Week 5 HW: Protein Design Part II
PART 1 Generate Binders with PepMLM
The human SOD1 protein sequence was retrieved from the UniProt database (P00441). To model a disease-associated variant, the A4V mutation was introduced by substituting alanine with valine at residue position 4 of the protein sequence. This mutation is known to be associated with amyotrophic lateral sclerosis (ALS). The resulting mutant SOD1 sequence was then used as the input for subsequent peptide binder generation using the PepMLM model.
In bioinformatics, the letter “X” in peptide sequences denotes an unknown or ambiguous amino acid, often arising from prediction model uncertainties during protein structure analysis. For instance, sequences like WRYYAYAIRWKX, HLVPAVAIEHKX, and WRYPAAAARLKX feature an unclear final residue, which disrupts processing in tools like AlphaFold3 as they do not support non-standard characters. To address this, X is typically replaced with the most contextually reasonable residue, such as K (lysine). This substitution is favored because lysine enhances electrostatic binding—via its positive charge that attracts negatively charged molecules—and improves overall peptide solubility, ensuring greater stability and compatibility for downstream simulations.
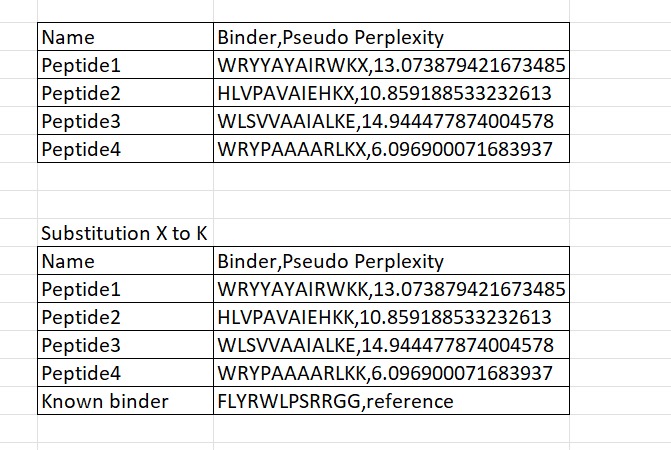
Four peptides of length 12 amino acids were generated using the PepMLM model conditioned on the mutant SOD1 A4V sequence. The generated peptides showed pseudo-perplexity scores ranging from 6.10 to 14.94, reflecting varying levels of model confidence. Among the generated candidates, peptide WRYPAAAARLKK displayed the lowest perplexity score (6.10), suggesting the highest likelihood of being a plausible binder according to the model. For comparison, the previously reported SOD1-binding peptide FLYRWLPSRRGG was included as a reference. These peptides were then used for further structural evaluation using AlphaFold3.
Part 2 Evaluate Binders with AlphaFold3
Peptide 1 : WRYYAYAIRWKK
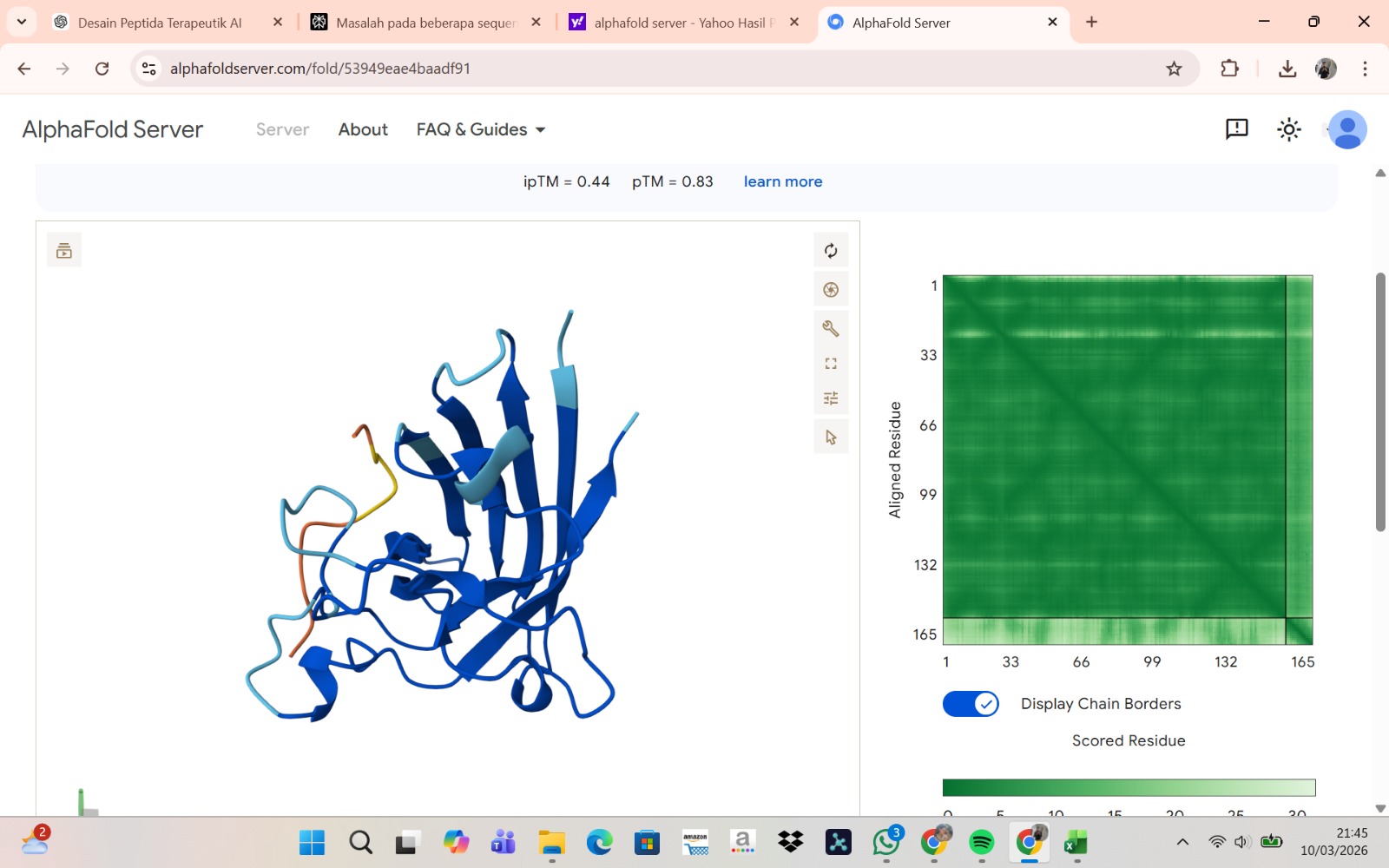
Peptide 2 : HLVPAVAIEHKK
Peptide 3 : WLSVVAAIALKE
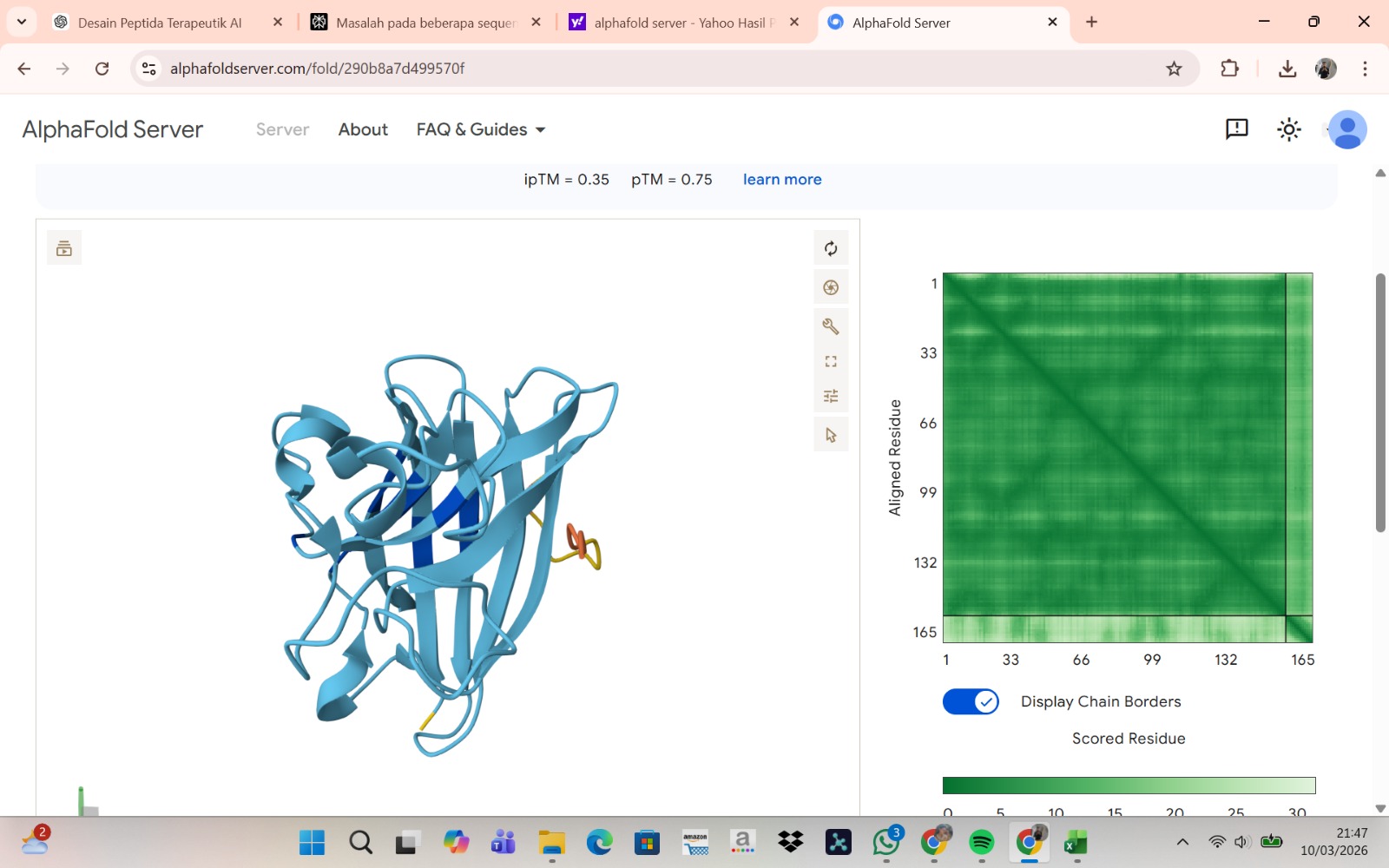
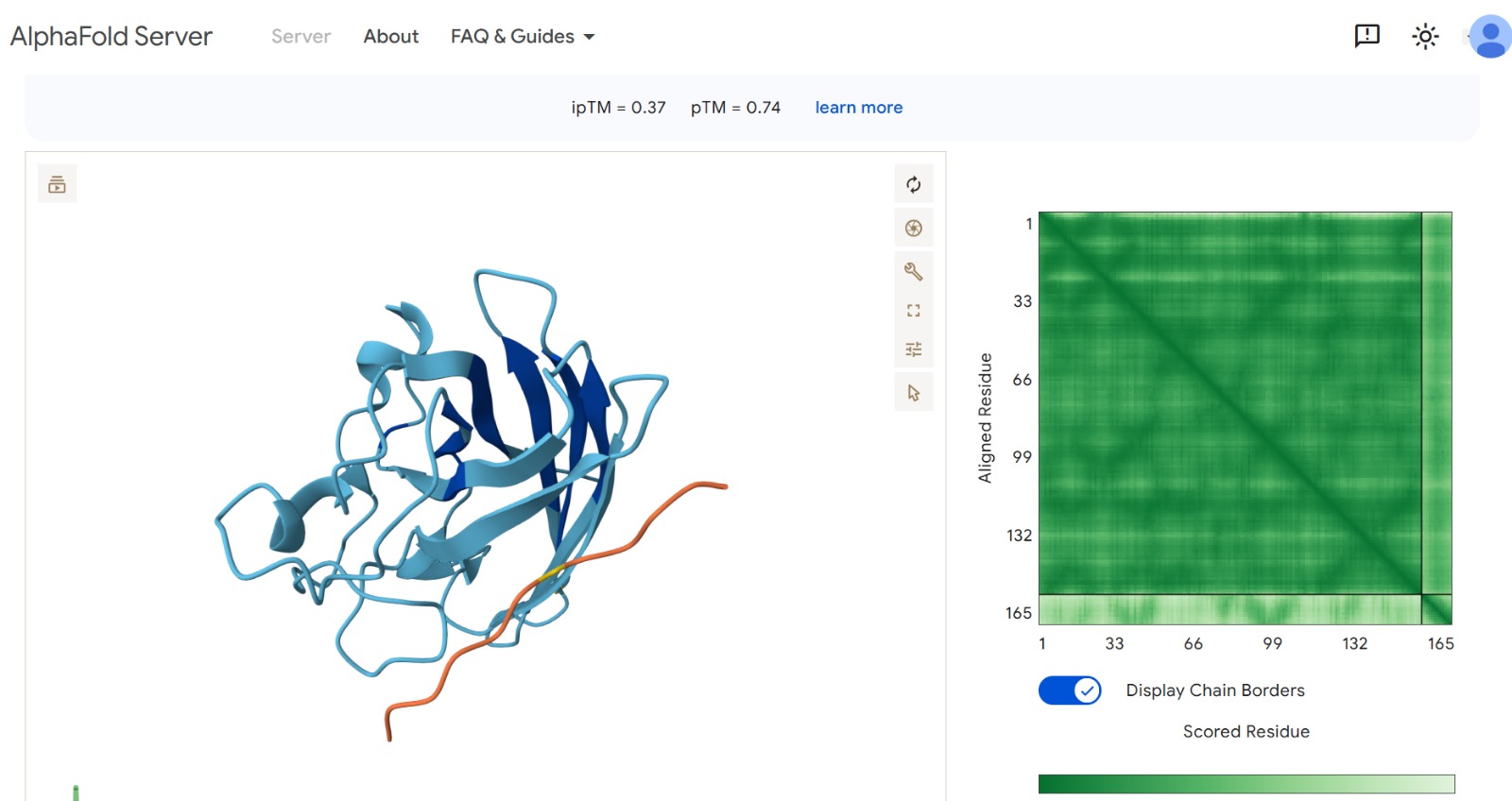
Peptide 4 : WRYPAAAARLKK
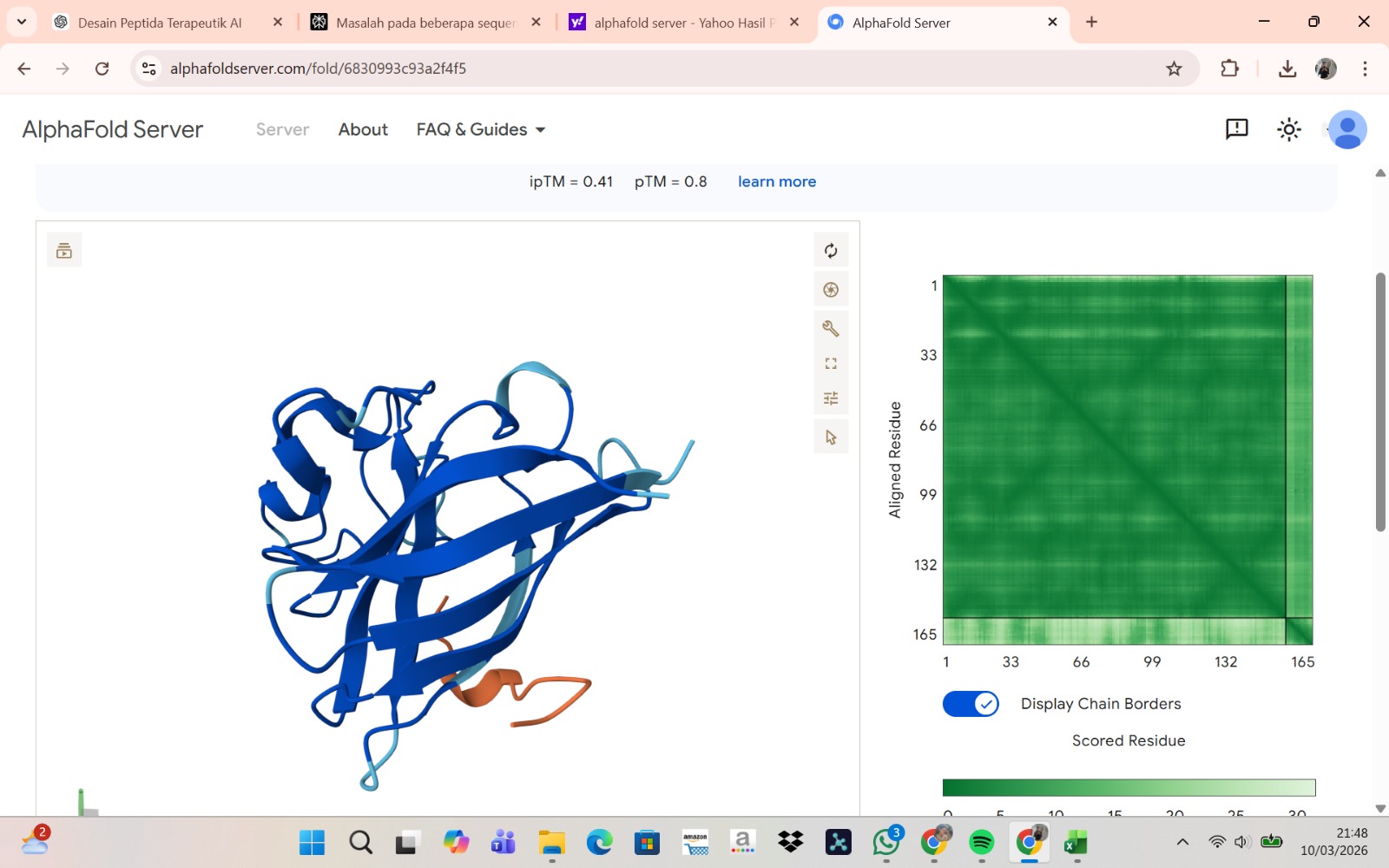
The peptide–protein complexes were modeled using AlphaFold3 by submitting the mutant SOD1 sequence as chain A and each peptide sequence as chain B. The predicted interface scores (ipTM) ranged from 0.35 to 0.44, indicating relatively weak but detectable interactions between the peptides and the SOD1 protein. Among the generated candidates, the peptide HLVPAVAIEHKK showed the highest ipTM value (0.44) and the highest pTM score (0.83), suggesting a more stable predicted complex compared to the other peptides. The remaining peptides displayed slightly lower ipTM scores, indicating weaker predicted interactions. Based on the predicted structures, the peptides appear to bind primarily to the surface of the SOD1 protein rather than being deeply buried within the structure. The interactions likely occur near exposed regions of the β-barrel or close to the N-terminal region where the A4V mutation is located. Although the binding confidence is modest, these results suggest that some of the PepMLM-generated peptides may interact with the mutant SOD1 surface and could serve as starting points for further optimization.
Part 3 Evaluate Properties with PeptiVerse
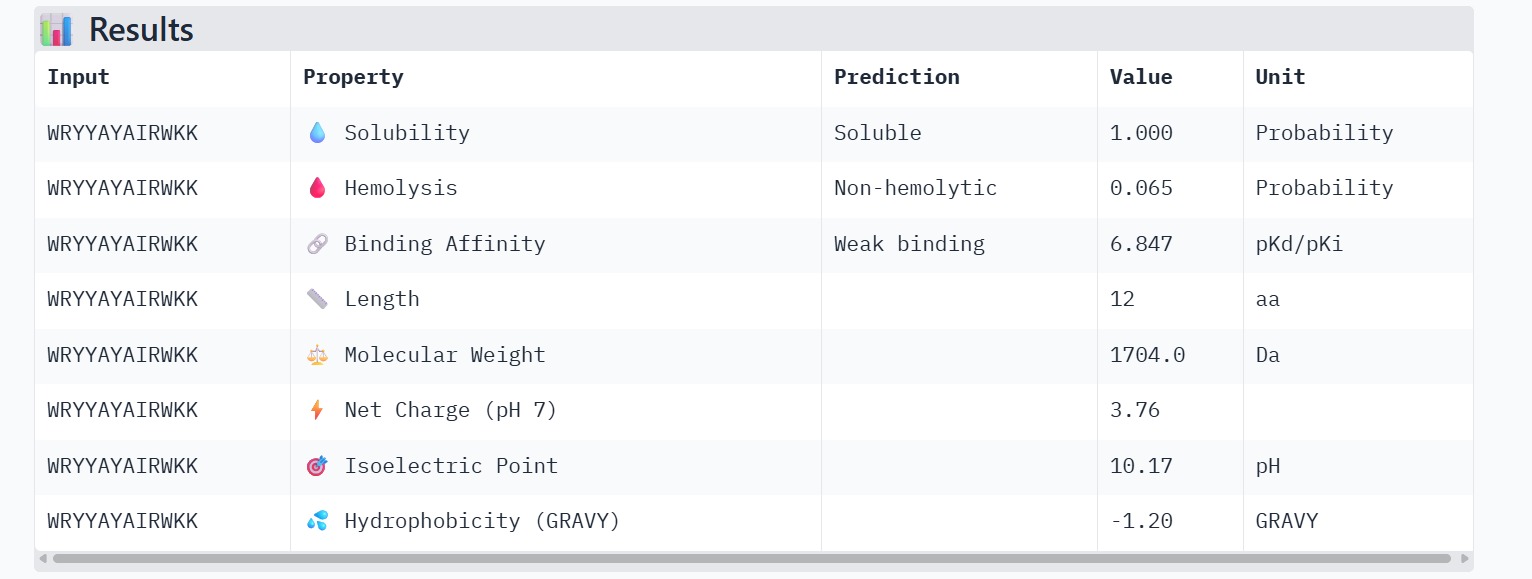
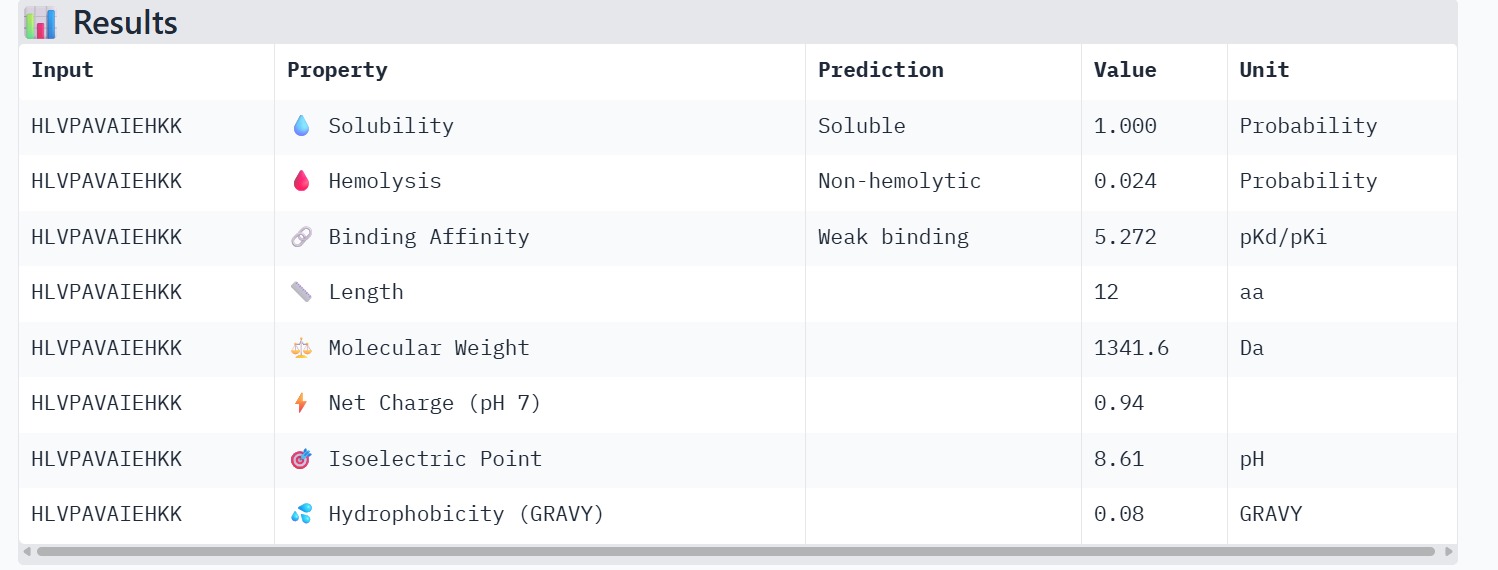
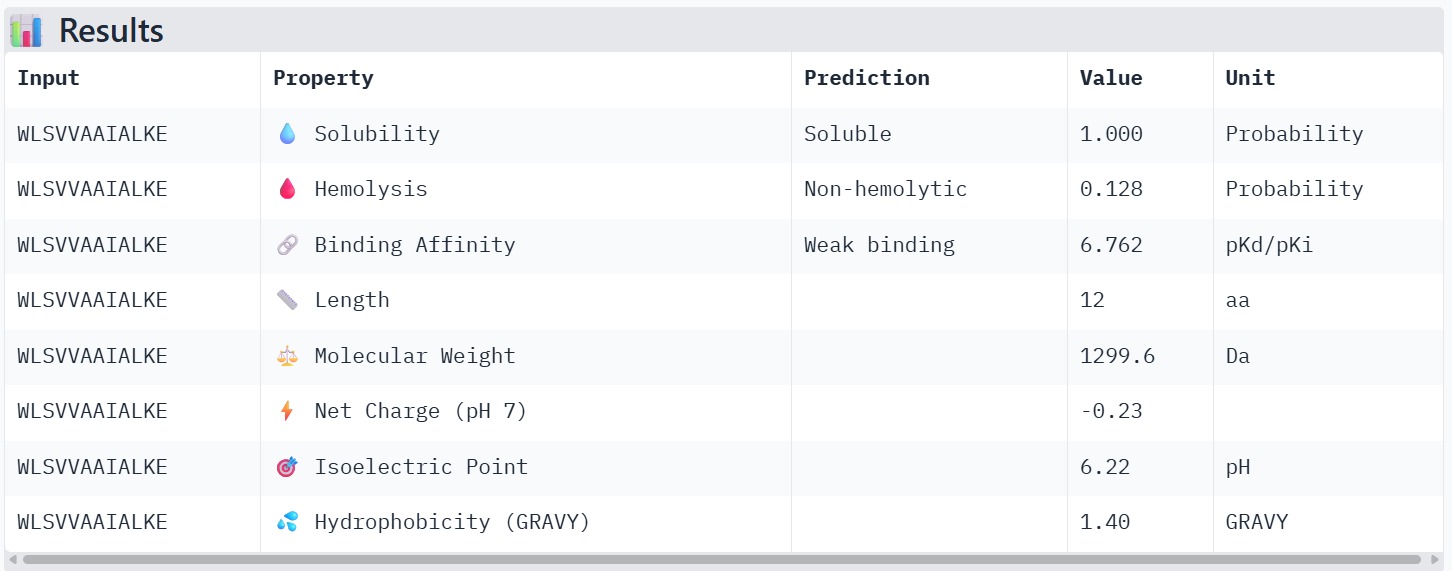
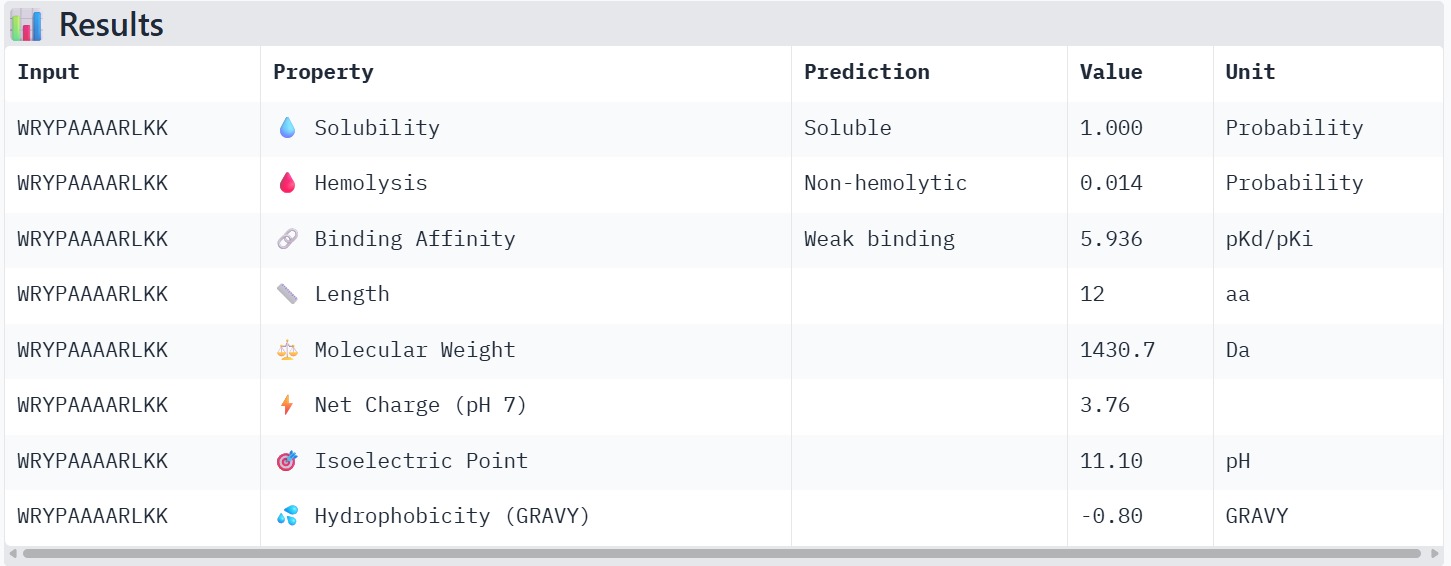
The therapeutic properties of the generated peptides were evaluated using PeptiVerse. All four peptides were predicted to be highly soluble with a probability of 1.000, indicating favorable physicochemical characteristics for biological applications. Hemolysis prediction suggested that all peptides are non-hemolytic, with WRYPAAAARLKK showing the lowest hemolysis probability (0.014). Binding affinity predictions ranged from pKd/pKi values of 5.27 to 6.85. Among the candidates, WRYYAYAIRWKK displayed the highest predicted binding affinity (6.847), suggesting stronger interaction with the A4V mutant SOD1 target. This peptide also exhibits a positive net charge (+3.76), which may enhance electrostatic interactions with the protein surface. Hydrophobicity analysis indicates that WRYYAYAIRWKK is moderately hydrophilic (GRAVY = −1.20), which supports solubility and structural stability. Overall, WRYYAYAIRWKK appears to provide the best balance between predicted binding affinity, solubility, and safety properties, making it the most promising candidate for further structural and experimental validation.
Part 4: Generate Optimized Peptides with moPPIt
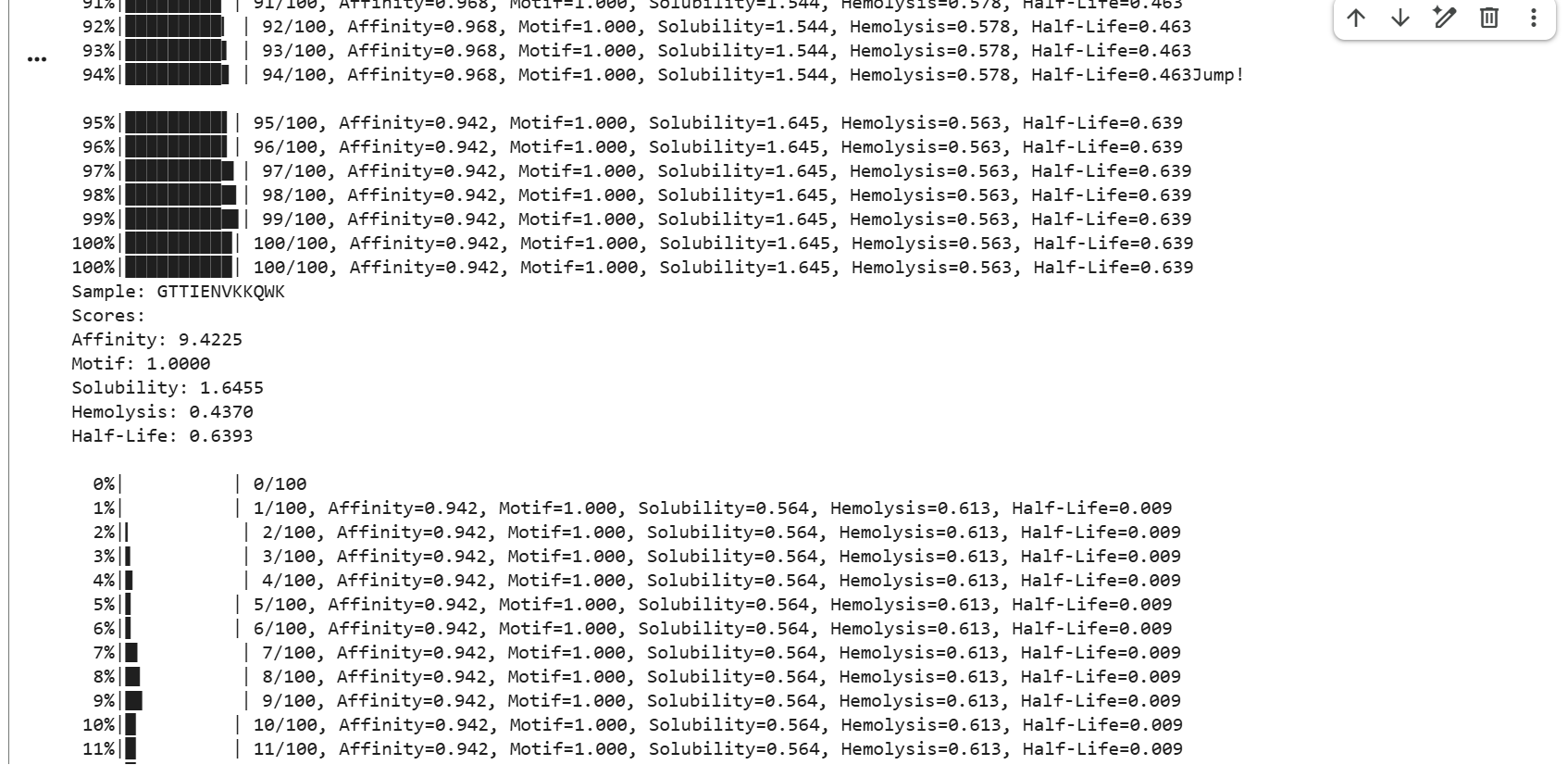
GTTIENVKKQWK showed strong affinity with a score of 9.4225, a perfect motif score of 1.0000, moderate solubility of 1.6455, hemolysis of 0.4370, and a half-life score of 0.6393.
ALWKWYRATAWQ showed strong affinity with a score of 9.4688, a perfect motif score of 1.0000, good solubility of 2.1135, low hemolysis of 0.3449, and a half-life score of 0.0134.
PSAAEWVEWLFK showed strong affinity with a score of 9.6628, a perfect motif score of 1.0000, good solubility of 2.0993, low hemolysis of 0.3619, and a half-life score of 0.0184.
LLAKIANPTQWK showed strong affinity with a score of 9.7345, a perfect motif score of 1.0000, moderate solubility of 1.5554, hemolysis of 0.4262, and a half-life score of 0.1850.
AWKPTALEFNWV showed strong affinity with a score of 9.5225, a perfect motif score of 1.0000, good solubility of 1.8301, hemolysis of 0.3823, and a half-life score of 0.0755.
ATETRFLPPWLW showed strong affinity with a score of 9.5070, a perfect motif score of 1.0000, moderate solubility of 1.4245, hemolysis of 0.3597, and a half-life score of 0.0264.
APTPEYEALFRF showed the highest affinity among these samples with a score of 9.8719, a perfect motif score of 1.0000, low solubility of 1.2404, hemolysis of 0.3469, and a half-life score of 0.0897.
TAKQFWDGWKWG showed strong affinity with a score of 9.6989, a perfect motif score of 1.0000, good solubility of 2.0194, hemolysis of 0.3731, and a half-life score of 0.2847.
NWKFAAWIHRPT showed strong affinity with a score of 9.6552, a near-perfect motif score of 0.9971, moderate solubility of 1.6367, hemolysis of 0.4038, and a very low half-life score of 0.0069.
FAGMFPLDAPTL showed the highest affinity in the set with a score of 9.8837, a near-perfect motif score of 0.9972, moderate solubility of 1.4887, hemolysis of 0.4193, and a half-life score of 0.0159.
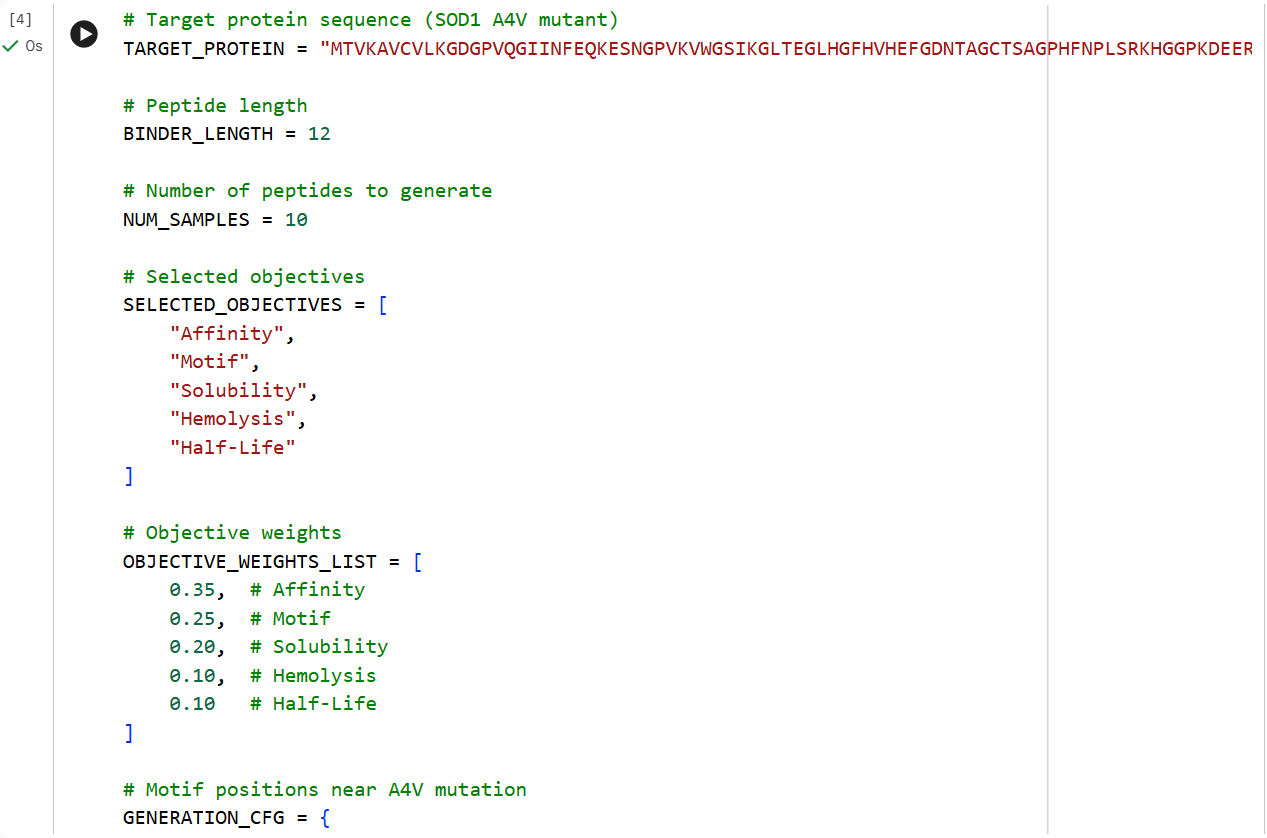
The A4V mutant SOD1 sequence was used as the target protein for controlled peptide generation using moPPIt. Residues near the N-terminal region (positions 1–10), where the ALS-associated A4V mutation is located, were selected as the target binding region. Peptide length was set to 12 amino acids, and multiple optimization objectives including affinity, motif guidance, solubility, hemolysis, and half-life were enabled.
The generated peptides showed consistently high predicted affinity scores (9.42–9.88), indicating strong predicted interactions with mutant SOD1. Most peptides also achieved perfect motif scores, demonstrating successful targeting of the selected residue region near the mutation site. In addition, several peptides displayed favorable solubility and relatively low hemolysis probabilities, suggesting improved therapeutic properties. Among the generated candidates, GTTIENVKKQWK showed one of the best overall balances between affinity, solubility, safety, and predicted biological stability due to its relatively high half-life score.
Compared to the PepMLM-generated peptides, the moPPIt peptides were more optimized and therapeutically oriented. PepMLM primarily generated plausible binders based on sequence patterns, whereas moPPIt optimized multiple objectives simultaneously using guided generation. As a result, the moPPIt peptides displayed improved balance between binding affinity and drug-like properties such as solubility, reduced hemolysis, and stability. The peptide sequences generated by moPPIt therefore differed substantially from the PepMLM outputs, which is expected and reflects the different underlying design strategies of the two models.
Before advancing these peptides toward clinical studies, additional computational and experimental validation would be required. Molecular docking and molecular dynamics simulations should be performed to evaluate binding stability and interaction specificity. Experimental assays such as surface plasmon resonance or isothermal titration calorimetry would then be necessary to confirm binding affinity. Furthermore, cytotoxicity, hemolysis, serum stability, and cellular uptake assays would be required to assess safety and pharmacological potential before proceeding to animal studies and eventual clinical development.