Week 4 HW: Protein Design Part I
A. Conceptual Question
- Molecules of Amino Acids in 500g Meat
A typical 500 g serving of lean meat contains about 125–150 g of protein (≈25–30% by weight). Taking 130 g as an average and assuming an average amino acid residue mass of ~110 g/mol, this corresponds to:
130 g ÷ 110 g/mol ≈ 1.18 mol of amino acid residues.
Multiplying by Avogadro’s number (6.022 × 10²³ mol⁻¹) gives:
≈ 7 × 10²³ amino acid molecules.
Therefore, a 500 g serving of lean meat provides on the order of 10²⁴ amino acid molecules
- Why Humans Eat Beef/Fish Without Becoming Them
Humans do not become cows or fish after eating them because dietary proteins are first degraded by proteolysis in the gastrointestinal tract into free amino acids and small peptides. The original protein sequence information is destroyed during digestion.These amino acids enter the bloodstream and are reused by ribosomes to synthesize human proteins according to mRNA transcribed from the human genome, following the central dogma (DNA → RNA → protein).Protein synthesis is directed exclusively by endogenous gene expression, and dietary nucleic acids are also degraded into nucleotides before absorption. Therefore, body structure and identity are determined by the host genome, not by consumed biomolecules.
- Why Only 20 Natural Amino Acids
The 20 canonical amino acids were likely selected through evolutionary processes because they provided sufficient chemical diversity while maintaining translational efficiency and genetic stability. Some, such as glycine and alanine, were probably abundant under prebiotic conditions.Together, the 20 amino acids span a broad range of chemical properties — including size, charge, hydrophobicity, and reactivity — enabling the formation of stable and functionally diverse protein structures.
They are encoded within a 64-codon triplet genetic system that provides redundancy and mutational robustness. Expanding the amino acid repertoire beyond 20 may not have offered sufficient selective advantage to offset the increased complexity of translation machinery. Additionally, once the genetic code became established, it likely became evolutionarily constrained (“frozen”), limiting further expansion. Rare additions such as selenocysteine require specialized recoding mechanisms and do not fundamentally alter the canonical set.
- Prebiotic Origin of Amino Acids
Before the emergence of life and enzymatic pathways, amino acids likely formed abiotically through several prebiotic chemical processes. Laboratory simulations such as the Miller–Urey experiment demonstrated that amino acids can be synthesized in reducing gas mixtures (e.g., CH₄, NH₃, H₂O, H₂) subjected to electrical discharges.Additional mechanisms include UV-driven photochemistry in atmospheric gases and geochemical reactions in hydrothermal environments, where mineral surfaces may have facilitated Strecker-type synthesis from aldehydes, hydrogen cyanide (HCN), and ammonia.
Extraterrestrial delivery also contributed to the prebiotic pool, as carbonaceous meteorites such as the Murchison meteorite contain more than 70 amino acids.Under certain environmental conditions, such as drying–wetting cycles or mineral-catalyzed reactions, these amino acids may have undergone condensation reactions to form short peptides, providing building blocks for early biochemical evolution.
- Handedness of D-Amino Acid α-Helix
An α-helix composed entirely of D-amino acids would adopt a left-handed helical conformation, which is the mirror image of the right-handed α-helix formed by L-amino acids.This occurs because chirality determines the energetically allowed φ and ψ backbone dihedral angles. In D-amino acids, the Ramachandran plot is effectively mirrored relative to L-amino acids, shifting the energy minimum to the opposite quadrant.As a result, the helical geometry is inverted while preserving the characteristic i→i+4 hydrogen bonding pattern of the α-helix. Experimental studies of synthetic D-peptides confirm the formation of stable left-handed α-helices.
- Additional Helices in Proteins
Beyond the canonical α-helix (3.6 residues per turn, i→i+4 hydrogen bonding), proteins can adopt additional helical conformations.The 3₁₀-helix contains approximately 3 residues per turn with i→i+3 hydrogen bonding and is often observed as short segments, frequently at α-helix termini. The π-helix has about 4.4 residues per turn with i→i+5 hydrogen bonding and typically appears as local insertions (π-bulges) within α-helices, sometimes contributing to functional or ligand-binding sites.
Additionally, the polyproline II (PPII) helix is an extended, left-handed helix lacking classical intrahelical hydrogen bonds. It is common in intrinsically disordered regions, and individual collagen chains adopt a PPII-like conformation before assembling into a triple helix. These examples demonstrate that protein secondary structure includes multiple helical geometries beyond the classical α-helix.
- Why Most Molecular Helices Right-Handed
Most biological helices are right-handed because proteins are composed almost exclusively of L-amino acids. The L-configuration at the α-carbon restricts the backbone φ and ψ dihedral angles such that the right-handed α-helix occupies the lowest-energy region of the Ramachandran plot (approximately φ ≈ −60°, ψ ≈ −45°). In contrast, a left-handed α-helix for L-amino acids falls into a higher-energy region due to unfavorable steric and torsional interactions. Thus, right-handed helices are energetically preferred. This molecular asymmetry arises from biological homochirality and contributes to the predominance of right-handed helices in proteins and many nucleic acid structures, although rare left-handed forms such as Z-DNA also exist.
- Why β-Sheets Aggregate
β-sheets aggregate as unsatisfied H-bond donors/acceptors at strand edges pair intermolecularly with adjacent sheets, propagating fibrils; flat sheet geometry exposes hydrophobic faces, driving lateral association in water. This is entropically favored by releasing structured water.
Driving Force
Primary force is hydrophobic burial of nonpolar side chains (e.g., valine, leucine) between sheets, supplemented by van der Waals packing and H-bonds; electrostatics (salt bridges) fine-tune. Thermodynamic stability (low free energy) from these non-covalent interactions exceeds soluble states.
- Why Amyloid Diseases Form β-Sheets
Many amyloid diseases arise because certain proteins can misfold into β-sheet–rich conformations under conditions that destabilize their native structure or impair proteostasis. For example, amyloid-β in Alzheimer’s disease and misfolded prion protein undergo nucleation-dependent polymerization, in which exposed aggregation-prone segments template further misfolding. The resulting cross-β architecture—where β-strands run perpendicular to the fibril axis and sheets stack along it—forms extensive intermolecular hydrogen-bonding networks. This structure confers high thermodynamic stability, protease resistance, and kinetic persistence.
As materials, amyloid fibrils self-assemble into highly ordered nanofibers with mechanical stiffness in the gigapascal range. Their nanoscale organization and robustness have inspired applications in biomaterials, hydrogels, tissue scaffolds, and bioelectronic nanowires. Engineered peptide variants can be designed to retain self-assembly properties while minimizing cytotoxicity, enabling safe material applications.
Part B: Protein Analysis and Visualization
The selected protein is the human Hemoglobin subunit beta, which plays a crucial role in transporting oxygen through the bloodstream as a key component of hemoglobin. This protein was chosen for its vital importance in human physiology, its availability of a high-quality 3D structure, and its frequent use as a model in protein structure studies.
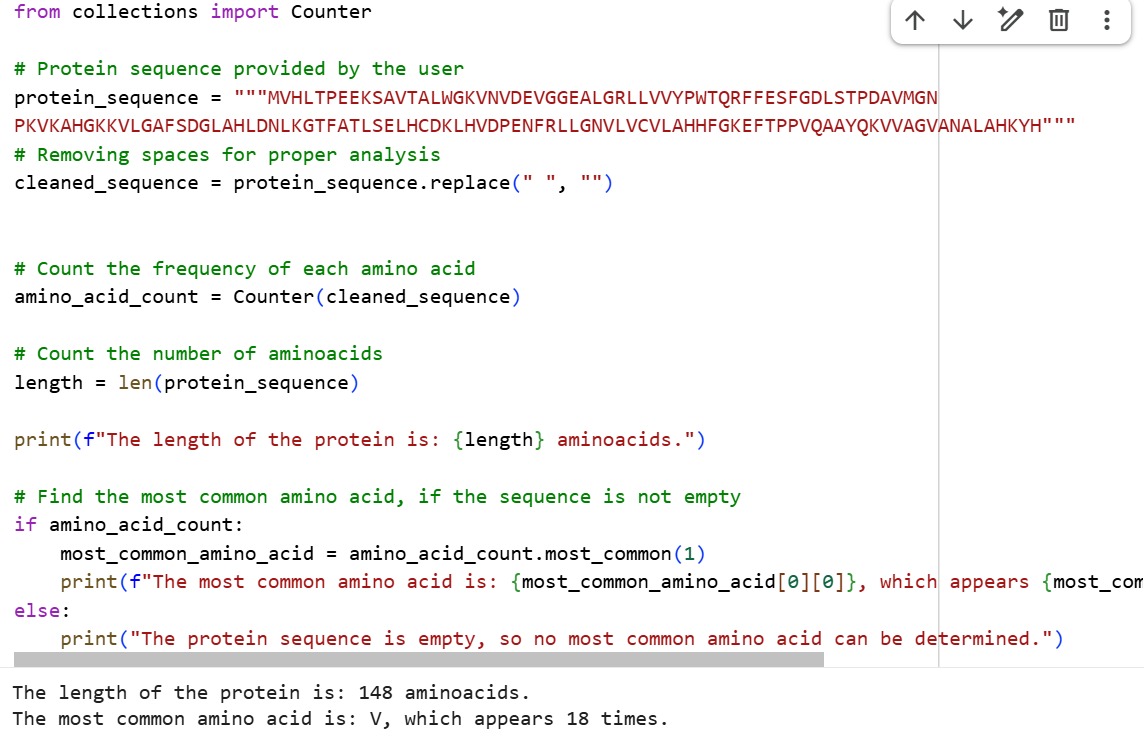
Amino Acid Sequence
The sequence was obtained from UniProt entry P68871
MVHLTPEEKSAVTALWGKVNVDEVGGEALGRLLVVYPWTQRFFESFGDLSTPDAVMGNPKVKAHGKKVLGAFSDGLAHL DNLKGTFATLSELHCDKLHVDPENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKVVAGVANALAHKYH
The length of the protein is: 148 aminoacids. The most common amino acid is: V, which appears 18 times.
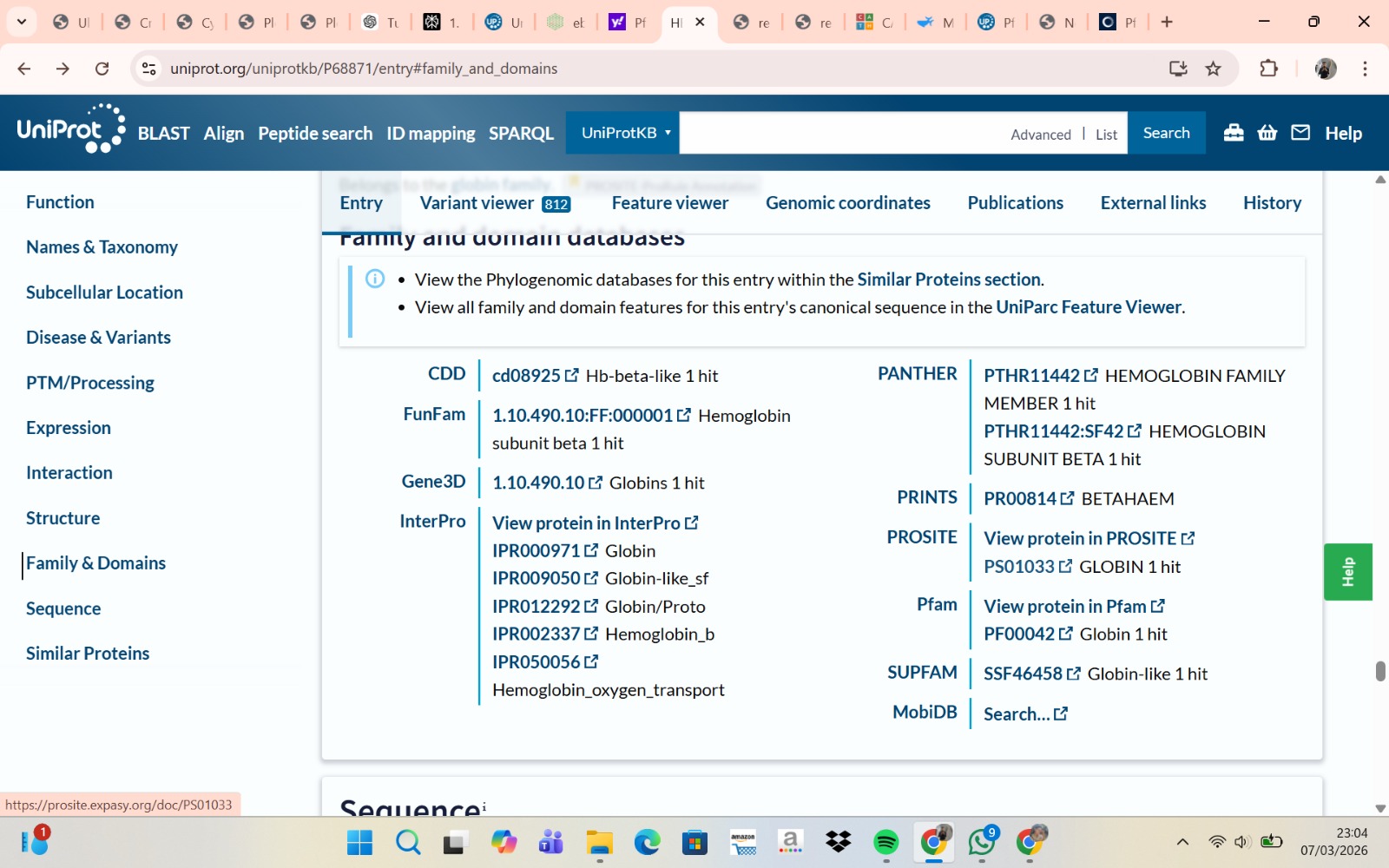
The human Hemoglobin subunit beta belongs to the hemoglobin protein family within the globin superfamily. Domain analysis from the UniProt protein database indicates that the protein contains a conserved globin domain spanning residues 1–147. Multiple protein classification databases consistently identify this protein as a member of the globin family, including Pfam (PF00042), InterPro (IPR000971), and Gene3D (Globins). InterPro further classifies it within the globin-like superfamily (IPR009050) and the hemoglobin beta subfamily (IPR002337). Additional databases such as PANTHER (PTHR11442) and SUPFAM (SSF46458) also support its classification within the globin-like structural superfamily. These annotations indicate that the protein adopts the characteristic globin fold, which enables oxygen binding through a heme prosthetic group.
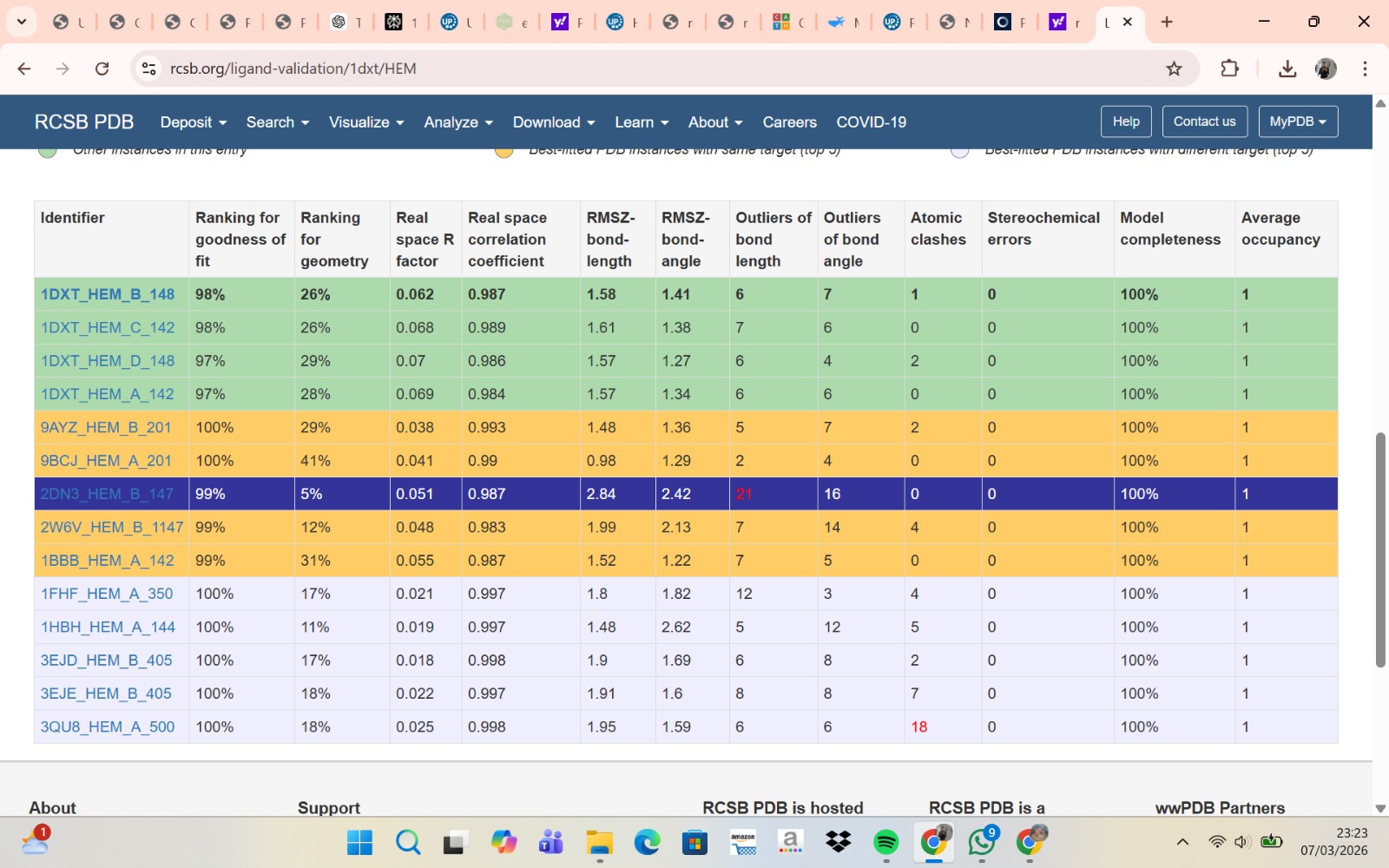
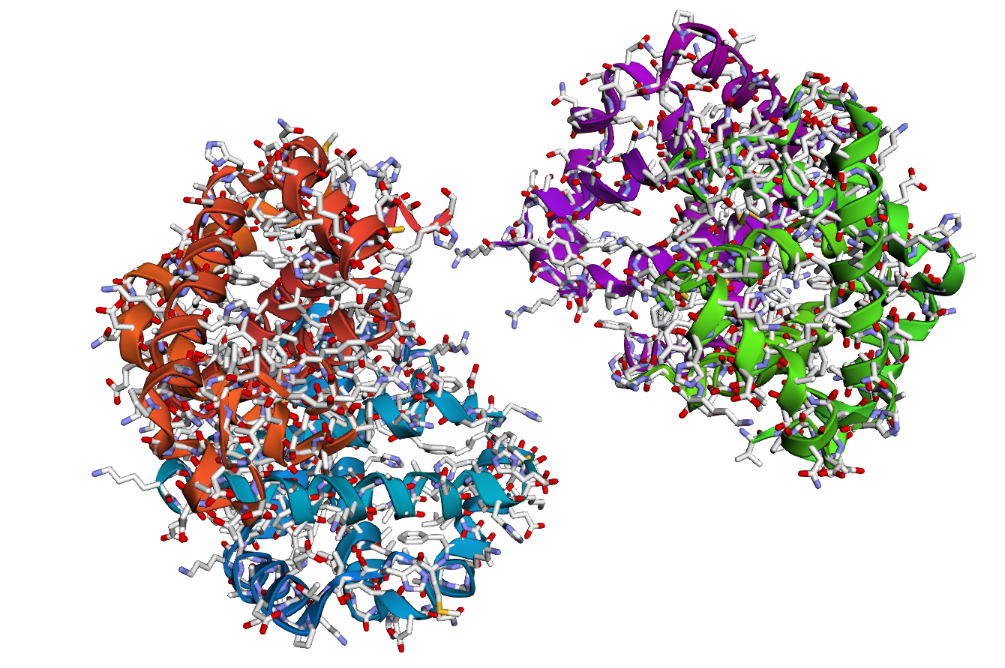
- The structure of Hemoglobin subunit beta from humans is available in the RCSB Protein Data Bank under the PDB ID 1DXT. The structure was determined using X-ray diffraction and deposited in 1992 and released in 1993. It has a resolution of 1.70 Å, indicating a high-quality structure with detailed atomic information. The biological assembly forms a hemoglobin tetramer (A2B2), consisting of two alpha and two beta subunits. In addition to the protein chains, the structure also contains a heme prosthetic group (HEM) that serves as the oxygen-binding site. Structurally, the protein belongs to the globin-like structural superfamily, characterized by a predominantly alpha-helical globin fold and the presence of a heme-binding pocket responsible for oxygen transport.
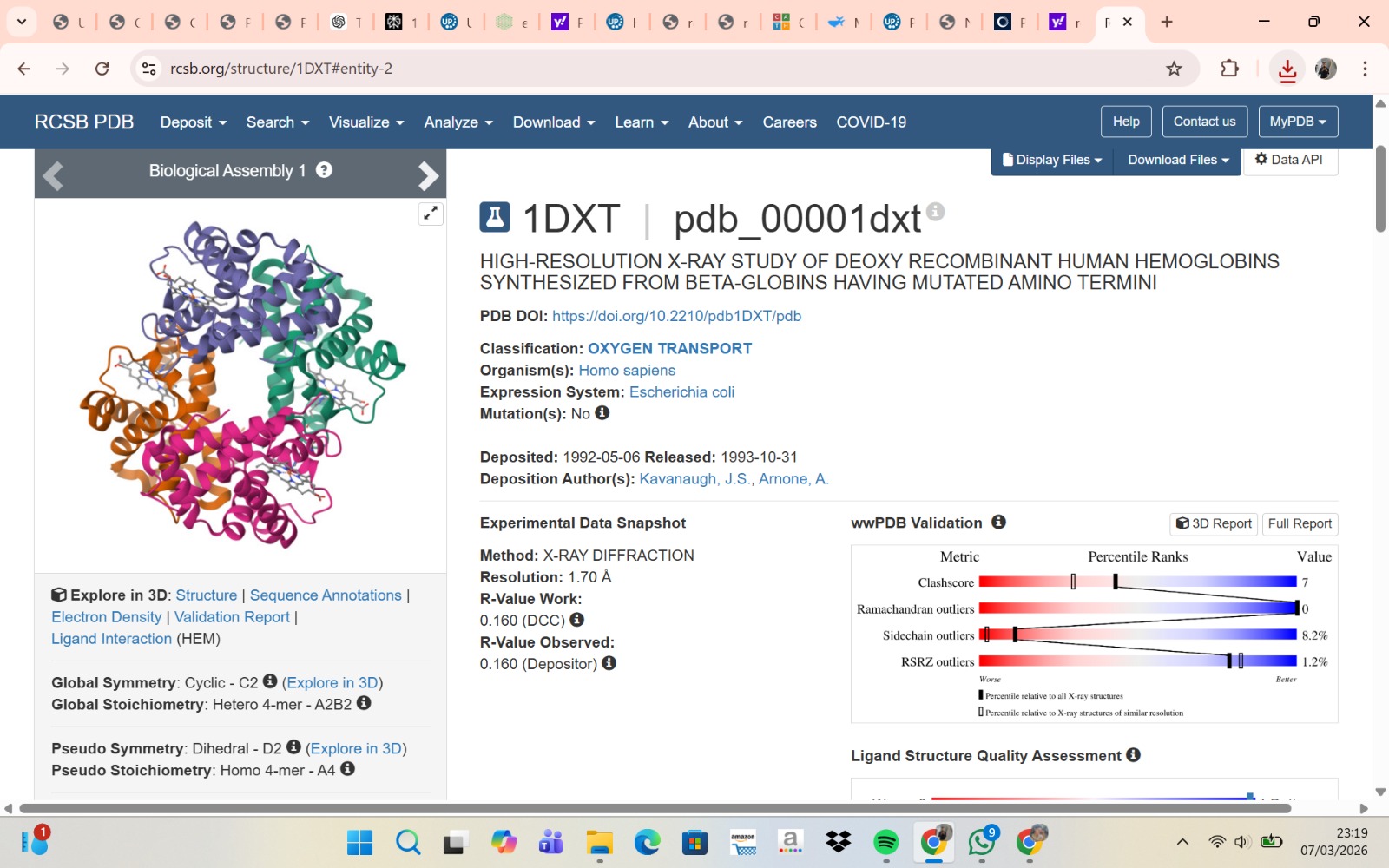
- PyMol Visualization
Visualization of the human Hemoglobin subunit beta protein using PyMOL software with stick representation and water removal.
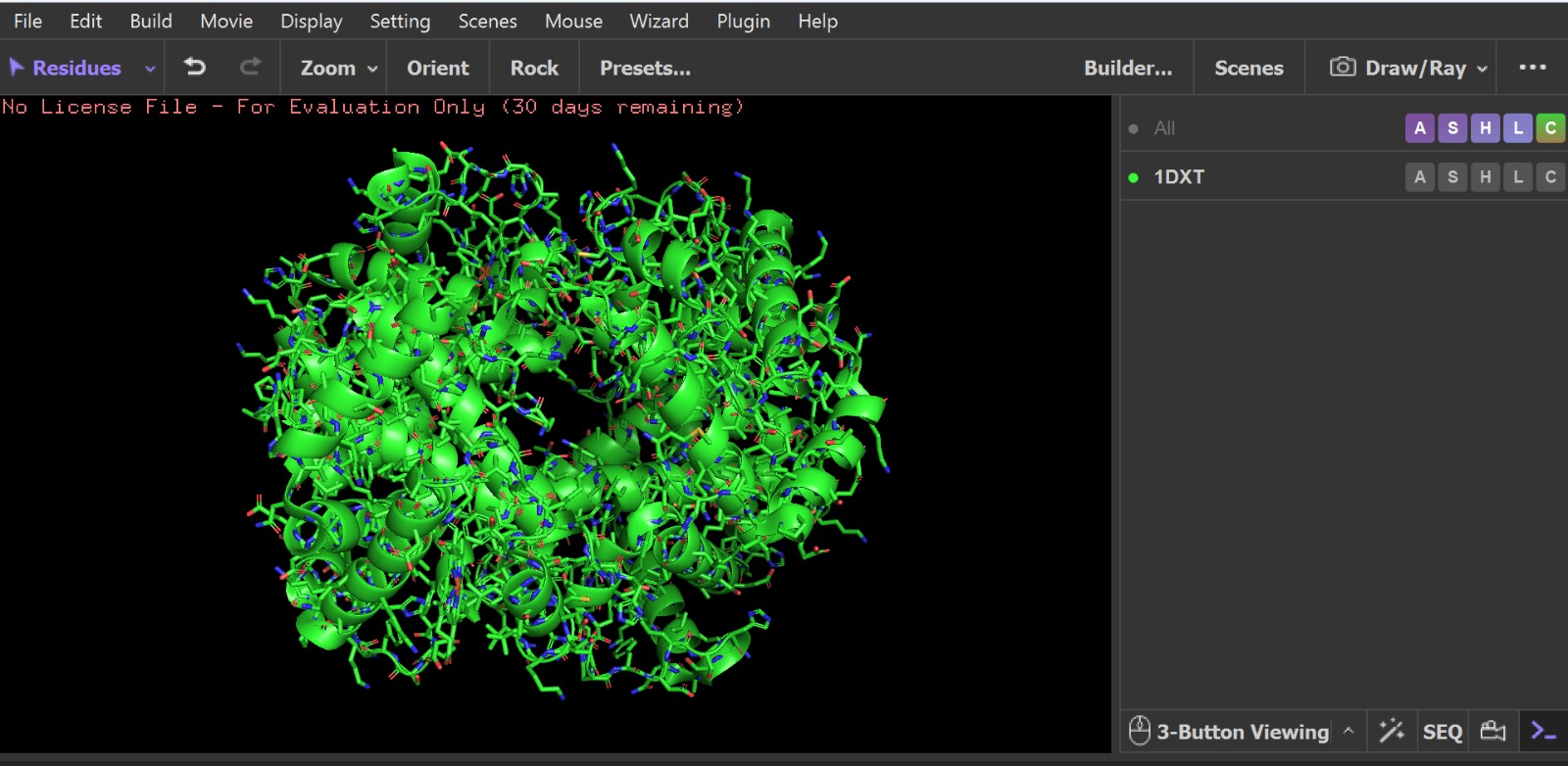
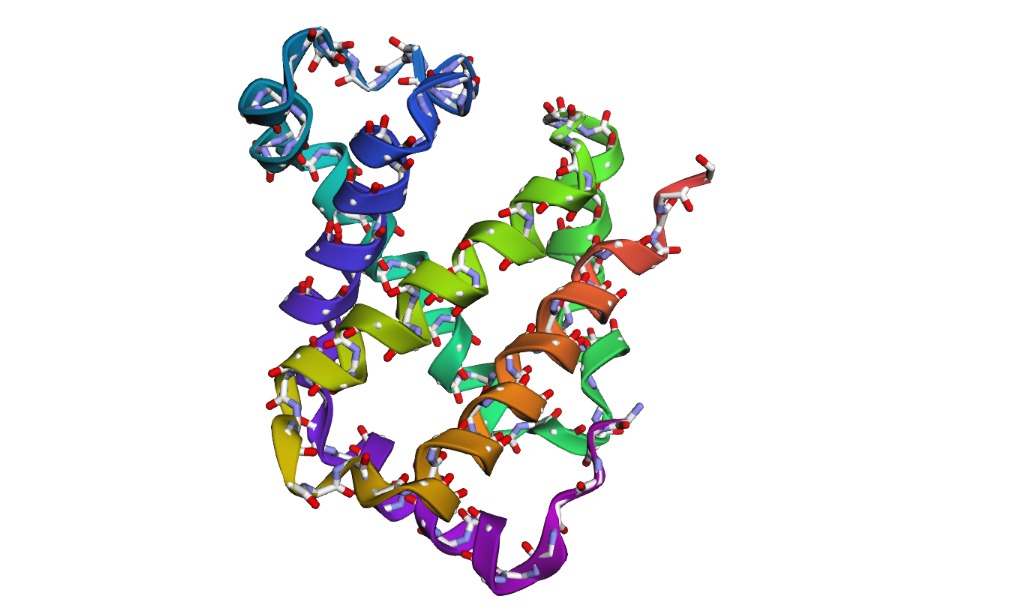
Visualization of the protein structure using PyMOL molecular visualization system colored by secondary structure shows that the structure is dominated by α-helices (red), while only a few regions correspond to loops or coils (green). No significant β-sheet structures are observed. Therefore, the protein contains many more α-helices than β-sheets. This observation is consistent with the typical globin fold of Hemoglobin subunit beta, where each subunit is primarily composed of multiple α-helices that form the structural framework around the heme-binding pocket.
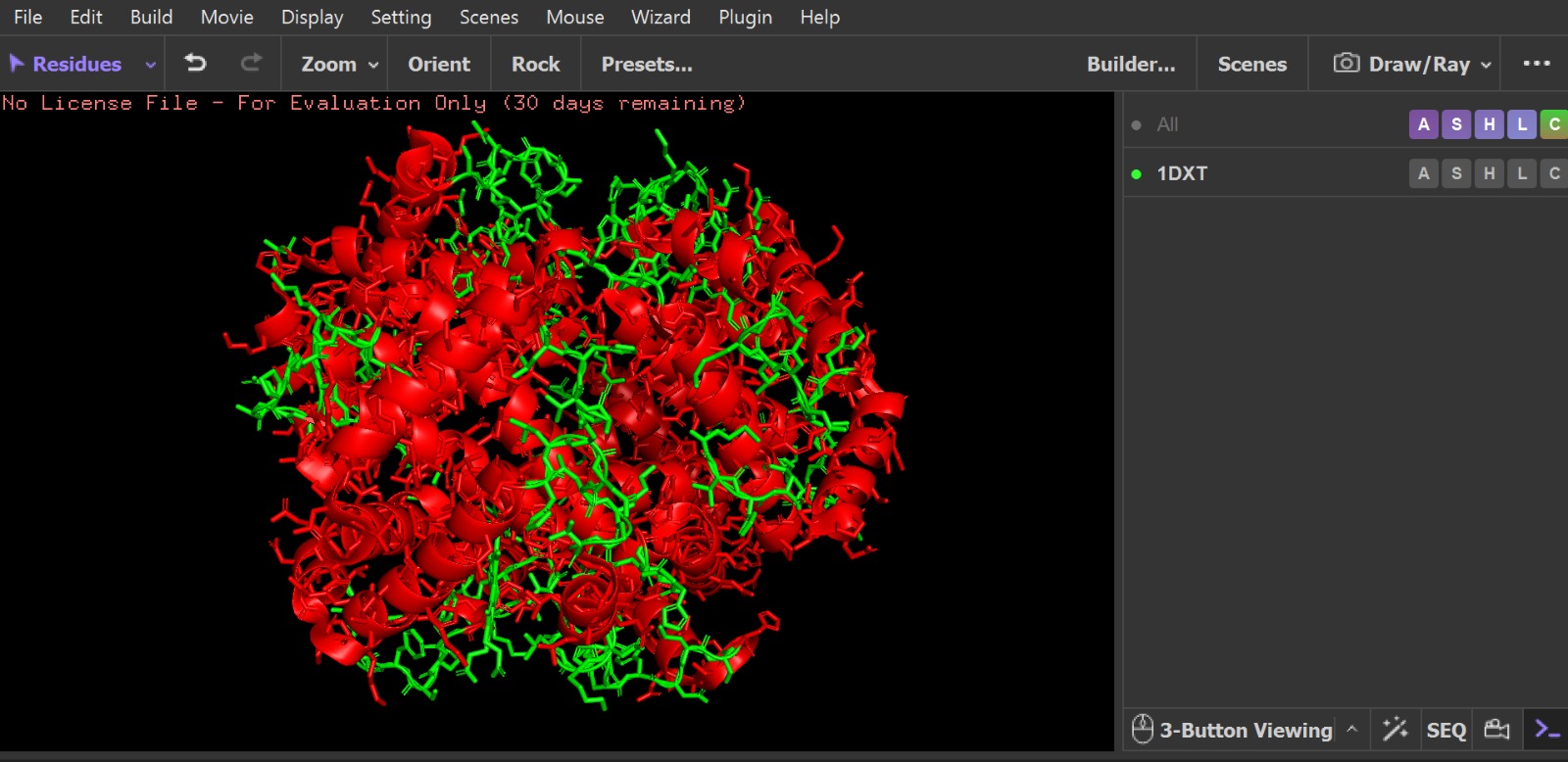
Visualization of the molecular structure of Human hemoglobin using PyMOL molecular visualization system reveals the presence of a distinct binding pocket within the protein. This pocket corresponds to the heme-binding site, where the heme prosthetic group containing an Fe²⁺ ion is located. The pocket is formed by several surrounding α-helices of the globin fold. This structural cavity allows oxygen molecules to bind to the Fe²⁺ ion in the heme group, enabling hemoglobin to perform its biological function in oxygen transport. Therefore, the protein clearly contains functional binding pockets on its surface.
Part C: Using ML-Based Protein Design Tools
C1. PROTEIN LANGUAGE MODELING
- Deep Mutational Scans
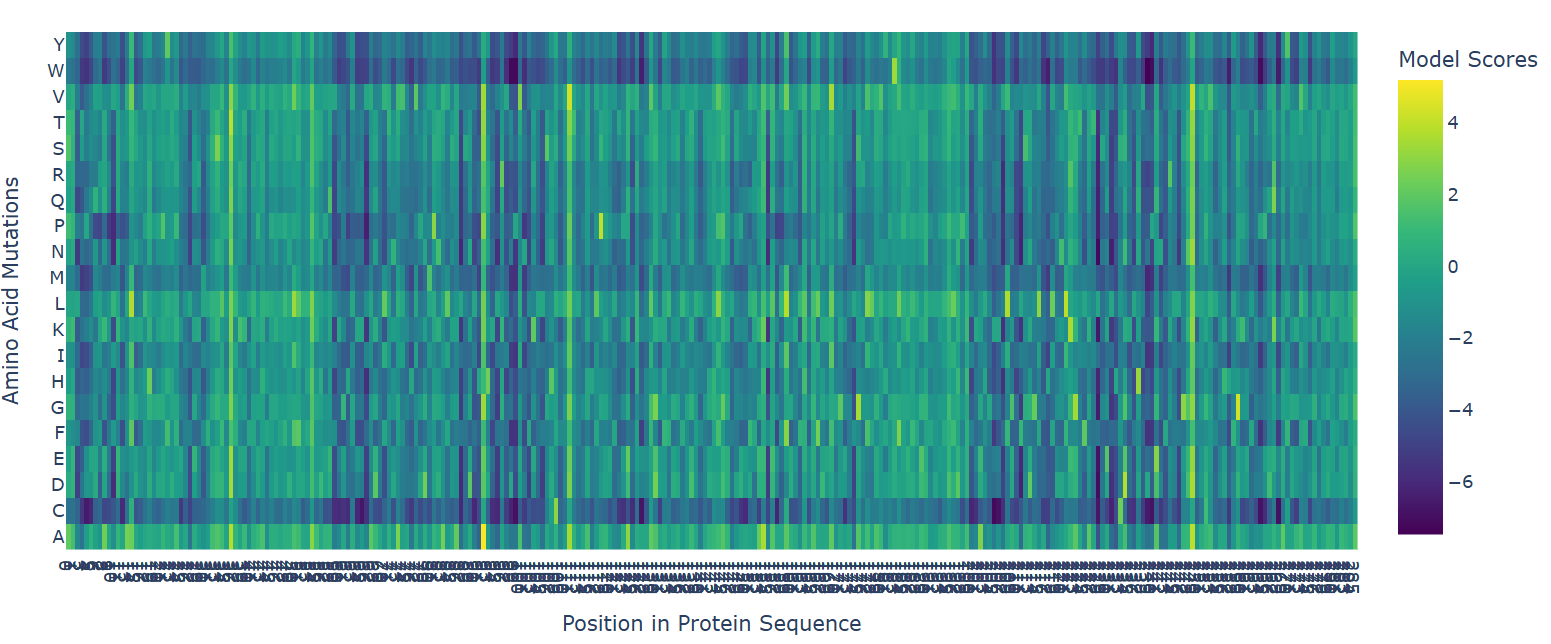
The deep mutational scan generated using the ESM2 protein language model reveals that many residues in human hemoglobin beta are highly conserved and sensitive to mutation. In the heatmap, mutations associated with strongly negative scores (dark blue/purple regions) are likely unfavorable and may disrupt protein stability or function. Since hemoglobin beta adopts a globin fold dominated by α-helices, mutations introducing structurally disruptive residues such as proline are often poorly tolerated.
One particularly important mutation is E6V, where glutamate at position 6 is replaced by valine. This mutation receives a relatively unfavorable model score, indicating reduced sequence likelihood. Biologically, E6V is the well-known mutation responsible for sickle cell disease. The substitution changes a negatively charged hydrophilic residue into a hydrophobic residue, promoting abnormal aggregation of hemoglobin molecules and deformation of red blood cells. The fact that ESM2 assigns a deleterious score to this mutation suggests that the language model successfully captures biologically meaningful constraints directly from protein sequence data.
- Latent Space Analysis
Before that, I would like to inform you that I made changes to the code you provided because I spent a lot of time fixing the errors that occurred while running it.
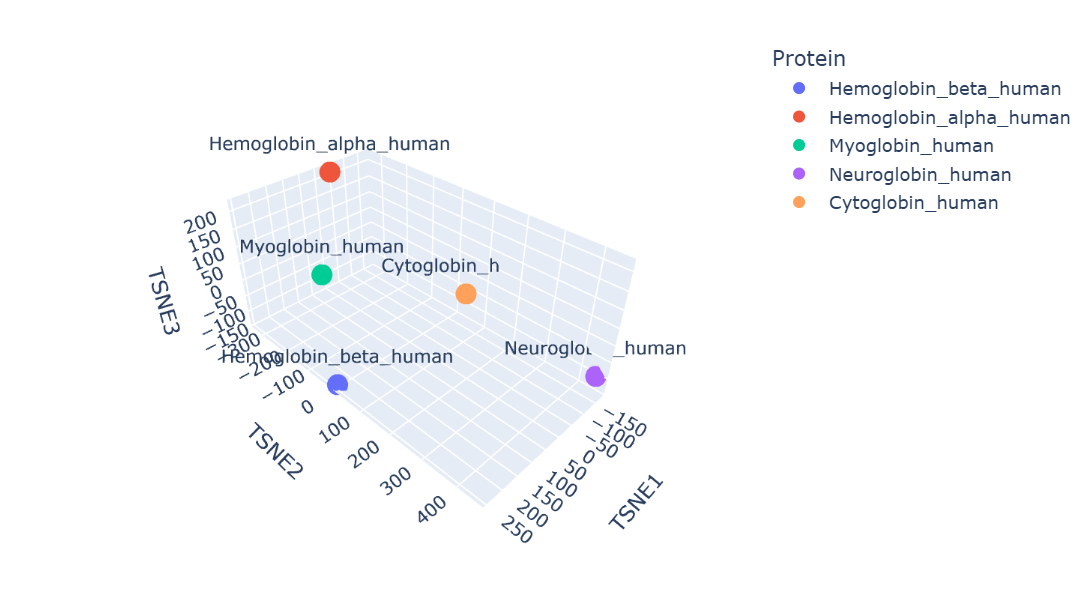
Protein embeddings generated using the ESM2 protein language model were reduced into three dimensions using t-SNE visualization. The resulting latent space demonstrates that proteins with similar sequence and structural characteristics tend to occupy nearby regions.
Human hemoglobin beta is located close to hemoglobin alpha and myoglobin, showing that these proteins have similar globin structures, oxygen-binding functions, and conserved sequence regions. Hemoglobin alpha is the closest because both proteins work together to form the hemoglobin tetramer in red blood cells. Myoglobin is also positioned nearby since it shares the globin domain and binds heme, although its role is mainly for oxygen storage in muscle. Neuroglobin and cytoglobin are placed farther away, indicating that their sequences and biological functions are more specialized and different from hemoglobin beta.
Overall, the latent space map shows that ESM2 can capture meaningful biological relationships between proteins directly from their amino acid sequences.
C2. PROTEIN FOLDING
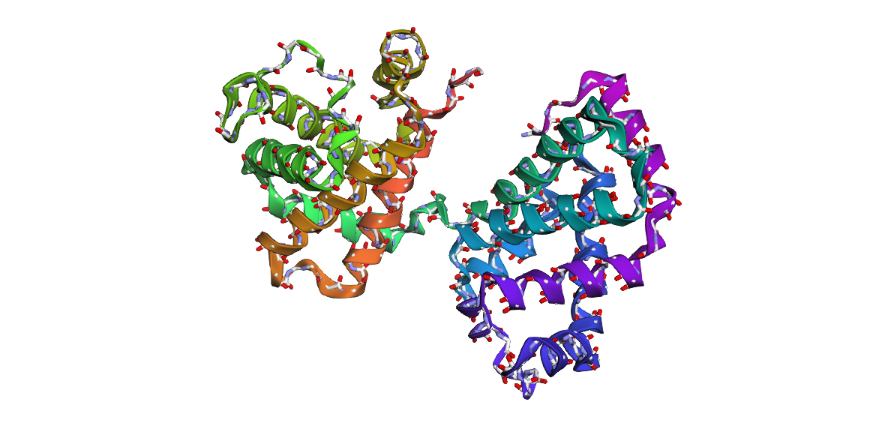
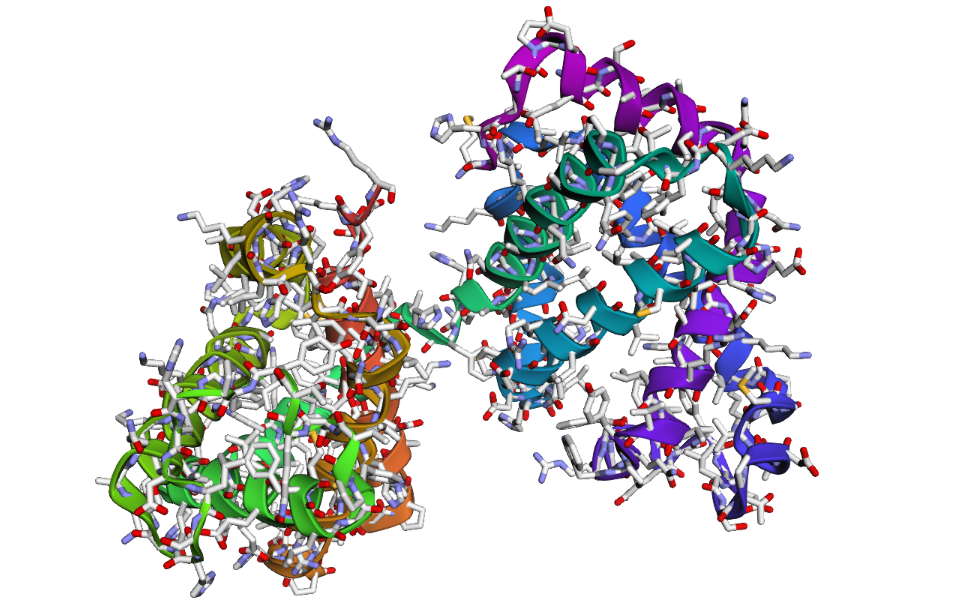
1 . The ESMFold prediction shows a structure that is highly similar to the original 1DXT hemoglobin beta structure. From the superposition images, both models have nearly the same overall globin fold, especially the dominant alpha-helical arrangement that is characteristic of hemoglobin proteins. Most helices overlap well between the experimental structure and the predicted model.
The predicted structure also preserves the general organization of the heme-binding region and the compact globin architecture. Small differences can be seen in several loop regions and flexible terminal parts, which is normal because these regions are usually more dynamic and harder to predict accurately compared to alpha-helices. Overall, the predicted coordinates match the original experimental structure quite well
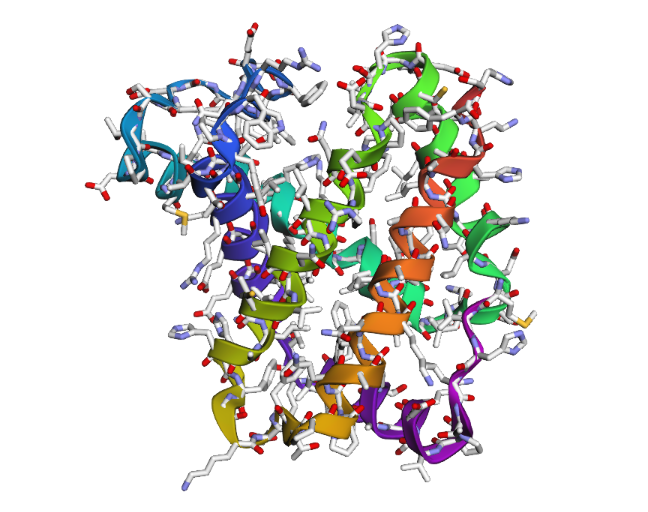
1DXT
1DXT_Mutan E6V
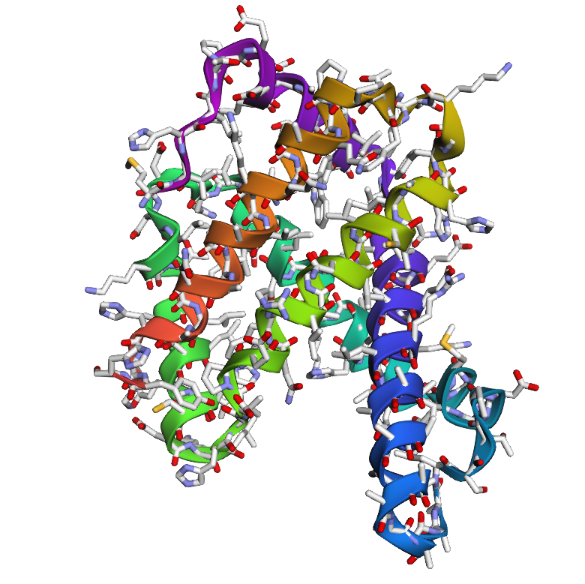
1DXT_Mutan 15-20A
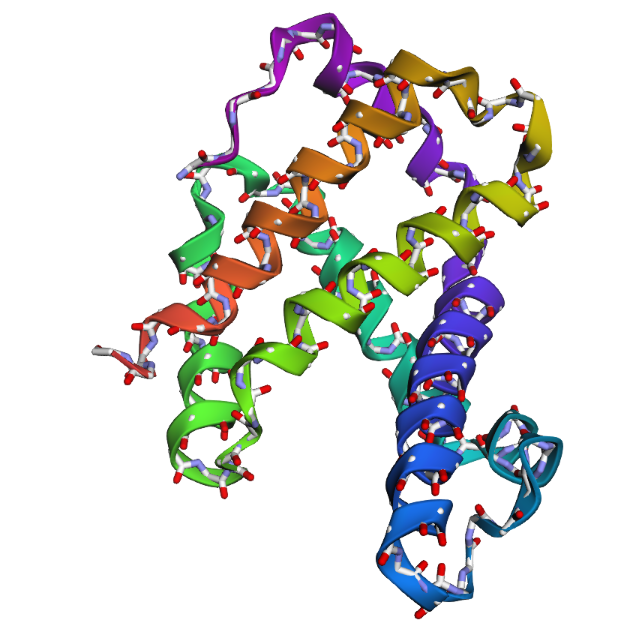
2 . After testing both types of mutations, the hemoglobin beta structure seems fairly resilient to small mutations but much less stable after large sequence changes. In the first mutation (E6V substitution), the overall structure still looks very similar to the original protein. Most of the alpha-helices are still maintained and the globin fold remains intact. The change mainly affects a local region around the mutation site, while the overall protein architecture is preserved. This suggests that hemoglobin beta can tolerate certain single amino acid mutations without major structural disruption, even though the mutation may still affect protein function biologically.
In the second mutation, where a larger segment of residues was replaced with alanine, the structure changes more noticeably. Several helices appear shifted or less organized, and the overall fold looks less compact compared to the original structure. Some regions become more flexible and lose their normal arrangement. This indicates that larger sequence modifications disrupt the stability of the protein much more strongly. Overall, the protein is relatively resilient to small point mutations, but not to large segment mutations. Conserved regions in the sequence are clearly important for maintaining the proper globin structure and stability.
C3. PROTEIN GENERATION
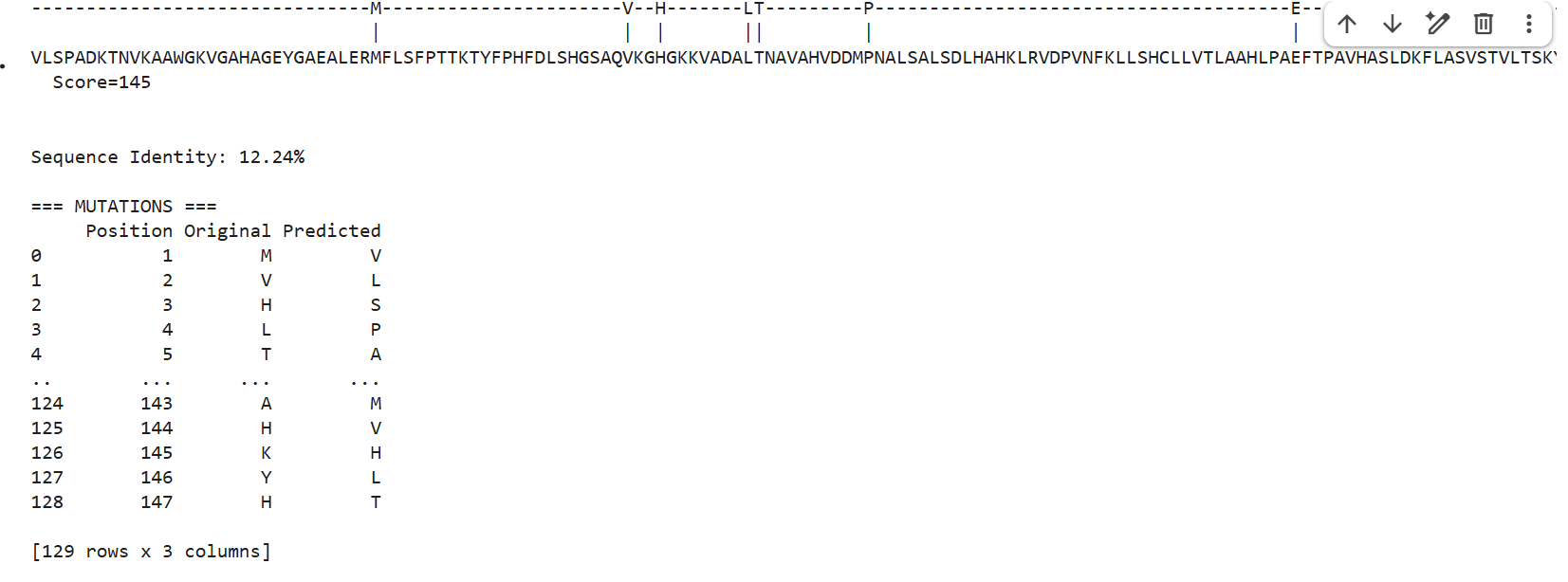
1 . ProteinMPNN generated several alternative sequences for the hemoglobin beta backbone. The predicted sequences showed relatively low sequence identity compared to the native sequence, ranging from approximately 9–12%. Despite these differences, several conserved structural patterns such as hydrophobic residues and globin-like motifs maybe were still maintained.
This suggests that multiple amino acid sequences may be compatible with the same overall globin fold. ProteinMPNN appears to preserve residues important for maintaining secondary structure and backbone stability while allowing substantial variation in less constrained positions.
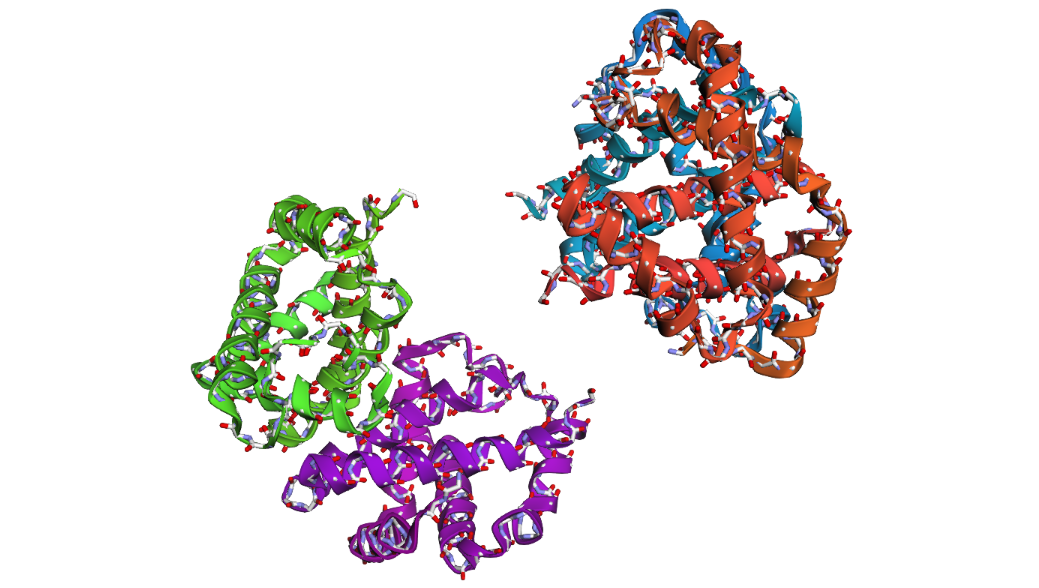
2 . After folding the ProteinMPNN-designed sequence with ESMFold, the predicted structure looked noticeably different from the original hemoglobin beta structure. Several helices were disrupted and the overall globin fold was less organized compared to the native protein. This suggests that although some sequence variation is possible, large changes in the amino acid sequence can strongly affect the stability and shape of the protein structure.