Week 10 HW: Advanced Imaging & Measurement Technology
Waters Part I — Molecular Weight
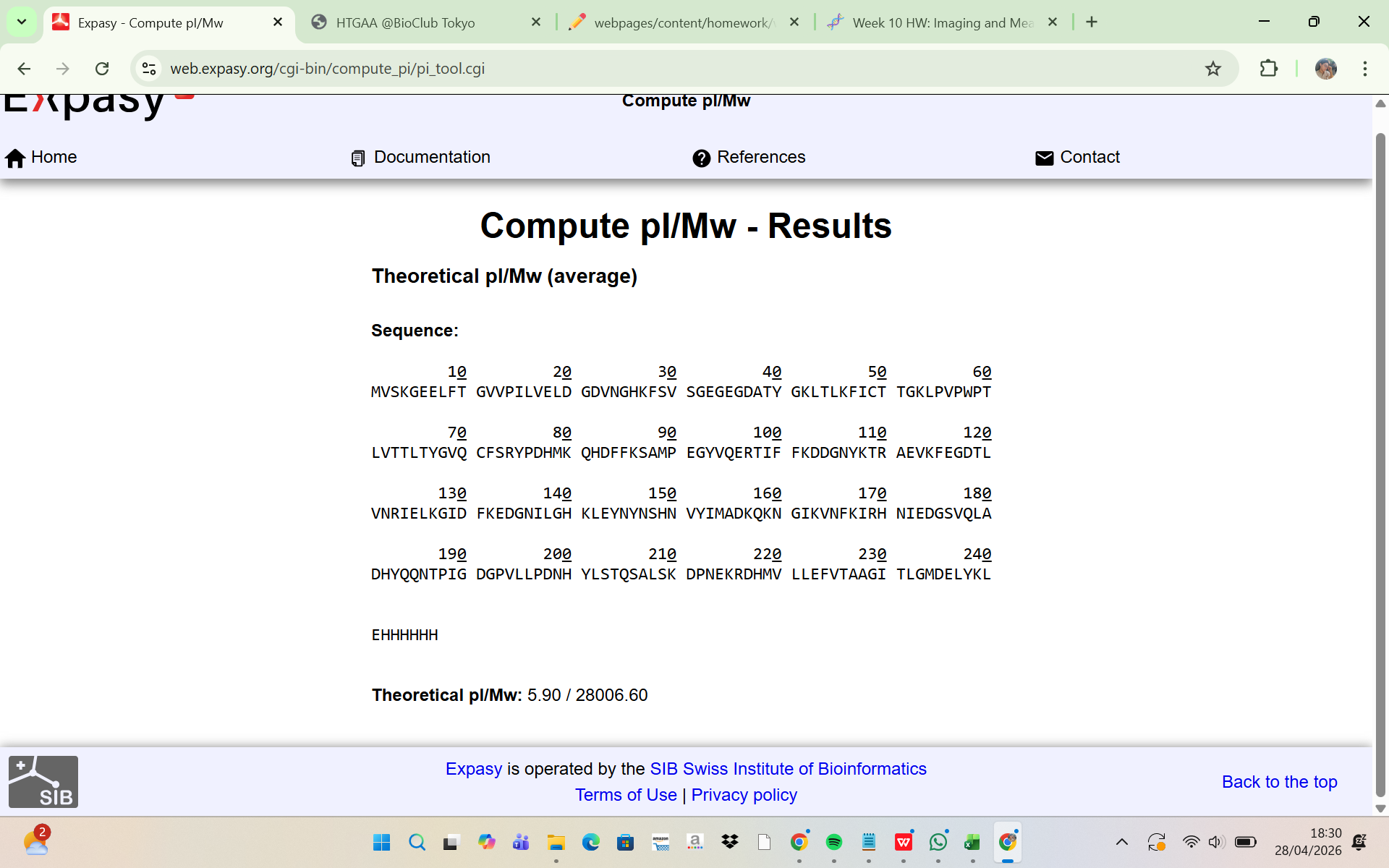
The amino acid sequence of the His-tagged eGFP was analyzed using the ExPASy Compute pI/Mw tool. The results showed:
Theoretical molecular weight (Mw) = 28,006.60 Da Theoretical pI = 5.90
This indicates that the predicted molecular mass of the intact eGFP protein, including the linker and His-tag, is approximately: 28.01 kDa This theoretical molecular weight will be used as the reference value for comparison with the experimentally determined mass from LC-MS analysis.
Two adjacent peaks were selected from the LC-MS spectrum:
m/z_n = 903.7148
m/z_n+1 = 933.8044
The charge state was determined using:
z = (m/z_n+1) / ((m/z_n) - (m/z_n+1))
Substituting the observed values:
z = 933.8044 / (933.8044 - 903.7148) = 31.03
Thus: z = 31
The experimental molecular weight was then calculated:
MW = z × (m/z)
MW = 31 × 903.7148 = 28,015.16 Da
Experimental MW = 28,015.16 Da
The measurement accuracy was determined by comparing the experimental molecular weight with the theoretical molecular weight:
Accuracy = |MW_experiment - MW_theory| / MW_theory
Accuracy = |28015.16 - 28006.60| / 28006.60 = 0.000306
Percent Error = 0.0306%
This very small error indicates that the experimentally measured mass is highly consistent with the theoretical mass of the His-tagged eGFP.
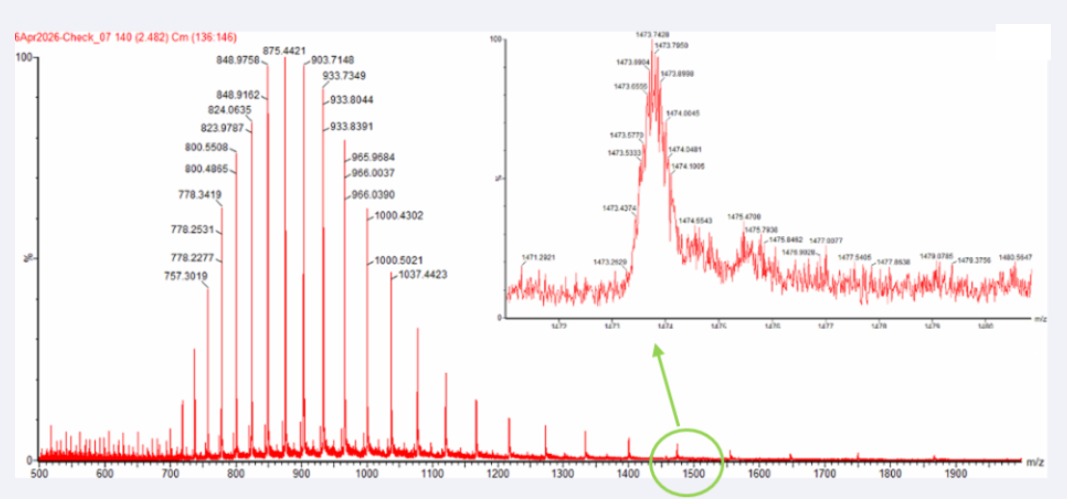
Yes, the charge state can be observed from the zoomed-in isotopic peak pattern.
The spacing between adjacent isotopic peaks is approximately:
Δ(m/z) ≈ 0.05
Using the relationship:
z = 1 / Δ(m/z)
The charge state is:
z = 1 / 0.05 = 20
Therefore: The zoomed-in peak has an approximate charge state of +20
This is consistent with the denatured intact eGFP ion observed in LC-MS, where the unfolded protein carries multiple charges.
Waters Part III — Peptide Mapping - primary structure
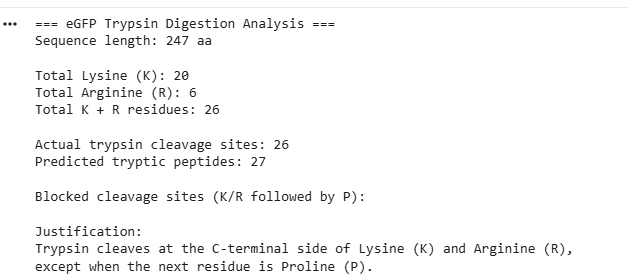
- The eGFP amino acid sequence was analyzed computationally to determine the number of potential trypsin cleavage sites. The analysis showed that the protein contains 20 lysine (K) residues and 6 arginine (R) residues, giving a total of 26 residues that can serve as cleavage sites for trypsin. Since trypsin specifically cleaves peptide bonds at the C-terminal side of lysine and arginine residues, each of these residues represents a potential digestion site unless the following residue is proline. In this sequence, no lysine or arginine residues were followed by proline, meaning that all 26 cleavage sites were available for digestion. Therefore, the predicted number of peptides generated after complete tryptic digestion was 27 peptides, calculated as the number of cleavage sites plus one. This theoretical number represents the maximum number of peptide fragments expected under ideal digestion conditions. However, the number of peptides detected experimentally by LC-MS may be lower because some peptides may be too small, may ionize poorly, or may not be detected under the selected analytical conditions.
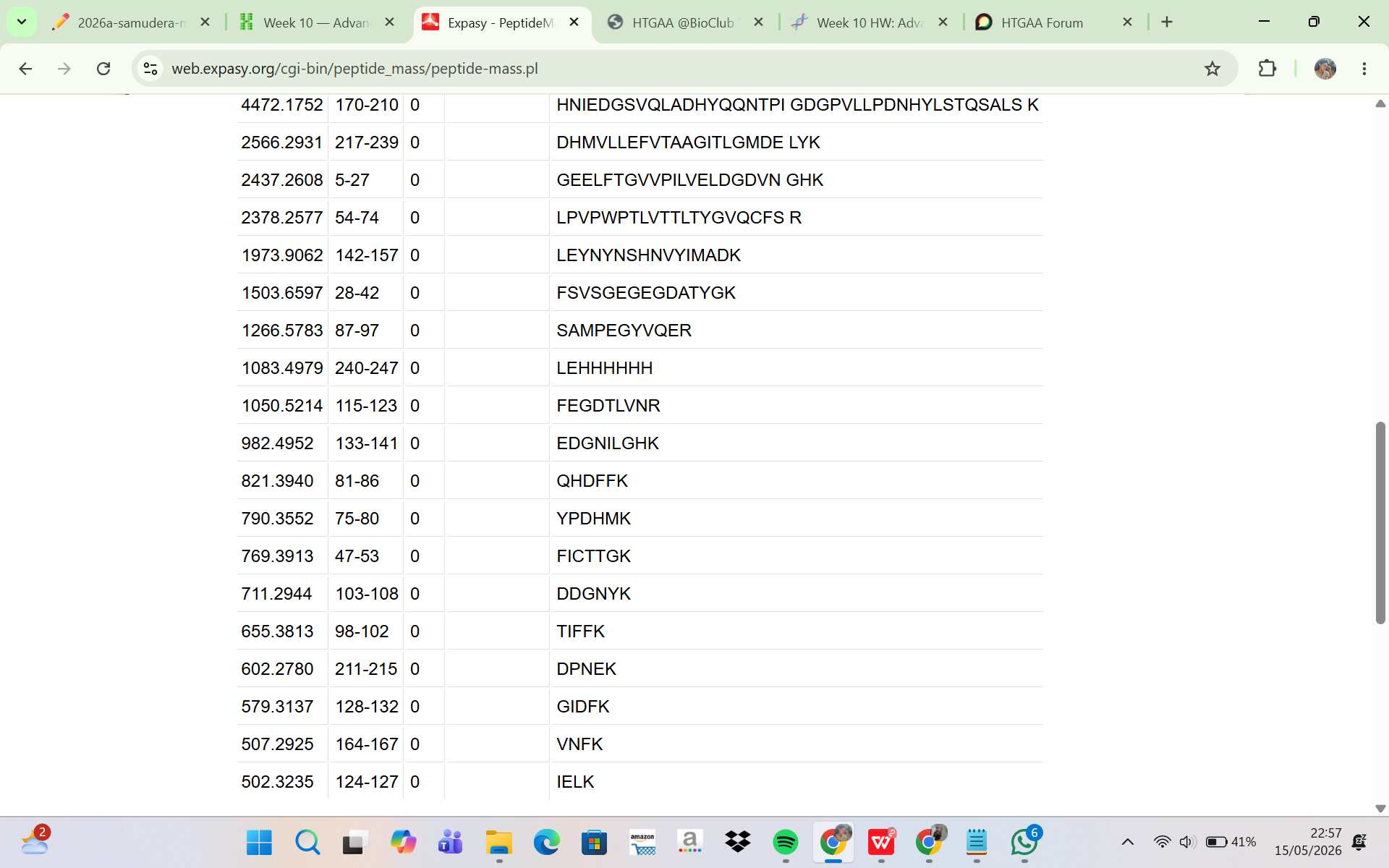
The eGFP sequence was analyzed using the ExPASy PeptideMass tool to predict the peptides generated after tryptic digestion. The analysis parameters included trypsin as the selected enzyme, zero missed cleavages, reduced cysteines, no methionine oxidation, and peptide masses greater than 500 Da. Based on these parameters, the digestion generated a total of 19 predicted peptides. Although complete theoretical digestion of eGFP would produce more peptide fragments, only peptides with masses above 500 Da were included in the output. This filtering step excludes very small peptides that are unlikely to be detected efficiently during LC-MS analysis. Therefore, the ExPASy peptide prediction represents the subset of peptides that are most relevant for experimental peptide mapping by LC-MS.
The LC-MS total ion chromatogram (TIC) shown in Figure 5a was analyzed to determine the number of chromatographic peaks present between 0.5 and 6 minutes. Only peaks with relative abundances greater than 10% were counted to avoid including noise or very minor signals. Based on visual inspection of the chromatogram, approximately 16 significant chromatographic peaks were observed within the selected retention time range. These peaks represent peptide ions generated from the tryptic digestion of eGFP and separated during liquid chromatography prior to mass spectrometric detection. The distribution of peaks across the chromatogram indicates the presence of multiple peptides with different hydrophobicities and retention behaviors.
Comparison of the theoretical peptide prediction with the experimental LC-MS chromatogram showed that the number of observed chromatographic peaks was lower than the number of peptides predicted computationally. ExPASy predicted 19 peptides, whereas the TIC chromatogram showed only approximately 16 major chromatographic peaks. This discrepancy is expected in peptide mapping experiments because not all peptides are detected equally during LC-MS analysis. Some peptides may have very low ionization efficiency, low abundance, or overlapping retention times that cause co-elution with other peptides. In addition, certain peptides may produce signals below the detection threshold of the instrument. Therefore, the experimentally observed chromatographic peaks do not necessarily correspond one-to-one with the total number of predicted tryptic peptides.
The mass spectrum shown in Figure 5b corresponds to the chromatographic peak at 2.78 minutes from the peptide map. The dominant precursor ion observed in the spectrum had an m/z value of approximately 525.767. To determine the charge state of this peptide ion, the isotopic peak spacing in the zoomed inset spectrum was analyzed. The difference between adjacent isotope peaks was approximately: 526.25918 − 525.76712 ≈ 0.492.
In electrospray ionization mass spectrometry, isotope spacing follows the relationship: Δ(m/z) = 1 / z. Thus, the charge state was calculated as: z ≈ 1 / 0.492 ≈ 2. This indicates that the precursor peptide ion carried a +2 charge state. The singly protonated peptide mass ([M+H]+) was then calculated using the relationship between m/z and charge: [M+H]+ = z(m/z) − (z − 1)(1.0073). Substituting the experimental values: [M+H]+ = 2(525.76712) − 1.0073. Resulting in: [M+H]+ ≈ 1050.53 Da. This calculated mass closely matched the theoretical monoisotopic mass predicted by ExPASy for the peptide FEGDTLVNR, which had a predicted mass of 1050.5214 Da.
Furthermore, Figure 5c provided fragmentation (MS/MS) data for the precursor ion at m/z 525.767. Several fragment ions were observed in the fragmentation spectrum, including peaks near m/z 774.41, 903.44, and 1050.52. These fragment ions are consistent with peptide backbone fragmentation patterns typically observed during collision-induced dissociation (CID). The fragmentation spectrum therefore supports the identification of the peptide sequence and confirms that the precursor ion detected at m/z 525.767 corresponds to the tryptic peptide FEGDTLVNR. The close agreement between theoretical peptide mass, precursor ion mass, isotope spacing, and fragmentation pattern demonstrates the high accuracy of the LC-MS/MS peptide mapping analysis.
- The peptide eluting at 2.78 min was identified by comparing the experimentally measured peptide mass to theoretical masses generated with the ExPASy PeptideMass tool. The experimentally determined singly protonated mass was: Experimental mass ([M+H]+) = 1050.53 Da
This matched the tryptic peptide FEGDTLVNR, which has a theoretical monoisotopic mass of Theoretical mass = 1050.5214 Da
The mass error in parts per million (ppm) was calculated as:
Mass error (ppm) = (|MW_experiment − MW_theory| / MW_theory) × 10^6
Substituting the values: Mass error (ppm) = (|1050.53 − 1050.5214| / 1050.5214) × 10^6
Difference between masses:
|1050.53 − 1050.5214| = 0.0086
Therefore: Mass error (ppm) ≈ 8.2 ppm
A mass error of about 8.2 ppm shows excellent agreement between the measured and theoretical masses, supporting the confident identification of the peptide FEGDTLVNR and demonstrating the high mass accuracy of the LC-MS/MS analysis.
- Based on Figure 6, the peptide mapping analysis confirmed approximately 88% sequence coverage of the eGFP protein. This means that most regions of the protein sequence were successfully identified through the detected tryptic peptides during LC-MS/MS analysis, while a small portion of the sequence was not detected or not confidently assigned.
Bonus Peptide Map Questions
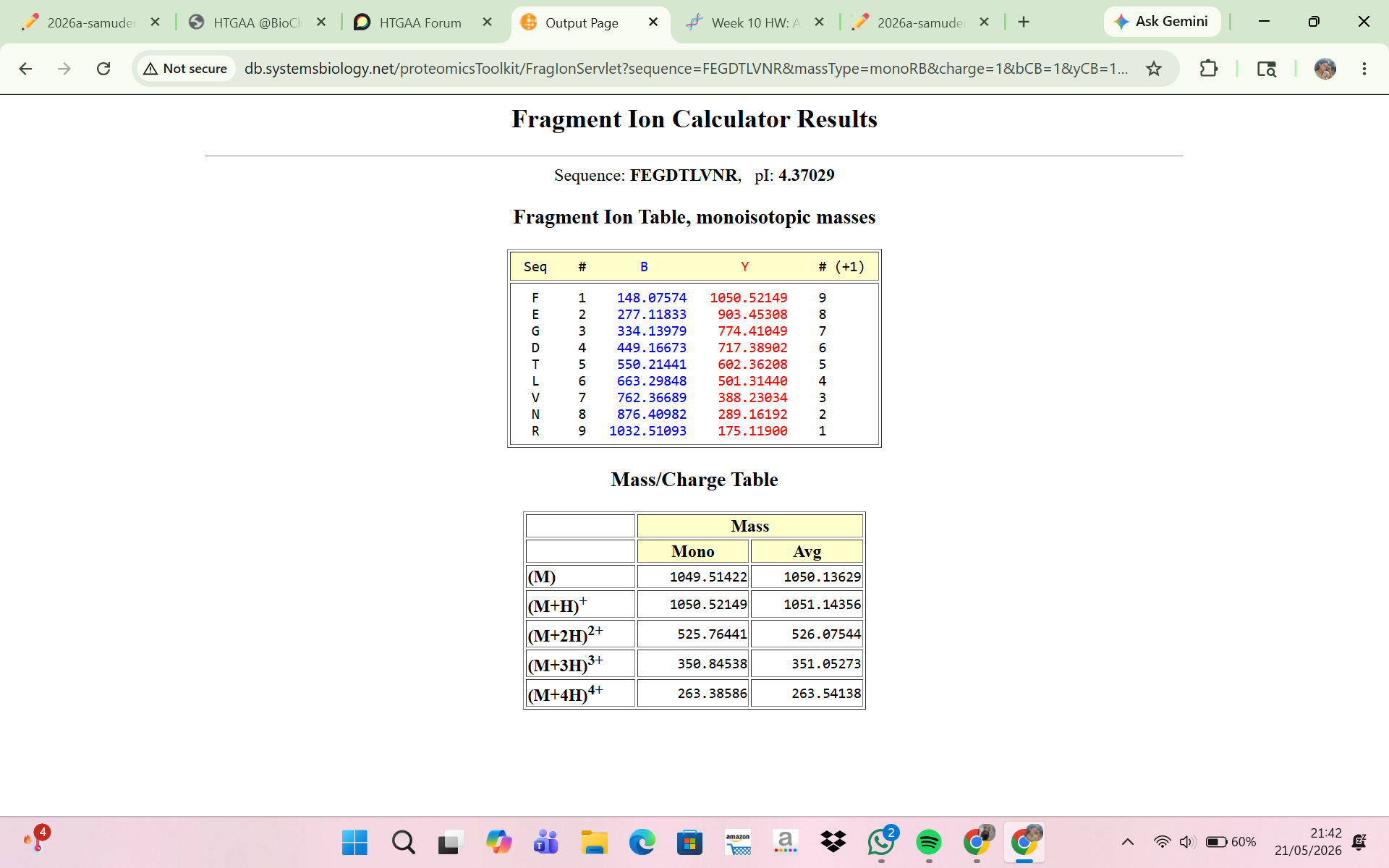
8 . Interpretation of the Fragment Ion Calculator Result
The Fragment Ion Calculator showed that the peptide FEGDTLVNR produced theoretical fragment ions that closely matched the experimental MS/MS spectrum in Figure 5c. Calculated peptide masses were:
[M+H]+ = 1050.52149 Da
[M+2H]2+ = 525.76441 m/z
These values agree with the experimentally detected precursor:
m/z ≈ 525.767
calculated peptide mass ≈ 1050.53 Da
The fragmentation table predicted several y-ions and b-ions that matched peaks in Figure 5c. Examples:
Experimental peak 903.44 — predicted ion y8
Experimental peak 774.41 — predicted ion y7
Experimental peak 602.35 — predicted ion y5
Experimental peak 501.31 — predicted ion y4
Experimental peak 388.22 — predicted ion y3
The close agreement between experimental fragments and predicted ions confirms that the peptide at 2.78 min is FEGDTLVNR. Thus, the MS/MS fragmentation data validate the peptide identity inferred from the peptide mass.
9 . Interpretation of the Peptide Map Data
The peptide mapping results strongly indicate the sample is the expected eGFP standard. LC-MS/MS produced peptide masses that closely matched theoretical tryptic peptides from the eGFP sequence, and MS/MS fragmentation confirmed specific peptides (notably FEGDTLVNR) by matching precursor masses and fragment patterns. Figure 6 shows approximately 88% sequence coverage, indicating most regions of eGFP were identified. The high sequence coverage, accurate peptide masses, and matching fragmentation patterns together demonstrate that the analyzed sample corresponds to the eGFP protein standard.
Waters Part IV — Oligomers
Using the known masses of the KLH subunits, the oligomeric species observed in the CDMS spectrum were assigned by calculating their expected molecular masses and comparing them with the peaks in Figure 7.
The 7FU subunit mass is 340 kDa (0.34 MDa). The mass of the 7FU decamer is:
10 × 0.34 = 3.4 MDa
This matches the peak observed around 3.4–4.0 MDa.
The 8FU subunit mass is 400 kDa (0.40 MDa). The expected mass of the 8FU didecamer is:
20 × 0.40 = 8.0 MDa
This corresponds to the major peak observed at approximately 8.33 MDa.
For the 8FU tridecamer:
30 × 0.40 = 12.0 MDa
This matches the peak near 12.67 MDa.
The expected mass of the 8FU tetradecamer is:
40 × 0.40 = 16.0 MDa
This corresponds to the broader low-intensity signal observed around 16–17 MDa.
Waters Part V — Did I make GFP?
| Theoretical | Observed/measured on the Intact LC-MS | PPM Mass Error | |
|---|---|---|---|
| Molecular weight (kDa) | 28.01 kDa | 28.00 kDa | 236 ppm |