Week 2 HW: DNA Read, Write & Edit
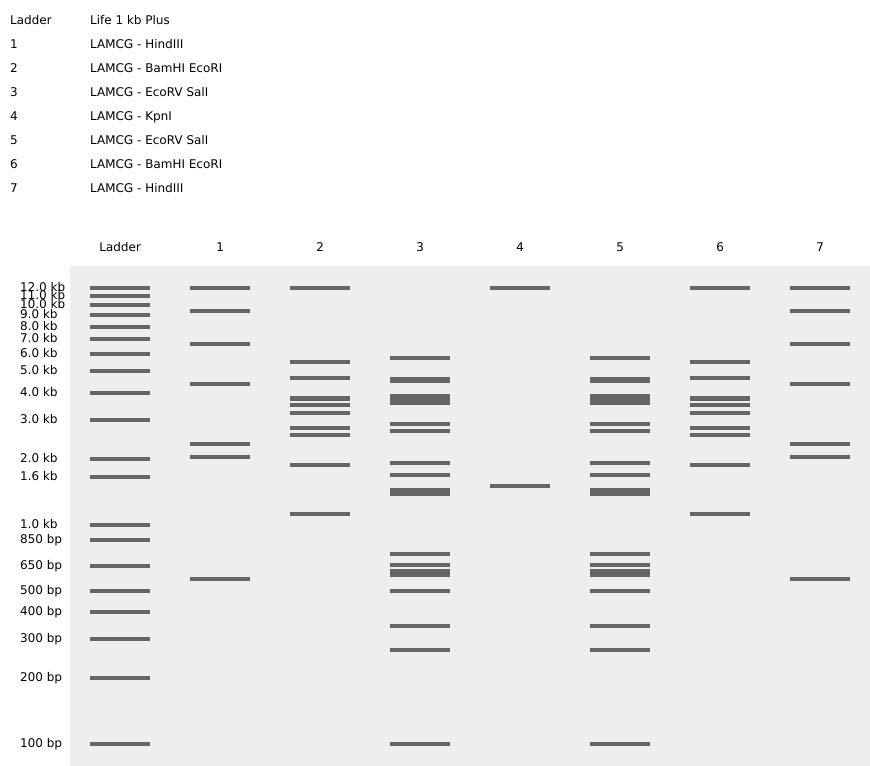
Part 1: Benchling & In-silico Gel Art

PART 3: DNA Design Challenge
3.1 Choose your protein
Erythropoietin is a hormone that stimulates red blood cell production. Selected because:
Vital in anemia therapy
High-value biotechnology protein
Relevant to the pharmaceutical industry
Erythroproietin :
sp|P01588|EPO_HUMAN Erythropoietin OS=Homo sapiens OX=9606 GN=EPO PE=1 SV=1 MGVHECPAWLWLLLSLLSLPLGLPVLGAPPRLICDSRVLERYLLEAKEAENITTGCAEHC SLNENITVPDTKVNFYAWKRMEVGQQAVEVWQGLALLSEAVLRGQALLVNSSQPWEPLQL HVDKAVSGLRSLTTLLRALGAQKEAISPPDAASAAPLRTITADTFRKLFRVYSNFLRGKL KLYTGEACRTGDR
3.2 Reverse Translate : Protein sequence to DNA sequence Erythroproietin DNA Sequence:
sp|P01588|EPO_HUMAN Erythropoietin OS=Homo sapiens OX=9606 GN=EPO PE=1 SV=1 ATGGGGGTGCACGAATGCCCAGCATGGTTGTGGCTACTATTGAGCCTTCTGTCCTTGCCCTTAGGTCTCCCTGTACTTGG GGCGCCCCCCCGACTAATATGTGACTCGCGGGTTTTAGAGCGGTACCTGTTGGAAGCAAAAGAAGCGGAAAATATCACTA CTGGCTGCGCTGAACATTGTTCCTTAAATGAGAATATCACAGTTCCCGACACCAAGGTAAATTTTTATGCGTGGAAACGC ATGGAGGTTGGCCAACAAGCAGTCGAAGTTTGGCAGGGGTTAGCGCTACTTTCTGAGGCAGTGCTTAGAGGCCAGGCATT GTTAGTAAATTCAAGCCAGCCTTGGGAGCCTCTACAACTTCATGTGGACAAAGCCGTGTCAGGCCTGAGATCCCTAACTA CGCTCCTCCGCGCGCTAGGAGCGCAAAAAGAGGCTATCAGTCCGCCCGACGCAGCTTCTGCCGCCCCACTCCGTACCATA ACAGCTGACACTTTCCGAAAACTTTTCAGAGTTTATTCAAACTTCCTACGAGGTAAATTGAAATTATACACTGGCGAAGC CTGCAGGACTGGGGATCGC
3.3 Codon Optimization
For this project, Escherichia coli has been chosen as the expression host.
ATGGGTGTGCACGAATGCCCAGCATGGTTGTGGCTACTGTTGAGCCTTCTGTCCTTGCCGTTAGGTCTCCCTGTACTTGGGGCGCCCCCGCGTCTTATTTGTGATTCGCGTGTTCTGGAGCGGTACCTGTTGGAAGCCAAAGAAGCGGAAAATATTACTACCGGCTGCGCTGAACATTGTTCCTTAAATGAAAACATCACAGTTCCGGACACCAAGGTCAACTTTTATGCGTGGAAACGCATGGAGGTCGGCCAACAGGCGGTCGAAGTGTGGCAGGGGCTGGCGCTACTGAGCGAGGCAGTGCTTCGTGGCCAGGCACTGTTAGTAAATAGTAGCCAGCCTTGGGAGCCGCTGCAACTGCATGTGGACAAAGCCGTGTCAGGCCTGCGCTCGCTGACGACGCTCCTCCGCGCGCTGGGAGCGCAGAAGGAAGCTATCAGTCCGCCGGATGCAGCCTCTGCCGCCCCACTGCGTACCATTACCGCTGATACATTCCGAAAACTGTTCCGTGTTTATTCAAACTTTCTGCGCGGTAAACTGAAATTATACACTGGTGAAGCCTGCAGAACGGGCGATCGC
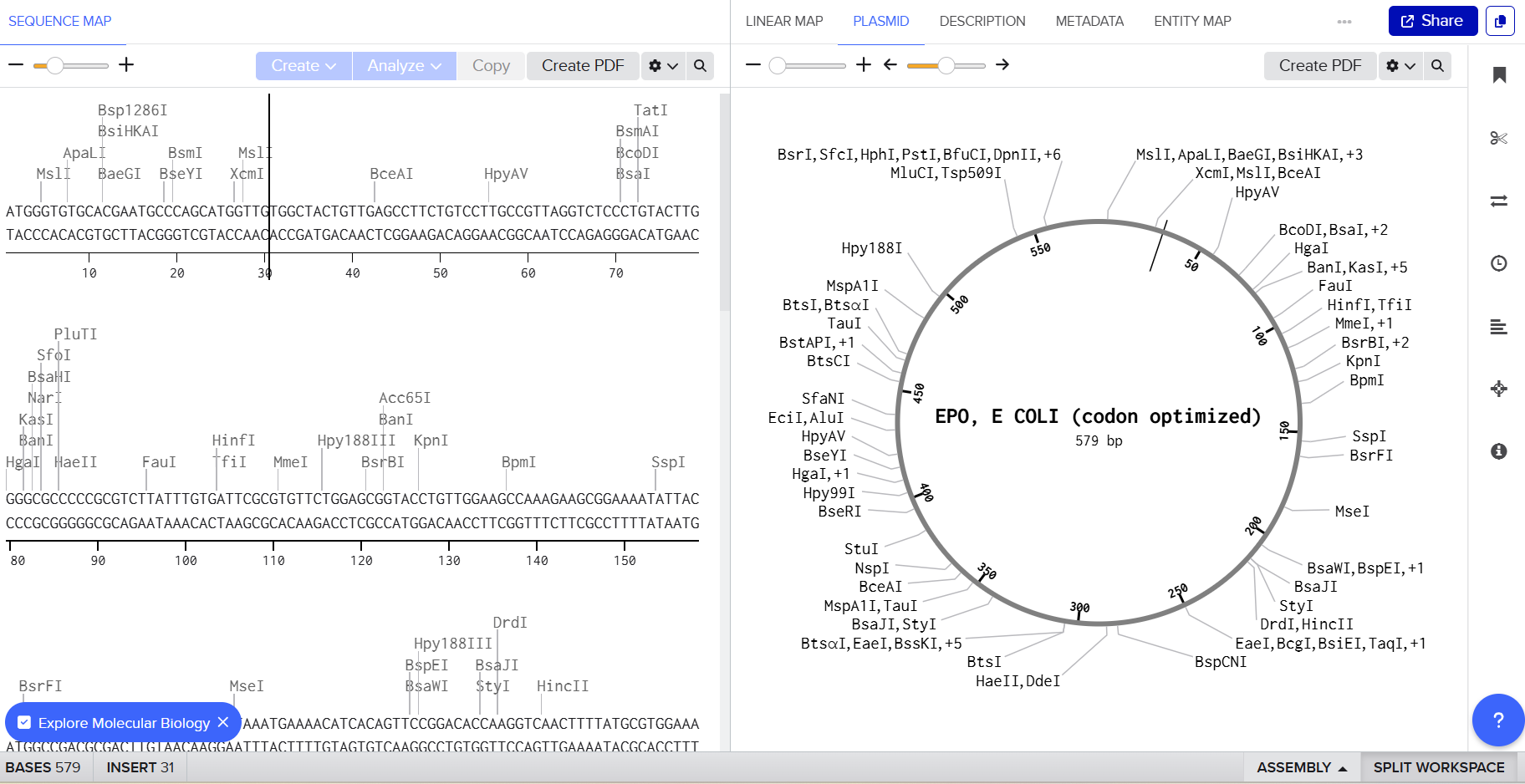
3.4 You have a sequence! Now what?
Once the DNA sequence encoding Erythropoietin (EPO) has been designed and codon-optimized for E. coli, the next step is to produce the protein using an appropriate expression system. Two major technological approaches can be used: cell-dependent expression systems and cell-free expression systems.
A. Cell-Dependent Expression System (Using E. coli)
In this project, a cell-dependent system is used with the pET-28a expression vector.
Step 1: Cloning into pET-28a
The optimized EPO gene is inserted into the Multiple Cloning Site (MCS) of the pET-28a plasmid. The construct includes:
a. T7 promoter
b. Ribosome Binding Site (RBS)
c. Start codon (ATG)
d. EPO coding sequence
e. Stop codon
f. Optional His-tag for purification
This produces the recombinant plasmid pET-28a-EPO.
Step 2: Transformation into E. coli
The recombinant plasmid is introduced into competent E. coli cells (e.g., BL21(DE3)). Transformed cells are selected using kanamycin resistance encoded by the plasmid.
Step 3: Transcription
Upon induction:
The T7 RNA polymerase recognizes the T7 promoter.
The DNA sequence is transcribed into messenger RNA (mRNA).
During transcription:
a. Adenine (A) pairs with Uracil (U)
b. Thymine (T) in DNA becomes Uracil (U) in RNA
The result is an mRNA molecule complementary to the DNA template strand
Step 4: Translation
The ribosome binds to the Ribosome Binding Site (RBS).
Translation begins at the start codon (AUG).
Each codon (three nucleotides) is translated into one amino acid.
Transfer RNA (tRNA) delivers the corresponding amino acids.
The polypeptide chain elongates until a stop codon is reached.
This process follows the Central Dogma of Molecular Biology:
DNA → RNA → Protein
The final product is recombinant EPO protein, typically fused with a His-tag for purification.
B. Cell-Free Expression System (Alternative Method)
Alternatively, the EPO protein can be produced using a cell-free expression system.
In this system:
a. The DNA template is added directly to a reaction mixture.
b. The mixture contains RNA polymerase, ribosomes, tRNAs, amino acids, and necessary cofactors.
c. Transcription and translation occur in vitro (outside living cells).
Advantages:
a. Faster protein production
b. No need for cell transformation
c. Suitable for rapid screening
However, for large-scale production, cell-dependent systems are generally preferred.
3.5 [Optional] How does it work in nature/biological systems?
In human cells:
a. Alternative splicing generates isoforms.
b.RNA editing may modify nucleotides post-transcription.
c. Post-translational modifications (e.g., glycosylation in EPO) alter protein stability and function.
In contrast, E. coli:
a. Does not perform alternative splicing.
b. Does not process introns.
c. Does not perform complex glycosylation.
Thus, recombinant EPO produced in E. coli may differ structurally from native human EPO.
Part 4: Prepare a Twist DNA Synthesis Order
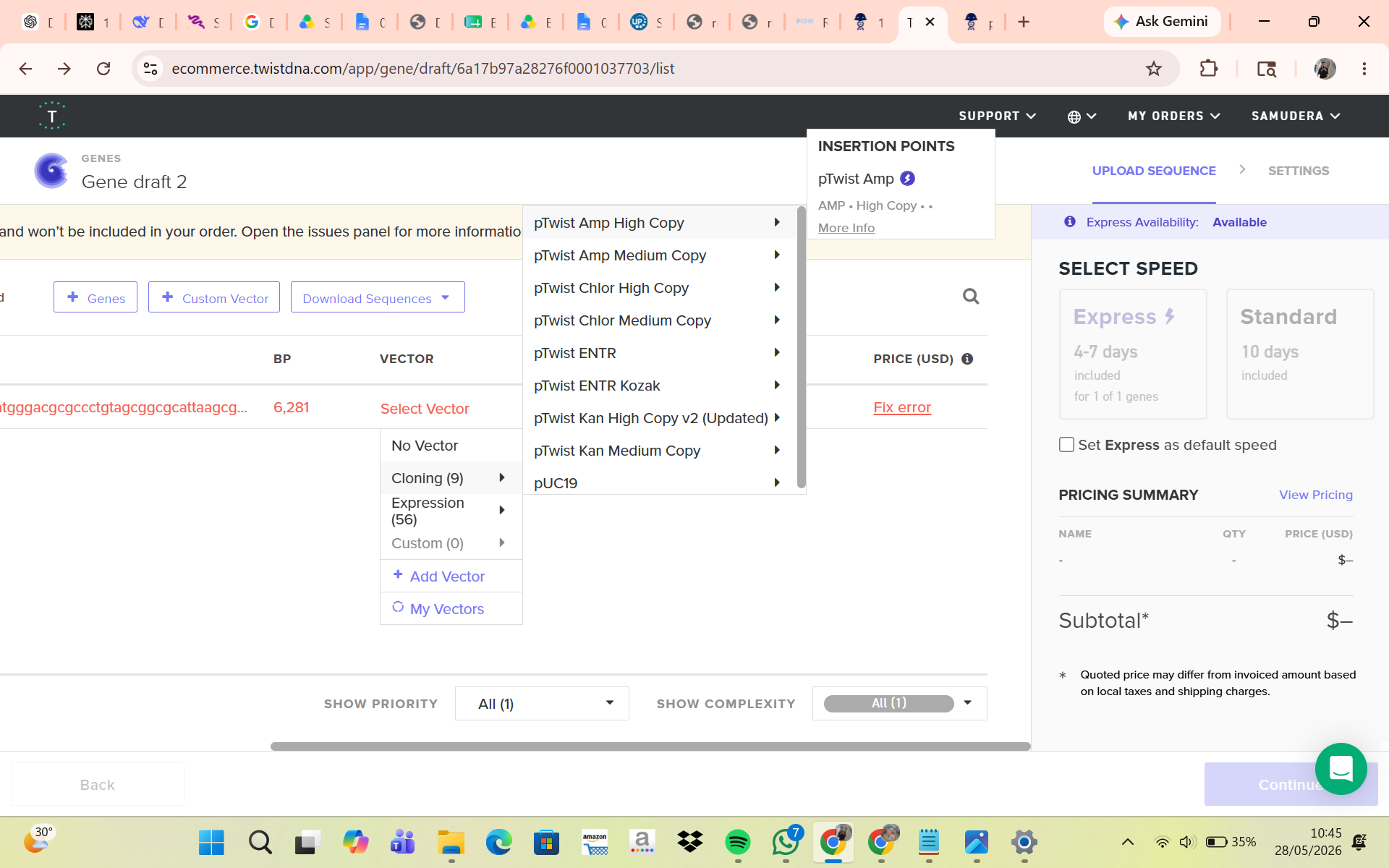
Part 5: DNA Read/Write/Edit
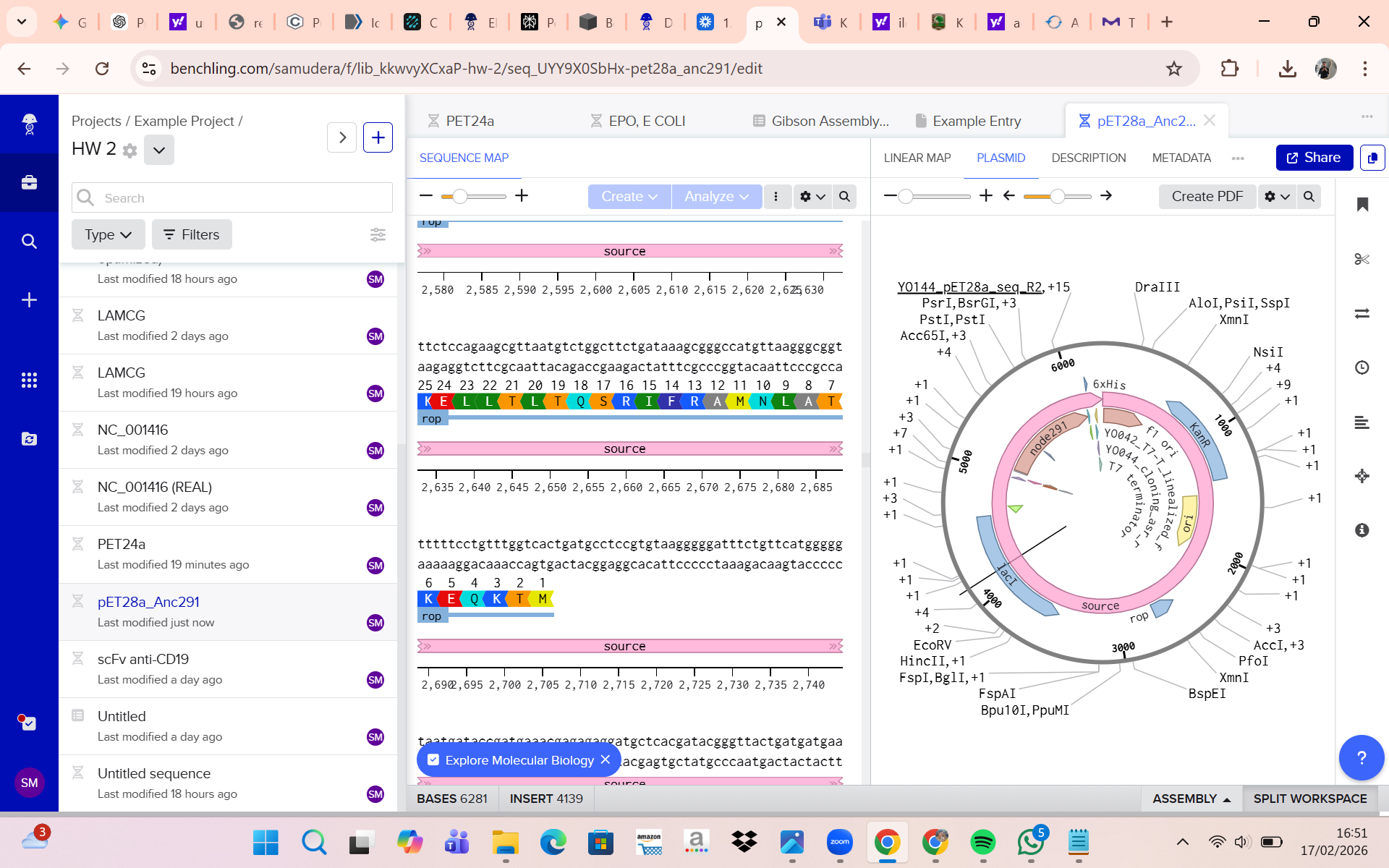
5.1 DNA Read
(i) What DNA would you want to sequence and why?
Primary DNA to Sequence in This Project
I would sequence:
The recombinant pET-28a-EPO plasmid
The EPO coding sequence (CDS)
The promoter–insert junction regions
Why?
a. To confirm that the EPO gene was inserted correctly.
b. To verify that no mutations occurred during gene synthesis or cloning.
c. To confirm the correct reading frame with the His-tag.
d. To ensure no premature stop codons or frameshifts are present.
Beyond this project, sequencing could be applied to sequencing disease-associated genes in Human health (e.g., cancer mutations).
(ii) What sequencing technology would you use and why?
For this project, I would use:
Sanger Sequencing Why?
a. The EPO gene is relatively short (~500–600 bp coding region).
b. Plasmid verification is well suited for Sanger sequencing.
c. High accuracy for single-gene validation.
d. Cost-effective for small constructs.
Classification
a. First-generation sequencing
b. Uses chain-termination chemistry
c. Produces highly accurate reads (~700–1000 bp per read)
Input: Purified plasmid DNA (pET-28a-EPO)
Preparation Steps:
Plasmid extraction from E. coli
Primer design (forward and reverse primers flanking insert)
PCR cycle sequencing reaction with labeled dideoxynucleotides (ddNTPs)
Essential Steps of Sanger Sequencing
DNA denaturation
Primer annealing
DNA polymerase extension
Random incorporation of fluorescently labeled ddNTPs
Chain termination
Capillary electrophoresis separation
Laser detection of fluorescent signals
Base Calling : Each ddNTP is labeled with a different fluorescent dye. When incorporated, elongation stops.Fragments of different lengths are separated and detected.The emitted fluorescence determines the base identity (A, T, C, or G).
Output : Chromatogram (electropherogram), DNA sequence file (.ab1 or .seq), Base quality scores
5.2 DNA Write
(i) What DNA would you want to synthesize and why?
In this project, I would synthesize:
Codon-optimized human EPO gene for E. coli
Purpose:
a. Produce recombinant EPO for research applications.
b. Study protein folding and expression optimization.
ii) What technology would you use for DNA synthesis?
Chosen Technology:
Chemical DNA synthesis followed by gene assembly & Commercial providers (e.g., gene synthesis companies) synthesize DNA de novo.
Essential Steps of DNA Synthesis
Oligonucleotide synthesis (phosphoramidite chemistry)
Assembly of short oligos into full-length gene
Error correction (if necessary)
Cloning into plasmid backbone (pET-28a)
Sequence verification
Limitations of DNA Synthesis
Speed: Synthesis of longer genes takes more time.
Accuracy: Errors can occur during chemical synthesis, Requires sequencing validation.
Scalability: Cost increases with gene length, Whole-genome synthesis remains complex and expensive.
However, for single-gene constructs like EPO, synthesis is efficient and practical.
5.3 DNA Edit
(i) What DNA would you want to edit and why?
In this project, I would edit:
The EPO coding sequence
Possible edits:
a. Improve solubility in E. coli
b. Reduce aggregation
c. Modify specific amino acids to increase stability
Beyond this project: i wish we can Correct disease-causing mutations in humans
(ii) What technology would you use?
For Plasmid Editing: Site-Directed Mutagenesis
For Genomic Editing: CRISPR-Cas systems
How CRISPR Edits DNA
Design guide RNA (gRNA) complementary to target DNA.
Cas enzyme binds to gRNA.
Complex locates target DNA.
Cas creates double-strand break.
Repair occurs via:
a. Non-homologous end joining (NHEJ)
b. Homology-directed repair (HDR)
Required Inputs : DNA template (target sequence), Guide RNA, Cas enzyme, Repair template (if precise edit desired), Host cells
Essential Design Steps
Identify target sequence.
Design guide RNA.
Check for off-target sites.
Prepare delivery system (plasmid or ribonucleoprotein).
Validate edits via sequencing.
Limitations of Editing Technologies
Efficiency: Editing efficiency may vary by cell type.
Precision: Off-target mutations may occur.
Delivery Challenges: Introducing CRISPR components into certain cells is difficult.
Ethical Considerations: Human genome editing raises significant ethical concerns.