different color Cycle process: Add all four labeled nucleotides Only ONE base incorporates (due to terminator block) Laser excites the flow cellCamera detects fluorescence color Software assigns base (A/T/C/G) Chemical cleavage removes terminator Repeat cycle Each cycle reads one base. After ~100–150 cycles → full read length obtained. How Base Calling Works Each cluster emits a fluorescent signal Intensity and wavelength correspond to base identity Imaging software converts signal to: Base (A/T/C/G) Quality score (Phred score) Base calling is essentially: Fluorescent color → digital base → quality score What is the Output? The output is:📁 FASTQ files Each read contains: Read ID DNA sequence (A/T/C/G) Quality scores per base For ancient DNA: Millions to billions of short reads (30–150 bp) Later assembled computationally Compared to reference genome (e.g., Asian elephant if reconstructing mammoth) Summary Table Stage What Happens Input Extracted fragmented ancient DNA Library Prep End repair → Adapter ligation → PCR Sequencing Bridge amplification → Sequencing by synthesis Base Calling Fluorescent detection per cycle Output FASTQ files with short reads + quality scores In One Sentence Using an Illumina platform: Your input is fragmented fossil DNA, prepared by adapter ligation and PCR into a sequencing library, decoded via fluorescent sequencing-by-synthesis, and output as millions of short reads in FASTQ format. If you’d like, I can also explain how those reads are assembled into a full extinct genome — which is actually the hardest part.
If your goal is to engineer cells to produce biodegradable plastics (like PHAs), you’d combine DNA synthesis + molecular cloning + metabolic engineering. Here’s a clear, structured answer. 🧬 Goal Design and synthesize genes encoding plastic-producing enzymes, insert them into a vector, and express them in a microbial host. A common biodegradable plastic: Polyhydroxyalkanoates (PHAs) Naturally produced by bacteria under nutrient stress. 1️⃣ DNA Synthesis Technology ✅ Solid-Phase Chemical DNA Synthesis Used to synthesize: Custom genes (codon-optimized) Promoters Regulatory sequences Modern providers use phosphoramidite chemistry to synthesize oligonucleotides, then assemble them into full genes. Why? Precise sequence design Codon optimization for host (e.g., E. coli) Ability to modify promoters and RBS strength Gene Assembly Methods After oligo synthesis: 🔹 Gibson Assembly Efficient, seamless DNA assembly method. Invented by: Daniel Gibson Why use it? Allows assembly of multiple DNA fragments in one reaction No restriction enzyme scars Ideal for building synthetic metabolic pathways 2️⃣ Vector Construction Plasmid Vectors Circular DNA that replicates in bacteria. Inserted genes: phaA phaB phaC (enzymes required for PHA synthesis) Host organism:Escherichia coli (commonly engineered lab strain) Why plasmids? High copy number Easy transformation Fast growth 3️⃣ Genome Editing (Optional but Advanced) Instead of plasmids, you could integrate genes into the chromosome using: CRISPR-Cas9 Gene editing system. Why? Stable expression No plasmid maintenance burden Industrial-scale stability 4️⃣ Metabolic Engineering Tools To improve yield: Promoter engineering Ribosome binding site tuning Knockout competing metabolic pathways Flux balance modeling This turns the host into a “cell factory.” 5️⃣ Fermentation Technology After genetic engineering: Large-scale bioreactors Controlled nutrient limitation Induces PHA accumulation inside cells Cells accumulate plastic granules → harvested → purified. Why These Technologies? Technology Why Use? Solid-phase DNA synthesis Precisely design custom genes Codon optimization software Maximize expression Gibson Assembly Seamless multi-gene cloning Plasmid vectors Fast, flexible expression CRISPR Stable genomic integration Fermentation systems Scale production Example of Industrial Relevance Companies producing biodegradable plastics via engineered microbes include: Danimer Scientific NatureWorks Concise Exam-Style Answer I would use solid-phase chemical DNA synthesis to design and synthesize codon-optimized genes for PHA biosynthesis. These genes would be assembled into plasmid vectors using Gibson Assembly and transformed into E. coli. Optionally, CRISPR could be used for stable genome integration. The engineered microbes would then be grown in bioreactors to produce biodegradable plastics at scale. If you want, I can also explain which specific enzymes are required in the PHA pathway and how carbon flux is redirected toward plastic production.
To fix genetic diseases at the DNA level, the leading technology today is CRISPR-based genome editing, particularly: CRISPR–Cas9 Base editing Prime editing CRISPR is already being used clinically (e.g., sickle cell disease therapy). 🧬 Technology of Choice: CRISPR–Cas9 CRISPR systems were adapted from bacterial immunity and developed for genome editing by researchers including: Jennifer Doudna Emmanuelle Charpentier A real clinical example: Casgevy – approved CRISPR therapy for sickle cell disease. 1️⃣ How Does CRISPR–Cas9 Edit DNA? Core Mechanism CRISPR–Cas9 uses: Cas9 enzyme → molecular scissors Guide RNA (gRNA) → directs Cas9 to specific DNA sequence Essential Steps of Editing Step 1: Target Recognition gRNA binds to complementary DNA sequence. Cas9 binds nearby PAM sequence (e.g., NGG). Step 2: DNA Cleavage Cas9 creates a double-strand break (DSB) at the target site. Step 3: DNA Repair (Cell’s Own Machinery) Two possible pathways: A) Non-Homologous End Joining (NHEJ) Error-prone Creates insertions/deletions (indels) Often used to knock out genes B) Homology-Directed Repair (HDR) Requires donor DNA template Allows precise correction Used to fix mutations 2️⃣ Preparation & Design Steps Before editing, careful design is required. 🔬 A) Target Identification Sequence patient DNA Identify mutation Choose exact target site 🧠 B) Guide RNA Design Design gRNA complementary to mutation region Ensure minimal off-target similarity Confirm PAM availability Software tools predict: Off-target risk Cutting efficiency 🧪 C) Donor Template (for precise correction) If correcting mutation: Provide synthetic DNA template Contains correct sequence Includes homology arms (~50–1000 bp) 3️⃣ What Is the Input? Depends on delivery method. Typical components: Component Purpose Cas9 protein or Cas9 mRNA DNA cutting Guide RNA Target specificity Donor DNA template (optional) Precise correction Delivery system Introduce into cells Delivery Methods Viral vectors (AAV, lentivirus) Lipid nanoparticles Electroporation (ex vivo editing) Example: For sickle cell: Stem cells are removed Edited ex vivo Reinfused into patient 4️⃣ Variants of CRISPR Editing 🧬 Base Editing No double-strand break. Converts single base (e.g., C → T) Higher precision Fewer indelsGood for: Point mutations 🧬 Prime Editing More flexible: Can insert, delete, or replace small DNA segments Uses reverse transcriptase More precise but technically complex 5️⃣ Limitations of CRISPR Editing ⚠️ 1. Off-Target Effects Cas9 may cut unintended sites Risk of mutations elsewhere Improved variants reduce this risk. ⚠️ 2. HDR Efficiency Is Low Precise repair (HDR) is inefficient Especially in non-dividing cells NHEJ often dominates. ⚠️ 3. Mosaicism Not all cells get edited Leads to mixed populations ⚠️ 4. Delivery Challenges Difficult to deliver into certain tissues Viral vectors have size limits ⚠️ 5. Immune Response Cas9 is bacterial protein Can trigger immune reactions 6️⃣ Efficiency & Precision Summary Method Precision Efficiency Best For CRISPR-Cas9 (NHEJ) Moderate High Gene knockouts CRISPR-Cas9 (HDR) High Low–Moderate Gene correction Base Editing Very high (point edits) Moderate–High Single base mutations Prime Editing Very high Lower Complex edits Concise Exam-Style Answer I would use CRISPR–Cas9 genome editing to correct disease-causing mutations. A guide RNA directs the Cas9 nuclease to a specific DNA sequence, where it induces a double-strand break. The cell repairs the break either by NHEJ (causing gene disruption) or HDR if a donor template is supplied for precise correction. Inputs include Cas9 protein or mRNA, guide RNA, and optionally a donor DNA template. Limitations include off-target effects, low HDR efficiency, delivery challenges, and possible immune responses. If you’d like, I can also explain when base editing or prime editing would be better than standard CRISPR for specific diseases.
Week 2: Lecture Prep
Homework Questions
I. From Professor Jacobson
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
The Error Rate of polymerase is 1:106 when the throughput is 10 mS per base addition. The length of the human genome is ~3.2 giga base pairs. This means that polymerase will make errors in ~3.2 kilo base pairs for a human genome. Biological DNA Synthesis has a process called Proofreading post the copying of the DNA, where most of these discrepancies are taken care of.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
An average human protein would have 1036 base pairs. This means there are ~345 amino acids. If every amino acid could be coded with 1 codon, there would be exactly 1 way (not usually possible) but if every amino acid had 6 codons, there would be a maximum of 6345 possible ways to code the protein. In my opinion, the reasons why many of these ways fail is the energy requirement. Only the most efficient route is preferred. Secondly, the error rates must also play a role here. Additionally, many of the methods would simply not have been tried since evolution followed a specific path.
II. From Dr. LeProust
What’s the most commonly used method for oligo synthesis currently?
The solid phase synthesis of oligos on inorganic support (CPG) by Caruthers is the most commonly used method for oligo synthesis.
Why is it difficult to make oligos longer than 200nt via direct synthesis?
I think it is because of yield losses at each step, which results in the final product having a large efficiency loss, as the errors add up.
Why can’t you make a 2000bp gene via direct oligo synthesis?
It would be the same reason, as the errors would pile up and the impurities will increase. The end product will not be as intended, especially with large number of base pairs (like 2000), where the number of errors at the end would also be a larger number. Hence, synthesizing smaller oligos and later assembling them is a much more viable option.
III. From Professor George Church
[Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The 10 essential amino acids are:
Arginine
Histidine
Methionine
Isoleucine
Leucine
Lysine
Phenylalanine
Threonine
Tryptophan
Valine
Lysine is a relatively rare amino acid in foods, compared to the others, so humans are in short supply of lysine with staple diets (usually relying on grains). Hence, in many parts of the world, lysine is the ’limiting’ amino acid. However, lysine also has a relatively high requirement in the body. Lysine is non-enzymatic, making it chemically vulnerable, furhtering the “lysine contingency”.
Chat-GPT prompts used in answering the questions from Professor George Church:
How is Lysine different from the other EAA?
Why does the body specifically lack Lysine?
What is Non-Enzymatic?
Week 3 HW: Lab Automation
Python Script
Having no prior experience in python, I decided to use TA Donovan’s website. I chose the mRFP1 Bacteria to design the Transformers Autobots logo on the agar plate. I manually drew the desired shape and manipulated using only the cursor. I ended up with the following:
After making the logo of one faction, I decided it would be fun to attempt again with the Transformers Decepticons logo, as well. I chose the Azurite bacteria for this one. Again after careful trial and error (especially to make diagonal lines), I finished with the following:
The paper describes that optimisation of drug delivery systems is a complex and multidimensional challenge that involves lots of factors, such asformulation composition, process parameters, and biological performance. The conventional approaches are limited, due to the high complexity, nonlinearity and multiple objective nature of the drug delivery problems of today's age. This article explores how Artificial Intelligence (AI) paired with Machine Learning (ML) can be used to improve formulation science by allowing data-driven, adaptive and efficient strategies. The paper lists both a conceptual and practical overview of ML-Guided optimisation workflows. The paper also discusses key challenges, such as data scarcity, experimental throughput, and model interpretability. Diverse Delivery applications are critically examined, highlighting how ML accelerates formulation development, reduce experimental burden, and uncover novel design spaces. The paper concludes, outlining the future of directions for integrating AI into pharmaceuticals and a focus on self-driving labs. The paper aims to aid drug delivery scientists with foundational knowledge and practical tools to harness AI and ML in the optimisation of advanced drug delivery systems.
My main objective is to use lab automation in large production of my ideas. Once I have developed a concrete final idea, I do not possess access to a lab in my vicinity that would allow R&D to minors like myself. The goal is to be able to utilize the remote lab automation and create novel bioproducts. Additionally, a major reason I plan to use it is due to its precision. With my prior experiences in a wet lab (High School courses in University Labs), I have come to understand the impact of human error in the experiments. I wish to erase (or at least diminish) the variable of error with the help of lab automation. Additionally, these robots can perform multiple experiments that may contain components toxic to humans. This ensures utmost safety of the researchers, which is another priority.
Final Project Ideas
I have added my Ideas on slides:
Week 4 HW: Protein Design Part I
Part A
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
In 500 grams of meat, ~100 grams is protein. That is equivalent to 100 grams of amino acids.
By the mol formula, the number of mols of amino acids = Mass/RMM = 100 grams/100 Daltons = 100 grams/100 gram mol^-1 = 1 mol
Therefore, number of molecules = Number of mols x Avogadro’s Number = 1 mol x 6.02214076×1023 mol−1 = 6.02214076×10^23
Hence, 6.02214076×10^23 molecules of amino acid are present in 500 grams of meat.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
The meat is usually cooked before consumption. This would lead to the denaturing of DNA.
Even bypassing that, the human digestive system causes the substances to be completely broken down to the constituent molecules (amino acids, water, etc.), with the help of multiple enzymes. The simpler molecules (nutrients) are then absorbed by the cells.
The DNA or RNA from the cells of the animal being consumed does not get a chance to enter our human cells. We also have our immune system to prevent the entry of foreign DNAs into the cells.
The human cells are also genetically coded to only produce more human cells. It cannot produce cells of other species (although viruses have the ability to alter this).
3. Why are there only 20 natural amino acids?
It is common knowledge that amino acids having 3 nitrogeneous bases, a total of 64 combinations (4^3) are possible. It is believed that the choice of 20 amino acids is to allow redundancy, where multiple codons code for the same amino acids, increasing efficiency and reducing mutations. However, this does not answer why the specific set of 20 amino acids. Any of the other 41 combinations (leaving 3 combinations for stop codons) could have been used instead, right?
The Frozen Accident Theory (Francis Crick, 1968) implies that these were the first 20 (or the most in amount) during the preliminary stages of biology, and later ended up being locked (frozen) as the default set, as changing the amino acids would not be energy-feasible, and these set of 20 seemed to be the most efficient.
Proteins can be made with a much smaller set of amino acids. A Japanese group headed by Satoshi Akanuma at Waseda University recently showed that a 13 amino acid alphabet can create folded, soluble, stable and catalytically active ‘proteins’, albeit not as active or stable as the parent proteins on which they were based (R Shibue et al. Sci. Rep., 2018).
It is therefore concluded, that after the initial 13-14 default amino acids, the rest were adaptive. They became “natural” amino acids, when oxygen started to become a part of the biochemical reactions on prehistoric Earth. That begs the question, “why not more?” Technically, there are 2 more amino acids, parttaking in protein synthesis, but not in genetic code. Hence, it can be hypothesized that the process had not stopped, it just reached a point where incorporating new amino acids was extremely hard.
The limitation is in the recognition of the tRNA. Each tRNA molecule has a well-defined tertiary structure that is recognized by the enzyme aminoacyl tRNA synthetase, which adds the correct amino acid. From studying tRNA structures, it was concluded that the problem is finding ways to make new tRNA molecules that could recognise a new amino acid without picking up existing ones (A Saint-Léger et al. Sci. Adv., 2016). This was the ultimate problem and answer to why there are only 20 amino acids.
4. Where did amino acids come from before enzymes that make them, and before life started?
In 1953, Miller and Urey attempted to re-create the conditions of primordial Earth. In a flask, they combined ammonia, hydrogen, methane, and water vapor plus electrical sparks (Miller 1953). They found that new molecules were formed, and they identified these molecules as eleven standard amino acids. From this observation, they posited that the first organisms likely arose in an environment similar to the one they constructed in their flask, widely regarded as the primordial soup.
This claim extended that in this soup, single-celled organisms evolved, while the necessary compounds depleted. In the competitive environment, only some organisms gained the ability to biosynthesize the required compounds (amino acids), while the rest died off, leading to the amino acids production by life we now know.
6. Can you discover additional helices in proteins?
Additional Helices like pi-helix and 310 helix have been identified through research.
7. Why are most molecular helices right-handed?
Despite the fact that both right-handed and left-handed α-helices are among the permitted conformations, the right-handed α-helix is energetically more favorable because of fewer steric clashes between the side chains and the main chain. Thus, all α-helices in proteins are right-handed (Robinson et al. Academic Press, 2014)
8. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets tend to aggregate due to 2 major reasons. The first, the hydrophobic effect (non-covalent interactions), which leads to face-to-face aggregation. The second, Van der Waals force (weak inter-molecular interactions), which lead to a sandwich-like, layered structure (Pham et al. Pub. Med., 2014).
9. Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Amyloids consist of a β-sheet motif that repeats almost indefinitely. This and other unique properties bestow on these aggregates many biological activities like template assistance, membrane binding, and infectivity. This means that the β-sheet structure allows stability to the amyloid and helps it perform many vital activities, making it a staple among amyloid diseases (Riek, Pub. Med., 2017).
The rigidity, chemical stability, high aspect ratio, and sequence programmability of amyloid fibrils have made them attractive candidates for functional materials with applications in environmental sciences, material engineering, and translational medicines (Zhang et al. Pub. Med., 2021).
Part B
1. Amino Acid Description
For this assignment, I have chosen Cryptochrome. This is because I find it very interesting that a protein is responsible for circadian rhythms across different organisms. I have obtained the sequence and information about the CRY1 (Cryptochrome) in Homo sapiens from UniProt.
2. Amino Acid Identification
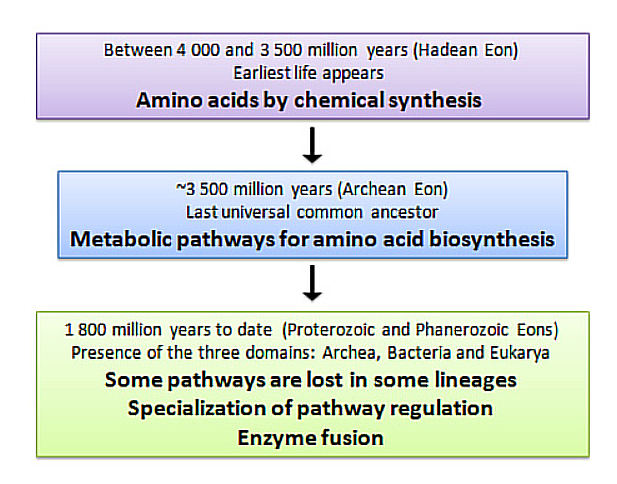
This is what I obtained from the Seqeunce section on the UniProt website:
After using the Colab code, I obtained the following results:
Length = 586 Amino Acids
Most Frequent Amino Acid = L (60 times)
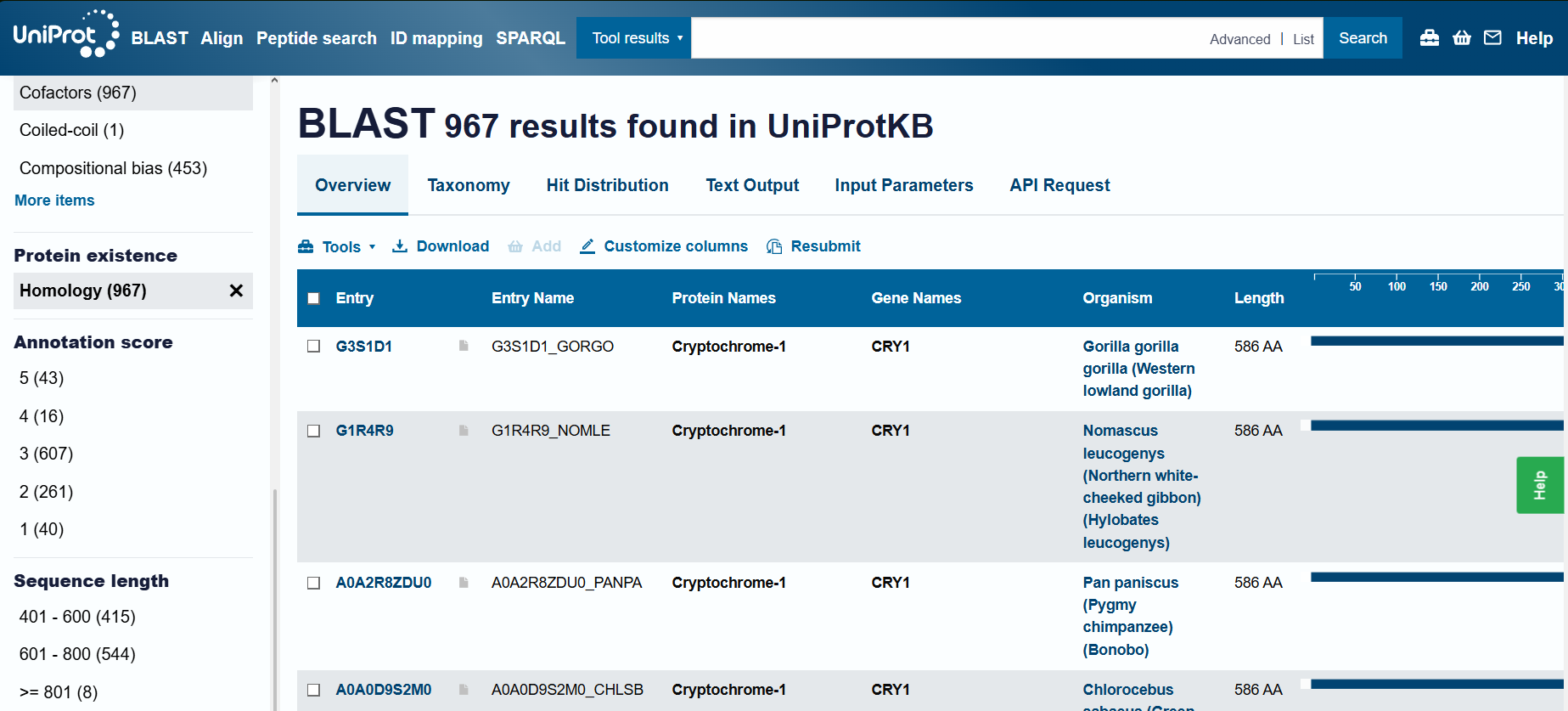
Number of Protein Sequence Homologs: BLAST found 967 results (I had increased the maximum limit to 1000).
Cryptochrome belongs to the DNA photolyase class-1 family, according to UniProt
3. Protein Structure
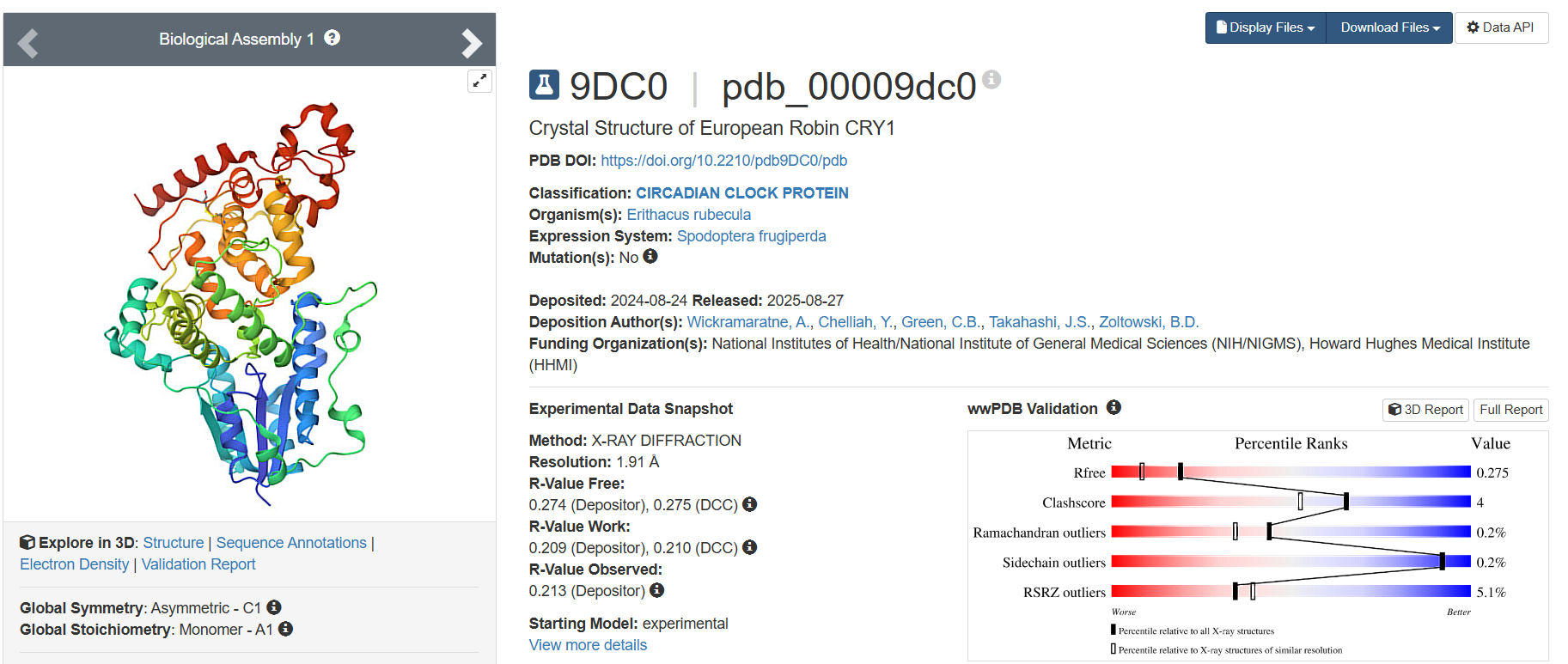
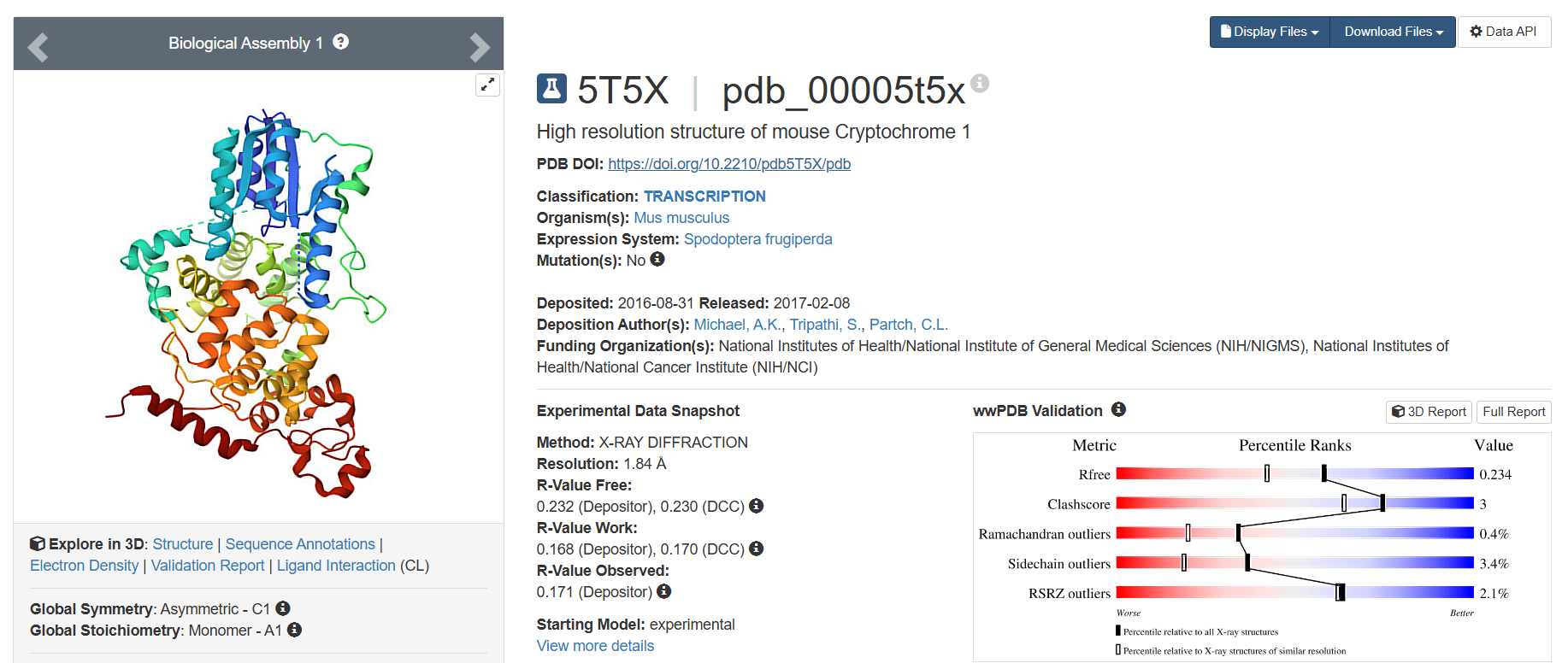
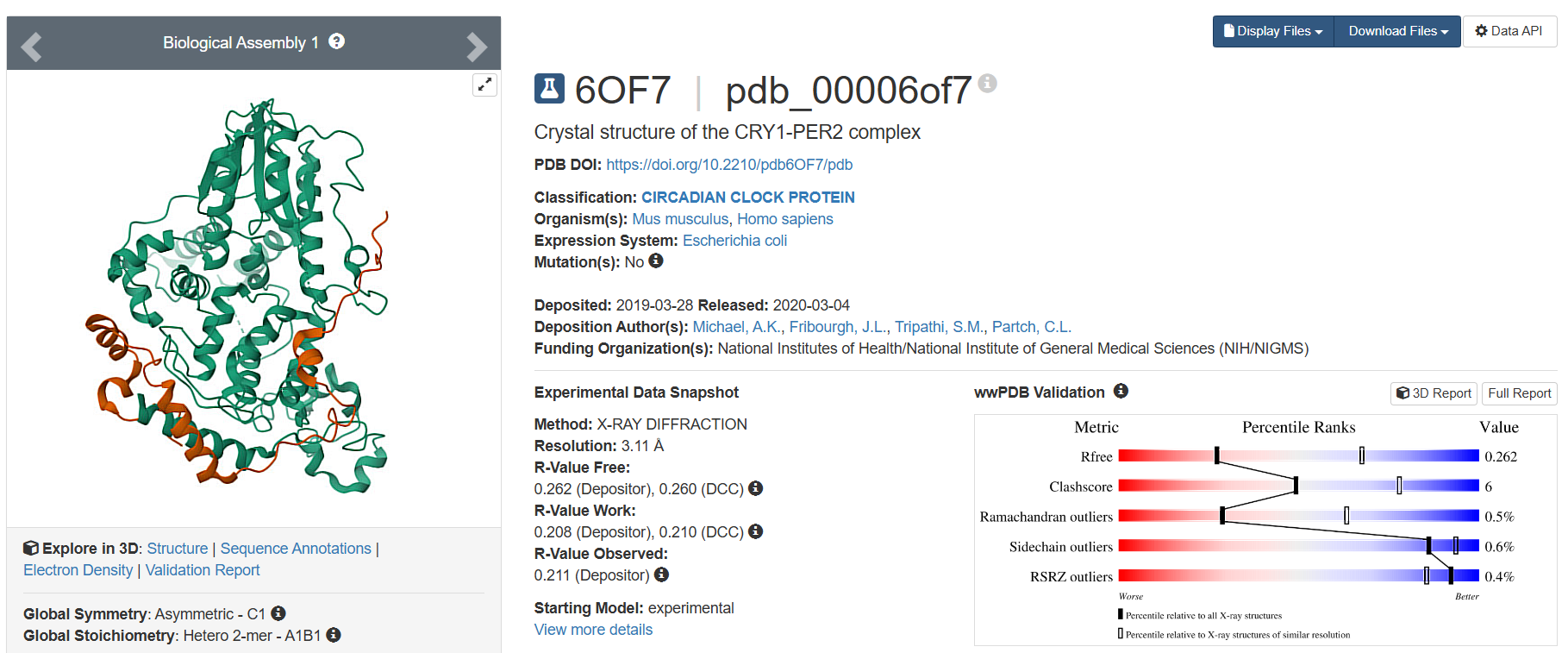
On RCSB, I found 19 results. However, only one was the Cryptochrome in Homo sapiens. The other results included Mus musculus (16) and 3 other organisms (1 each). I have listed 3 of these 19:
The latest one, deposited on 2024-08-24, having 1.91 Å resolution, is one of the finer results.
The one with best resolution, deposited on 2017-02-08, having 1.84 Å resolution, has the lowest resolution and therefore, is the best quality structure.
The only one belonging to Homo sapiens, deposited on 2019-03-28, having 3.11 Å resolution, has the highest resolution and therefore, is not a very good quality structure.
According to the 3D Structures, only water molecules and polymer molecules are present.
I was unable to find anything on SCOP related to the structural family of this protein.
4. 3D Molecule Visualization Software
I chose to use UCSF Chimera.
I downloaded and opened the latest deposited structure of Cryptochrome on RCSB.
I tried visualization according to the tools and actions in Chimera.
The visualization using both ‘Ribbon’ and ‘Ball and Stick.’
The visualization using only ‘Ribbon.’
The visualization using only ‘Ball and Stick.’
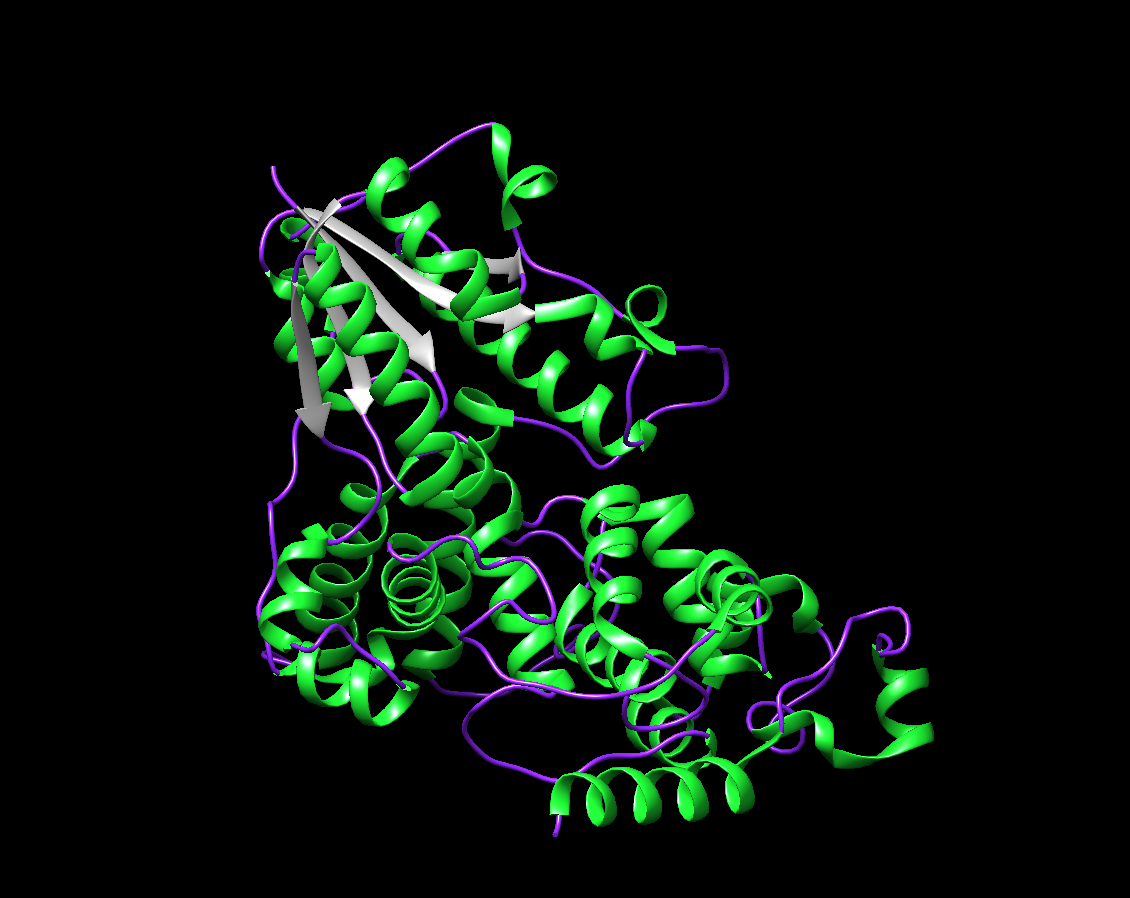
The visualization by colored Secondary Structure. I chose Green for the Helix, Purple for the Coil, and Gray for the Strand. There are around 4 sheets, and many more numerous helices.
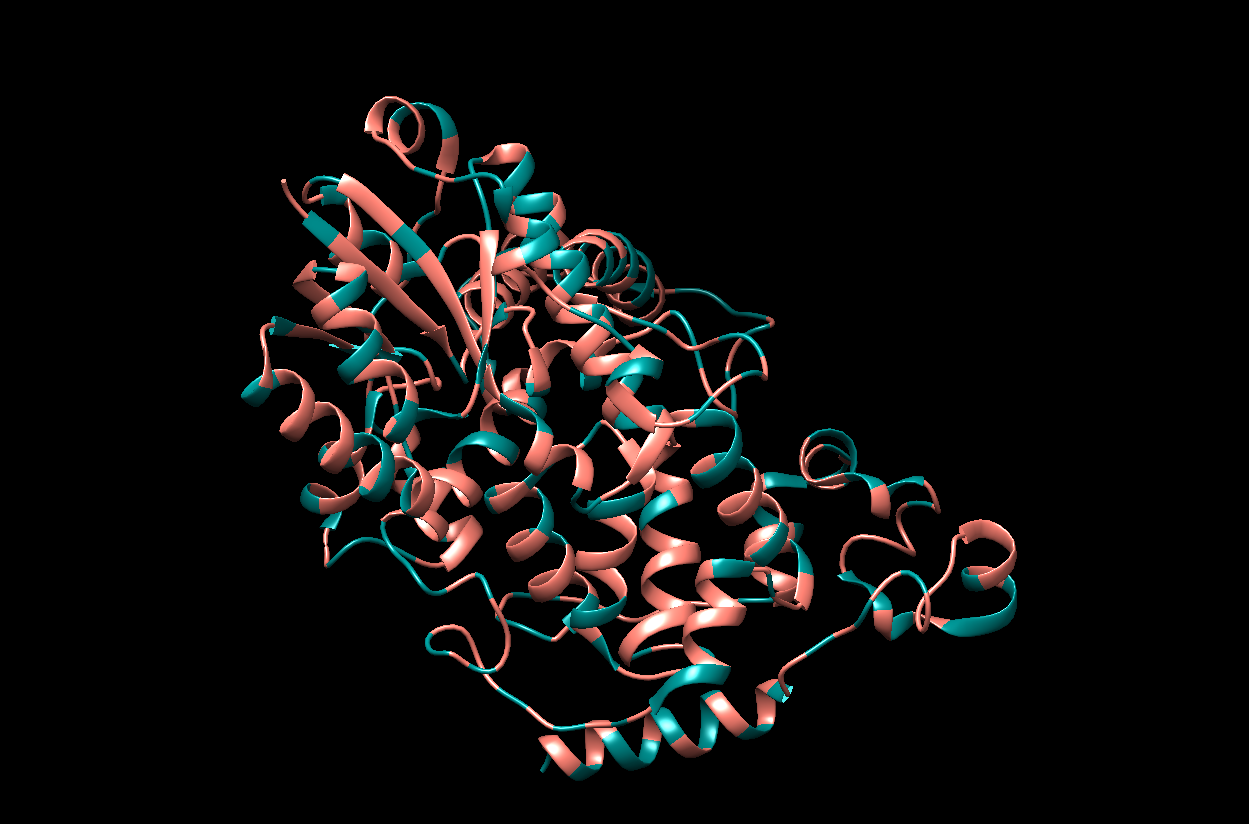
The visualization by colored Residue Type. I chose Salmon for hydrophobic residues and Dark Cyan for the remaining (hydrophillic residues).
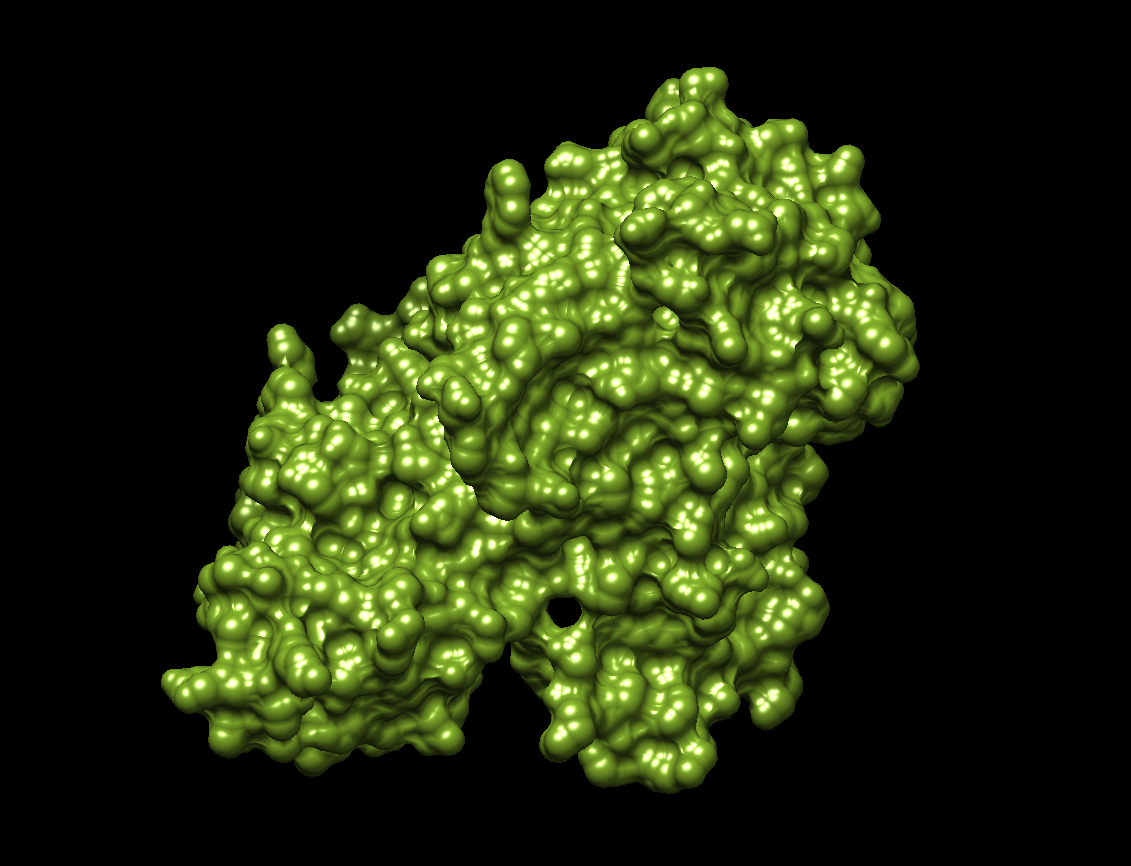
The visualization of the surface. The protein surface seems to have multiple holes, grooves and depressions, meaning it should have multiple binding sites.
Part C
Week 5 HW: Protein Design Part II
Part A
Part 1
I added the protein sequence from UniProt after mutating it, and changed the parameters, as needed.
The Sequence: MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
I ended up with these 4 results.
I added the given peptide sequence and calculated the Pseudo Perplexity for the same.