Subsections of Homework
Week 1 HW: Principles and Practices
1. Bioengineering Application / Product to Be Developed
Bioengineering Product
A bioplastic derived from organic waste that is composed of 100% biopolymers, food-grade, and naturally biodegradable.
This product is designed as an alternative to fossil-based polymer plastics that are widely used today, particularly for single-use packaging applications.
Key Product Specifications
• Main raw materials
Sourced from underutilized organic waste, such as:
o cassava peels
o sugarcane bagasse
o coconut coir
o other organic biomass rich in starch, cellulose, or lignin
• Material composition
o The matrix, plasticizer, and filler are entirely derived from natural biopolymers
o No fossil-based synthetic additives are used
• Product characteristics
o Mechanical and physical properties comparable to conventional plastics
o Safe for human health and compliant with food-grade standards
o Fully bio-based
o Easily biodegradable without leaving harmful residues
Rationale for Product Development
• Single-use plastic remains one of the most significant environmental challenges, and effective solutions to reduce its use are still limited.
• Indonesia has abundant organic waste and rich biodiversity that have not been optimally utilized as sustainable raw materials.
• The development of this bioplastic is expected to:
o reduce dependence on fossil-based plastics
o increase the value of organic waste
o promote a circular economy based on local resources.
2. Governance Goals
Primary Governance Goal
To ensure that this bioplastic serves as a viable alternative to fossil-based polymer plastics without creating biological, social, or environmental risks, while supporting an ethical and sustainable future.
Sub-Goals
1) Ensuring the Principle of Non-Maleficence
• Ensuring that all bioplastic components:
o are non-toxic
o do not release harmful substances
o safe for use, particularly in food packaging.
• Establishing clear biodegradability standards to ensure complete natural degradation without harmful residues.
2) Ensuring Safety and Security
• Establishing occupational safety standards in the bioplastic production process.
• Requiring transparent labeling related to:
o material composition,
o product properties, and
o post-use disposal methods,
to prevent misuse.
3) Ensuring Environmental and Ecosystem Sustainability
• Ensuring that raw materials (starch, cellulose, lignin) are sourced sustainably without:
o deforestation,
o excessive resource exploitation, or
o ecosystem disruption.
• Limiting agricultural land use for bioplastic production to a maximum of 5% of total national food-producing land.

3. Governance Actions
Governance Action 1 : Mandatory Standards for Bioplastic Production to Prevent Biological Risks
Key Actors
Government regulators, National Agency of Drug and Food Control (BPOM), Ministry of Health, Ministry of Industry.
Objective
To ensure that bioplastics produced and distributed are safe for human health, leave no harmful residues, and are genuinely biodegradable.
Design
• Establishment of national standards covering:
o toxicity testing,
o biological degradation testing,
o post-use residue testing.
• Mandatory laboratory testing and certification prior to market distribution.
• Audits and compliance enforcement by BPOM and the Ministry of Health.
• Mandatory labeling indicating certification status and estimated degradation time.
• Sanctions, ranging from fines to revocation of production licenses, for non-compliance.
Assumptions
• Mandatory standards will encourage the development of safe and fully biodegradable bioplastics.
• Strict oversight will minimize harmful residues.
• Certification costs may increase product prices and pose challenges for small and medium enterprises (SMEs).
Risks of Failure and “Success”
• Risk of failure:
o overly lenient standards may lead to greenwashing
o overly strict standards may hinder innovation and adoption.
• Risk of “success”:
o certified products may be perceived as completely safe, leading to neglect of proper post-use waste management.
Governance Action 2 : Sustainable Bioplastic Adoption and Economic Incentives
Key Actors
Government, Ministry of Finance, SMEs, manufacturing industries, Ministry of Creative Economy, Ministry of Industry, schools, universities, and the public.
Objective
Bioplastics remain relatively unfamiliar to the public. This policy aims to introduce the advantages of bioplastics while encouraging reduced use of single-use plastics. Economic incentives are provided because bioplastic production costs remain higher than fossil-based plastics and to support long-term adoption.
Design
• Development of subsidy programs, tax incentives, or research grants for bioplastic producers.
• Training and skill development programs for SMEs to support innovation in bioplastic-based products.
• Public awareness campaigns and initiatives promoting the downstream transition from single-use plastics to bioplastics.
Assumptions
• Incentives can shift industrial and market behavior.
• Government oversight can prevent misuse of incentives.
• New bioplastic-based innovations will emerge.
• Reduced consumption of single-use plastics will support sustainable adoption.
Risks of Failure and “Success”
• Risk of failure: unequal incentive distribution may widen gaps between large industries and SMEs.
• Risk of “success”: rapid production growth without adequate waste management systems may create new waste-related challenges.
Governance Action 3 : Standards for Environmentally Sustainable Raw Material Use
Key Actors
Government, Ministry of Environment and Forestry (KLHK), agricultural and food experts, manufacturing industries, environmental experts.
Objective
To prevent land exploitation, deforestation, and threats to food security arising from bioplastic raw material sourcing.
Design
• Prioritization of organic waste as raw materials.
• Establishment of maximum limits on the use of primary food crops.
• Prohibition of deforestation for raw material supply.
• Sustainability certification (zero deforestation, 5–10% crop rotation, zero hazardous waste).
• Supply chain traceability systems using blockchain technology to prevent greenwashing.
Assumptions
• Organic waste availability is sufficient and can be consistently supplied at industrial scale.
• Regulatory enforcement and traceability systems function effectively and transparently.
Risk of Failure
• Insufficient organic waste supply or weak enforcement leads to continued land exploitation.
• High compliance costs reduce industry participation, especially among small producers.
Risk of “Success”
• Increased production costs raise bioplastic prices and limit market accessibility.
• Strict standards shift unsustainable practices to informal or unregulated sectors.
4. Government governance assessment
Option 1 : Mandatory production standards that do not pose biological risks
Option 2 : Adoption of sustainable bioplastics and provision of economic incestives
Option 3 : Standards for raw material use that do not harm the environment
| Does the option: | Option 1 | Option 2 | Option 3 |
|---|
| Enhance Biosecurity | | | |
| • By preventing incidents | 1 | 2 | 3 |
| • By helping respond | 1 | 3 | 2 |
| Foster Lab Safety | | | |
| • By preventing incident | 1 | 2 | 3 |
| • By helping respond | 2 | 1 | n/a |
| Protect the environment | | | |
| • By preventing incidents | 2 | 3 | 1 |
| • By helping respond | 3 | 2 | 1 |
| Other considerations | | | |
| • Minimizing costs and burdens to stakeholders | 3 | 1 | 2 |
| • Feasibility? | 2 | 1 | 3 |
| • Not impede research | 3 | 1 | 1 |
| • Promote constructive applications | 2 | 2 | 1 |
5. Prioritized Governance Options
Main Recommendation
A combination of Option 1 (mandatory production standards) and Option 3 (sustainable raw material standards).
Rationale
• Option 1 is critical for preventing biological and health risks.
• Option 3 ensures environmental sustainability and balance between the bioplastic industry and food security.
• Option 2 remains important but is more effective if implemented gradually to avoid excessive fiscal burden.
6. Ethical Reflection
Although these bioethical issues are not directly related to bioplastics, they are conceptually relevant as they reflect broader ethical challenges in bioengineering innovation.
Bioethical Issues
• Risks to mass genetic data privacy
• Gene editing risks related to off-target effects and permanent germline modifications
• Inequality in access to biotechnology
• Risks of GMO contamination
Governance Responses
• Establishment of an independent genetic data oversight body
• Mandatory gene-editing safety testing standards at the cellular, model organism, and bioinformatics simulation levels
• Technology transfer and capacity-building cooperation
• Long-term ecological testing prior to GMO release into the environment
Final Reflection
• Bioethics is essential because biological innovation has wide-ranging impacts beyond scientific outcomes, affecting human health, the environment, and social structures.
• Bioethics cannot be addressed from a single perspective, as innovation involves diverse interests, including researchers, governments, industries, and affected communities.
• Stakeholder collaboration is necessary to establish balanced governance, ensuring that bioethical regulations are technically sound, socially fair, and environmentally sustainable.
Assignment (Final Project)
To ensure this project supports an ethical biological future, the development of the humanized chimeric ADI should focus on safety, transparency, and responsible use. All predicted results need proper validation to avoid harmful side effects or unexpected immune responses. The engineered enzyme should only be used for medical and research purposes under ethical and biosafety guidelines. In addition, research data and methods should be clearly documented so the work can be reproduced and evaluated by other researchers. Using in silico approaches can also reduce unnecessary animal testing in early research stages. Future development should also consider affordability and equal access so the therapy can benefit more patients.
QUIZ PRE CLASS 2
Questions from Professor Jacobson
1. What is the error rate of polymerase? How does this compare to the length of the human genome? How does biology deal with that discrepancy?
DNA polymerase has an intrinsic error rate of approximately 10⁻⁵ to 10⁻⁷ errors per nucleotide during initial DNA synthesis. With the presence of 3’→5’ exonuclease proofreading activity, the error rate is reduced to around 10⁻⁷–10⁻⁸. When combined with DNA mismatch repair mechanisms, the final error rate can be as low as ~10⁻¹⁰ errors per base per replication.
In comparison, the human genome consists of approximately 3 × 10⁹ base pairs. Without error-correction mechanisms, this would result in an unsustainable number of mutations per cell division.
Biology addresses this discrepancy through:
1).Proofreading by DNA polymerase, which corrects errors immediately during replication.
2).Mismatch repair systems, which detect and repair distortions in the DNA helix after replication.
3).Genetic redundancy and tolerance, particularly in non-coding regions of the genome.
Together, these mechanisms ensure highly accurate DNA replication despite the large size of the human genome.
2. How many different ways are there to code for an average human protein? Why don’t all of these codes work in practice?
In theory, an average human protein (~300 amino acids long) can be encoded by an astronomically large number of different DNA sequences due to the degeneracy of the genetic code, where most amino acids are encoded by multiple synonymous codons.
If each amino acid has, on average, three synonymous codons, the total number of possible coding sequences is approximately 3³⁰⁰.
However, in practice, not all of these sequences function effectively due to:
1).Codon usage bias, where organisms preferentially use specific codons.
2).mRNA stability and secondary structure, which can interfere with transcription and translation.
3).Regulatory constraints, including ribosome binding efficiency and splice-site recognition.
4).Protein folding and toxicity issues, which can arise from altered translation kinetics.
Thus, while the genetic code allows many theoretical possibilities, biological function severely constrains which coding sequences are viable.
Questions from Dr. LeProust
1. What’s the most commonly used method for oligo synthesis currently?
The most commonly used method for oligonucleotide synthesis today is solid-phase phosphoramidite DNA synthesis.
This method involves:
1).Sequential addition of nucleotides to a growing chain attached to a solid support
2).Highly controlled chemical reactions
3).Automation, enabling high-throughput synthesis of short DNA oligos
2. Why is it difficult to make oligos longer than 200 nt via direct synthesis?
Producing oligonucleotides longer than ~200 nucleotides via direct chemical synthesis is difficult because:
1).Each coupling step has less than 100% efficiency
2).Errors such as deletions and truncations accumulate with length
3).The yield of full-length product decreases exponentially as oligo length increases
As a result, purification of accurate, full-length oligos becomes increasingly impractical beyond this length.
3. Why can’t you make a 2000 bp gene via direct oligo synthesis?
Direct synthesis of a 2000 bp gene is not feasible because:
1).The cumulative error rate would be extremely high
2).The yield of full-length product would be nearly zero
3).Purification would be technically and economically impractical
Therefore, long genes are constructed using assembly of shorter oligonucleotides through methods such as PCR-based assembly, Gibson assembly, or Golden Gate cloning.
Homework Question from George Church
1. What are the 10 essential amino acids in all animals, and how does this affect your view of the “Lysine Contingency”?
The 10 essential amino acids in animals are:
1).Histidine
2).Isoleucine
3).Leucine
4).Lysine
5).Methionine
6).Phenylalanine
7).Threonine
8).Tryptophan
9).Valine
10).Arginine (essential in many animals, particularly during growth)
The lysine contingency refers to the fact that animals cannot synthesize lysine de novo and must obtain it from dietary or microbial sources.
This has important implications:
Ecosystems and food chains depend on lysine-producing organisms such as plants and microbes.
Lysine represents a metabolic and evolutionary bottleneck.
This supports Professor Church’s view that biochemical dependencies can be exploited for biocontainment, for example by engineering organisms that require an external lysine supply.
This perspective reframes the genetic code not only as an information system, but also as a tool for ecological control and biosafety.
References / AI use:
Lecture 2 slides; standard molecular biology textbooks (e.g., Alberts et al., Molecular Biology of the Cell). AI (ChatGPT) was used for synthesis and conceptual explanation.
Week 10 HW: Advanced Imaging & Measurement Technology
Waters Part I — Molecular Weight
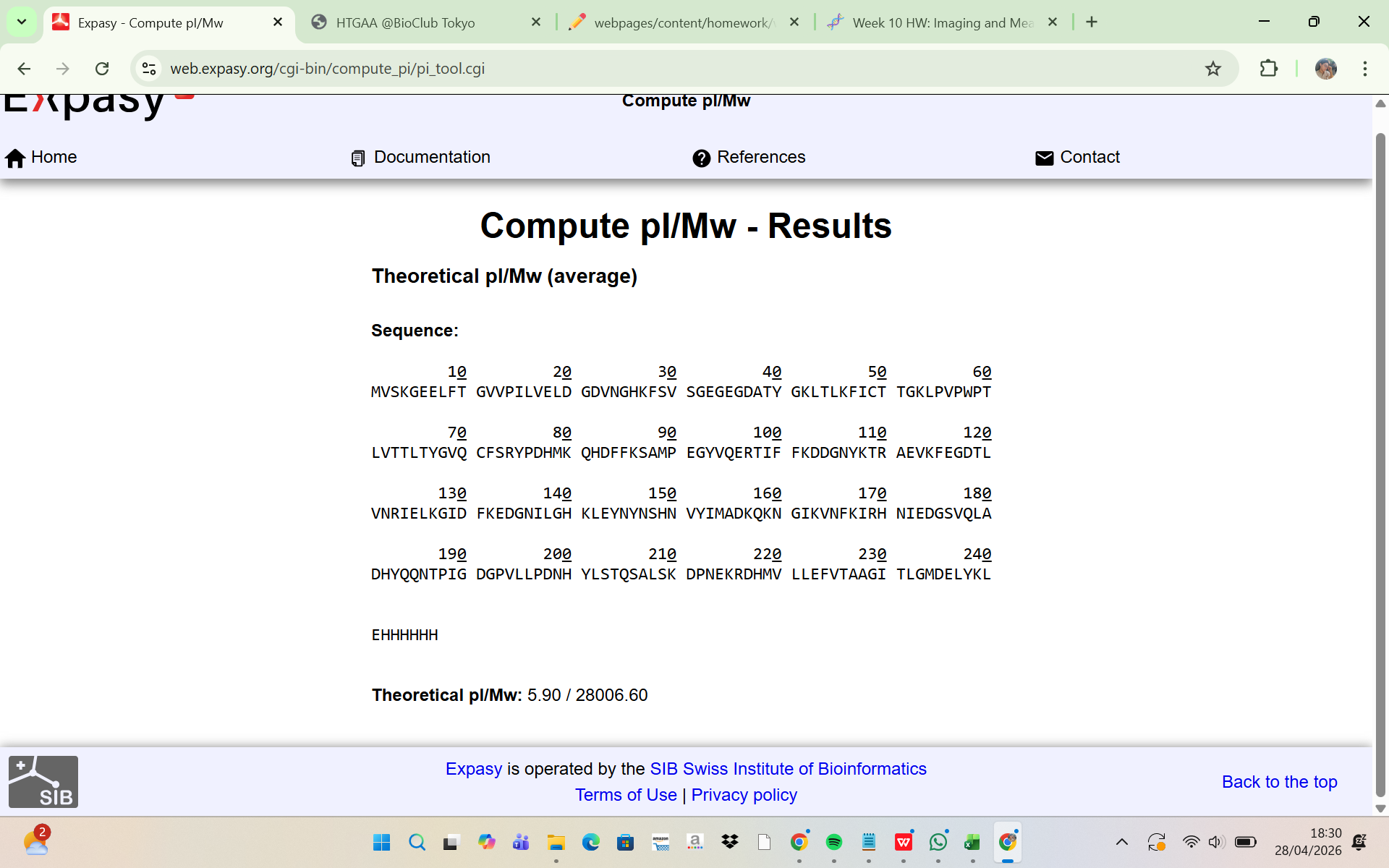
The amino acid sequence of the His-tagged eGFP was analyzed using the ExPASy Compute pI/Mw tool. The results showed:
Theoretical molecular weight (Mw) = 28,006.60 Da Theoretical pI = 5.90
This indicates that the predicted molecular mass of the intact eGFP protein, including the linker and His-tag, is approximately: 28.01 kDa This theoretical molecular weight will be used as the reference value for comparison with the experimentally determined mass from LC-MS analysis.
Two adjacent peaks were selected from the LC-MS spectrum:
m/z_n = 903.7148
m/z_n+1 = 933.8044
The charge state was determined using:
z = (m/z_n+1) / ((m/z_n) - (m/z_n+1))
Substituting the observed values:
z = 933.8044 / (933.8044 - 903.7148) = 31.03
Thus: z = 31
The experimental molecular weight was then calculated:
MW = z × (m/z)
MW = 31 × 903.7148 = 28,015.16 Da
Experimental MW = 28,015.16 Da
The measurement accuracy was determined by comparing the experimental molecular weight with the theoretical molecular weight:
Accuracy = |MW_experiment - MW_theory| / MW_theory
Accuracy = |28015.16 - 28006.60| / 28006.60 = 0.000306
Percent Error = 0.0306%
This very small error indicates that the experimentally measured mass is highly consistent with the theoretical mass of the His-tagged eGFP.
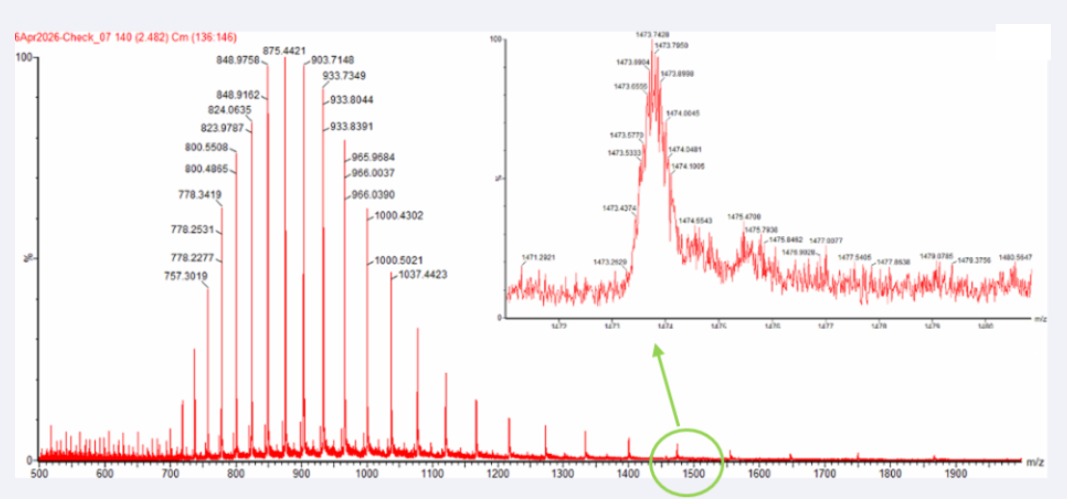
Yes, the charge state can be observed from the zoomed-in isotopic peak pattern.
The spacing between adjacent isotopic peaks is approximately:
Δ(m/z) ≈ 0.05
Using the relationship:
z = 1 / Δ(m/z)
The charge state is:
z = 1 / 0.05 = 20
Therefore:
The zoomed-in peak has an approximate charge state of +20
This is consistent with the denatured intact eGFP ion observed in LC-MS, where the unfolded protein carries multiple charges.
Waters Part III — Peptide Mapping - primary structure
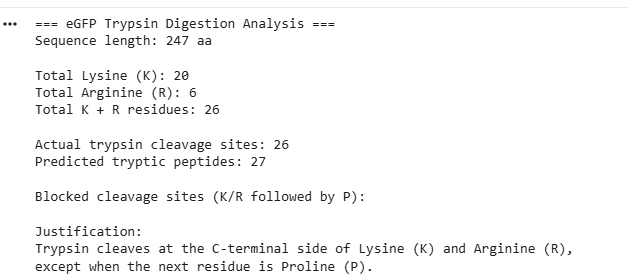
- The eGFP amino acid sequence was analyzed computationally to determine the number of potential trypsin cleavage sites. The analysis showed that the protein contains 20 lysine (K) residues and 6 arginine (R) residues, giving a total of 26 residues that can serve as cleavage sites for trypsin. Since trypsin specifically cleaves peptide bonds at the C-terminal side of lysine and arginine residues, each of these residues represents a potential digestion site unless the following residue is proline. In this sequence, no lysine or arginine residues were followed by proline, meaning that all 26 cleavage sites were available for digestion. Therefore, the predicted number of peptides generated after complete tryptic digestion was 27 peptides, calculated as the number of cleavage sites plus one. This theoretical number represents the maximum number of peptide fragments expected under ideal digestion conditions. However, the number of peptides detected experimentally by LC-MS may be lower because some peptides may be too small, may ionize poorly, or may not be detected under the selected analytical conditions.
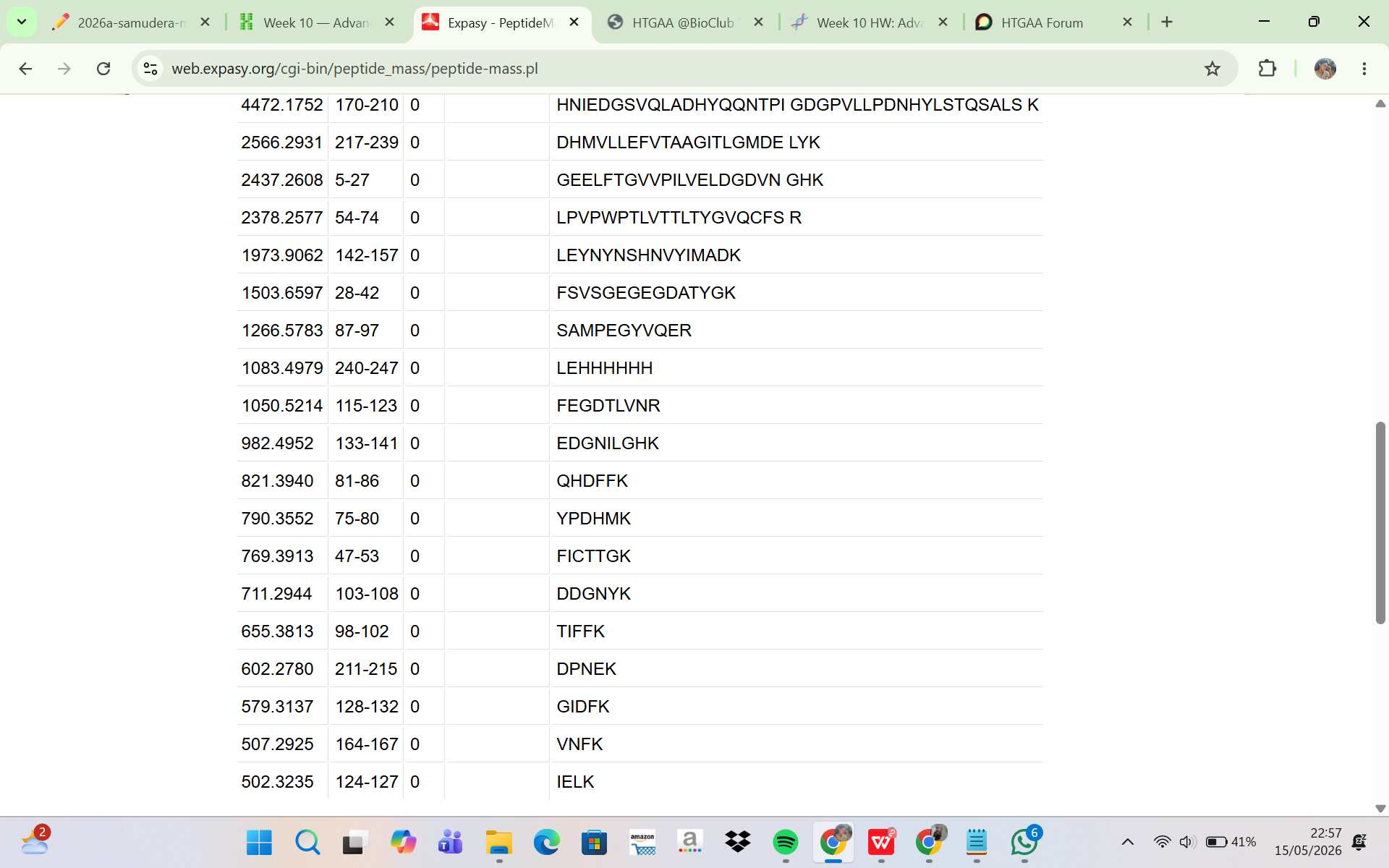
The eGFP sequence was analyzed using the ExPASy PeptideMass tool to predict the peptides generated after tryptic digestion. The analysis parameters included trypsin as the selected enzyme, zero missed cleavages, reduced cysteines, no methionine oxidation, and peptide masses greater than 500 Da. Based on these parameters, the digestion generated a total of 19 predicted peptides. Although complete theoretical digestion of eGFP would produce more peptide fragments, only peptides with masses above 500 Da were included in the output. This filtering step excludes very small peptides that are unlikely to be detected efficiently during LC-MS analysis. Therefore, the ExPASy peptide prediction represents the subset of peptides that are most relevant for experimental peptide mapping by LC-MS.
The LC-MS total ion chromatogram (TIC) shown in Figure 5a was analyzed to determine the number of chromatographic peaks present between 0.5 and 6 minutes. Only peaks with relative abundances greater than 10% were counted to avoid including noise or very minor signals. Based on visual inspection of the chromatogram, approximately 16 significant chromatographic peaks were observed within the selected retention time range. These peaks represent peptide ions generated from the tryptic digestion of eGFP and separated during liquid chromatography prior to mass spectrometric detection. The distribution of peaks across the chromatogram indicates the presence of multiple peptides with different hydrophobicities and retention behaviors.
Comparison of the theoretical peptide prediction with the experimental LC-MS chromatogram showed that the number of observed chromatographic peaks was lower than the number of peptides predicted computationally. ExPASy predicted 19 peptides, whereas the TIC chromatogram showed only approximately 16 major chromatographic peaks. This discrepancy is expected in peptide mapping experiments because not all peptides are detected equally during LC-MS analysis. Some peptides may have very low ionization efficiency, low abundance, or overlapping retention times that cause co-elution with other peptides. In addition, certain peptides may produce signals below the detection threshold of the instrument. Therefore, the experimentally observed chromatographic peaks do not necessarily correspond one-to-one with the total number of predicted tryptic peptides.
The mass spectrum shown in Figure 5b corresponds to the chromatographic peak at 2.78 minutes from the peptide map. The dominant precursor ion observed in the spectrum had an m/z value of approximately 525.767. To determine the charge state of this peptide ion, the isotopic peak spacing in the zoomed inset spectrum was analyzed. The difference between adjacent isotope peaks was approximately: 526.25918 − 525.76712 ≈ 0.492.
In electrospray ionization mass spectrometry, isotope spacing follows the relationship: Δ(m/z) = 1 / z. Thus, the charge state was calculated as: z ≈ 1 / 0.492 ≈ 2. This indicates that the precursor peptide ion carried a +2 charge state. The singly protonated peptide mass ([M+H]+) was then calculated using the relationship between m/z and charge: [M+H]+ = z(m/z) − (z − 1)(1.0073). Substituting the experimental values: [M+H]+ = 2(525.76712) − 1.0073. Resulting in: [M+H]+ ≈ 1050.53 Da. This calculated mass closely matched the theoretical monoisotopic mass predicted by ExPASy for the peptide FEGDTLVNR, which had a predicted mass of 1050.5214 Da.
Furthermore, Figure 5c provided fragmentation (MS/MS) data for the precursor ion at m/z 525.767. Several fragment ions were observed in the fragmentation spectrum, including peaks near m/z 774.41, 903.44, and 1050.52. These fragment ions are consistent with peptide backbone fragmentation patterns typically observed during collision-induced dissociation (CID). The fragmentation spectrum therefore supports the identification of the peptide sequence and confirms that the precursor ion detected at m/z 525.767 corresponds to the tryptic peptide FEGDTLVNR. The close agreement between theoretical peptide mass, precursor ion mass, isotope spacing, and fragmentation pattern demonstrates the high accuracy of the LC-MS/MS peptide mapping analysis.
- The peptide eluting at 2.78 min was identified by comparing the experimentally measured peptide mass to theoretical masses generated with the ExPASy PeptideMass tool. The experimentally determined singly protonated mass was:
Experimental mass ([M+H]+) = 1050.53 Da
This matched the tryptic peptide FEGDTLVNR, which has a theoretical monoisotopic mass of Theoretical mass = 1050.5214 Da
The mass error in parts per million (ppm) was calculated as:
Mass error (ppm) = (|MW_experiment − MW_theory| / MW_theory) × 10^6
Substituting the values:
Mass error (ppm) = (|1050.53 − 1050.5214| / 1050.5214) × 10^6
Difference between masses:
|1050.53 − 1050.5214| = 0.0086
Therefore:
Mass error (ppm) ≈ 8.2 ppm
A mass error of about 8.2 ppm shows excellent agreement between the measured and theoretical masses, supporting the confident identification of the peptide FEGDTLVNR and demonstrating the high mass accuracy of the LC-MS/MS analysis.
- Based on Figure 6, the peptide mapping analysis confirmed approximately 88% sequence coverage of the eGFP protein. This means that most regions of the protein sequence were successfully identified through the detected tryptic peptides during LC-MS/MS analysis, while a small portion of the sequence was not detected or not confidently assigned.
Bonus Peptide Map Questions
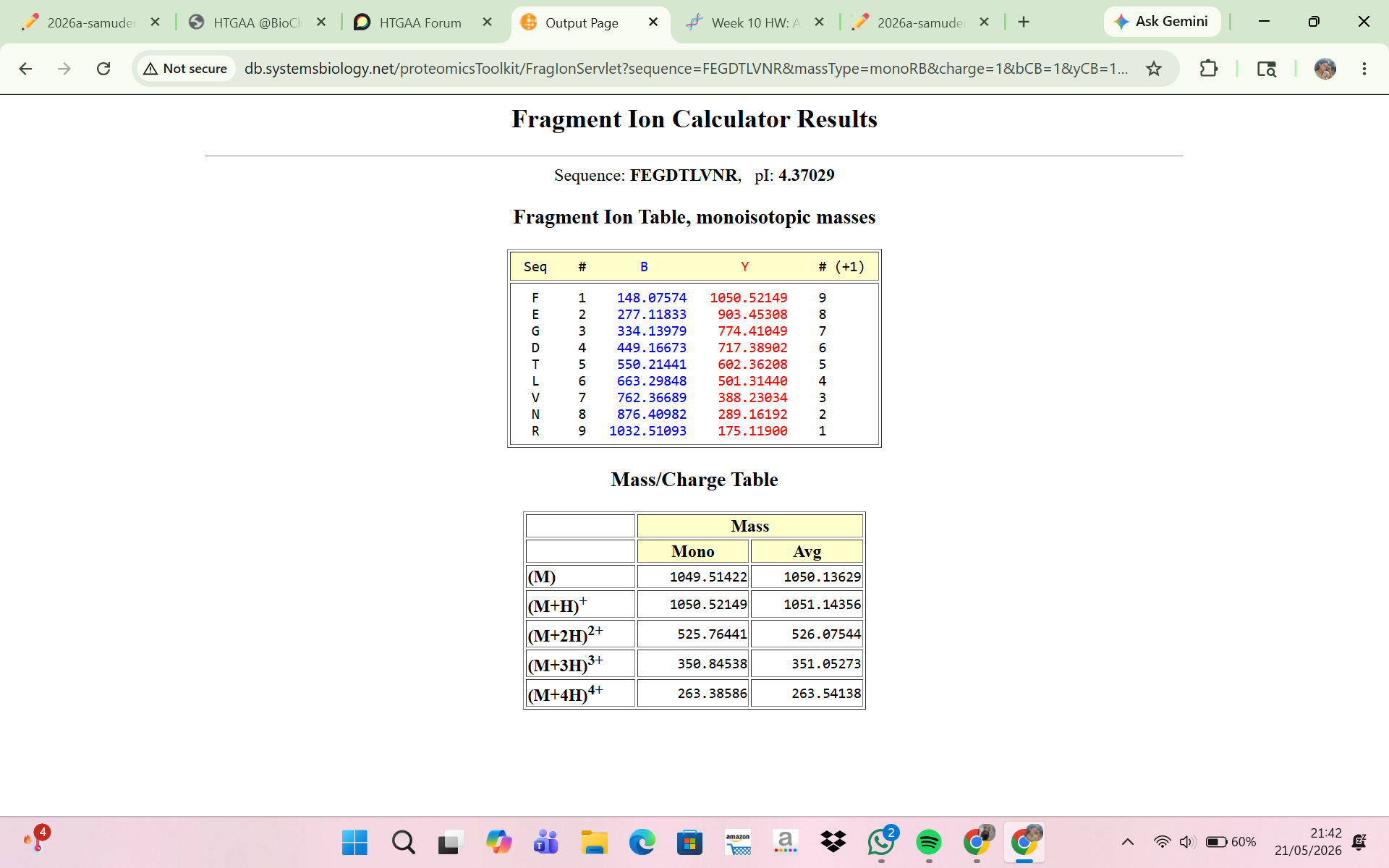
8 . Interpretation of the Fragment Ion Calculator Result
The Fragment Ion Calculator showed that the peptide FEGDTLVNR produced theoretical fragment ions that closely matched the experimental MS/MS spectrum in Figure 5c. Calculated peptide masses were:
[M+H]+ = 1050.52149 Da
[M+2H]2+ = 525.76441 m/z
These values agree with the experimentally detected precursor:
m/z ≈ 525.767
calculated peptide mass ≈ 1050.53 Da
The fragmentation table predicted several y-ions and b-ions that matched peaks in Figure 5c. Examples:
Experimental peak 903.44 — predicted ion y8
Experimental peak 774.41 — predicted ion y7
Experimental peak 602.35 — predicted ion y5
Experimental peak 501.31 — predicted ion y4
Experimental peak 388.22 — predicted ion y3
The close agreement between experimental fragments and predicted ions confirms that the peptide at 2.78 min is FEGDTLVNR. Thus, the MS/MS fragmentation data validate the peptide identity inferred from the peptide mass.
9 . Interpretation of the Peptide Map Data
The peptide mapping results strongly indicate the sample is the expected eGFP standard. LC-MS/MS produced peptide masses that closely matched theoretical tryptic peptides from the eGFP sequence, and MS/MS fragmentation confirmed specific peptides (notably FEGDTLVNR) by matching precursor masses and fragment patterns. Figure 6 shows approximately 88% sequence coverage, indicating most regions of eGFP were identified. The high sequence coverage, accurate peptide masses, and matching fragmentation patterns together demonstrate that the analyzed sample corresponds to the eGFP protein standard.
Waters Part IV — Oligomers
Using the known masses of the KLH subunits, the oligomeric species observed in the CDMS spectrum were assigned by calculating their expected molecular masses and comparing them with the peaks in Figure 7.
The 7FU subunit mass is 340 kDa (0.34 MDa). The mass of the 7FU decamer is:
10 × 0.34 = 3.4 MDa
This matches the peak observed around 3.4–4.0 MDa.
The 8FU subunit mass is 400 kDa (0.40 MDa). The expected mass of the 8FU didecamer is:
20 × 0.40 = 8.0 MDa
This corresponds to the major peak observed at approximately 8.33 MDa.
For the 8FU tridecamer:
30 × 0.40 = 12.0 MDa
This matches the peak near 12.67 MDa.
The expected mass of the 8FU tetradecamer is:
40 × 0.40 = 16.0 MDa
This corresponds to the broader low-intensity signal observed around 16–17 MDa.
Waters Part V — Did I make GFP?
| Theoretical | Observed/measured on the Intact LC-MS | PPM Mass Error |
|---|
| Molecular weight (kDa) | 28.01 kDa | 28.00 kDa | 236 ppm |
Week 11 HW: Bioproduction & Cloud labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
1. Contribution
Honestly, I did not contribute anything to the canvas, as I was already too late to participate in the artwork. However, that is okay. I feel that I still have a lot to learn about this kind of project, and I was genuinely excited to see how enthusiastic everyone was while working on it.
2. What I liked about the project
This project provided us with the freedom to create unique and creative artwork. I was also truly impressed by how beautiful artistic outcomes can emerge from science, which broadened my perspective that science is not only intellectually engaging but also aesthetically inspiring. The global collaboration allowed me to connect with people from diverse backgrounds, and I learned a lot from that experience.
3. What could be improved
I hope that access to editing cell-free reaction compositions could still be maintained, but with some modifications. For example, it would be better if participants could create and explore their own versions without altering the collaborative work of others. Overall, however, the project was conducted very well.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
A. Functional Roles of Each Component
- Lysate
E. coli Lysate: The E. coli lysate provides the essential cellular machinery required for protein synthesis, including ribosomes, tRNAs, aminoacyl-tRNA synthetases, and various transcription and translation factors.
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase): This lysate contains T7 RNA polymerase, enabling efficient transcription of DNA templates into mRNA under the control of a T7 promoter.
- Salts and Buffer
Potassium Glutamate: Maintains ionic strength and mimics intracellular conditions, supporting optimal enzyme activity and protein stability.
HEPES-KOH (pH 7.5): Acts as a buffering agent to maintain a stable pH environment during the reaction.
Magnesium Glutamate: Provides magnesium ions, which are essential cofactors for ribosomal function and enzymatic activities in transcription and translation.
Potassium Phosphate Monobasic and Dibasic: Together form a phosphate buffer system that helps stabilize pH and supports metabolic reactions.
- Energy / Nucleotide System
Ribose: Serves as a precursor for nucleotide biosynthesis, enabling in situ generation of nucleotides.
Glucose: Functions as an energy source to regenerate ATP through metabolic pathways present in the lysate.
AMP, CMP, GMP, UMP: These nucleoside monophosphates act as precursors that are enzymatically converted into nucleoside triphosphates (NTPs) required for RNA synthesis.
Guanine: Serves as a substrate in the nucleotide salvage pathway, allowing the synthesis of GMP and subsequently GTP.
- Translation Mix (Amino Acids)
17 Amino Acid Mix: Provides the majority of amino acids necessary for protein synthesis.
Tyrosine: Supplied separately due to its specific solubility and stability requirements.
Cysteine: Added individually because it is prone to oxidation and requires controlled conditions.
- Additives
Nicotinamide: Acts as a precursor for NAD⁺, supporting redox reactions and cellular metabolism within the lysate.
- Backfill
Nuclease-Free Water: Used to adjust the final reaction volume without introducing nucleases that could degrade DNA or RNA.
B. Differences Between the 1-Hour and 20-Hour Systems
The 1-hour PEP-NTP system relies on the direct addition of high-energy molecules such as phosphoenolpyruvate (PEP) and nucleoside triphosphates (NTPs), enabling rapid protein synthesis but limiting reaction duration. In contrast, the 20-hour NMP-Ribose-Glucose system utilizes metabolic precursors like ribose and glucose to regenerate nucleotides and energy over time, resulting in a more sustainable and cost-effective system. This allows for prolonged protein production and improved reaction longevity.
C. Bonus Question
Transcription can still occur because guanine can be converted into GMP via the nucleotide salvage pathway. The GMP is then phosphorylated into GTP, which is required as a substrate for RNA synthesis during transcription.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
A. Biophysical and Functional Properties
- sfGFP (superfolder GFP)
sfGFP exhibits highly efficient folding and structural stability, allowing it to maintain strong fluorescence even under suboptimal conditions in cell-free systems.
- mRFP1
mRFP1 has relatively slow chromophore maturation, which can delay fluorescence development during shorter or suboptimal incubation periods.
- mKO2
mKO2 is sensitive to pH changes, and variations in pH can significantly affect its fluorescence intensity and stability.
- mTurquoise2
mTurquoise2 has a high quantum yield but requires proper folding conditions to achieve optimal fluorescence output.
- mScarlet-I
mScarlet-I displays rapid maturation and high brightness, making it well-suited for sustained fluorescence over long incubation periods.
- Electra2
Electra2 likely depends on sufficient oxygen availability for chromophore formation, which can influence fluorescence efficiency in cell-free reactions.
B. Hypothesis for Master Mix Optimization
To improve fluorescence of mRFP1, increasing magnesium ion concentration and optimizing energy sources such as glucose in the master mix may enhance protein synthesis and folding efficiency. This is expected to accelerate chromophore maturation and increase overall fluorescence intensity over a 36-hour incubation.
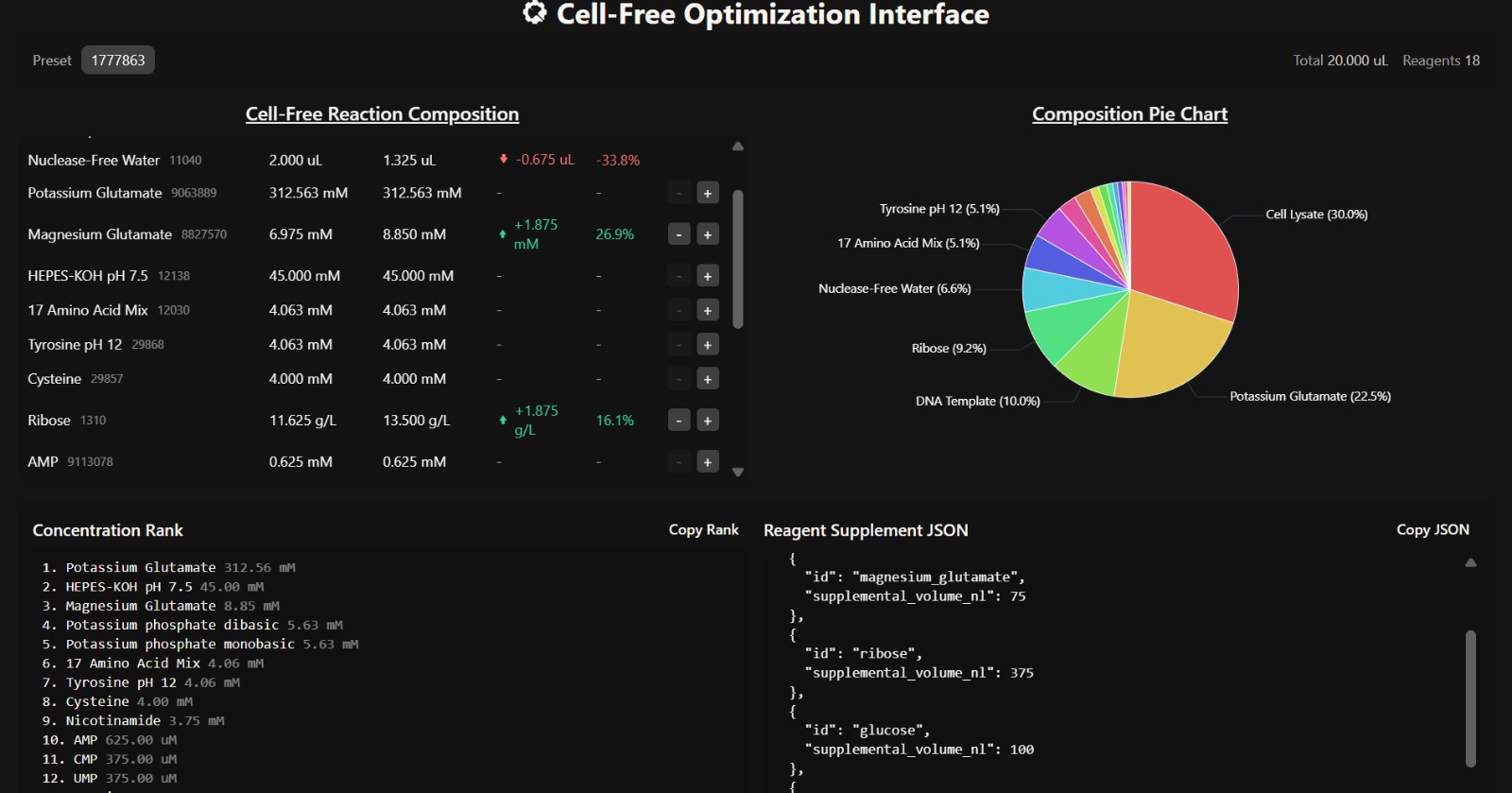
C. Interpretation of Cell-Free Reaction Composition
- Overall Composition Insight
The composition is dominated by cell lysate (30%) and potassium glutamate (22.5%), indicating that the system strongly mimics intracellular conditions and provides sufficient enzymatic machinery for transcription and translation. The presence of ribose (9.2%) and glucose supports a metabolically sustained system, enabling prolonged protein synthesis over a 36-hour incubation.
- Translation and Folding Efficiency
The increased magnesium glutamate concentration (8.85 mM) suggests enhanced ribosomal activity and improved translation efficiency. Magnesium ions are critical cofactors for ribosome stability and enzymatic reactions, meaning this adjustment likely promotes better protein folding and higher protein yield.
- Energy Sustainability
The elevated glucose (2.25 g/L) and ribose (13.5 g/L) concentrations indicate a system optimized for long-term energy regeneration and nucleotide synthesis. This supports continuous ATP production and RNA synthesis, which are essential for sustained protein expression.
- Metabolic Support
The presence of nicotinamide (3.75 mM) enhances metabolic stability by supporting NAD⁺ regeneration, which is essential for redox balance and energy metabolism within the lysate.
- Limitation in Supplement Strategy
Although key reagents were added, the supplemental volumes are relatively low, and a significant portion (1.325 µL) is still allocated to nuclease-free water. This may dilute the effect of critical reagents such as magnesium and glucose, potentially limiting the overall improvement in fluorescence.
- Expected Outcome
This composition is expected to produce moderate to high fluorescence output, particularly for proteins that benefit from sustained energy supply and improved folding conditions (e.g., mRFP1). However, the relatively small increase in key supplements may prevent the system from reaching maximum fluorescence potential.
Week 12 HW: Building Genomes
Week 13 HW: AI, SynBio, and Scaling Health Innovation (ARPA-H)
Week 14 HW: Bio Design & Bio Fabrication
Week 2 HW: DNA Read, Write & Edit
Part 1: Benchling & In-silico Gel Art

PART 3: DNA Design Challenge
3.1 Choose your protein
Erythropoietin is a hormone that stimulates red blood cell production. Selected because:
Vital in anemia therapy
High-value biotechnology protein
Relevant to the pharmaceutical industry
Erythroproietin :
sp|P01588|EPO_HUMAN Erythropoietin OS=Homo sapiens OX=9606 GN=EPO PE=1 SV=1
MGVHECPAWLWLLLSLLSLPLGLPVLGAPPRLICDSRVLERYLLEAKEAENITTGCAEHC
SLNENITVPDTKVNFYAWKRMEVGQQAVEVWQGLALLSEAVLRGQALLVNSSQPWEPLQL
HVDKAVSGLRSLTTLLRALGAQKEAISPPDAASAAPLRTITADTFRKLFRVYSNFLRGKL
KLYTGEACRTGDR
3.2 Reverse Translate : Protein sequence to DNA sequence
Erythroproietin DNA Sequence:
sp|P01588|EPO_HUMAN Erythropoietin OS=Homo sapiens OX=9606 GN=EPO PE=1 SV=1
ATGGGGGTGCACGAATGCCCAGCATGGTTGTGGCTACTATTGAGCCTTCTGTCCTTGCCCTTAGGTCTCCCTGTACTTGG
GGCGCCCCCCCGACTAATATGTGACTCGCGGGTTTTAGAGCGGTACCTGTTGGAAGCAAAAGAAGCGGAAAATATCACTA
CTGGCTGCGCTGAACATTGTTCCTTAAATGAGAATATCACAGTTCCCGACACCAAGGTAAATTTTTATGCGTGGAAACGC
ATGGAGGTTGGCCAACAAGCAGTCGAAGTTTGGCAGGGGTTAGCGCTACTTTCTGAGGCAGTGCTTAGAGGCCAGGCATT
GTTAGTAAATTCAAGCCAGCCTTGGGAGCCTCTACAACTTCATGTGGACAAAGCCGTGTCAGGCCTGAGATCCCTAACTA
CGCTCCTCCGCGCGCTAGGAGCGCAAAAAGAGGCTATCAGTCCGCCCGACGCAGCTTCTGCCGCCCCACTCCGTACCATA
ACAGCTGACACTTTCCGAAAACTTTTCAGAGTTTATTCAAACTTCCTACGAGGTAAATTGAAATTATACACTGGCGAAGC
CTGCAGGACTGGGGATCGC
3.3 Codon Optimization
For this project, Escherichia coli has been chosen as the expression host.
ATGGGTGTGCACGAATGCCCAGCATGGTTGTGGCTACTGTTGAGCCTTCTGTCCTTGCCGTTAGGTCTCCCTGTACTTGGGGCGCCCCCGCGTCTTATTTGTGATTCGCGTGTTCTGGAGCGGTACCTGTTGGAAGCCAAAGAAGCGGAAAATATTACTACCGGCTGCGCTGAACATTGTTCCTTAAATGAAAACATCACAGTTCCGGACACCAAGGTCAACTTTTATGCGTGGAAACGCATGGAGGTCGGCCAACAGGCGGTCGAAGTGTGGCAGGGGCTGGCGCTACTGAGCGAGGCAGTGCTTCGTGGCCAGGCACTGTTAGTAAATAGTAGCCAGCCTTGGGAGCCGCTGCAACTGCATGTGGACAAAGCCGTGTCAGGCCTGCGCTCGCTGACGACGCTCCTCCGCGCGCTGGGAGCGCAGAAGGAAGCTATCAGTCCGCCGGATGCAGCCTCTGCCGCCCCACTGCGTACCATTACCGCTGATACATTCCGAAAACTGTTCCGTGTTTATTCAAACTTTCTGCGCGGTAAACTGAAATTATACACTGGTGAAGCCTGCAGAACGGGCGATCGC
3.4 You have a sequence! Now what?
Once the DNA sequence encoding Erythropoietin (EPO) has been designed and codon-optimized for E. coli, the next step is to produce the protein using an appropriate expression system. Two major technological approaches can be used: cell-dependent expression systems and cell-free expression systems.
A. Cell-Dependent Expression System (Using E. coli)
In this project, a cell-dependent system is used with the pET-28a expression vector.
Step 1: Cloning into pET-28a
The optimized EPO gene is inserted into the Multiple Cloning Site (MCS) of the pET-28a plasmid. The construct includes:
a. T7 promoter
b. Ribosome Binding Site (RBS)
c. Start codon (ATG)
d. EPO coding sequence
e. Stop codon
f. Optional His-tag for purification
This produces the recombinant plasmid pET-28a-EPO.
Step 2: Transformation into E. coli
The recombinant plasmid is introduced into competent E. coli cells (e.g., BL21(DE3)). Transformed cells are selected using kanamycin resistance encoded by the plasmid.
Step 3: Transcription
Upon induction:
The T7 RNA polymerase recognizes the T7 promoter.
The DNA sequence is transcribed into messenger RNA (mRNA).
During transcription:
a. Adenine (A) pairs with Uracil (U)
b. Thymine (T) in DNA becomes Uracil (U) in RNA
The result is an mRNA molecule complementary to the DNA template strand
Step 4: Translation
The ribosome binds to the Ribosome Binding Site (RBS).
Translation begins at the start codon (AUG).
Each codon (three nucleotides) is translated into one amino acid.
Transfer RNA (tRNA) delivers the corresponding amino acids.
The polypeptide chain elongates until a stop codon is reached.
This process follows the Central Dogma of Molecular Biology:
DNA → RNA → Protein
The final product is recombinant EPO protein, typically fused with a His-tag for purification.
B. Cell-Free Expression System (Alternative Method)
Alternatively, the EPO protein can be produced using a cell-free expression system.
In this system:
a. The DNA template is added directly to a reaction mixture.
b. The mixture contains RNA polymerase, ribosomes, tRNAs, amino acids, and necessary cofactors.
c. Transcription and translation occur in vitro (outside living cells).
Advantages:
a. Faster protein production
b. No need for cell transformation
c. Suitable for rapid screening
However, for large-scale production, cell-dependent systems are generally preferred.
3.5 [Optional] How does it work in nature/biological systems?
In human cells:
a. Alternative splicing generates isoforms.
b.RNA editing may modify nucleotides post-transcription.
c. Post-translational modifications (e.g., glycosylation in EPO) alter protein stability and function.
In contrast, E. coli:
a. Does not perform alternative splicing.
b. Does not process introns.
c. Does not perform complex glycosylation.
Thus, recombinant EPO produced in E. coli may differ structurally from native human EPO.
Part 4: Prepare a Twist DNA Synthesis Order
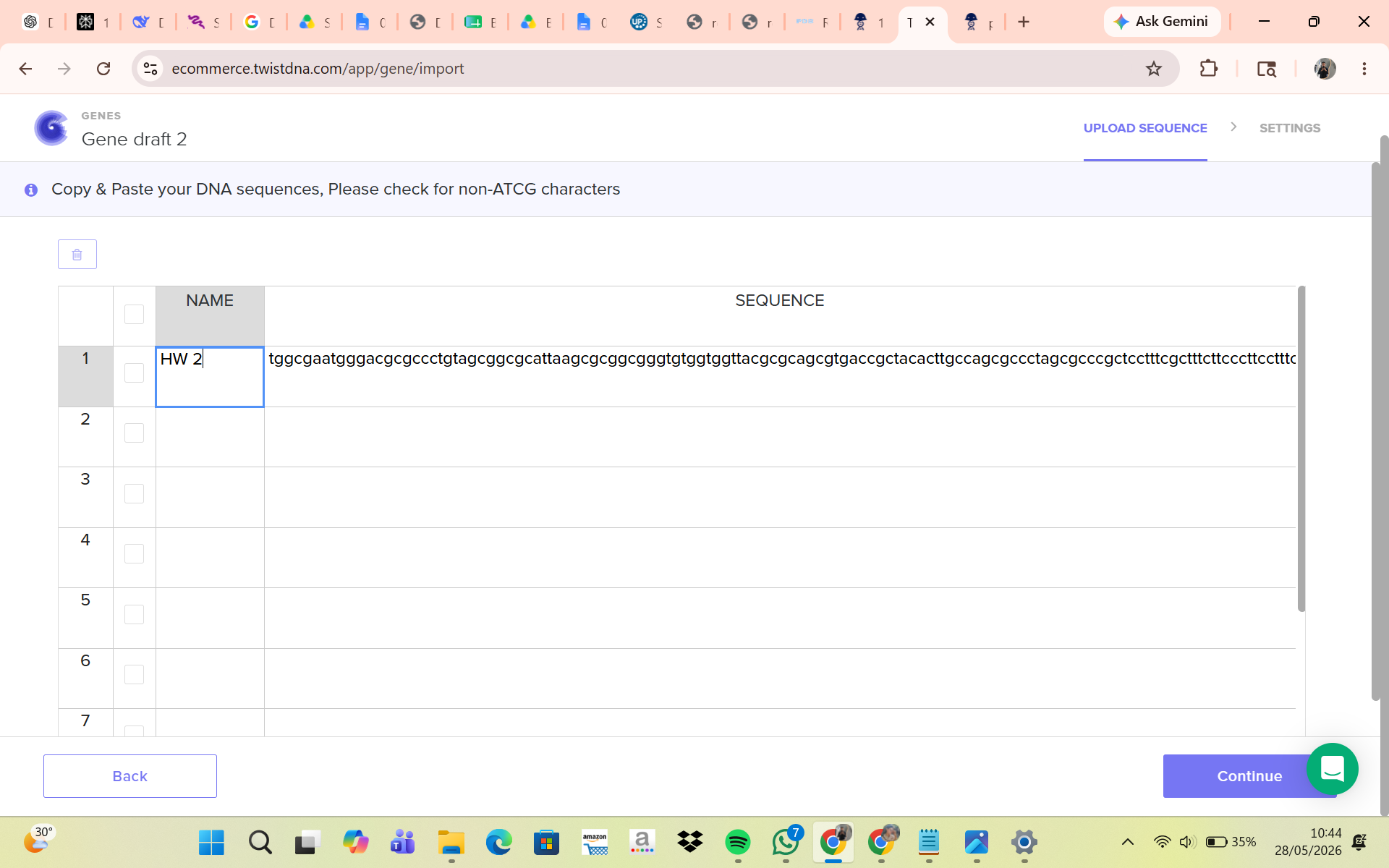
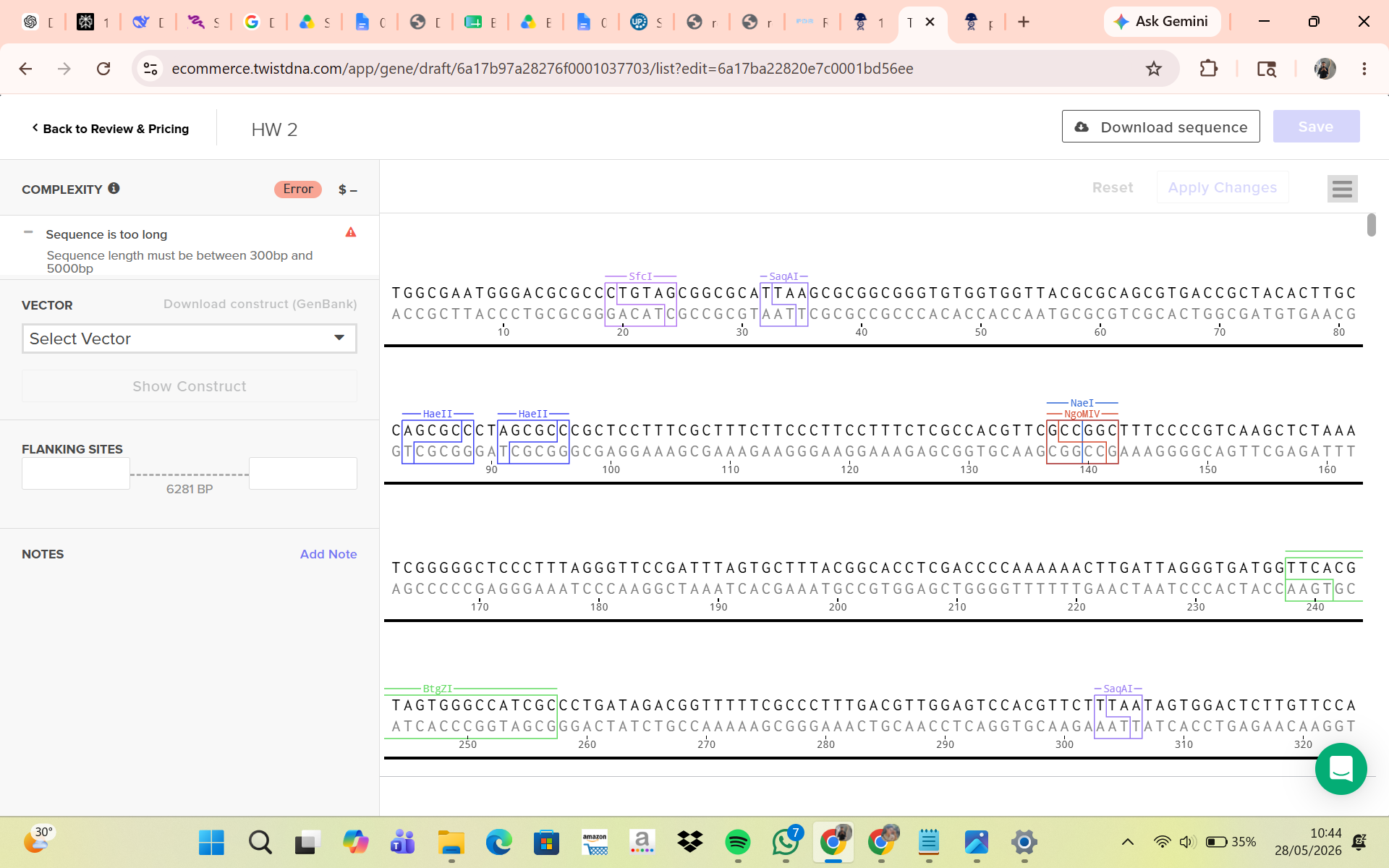
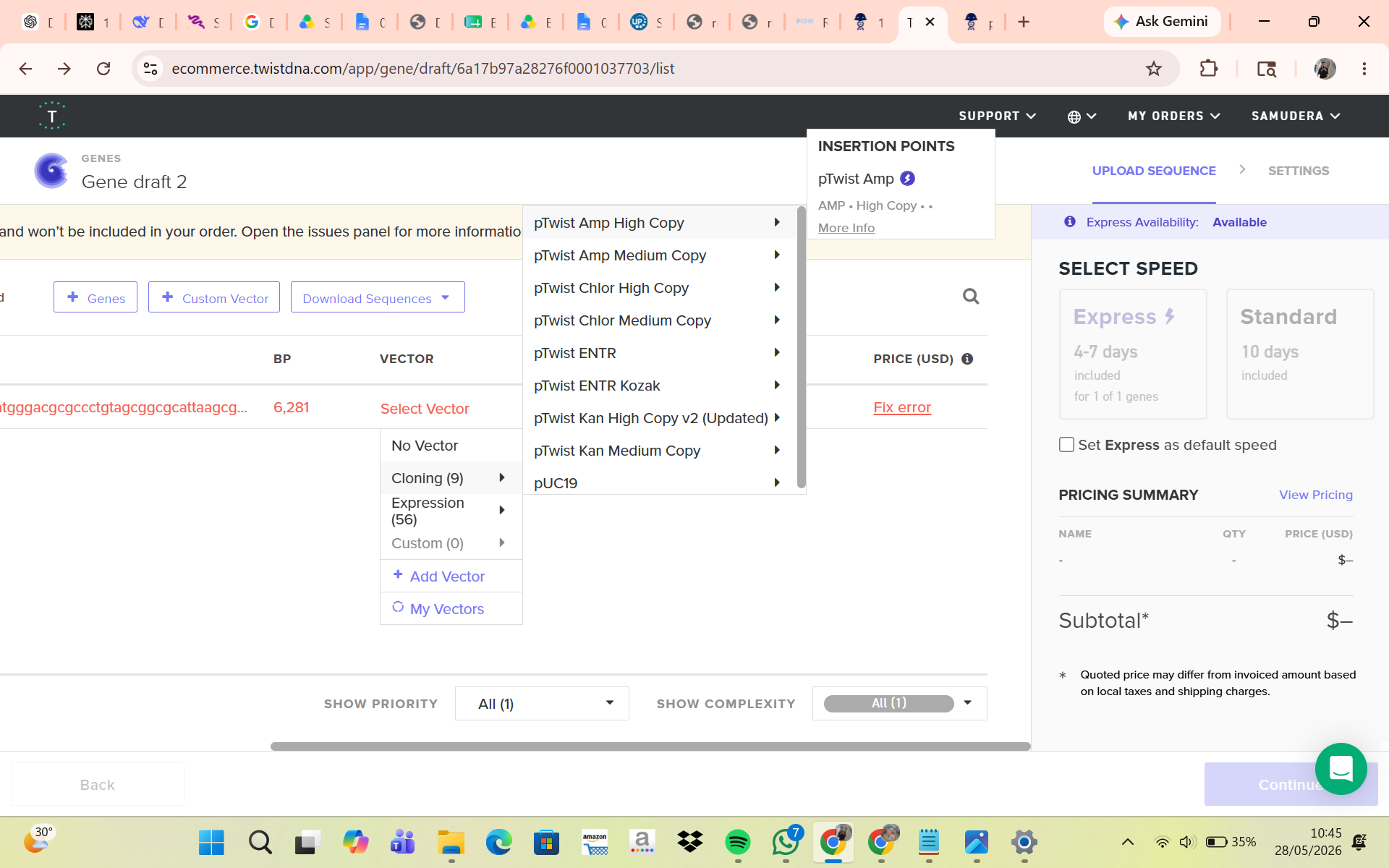
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence and why?
Primary DNA to Sequence in This Project
I would sequence:
The recombinant pET-28a-EPO plasmid
The EPO coding sequence (CDS)
The promoter–insert junction regions
Why?
a. To confirm that the EPO gene was inserted correctly.
b. To verify that no mutations occurred during gene synthesis or cloning.
c. To confirm the correct reading frame with the His-tag.
d. To ensure no premature stop codons or frameshifts are present.
Beyond this project, sequencing could be applied to sequencing disease-associated genes in Human health (e.g., cancer mutations).
(ii) What sequencing technology would you use and why?
For this project, I would use:
Sanger Sequencing
Why?
a. The EPO gene is relatively short (~500–600 bp coding region).
b. Plasmid verification is well suited for Sanger sequencing.
c. High accuracy for single-gene validation.
d. Cost-effective for small constructs.
Classification
a. First-generation sequencing
b. Uses chain-termination chemistry
c. Produces highly accurate reads (~700–1000 bp per read)
Input: Purified plasmid DNA (pET-28a-EPO)
Preparation Steps:
Plasmid extraction from E. coli
Primer design (forward and reverse primers flanking insert)
PCR cycle sequencing reaction with labeled dideoxynucleotides (ddNTPs)
Essential Steps of Sanger Sequencing
DNA denaturation
Primer annealing
DNA polymerase extension
Random incorporation of fluorescently labeled ddNTPs
Chain termination
Capillary electrophoresis separation
Laser detection of fluorescent signals
Base Calling : Each ddNTP is labeled with a different fluorescent dye. When incorporated, elongation stops.Fragments of different lengths are separated and detected.The emitted fluorescence determines the base identity (A, T, C, or G).
Output : Chromatogram (electropherogram), DNA sequence file (.ab1 or .seq), Base quality scores
5.2 DNA Write
(i) What DNA would you want to synthesize and why?
In this project, I would synthesize:
Codon-optimized human EPO gene for E. coli
Purpose:
a. Produce recombinant EPO for research applications.
b. Study protein folding and expression optimization.
ii) What technology would you use for DNA synthesis?
Chosen Technology:
Chemical DNA synthesis followed by gene assembly & Commercial providers (e.g., gene synthesis companies) synthesize DNA de novo.
Essential Steps of DNA Synthesis
Oligonucleotide synthesis (phosphoramidite chemistry)
Assembly of short oligos into full-length gene
Error correction (if necessary)
Cloning into plasmid backbone (pET-28a)
Sequence verification
Limitations of DNA Synthesis
Speed: Synthesis of longer genes takes more time.
Accuracy: Errors can occur during chemical synthesis, Requires sequencing validation.
Scalability: Cost increases with gene length, Whole-genome synthesis remains complex and expensive.
However, for single-gene constructs like EPO, synthesis is efficient and practical.
5.3 DNA Edit
(i) What DNA would you want to edit and why?
In this project, I would edit:
The EPO coding sequence
Possible edits:
a. Improve solubility in E. coli
b. Reduce aggregation
c. Modify specific amino acids to increase stability
Beyond this project: i wish we can Correct disease-causing mutations in humans
(ii) What technology would you use?
For Plasmid Editing: Site-Directed Mutagenesis
For Genomic Editing: CRISPR-Cas systems
How CRISPR Edits DNA
Design guide RNA (gRNA) complementary to target DNA.
Cas enzyme binds to gRNA.
Complex locates target DNA.
Cas creates double-strand break.
Repair occurs via:
a. Non-homologous end joining (NHEJ)
b. Homology-directed repair (HDR)
Required Inputs : DNA template (target sequence), Guide RNA, Cas enzyme, Repair template (if precise edit desired), Host cells
Essential Design Steps
Identify target sequence.
Design guide RNA.
Check for off-target sites.
Prepare delivery system (plasmid or ribonucleoprotein).
Validate edits via sequencing.
Limitations of Editing Technologies
Efficiency: Editing efficiency may vary by cell type.
Precision: Off-target mutations may occur.
Delivery Challenges: Introducing CRISPR components into certain cells is difficult.
Ethical Considerations: Human genome editing raises significant ethical concerns.
Week 3 HW : Lab Automation
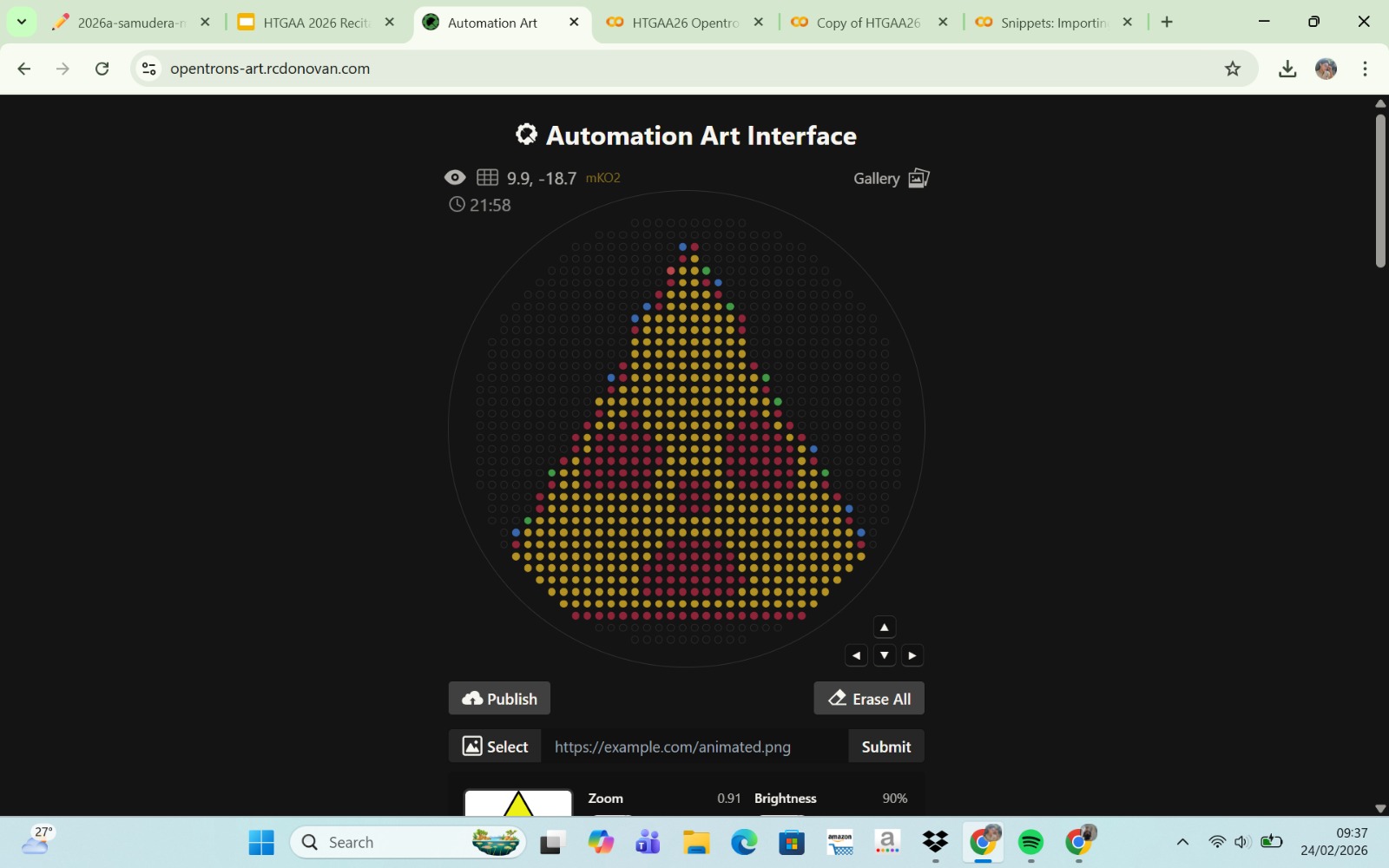
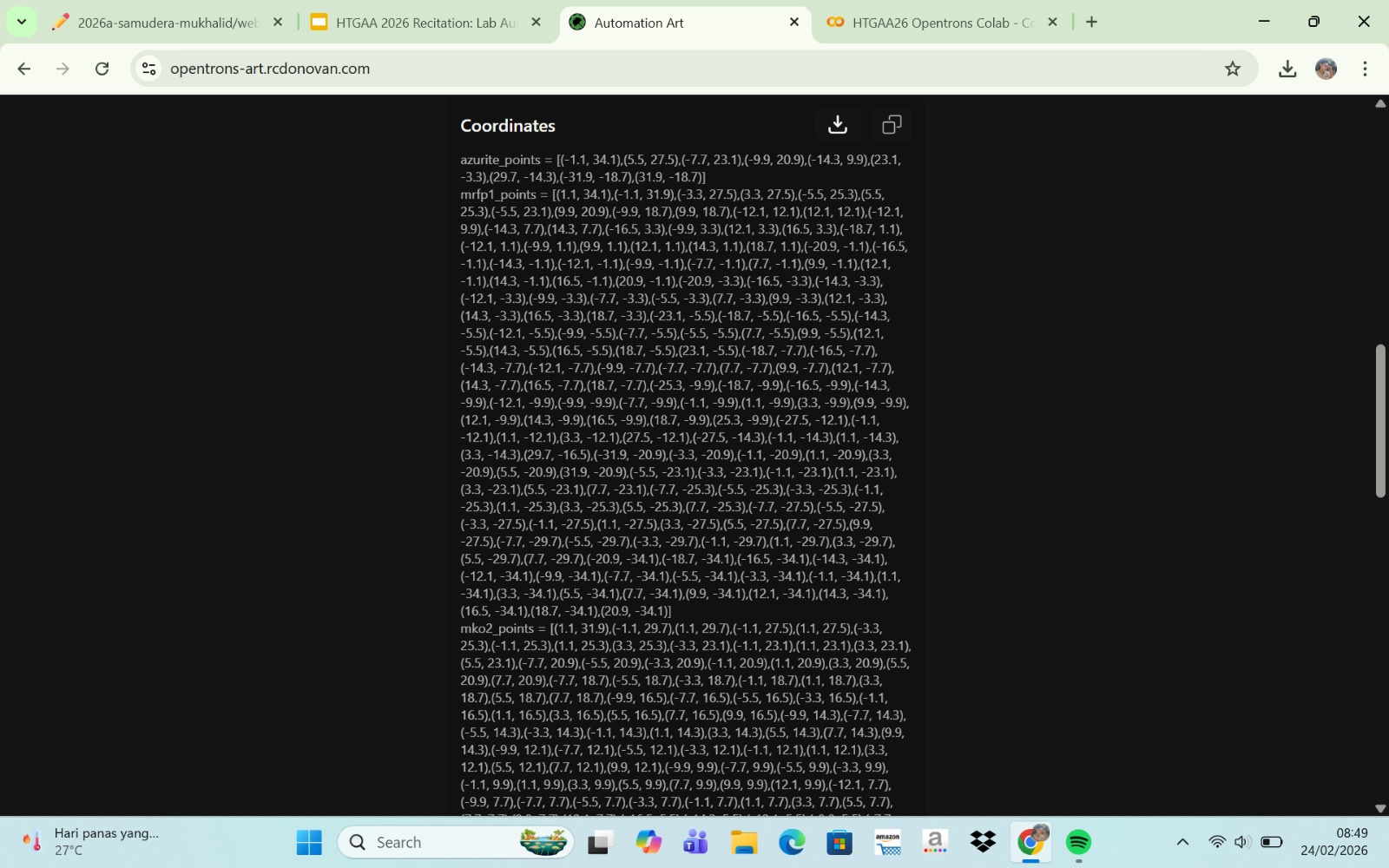

Published Paper on Opentrons Automation
The paper entitled AssemblyTron: flexible automation of DNA assembly with Opentrons OT-2 lab robots, published in the journal Synthetic Biology (2023), reports the development of AssemblyTron, a software platform that automates DNA assembly workflows using the Opentrons OT-2 liquid-handling robot. The system integrates DNA construct design (for example, from design software such as j5) with automated execution on the OT-2 to perform molecular biology procedures in a precise and standardized manner. AssemblyTron is designed to support the Design–Build–Test–Learn (DBTL) cycle in synthetic biology by automating PCR optimization, Golden Gate assembly, and in vivo assembly (IVA).
In the study, the authors demonstrated the system’s capabilities by constructing four-fragment chromoprotein plasmids and performing site-directed mutagenesis. The results showed that the assembly fidelity achieved by the automated system was comparable to that of manual methods. In addition, the use of AssemblyTron reduced the likelihood of human error, minimized the need for extensive technical training, and decreased reagent waste, while improving experimental reproducibility and throughput. Overall, this platform enables researchers to accelerate the “build” phase of the DBTL cycle and allocate more time to the design and analysis stages in synthetic biology research.
Final Project Automation Plan
I plan to automate cell-free protein synthesis (CFPS) screening for engineered fluorescent biosensors detecting environmental analytes like heavy metals, using Opentrons OT-2 integrated with Ginkgo Nebula for remote design and deployment.
Core Workflow :
A. Design Phase: Use Ginkgo Nebula to design and order biosensor genetic constructs (e.g., GFP variants fused to metal-binding domains), targeting 96 variants with randomized promoter strengths.
B. Prep and Deposition: 3D-print custom PCR tube racks (inspired by Opentrons directory) to hold construct templates; Opentrons pipets DNA/cofactors into a 96-well plate.
C. Reaction Setup: Echo transfers templates; dispense CFPS master mix (lysate, T7 RNA pol, NTPs); Multiflo adds lysate to initiate expression; PlateLoc seals; Inheco incubates at 30°C for 16h.
D. Readout: XPeel removes seal; PHERAstar measures fluorescence/excitation spectra to score sensor performance.
Example Python Pseudocode (Opentrons API) :
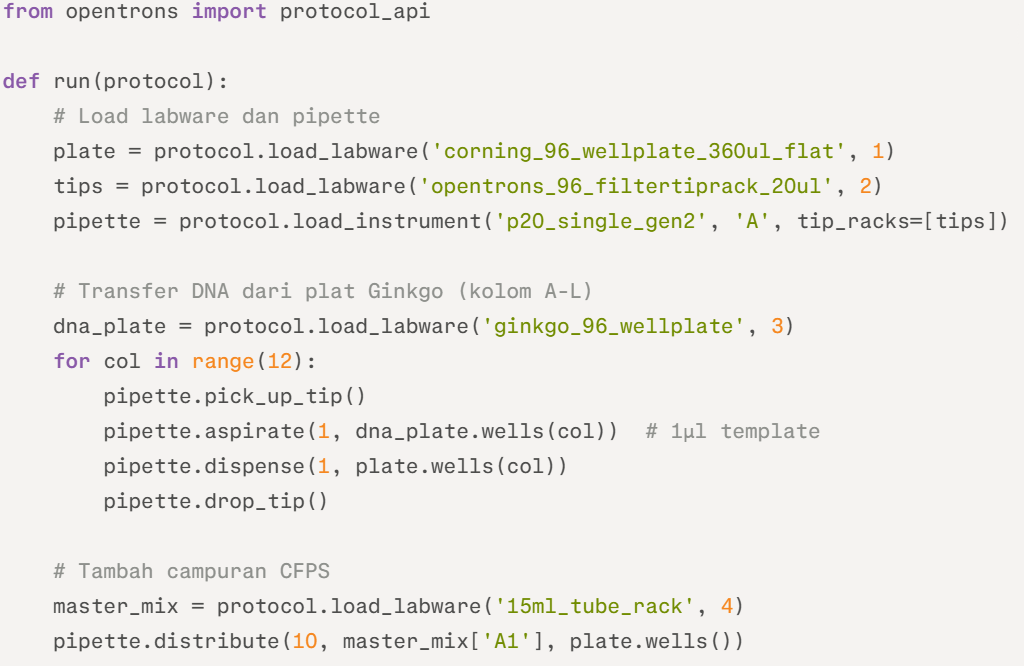
This script handles initial deposition; full protocol chains with temperature modules per recitation slides.
Hardware Additions :
A. 3D-printed biosensor plate holder for precise alignment during imaging.
B. Remote monitoring via Opentrons App, synced to Ginkgo for iterative DBTL (e.g., top 10% sensors resynthesized).
This setup enables 100+ parallel reactions weekly, identifying hits for in vivo validation, all deployable remotely without intervention.
Week 4 HW: Protein Design Part I
A. Conceptual Question
- Molecules of Amino Acids in 500g Meat
A typical 500 g serving of lean meat contains about 125–150 g of protein (≈25–30% by weight). Taking 130 g as an average and assuming an average amino acid residue mass of ~110 g/mol, this corresponds to:
130 g ÷ 110 g/mol ≈ 1.18 mol of amino acid residues.
Multiplying by Avogadro’s number (6.022 × 10²³ mol⁻¹) gives:
≈ 7 × 10²³ amino acid molecules.
Therefore, a 500 g serving of lean meat provides on the order of 10²⁴ amino acid molecules
- Why Humans Eat Beef/Fish Without Becoming Them
Humans do not become cows or fish after eating them because dietary proteins are first degraded by proteolysis in the gastrointestinal tract into free amino acids and small peptides. The original protein sequence information is destroyed during digestion.These amino acids enter the bloodstream and are reused by ribosomes to synthesize human proteins according to mRNA transcribed from the human genome, following the central dogma (DNA → RNA → protein).Protein synthesis is directed exclusively by endogenous gene expression, and dietary nucleic acids are also degraded into nucleotides before absorption. Therefore, body structure and identity are determined by the host genome, not by consumed biomolecules.
- Why Only 20 Natural Amino Acids
The 20 canonical amino acids were likely selected through evolutionary processes because they provided sufficient chemical diversity while maintaining translational efficiency and genetic stability. Some, such as glycine and alanine, were probably abundant under prebiotic conditions.Together, the 20 amino acids span a broad range of chemical properties — including size, charge, hydrophobicity, and reactivity — enabling the formation of stable and functionally diverse protein structures.
They are encoded within a 64-codon triplet genetic system that provides redundancy and mutational robustness. Expanding the amino acid repertoire beyond 20 may not have offered sufficient selective advantage to offset the increased complexity of translation machinery. Additionally, once the genetic code became established, it likely became evolutionarily constrained (“frozen”), limiting further expansion. Rare additions such as selenocysteine require specialized recoding mechanisms and do not fundamentally alter the canonical set.
- Prebiotic Origin of Amino Acids
Before the emergence of life and enzymatic pathways, amino acids likely formed abiotically through several prebiotic chemical processes. Laboratory simulations such as the Miller–Urey experiment demonstrated that amino acids can be synthesized in reducing gas mixtures (e.g., CH₄, NH₃, H₂O, H₂) subjected to electrical discharges.Additional mechanisms include UV-driven photochemistry in atmospheric gases and geochemical reactions in hydrothermal environments, where mineral surfaces may have facilitated Strecker-type synthesis from aldehydes, hydrogen cyanide (HCN), and ammonia.
Extraterrestrial delivery also contributed to the prebiotic pool, as carbonaceous meteorites such as the Murchison meteorite contain more than 70 amino acids.Under certain environmental conditions, such as drying–wetting cycles or mineral-catalyzed reactions, these amino acids may have undergone condensation reactions to form short peptides, providing building blocks for early biochemical evolution.
- Handedness of D-Amino Acid α-Helix
An α-helix composed entirely of D-amino acids would adopt a left-handed helical conformation, which is the mirror image of the right-handed α-helix formed by L-amino acids.This occurs because chirality determines the energetically allowed φ and ψ backbone dihedral angles. In D-amino acids, the Ramachandran plot is effectively mirrored relative to L-amino acids, shifting the energy minimum to the opposite quadrant.As a result, the helical geometry is inverted while preserving the characteristic i→i+4 hydrogen bonding pattern of the α-helix. Experimental studies of synthetic D-peptides confirm the formation of stable left-handed α-helices.
- Additional Helices in Proteins
Beyond the canonical α-helix (3.6 residues per turn, i→i+4 hydrogen bonding), proteins can adopt additional helical conformations.The 3₁₀-helix contains approximately 3 residues per turn with i→i+3 hydrogen bonding and is often observed as short segments, frequently at α-helix termini. The π-helix has about 4.4 residues per turn with i→i+5 hydrogen bonding and typically appears as local insertions (π-bulges) within α-helices, sometimes contributing to functional or ligand-binding sites.
Additionally, the polyproline II (PPII) helix is an extended, left-handed helix lacking classical intrahelical hydrogen bonds. It is common in intrinsically disordered regions, and individual collagen chains adopt a PPII-like conformation before assembling into a triple helix. These examples demonstrate that protein secondary structure includes multiple helical geometries beyond the classical α-helix.
- Why Most Molecular Helices Right-Handed
Most biological helices are right-handed because proteins are composed almost exclusively of L-amino acids. The L-configuration at the α-carbon restricts the backbone φ and ψ dihedral angles such that the right-handed α-helix occupies the lowest-energy region of the Ramachandran plot (approximately φ ≈ −60°, ψ ≈ −45°). In contrast, a left-handed α-helix for L-amino acids falls into a higher-energy region due to unfavorable steric and torsional interactions. Thus, right-handed helices are energetically preferred. This molecular asymmetry arises from biological homochirality and contributes to the predominance of right-handed helices in proteins and many nucleic acid structures, although rare left-handed forms such as Z-DNA also exist.
- Why β-Sheets Aggregate
β-sheets aggregate as unsatisfied H-bond donors/acceptors at strand edges pair intermolecularly with adjacent sheets, propagating fibrils; flat sheet geometry exposes hydrophobic faces, driving lateral association in water. This is entropically favored by releasing structured water.
Driving Force
Primary force is hydrophobic burial of nonpolar side chains (e.g., valine, leucine) between sheets, supplemented by van der Waals packing and H-bonds; electrostatics (salt bridges) fine-tune. Thermodynamic stability (low free energy) from these non-covalent interactions exceeds soluble states.
- Why Amyloid Diseases Form β-Sheets
Many amyloid diseases arise because certain proteins can misfold into β-sheet–rich conformations under conditions that destabilize their native structure or impair proteostasis. For example, amyloid-β in Alzheimer’s disease and misfolded prion protein undergo nucleation-dependent polymerization, in which exposed aggregation-prone segments template further misfolding. The resulting cross-β architecture—where β-strands run perpendicular to the fibril axis and sheets stack along it—forms extensive intermolecular hydrogen-bonding networks. This structure confers high thermodynamic stability, protease resistance, and kinetic persistence.
As materials, amyloid fibrils self-assemble into highly ordered nanofibers with mechanical stiffness in the gigapascal range. Their nanoscale organization and robustness have inspired applications in biomaterials, hydrogels, tissue scaffolds, and bioelectronic nanowires. Engineered peptide variants can be designed to retain self-assembly properties while minimizing cytotoxicity, enabling safe material applications.
Part B: Protein Analysis and Visualization
The selected protein is the human Hemoglobin subunit beta, which plays a crucial role in transporting oxygen through the bloodstream as a key component of hemoglobin. This protein was chosen for its vital importance in human physiology, its availability of a high-quality 3D structure, and its frequent use as a model in protein structure studies.
Amino Acid Sequence
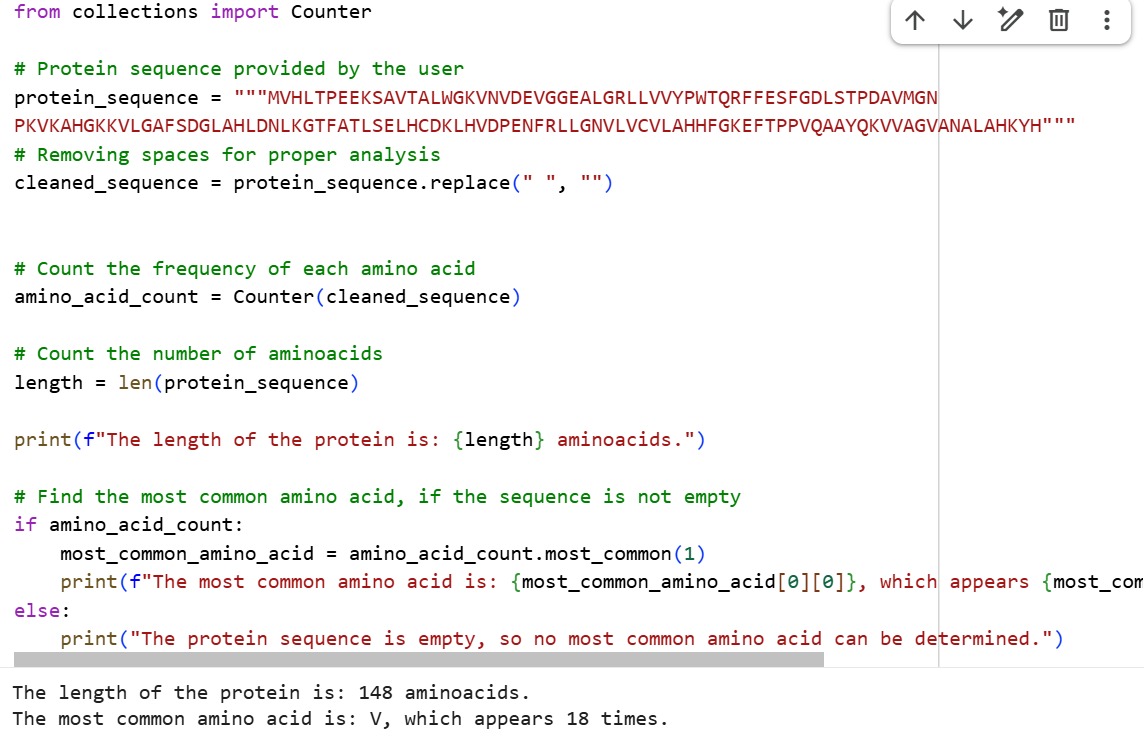
The sequence was obtained from UniProt entry P68871
MVHLTPEEKSAVTALWGKVNVDEVGGEALGRLLVVYPWTQRFFESFGDLSTPDAVMGNPKVKAHGKKVLGAFSDGLAHL
DNLKGTFATLSELHCDKLHVDPENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKVVAGVANALAHKYH
The length of the protein is: 148 aminoacids. The most common amino acid is: V, which appears 18 times.
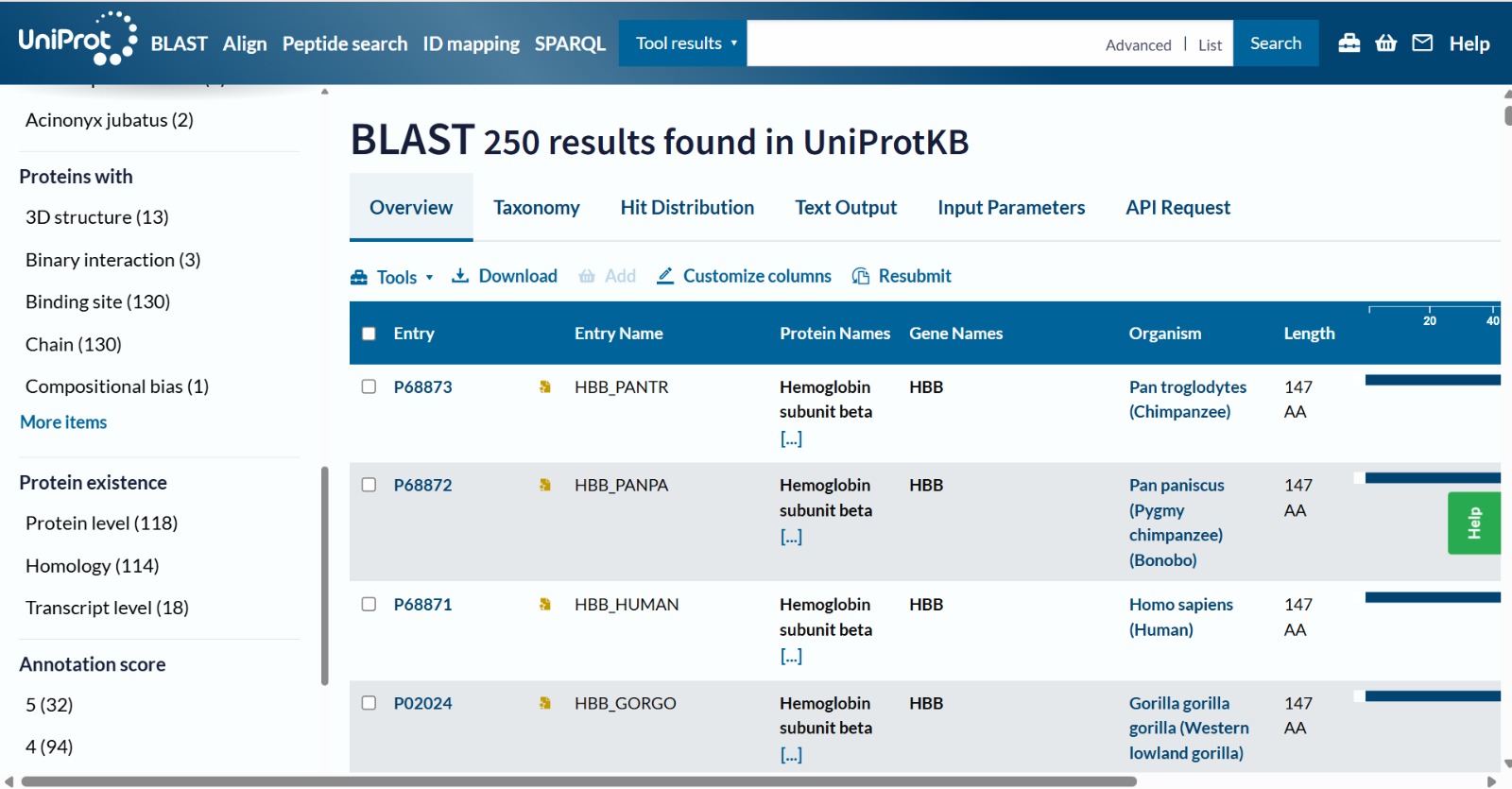
The human Hemoglobin subunit beta belongs to the hemoglobin protein family within the globin superfamily. Domain analysis from the UniProt protein database indicates that the protein contains a conserved globin domain spanning residues 1–147. Multiple protein classification databases consistently identify this protein as a member of the globin family, including Pfam (PF00042), InterPro (IPR000971), and Gene3D (Globins). InterPro further classifies it within the globin-like superfamily (IPR009050) and the hemoglobin beta subfamily (IPR002337). Additional databases such as PANTHER (PTHR11442) and SUPFAM (SSF46458) also support its classification within the globin-like structural superfamily. These annotations indicate that the protein adopts the characteristic globin fold, which enables oxygen binding through a heme prosthetic group.
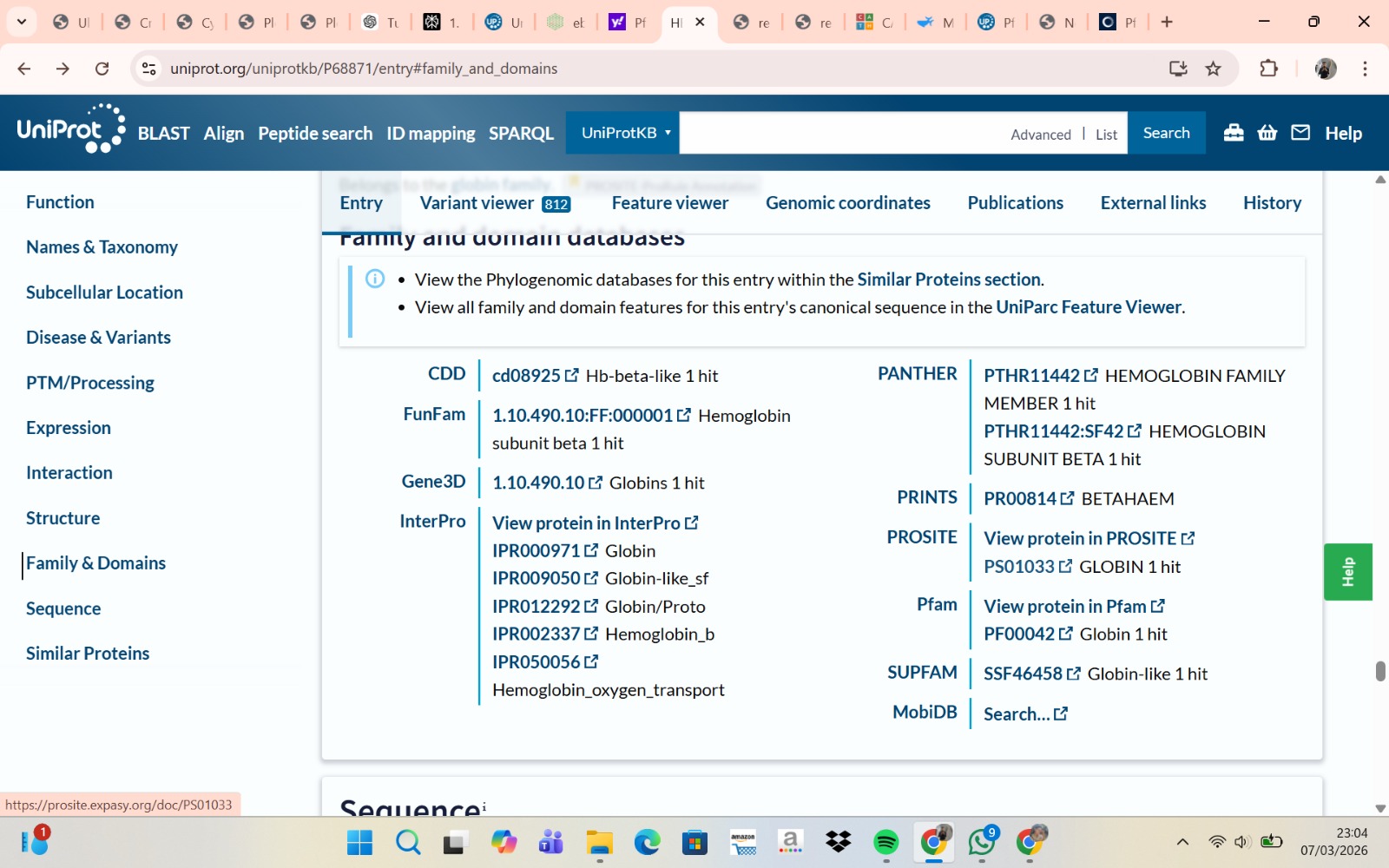
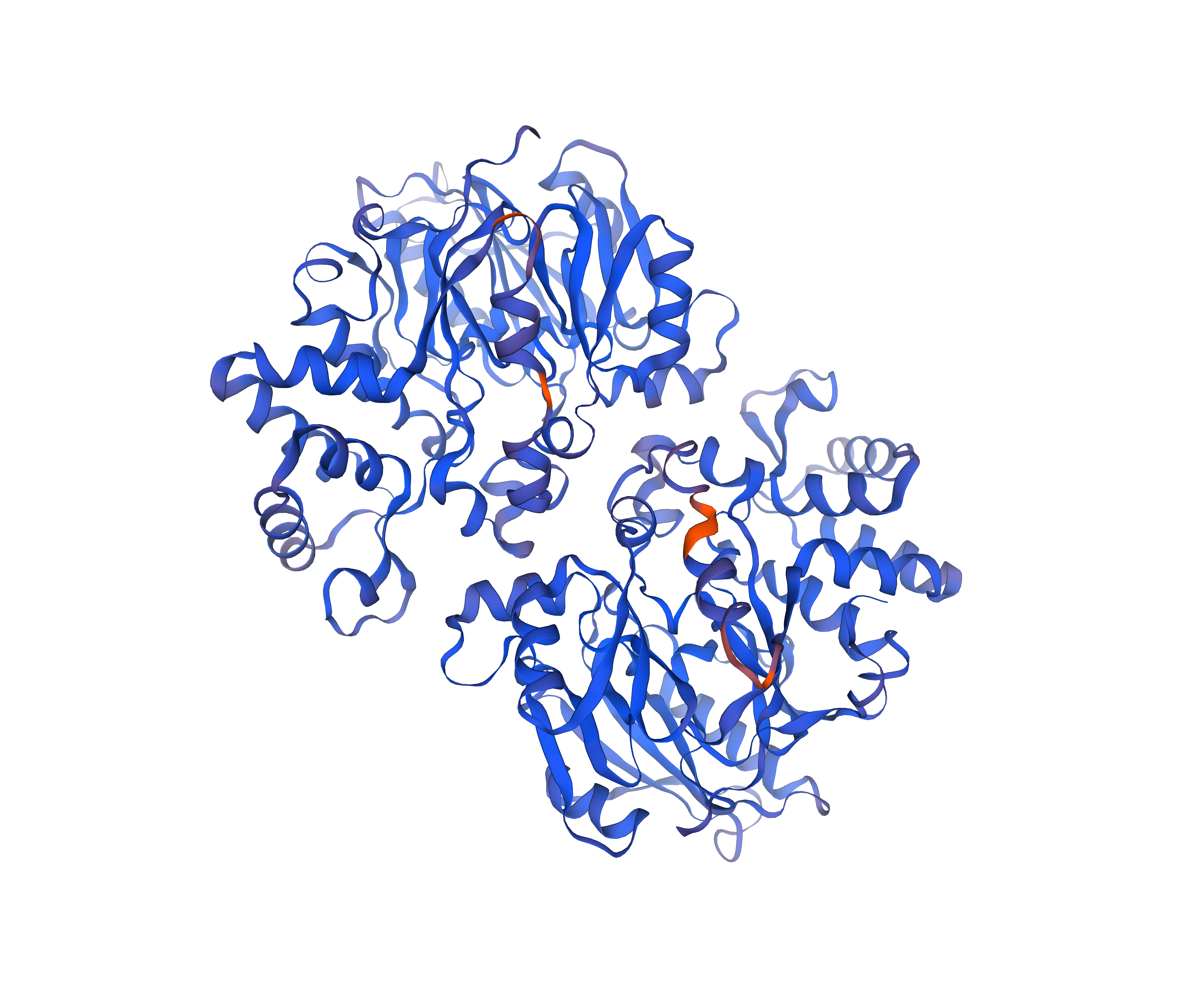
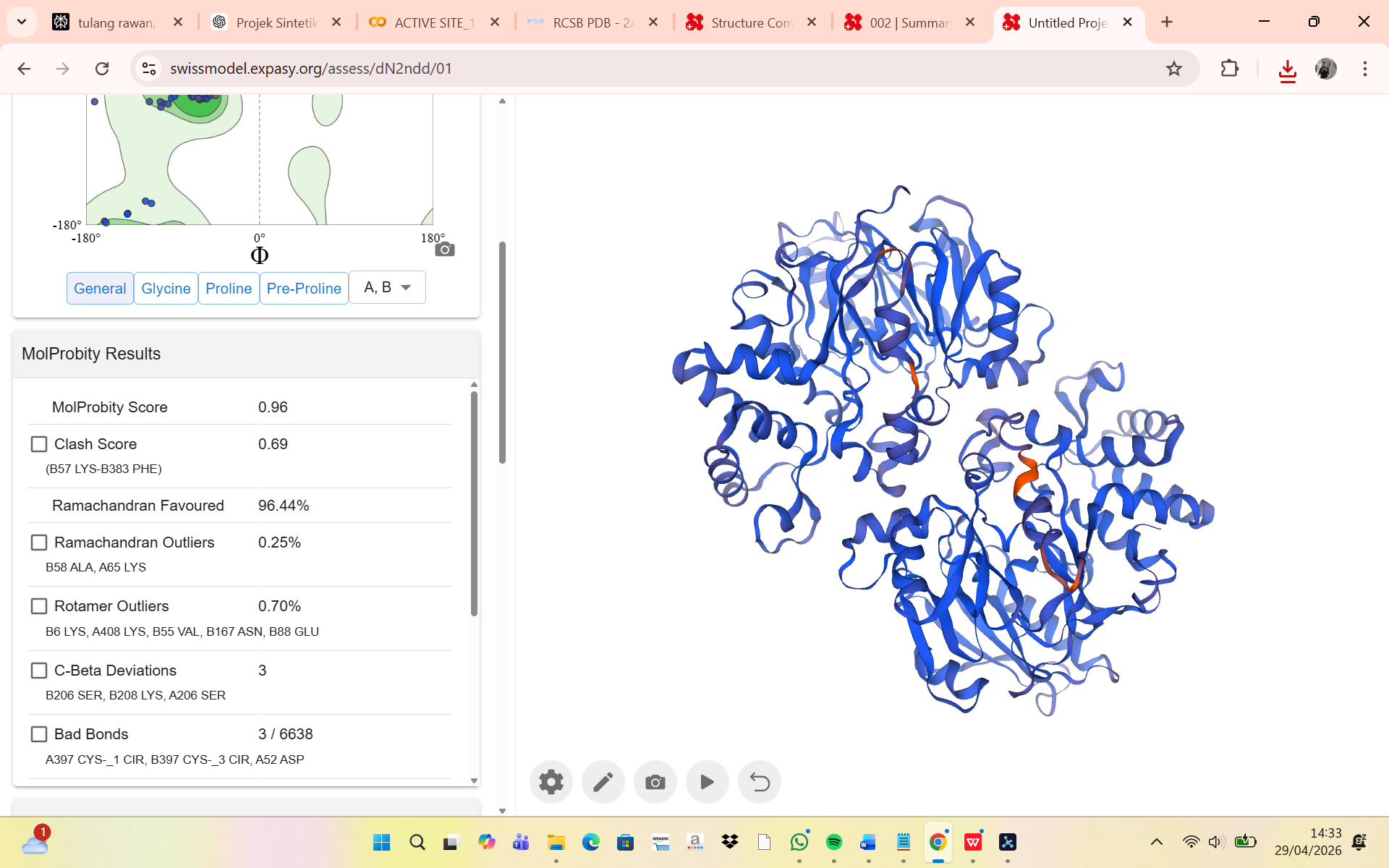
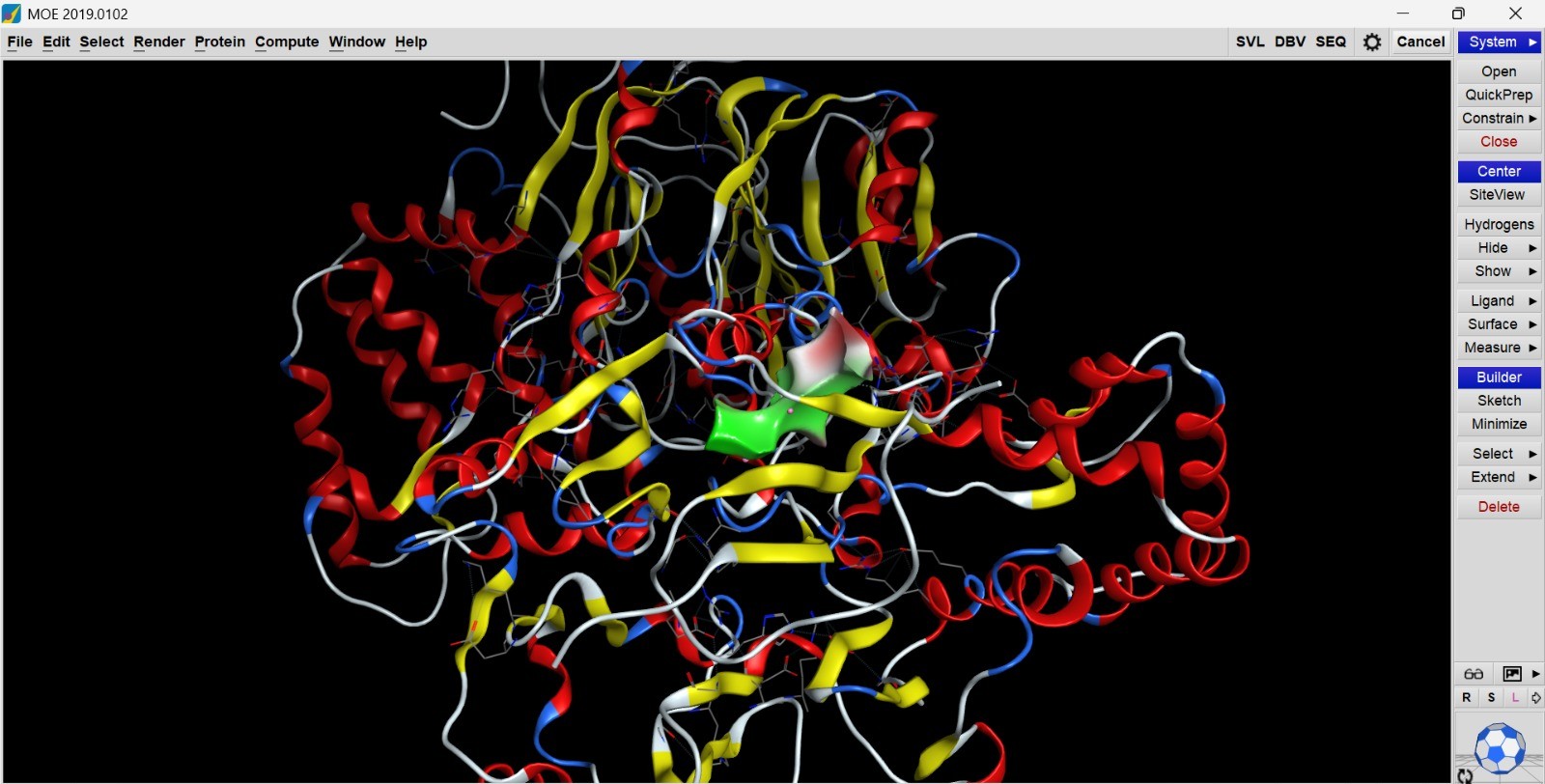
- The structure of Hemoglobin subunit beta from humans is available in the RCSB Protein Data Bank under the PDB ID 1DXT. The structure was determined using X-ray diffraction and deposited in 1992 and released in 1993. It has a resolution of 1.70 Å, indicating a high-quality structure with detailed atomic information. The biological assembly forms a hemoglobin tetramer (A2B2), consisting of two alpha and two beta subunits. In addition to the protein chains, the structure also contains a heme prosthetic group (HEM) that serves as the oxygen-binding site. Structurally, the protein belongs to the globin-like structural superfamily, characterized by a predominantly alpha-helical globin fold and the presence of a heme-binding pocket responsible for oxygen transport.
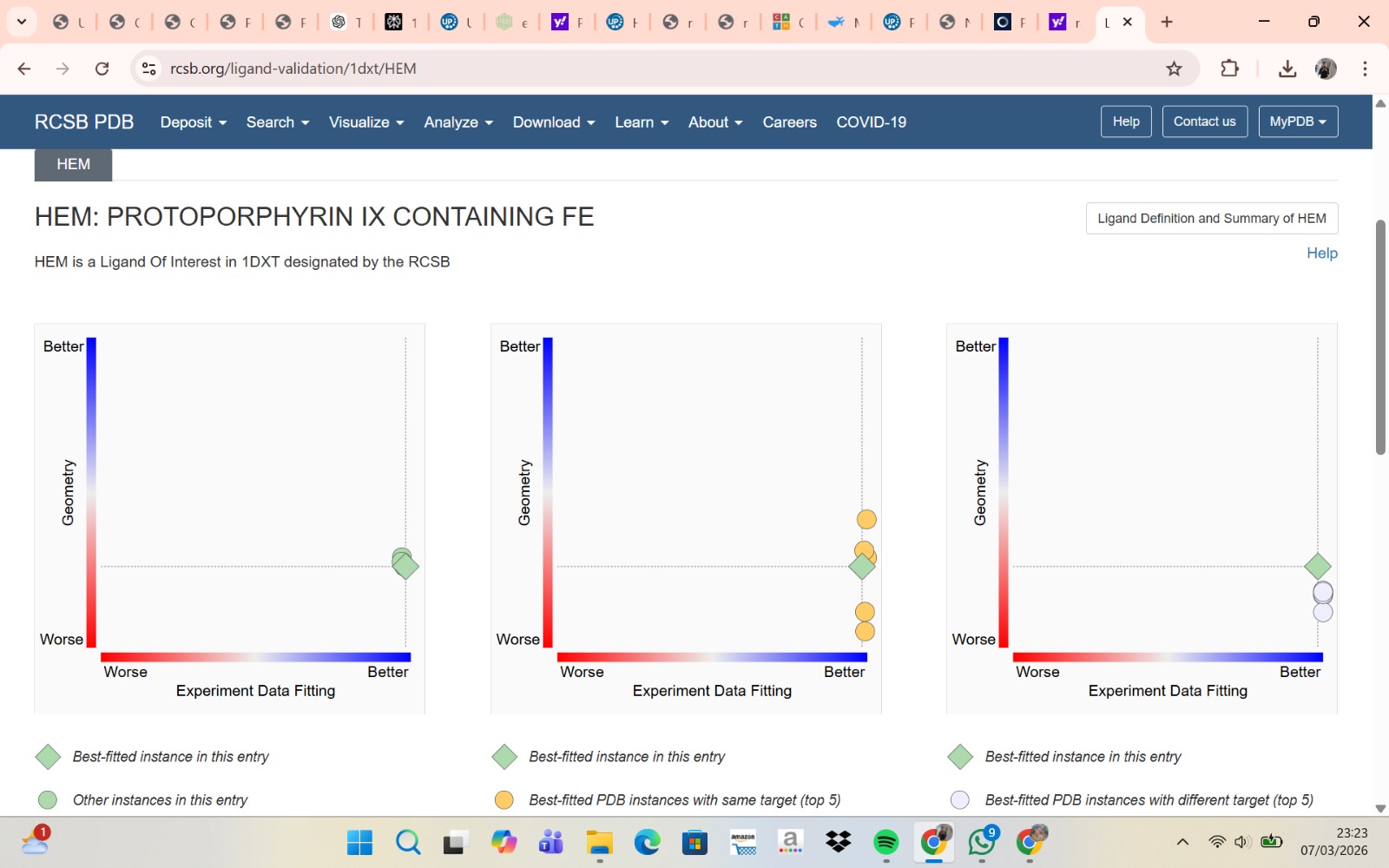
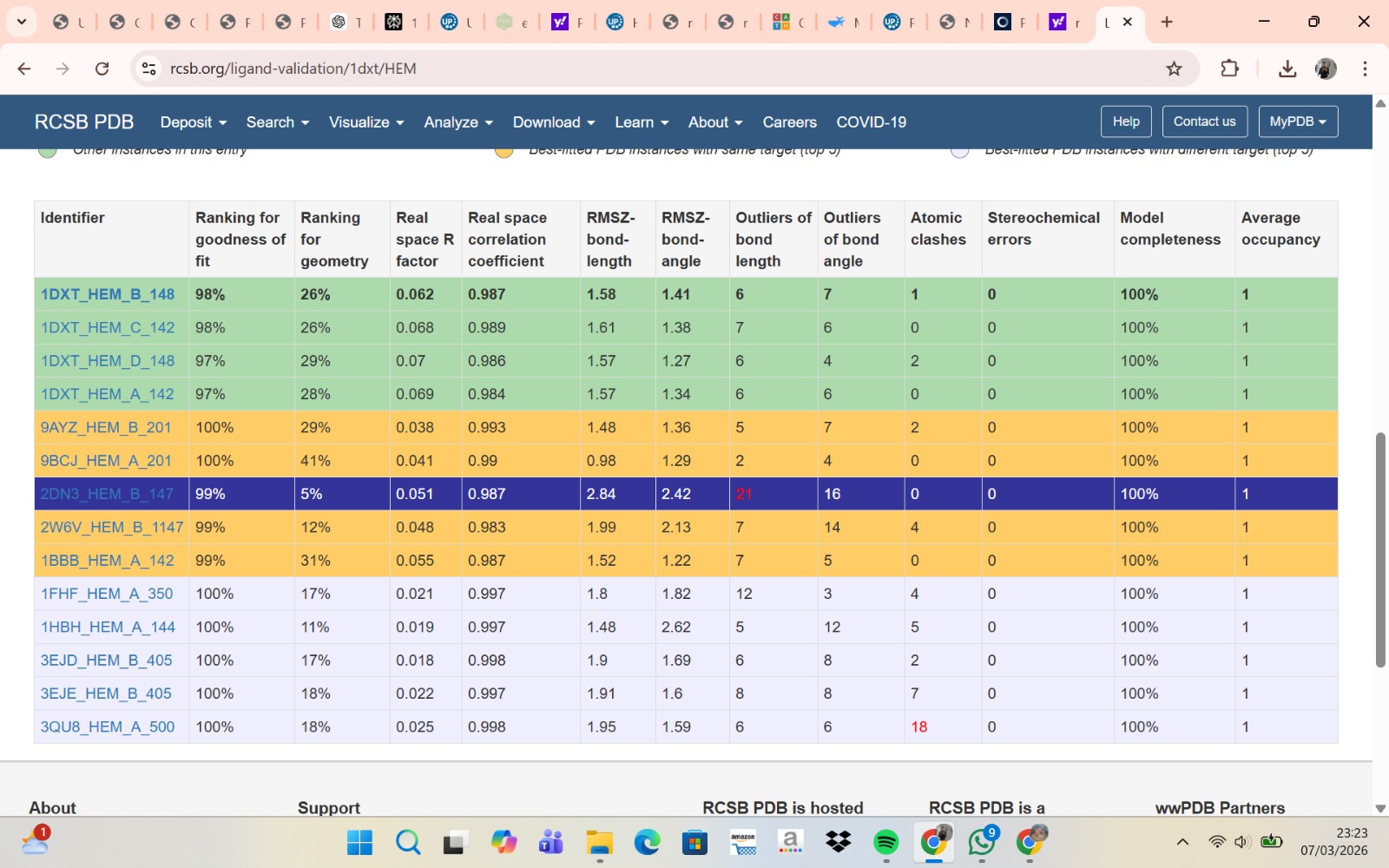
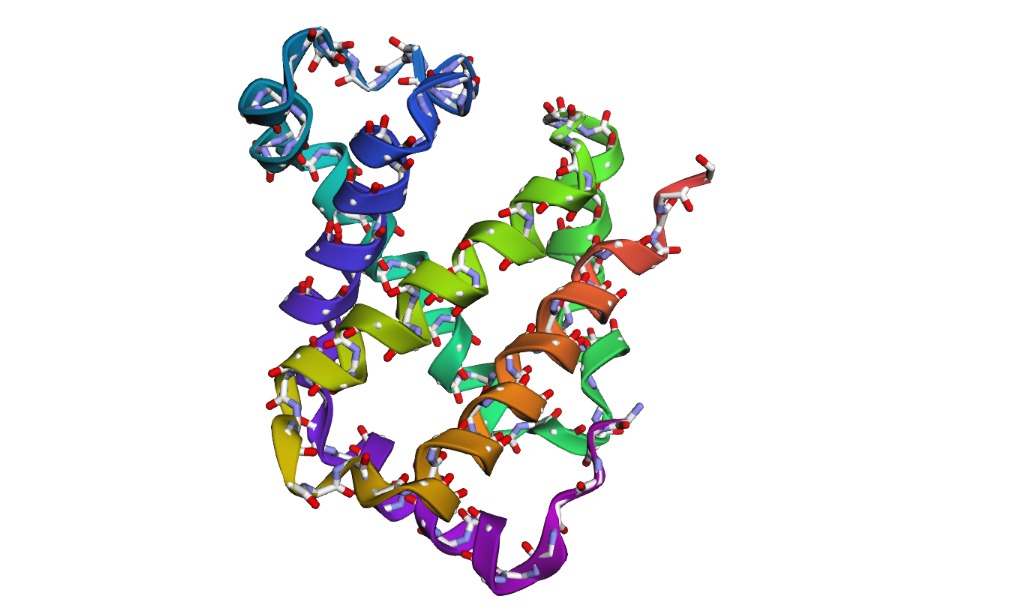
- PyMol Visualization
Visualization of the human Hemoglobin subunit beta protein using PyMOL software with stick representation and water removal.
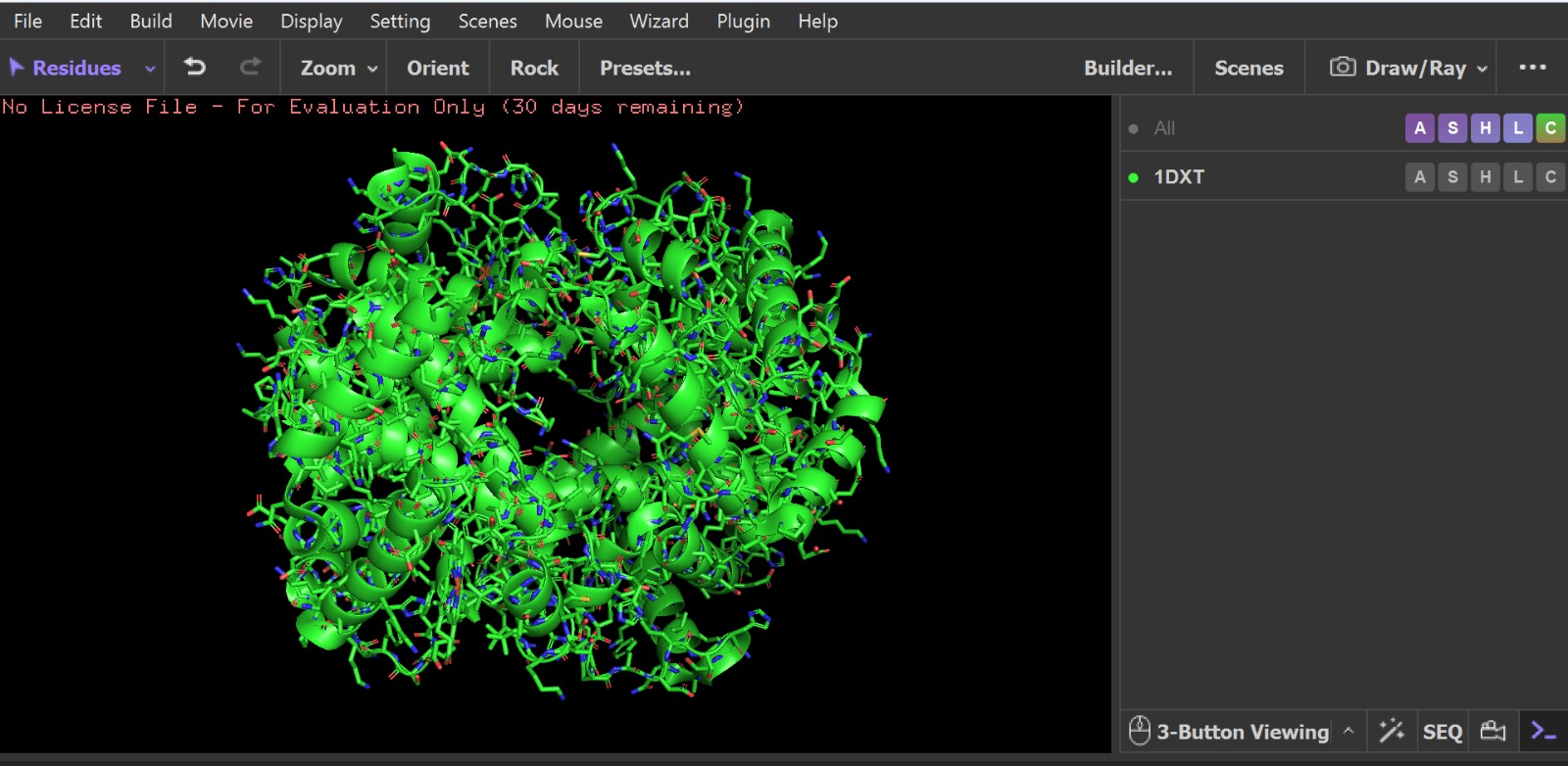
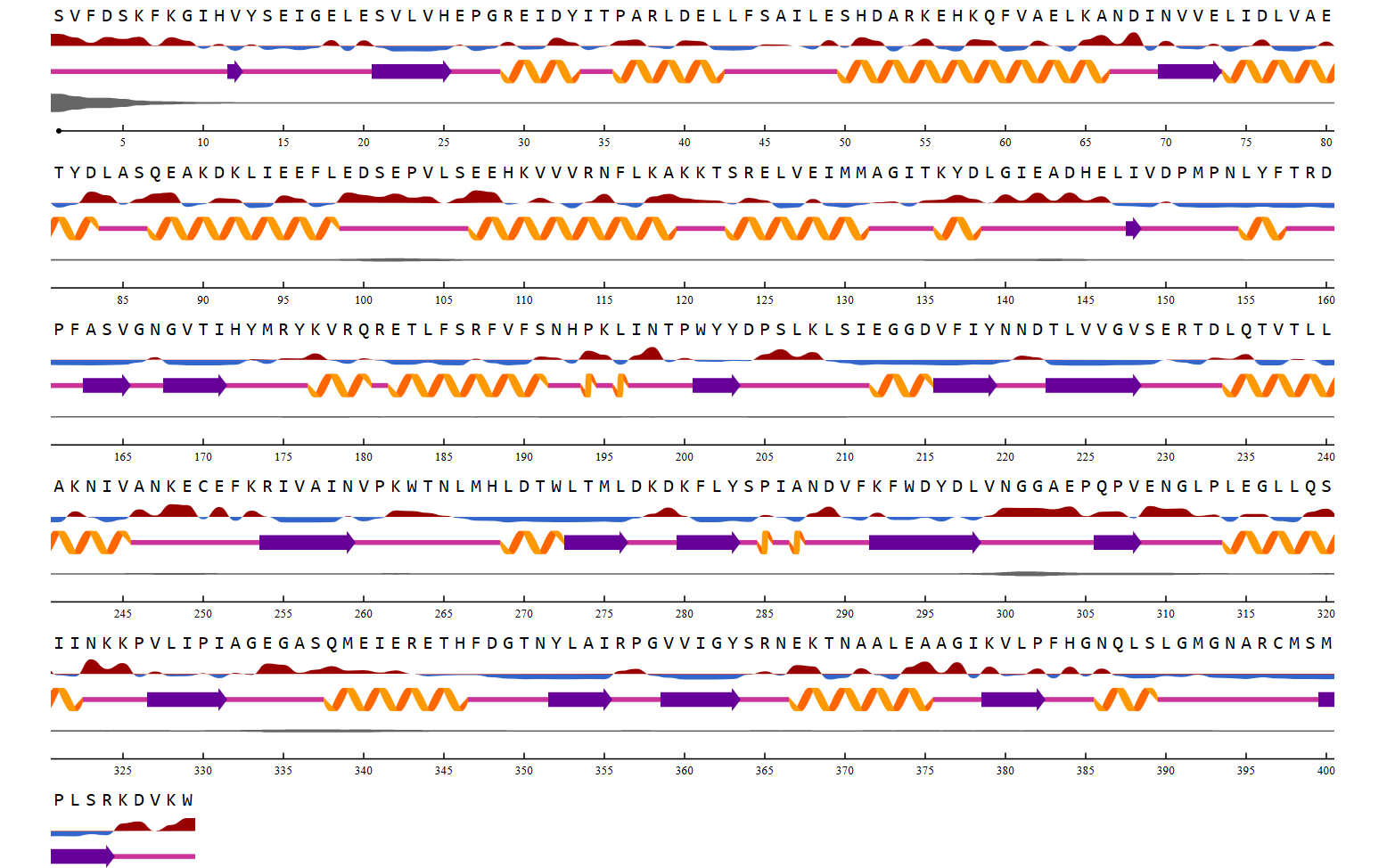
Visualization of the protein structure using PyMOL molecular visualization system colored by secondary structure shows that the structure is dominated by α-helices (red), while only a few regions correspond to loops or coils (green). No significant β-sheet structures are observed. Therefore, the protein contains many more α-helices than β-sheets. This observation is consistent with the typical globin fold of Hemoglobin subunit beta, where each subunit is primarily composed of multiple α-helices that form the structural framework around the heme-binding pocket.
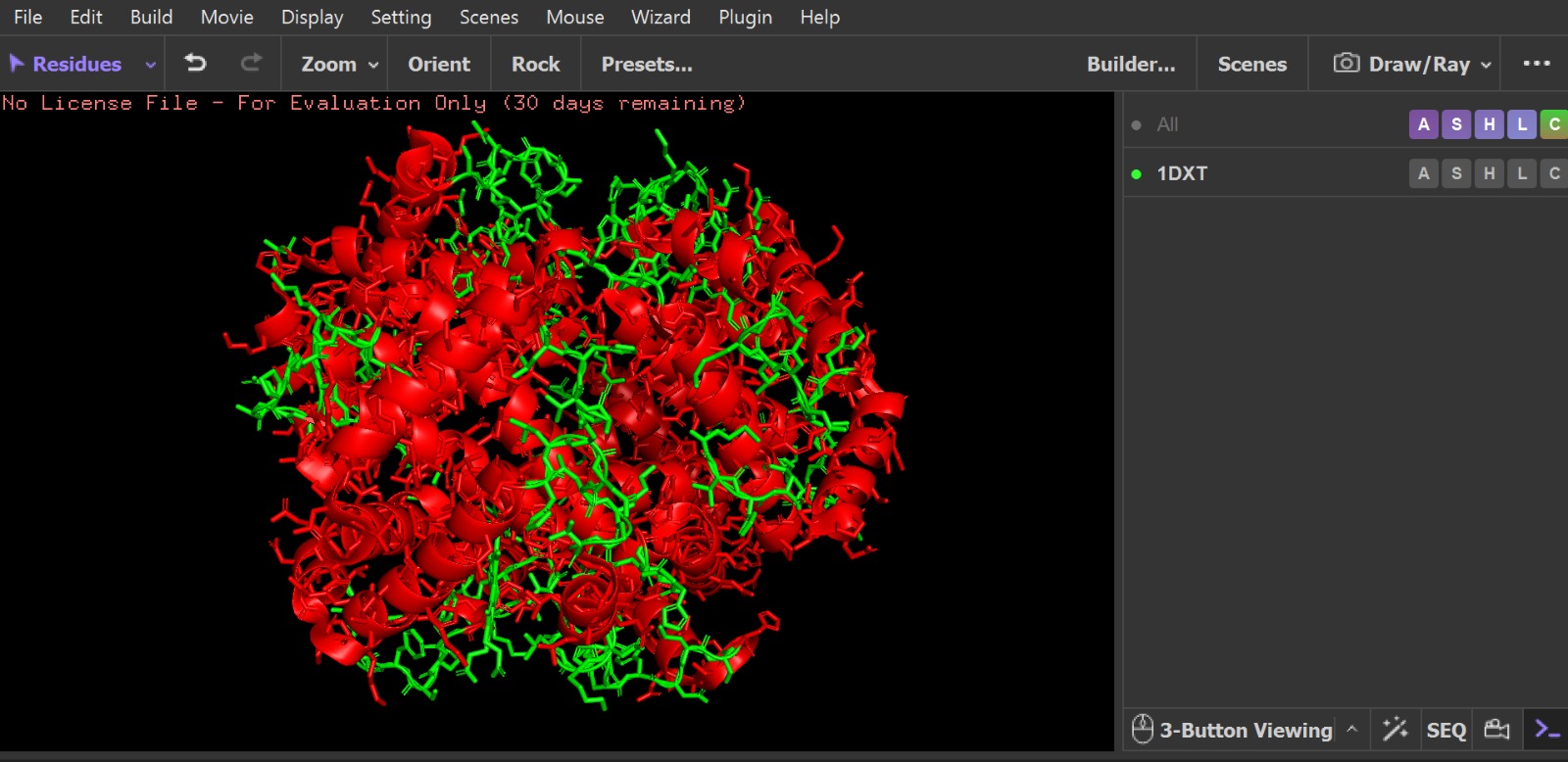
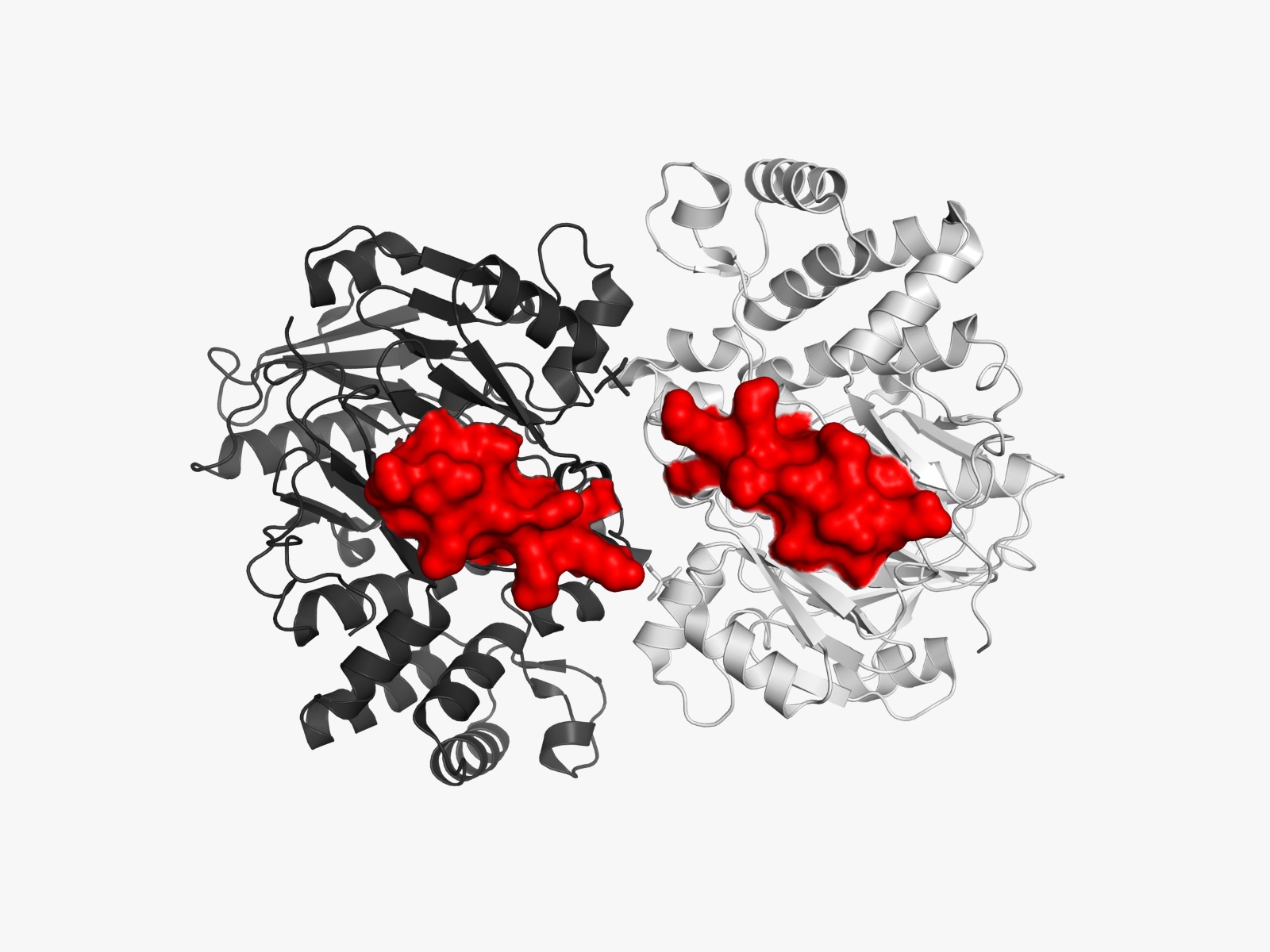
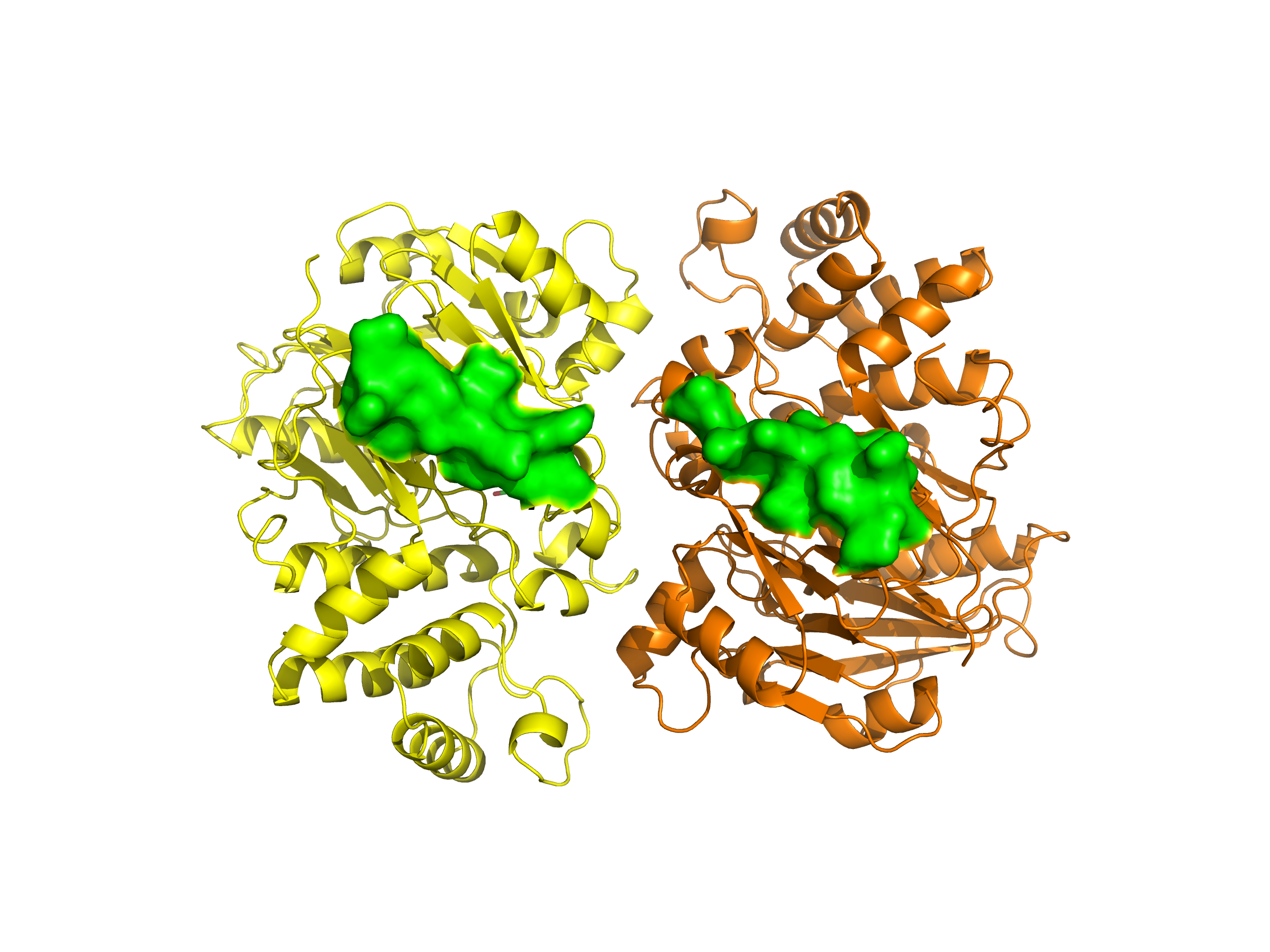
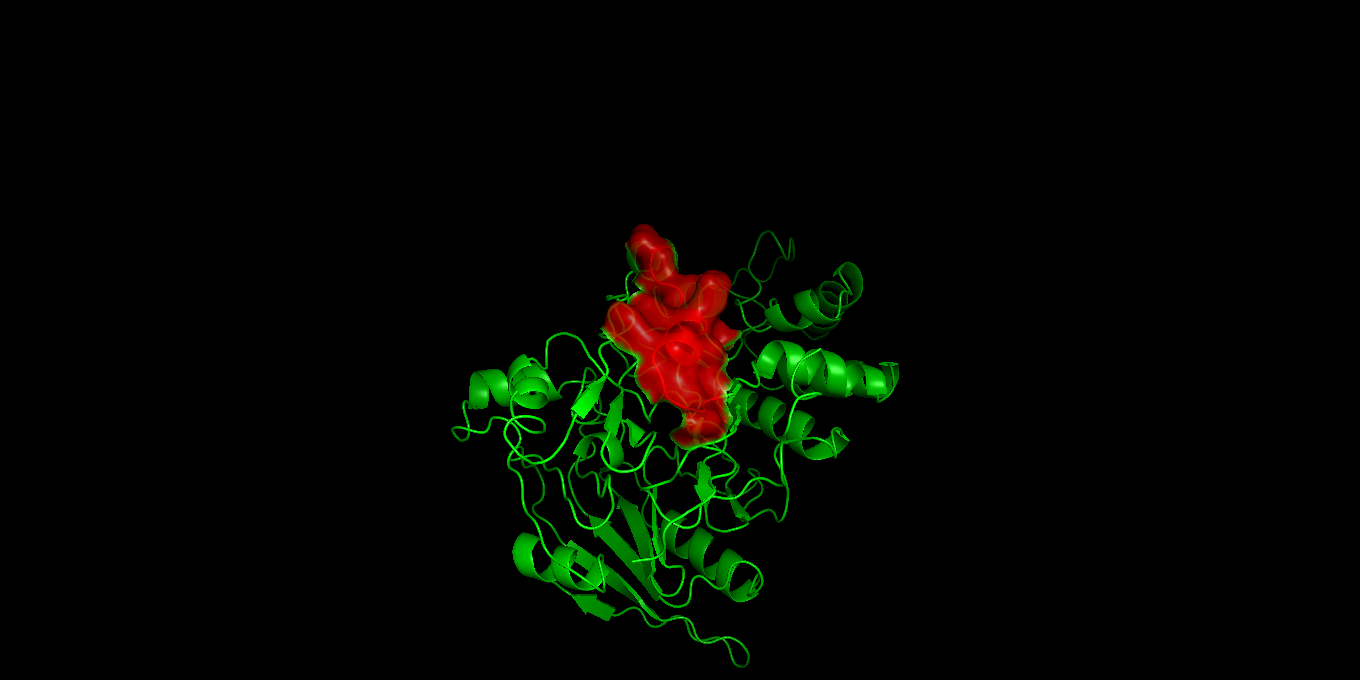
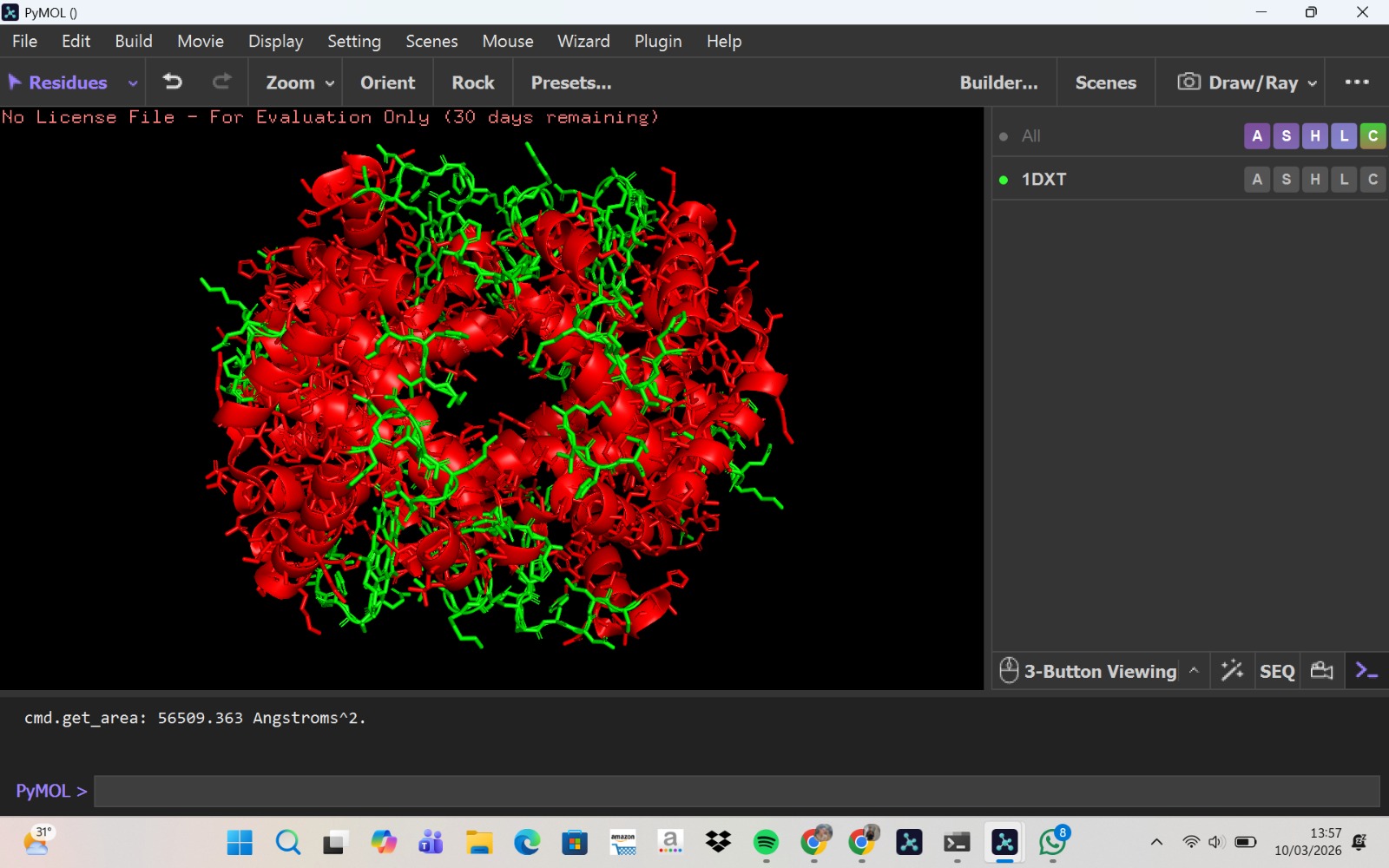
Visualization of the molecular structure of Human hemoglobin using PyMOL molecular visualization system reveals the presence of a distinct binding pocket within the protein. This pocket corresponds to the heme-binding site, where the heme prosthetic group containing an Fe²⁺ ion is located. The pocket is formed by several surrounding α-helices of the globin fold. This structural cavity allows oxygen molecules to bind to the Fe²⁺ ion in the heme group, enabling hemoglobin to perform its biological function in oxygen transport. Therefore, the protein clearly contains functional binding pockets on its surface.
Part C: Using ML-Based Protein Design Tools
C1. PROTEIN LANGUAGE MODELING
- Deep Mutational Scans
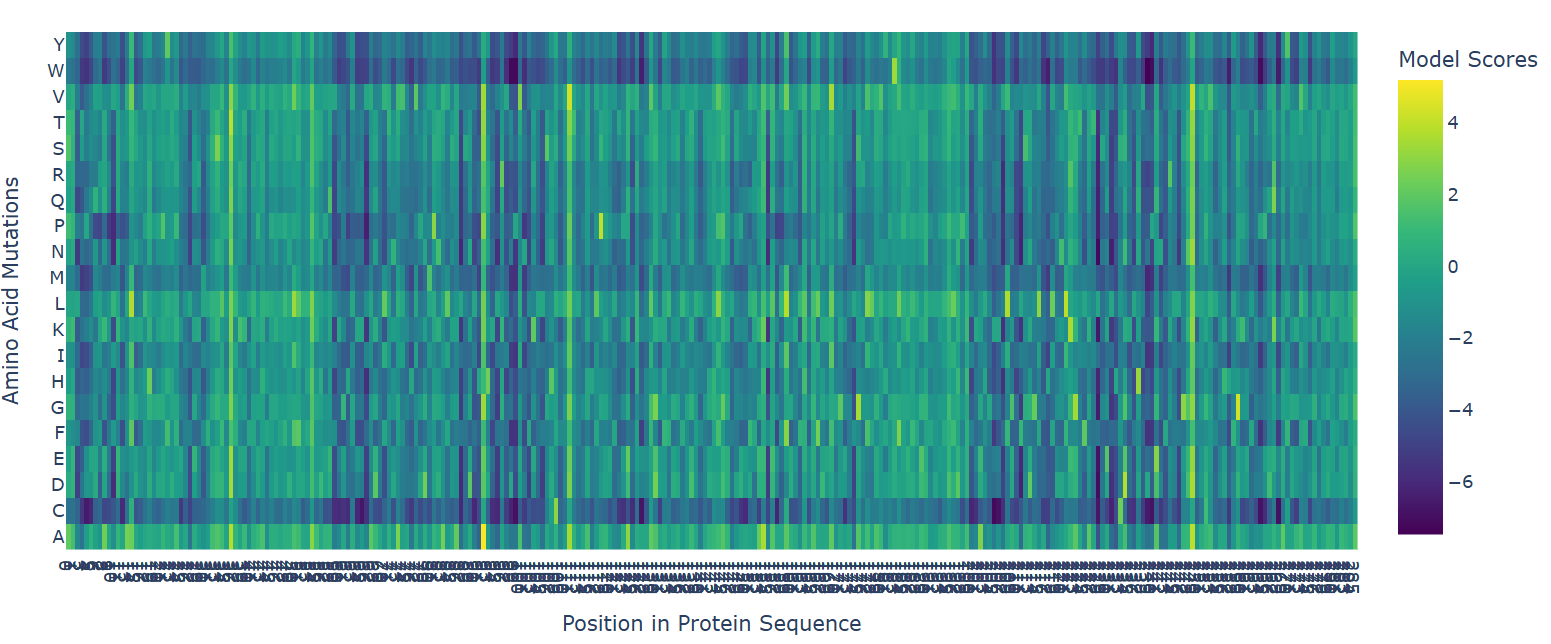
The deep mutational scan generated using the ESM2 protein language model reveals that many residues in human hemoglobin beta are highly conserved and sensitive to mutation. In the heatmap, mutations associated with strongly negative scores (dark blue/purple regions) are likely unfavorable and may disrupt protein stability or function. Since hemoglobin beta adopts a globin fold dominated by α-helices, mutations introducing structurally disruptive residues such as proline are often poorly tolerated.
One particularly important mutation is E6V, where glutamate at position 6 is replaced by valine. This mutation receives a relatively unfavorable model score, indicating reduced sequence likelihood. Biologically, E6V is the well-known mutation responsible for sickle cell disease. The substitution changes a negatively charged hydrophilic residue into a hydrophobic residue, promoting abnormal aggregation of hemoglobin molecules and deformation of red blood cells. The fact that ESM2 assigns a deleterious score to this mutation suggests that the language model successfully captures biologically meaningful constraints directly from protein sequence data.
- Latent Space Analysis
Before that, I would like to inform you that I made changes to the code you provided because I spent a lot of time fixing the errors that occurred while running it.
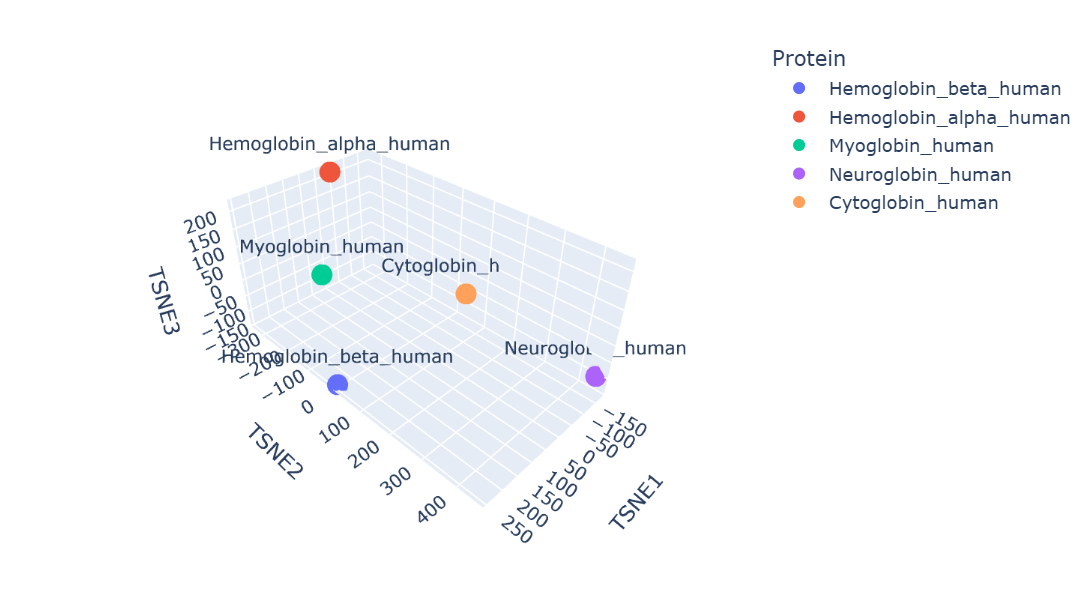
Protein embeddings generated using the ESM2 protein language model were reduced into three dimensions using t-SNE visualization. The resulting latent space demonstrates that proteins with similar sequence and structural characteristics tend to occupy nearby regions.
Human hemoglobin beta is located close to hemoglobin alpha and myoglobin, showing that these proteins have similar globin structures, oxygen-binding functions, and conserved sequence regions. Hemoglobin alpha is the closest because both proteins work together to form the hemoglobin tetramer in red blood cells. Myoglobin is also positioned nearby since it shares the globin domain and binds heme, although its role is mainly for oxygen storage in muscle. Neuroglobin and cytoglobin are placed farther away, indicating that their sequences and biological functions are more specialized and different from hemoglobin beta.
Overall, the latent space map shows that ESM2 can capture meaningful biological relationships between proteins directly from their amino acid sequences.
C2. PROTEIN FOLDING
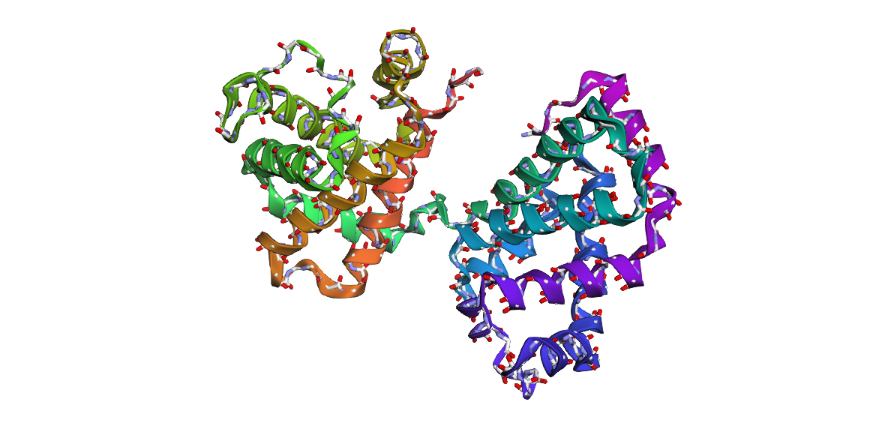
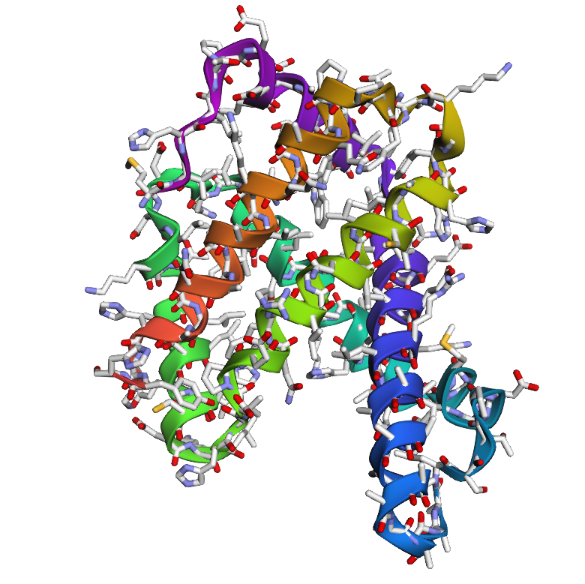
1 . The ESMFold prediction shows a structure that is highly similar to the original 1DXT hemoglobin beta structure. From the superposition images, both models have nearly the same overall globin fold, especially the dominant alpha-helical arrangement that is characteristic of hemoglobin proteins. Most helices overlap well between the experimental structure and the predicted model.
The predicted structure also preserves the general organization of the heme-binding region and the compact globin architecture. Small differences can be seen in several loop regions and flexible terminal parts, which is normal because these regions are usually more dynamic and harder to predict accurately compared to alpha-helices. Overall, the predicted coordinates match the original experimental structure quite well
1DXT
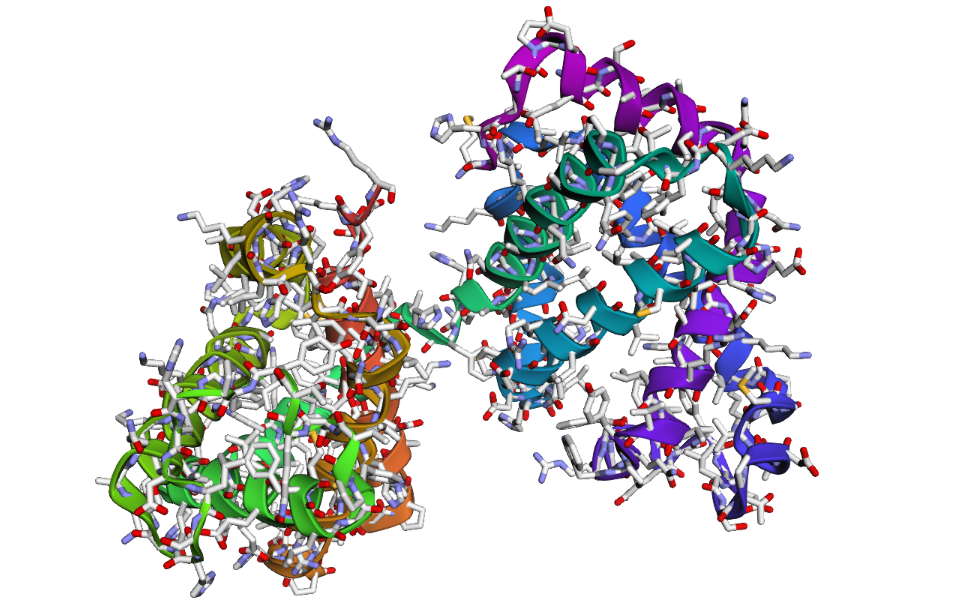
1DXT_Mutan E6V
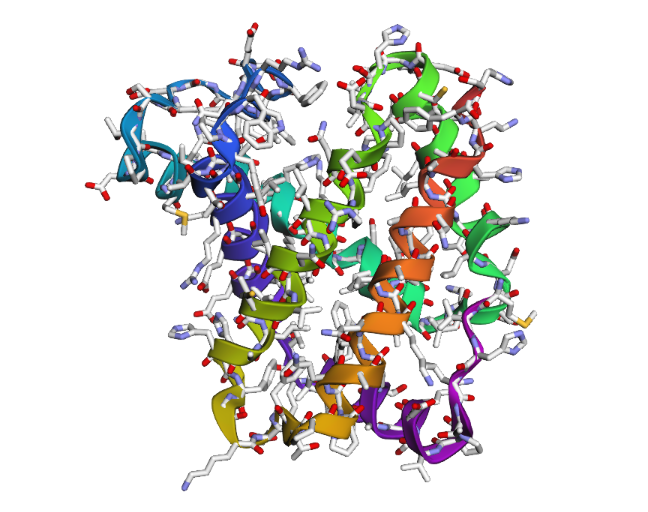
1DXT_Mutan 15-20A
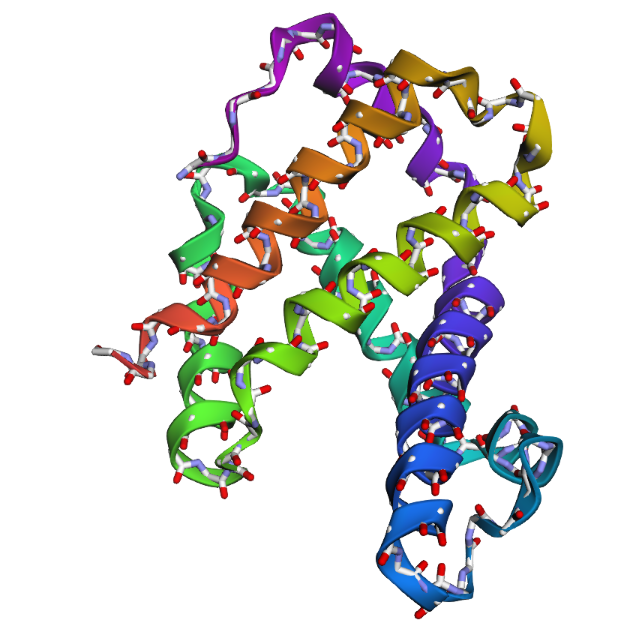
2 . After testing both types of mutations, the hemoglobin beta structure seems fairly resilient to small mutations but much less stable after large sequence changes. In the first mutation (E6V substitution), the overall structure still looks very similar to the original protein. Most of the alpha-helices are still maintained and the globin fold remains intact. The change mainly affects a local region around the mutation site, while the overall protein architecture is preserved. This suggests that hemoglobin beta can tolerate certain single amino acid mutations without major structural disruption, even though the mutation may still affect protein function biologically.
In the second mutation, where a larger segment of residues was replaced with alanine, the structure changes more noticeably. Several helices appear shifted or less organized, and the overall fold looks less compact compared to the original structure. Some regions become more flexible and lose their normal arrangement. This indicates that larger sequence modifications disrupt the stability of the protein much more strongly. Overall, the protein is relatively resilient to small point mutations, but not to large segment mutations. Conserved regions in the sequence are clearly important for maintaining the proper globin structure and stability.
C3. PROTEIN GENERATION
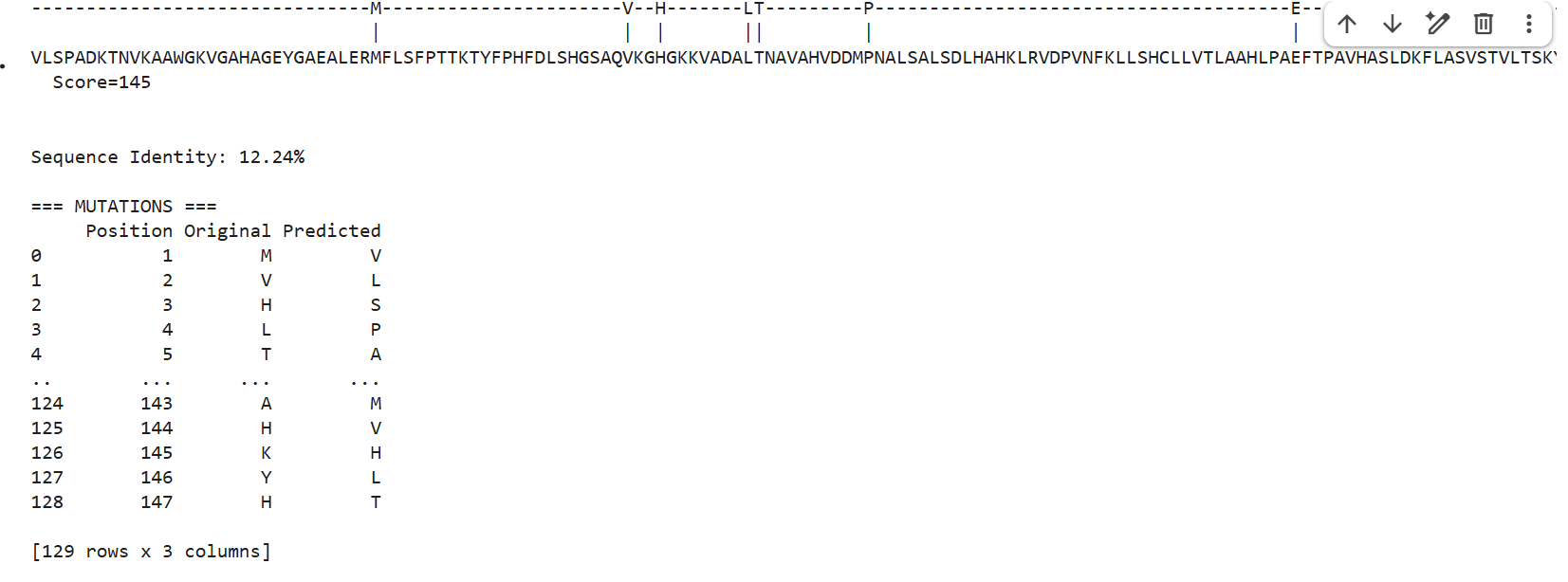
1 . ProteinMPNN generated several alternative sequences for the hemoglobin beta backbone. The predicted sequences showed relatively low sequence identity compared to the native sequence, ranging from approximately 9–12%. Despite these differences, several conserved structural patterns such as hydrophobic residues and globin-like motifs maybe were still maintained.
This suggests that multiple amino acid sequences may be compatible with the same overall globin fold. ProteinMPNN appears to preserve residues important for maintaining secondary structure and backbone stability while allowing substantial variation in less constrained positions.
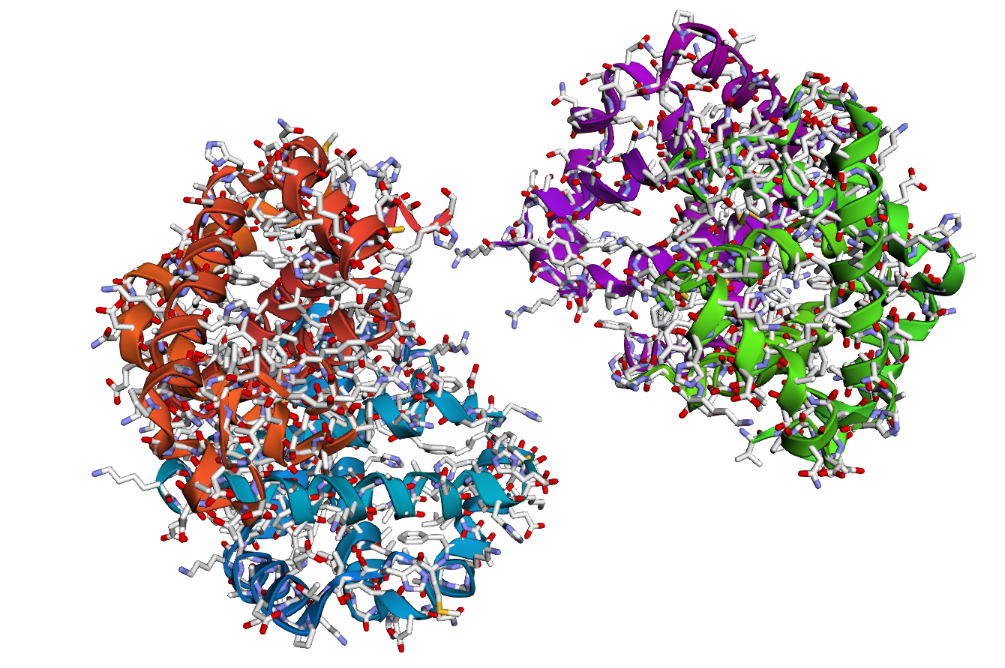
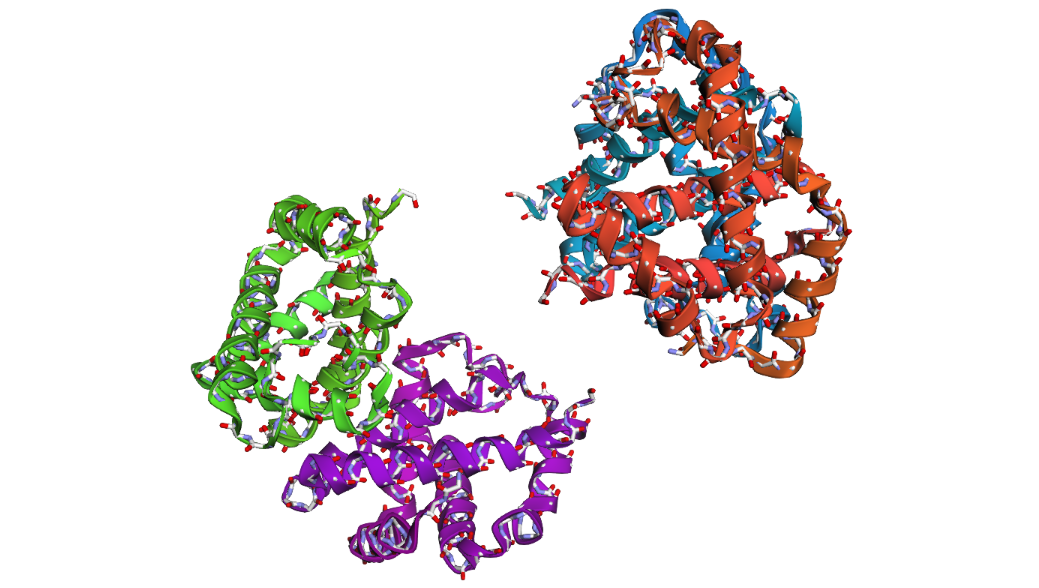
2 . After folding the ProteinMPNN-designed sequence with ESMFold, the predicted structure looked noticeably different from the original hemoglobin beta structure. Several helices were disrupted and the overall globin fold was less organized compared to the native protein. This suggests that although some sequence variation is possible, large changes in the amino acid sequence can strongly affect the stability and shape of the protein structure.
Week 5 HW: Protein Design Part II
PART 1 Generate Binders with PepMLM
The human SOD1 protein sequence was retrieved from the UniProt database (P00441). To model a disease-associated variant, the A4V mutation was introduced by substituting alanine with valine at residue position 4 of the protein sequence. This mutation is known to be associated with amyotrophic lateral sclerosis (ALS). The resulting mutant SOD1 sequence was then used as the input for subsequent peptide binder generation using the PepMLM model.
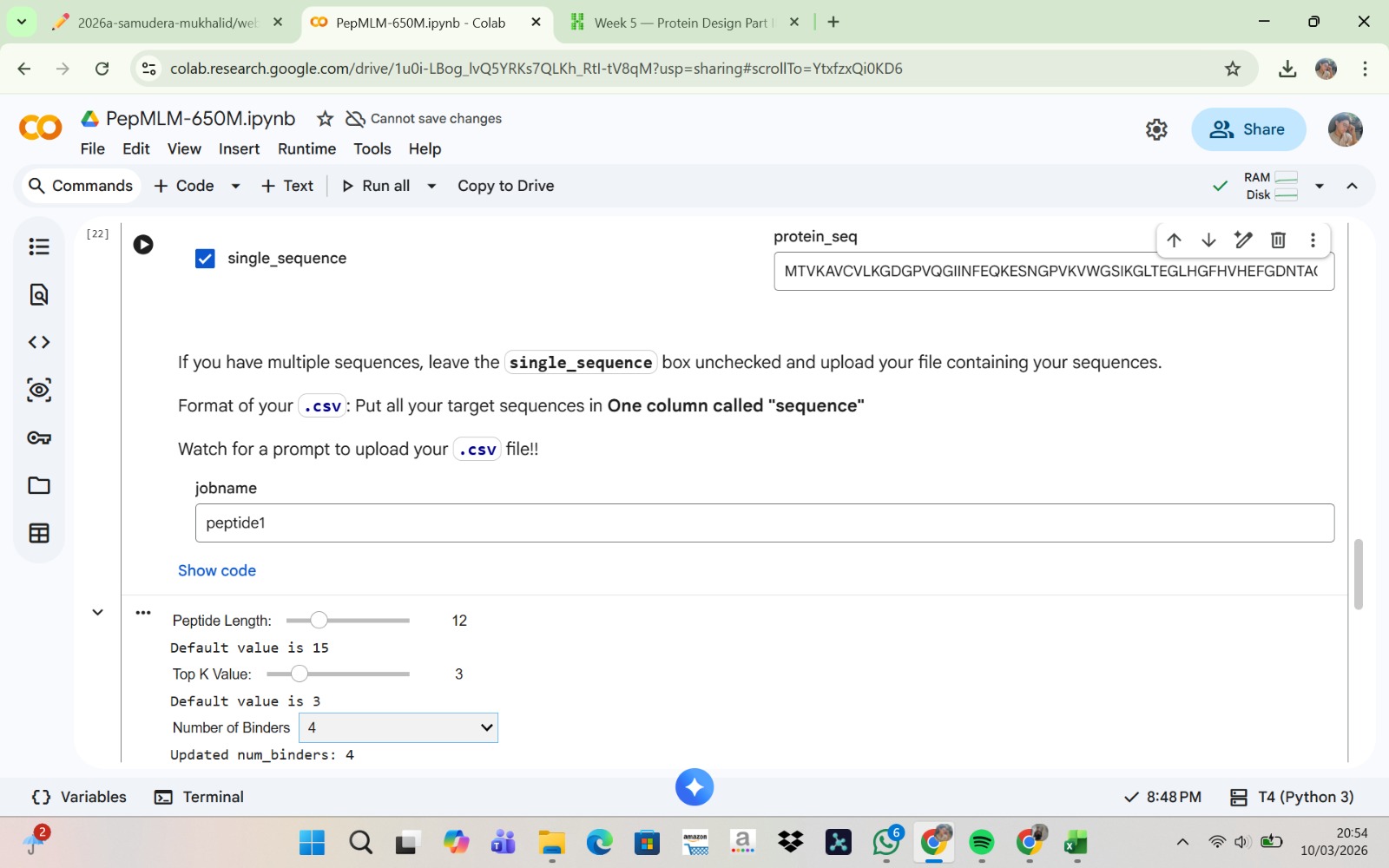
In bioinformatics, the letter “X” in peptide sequences denotes an unknown or ambiguous amino acid, often arising from prediction model uncertainties during protein structure analysis. For instance, sequences like WRYYAYAIRWKX, HLVPAVAIEHKX, and WRYPAAAARLKX feature an unclear final residue, which disrupts processing in tools like AlphaFold3 as they do not support non-standard characters. To address this, X is typically replaced with the most contextually reasonable residue, such as K (lysine). This substitution is favored because lysine enhances electrostatic binding—via its positive charge that attracts negatively charged molecules—and improves overall peptide solubility, ensuring greater stability and compatibility for downstream simulations.
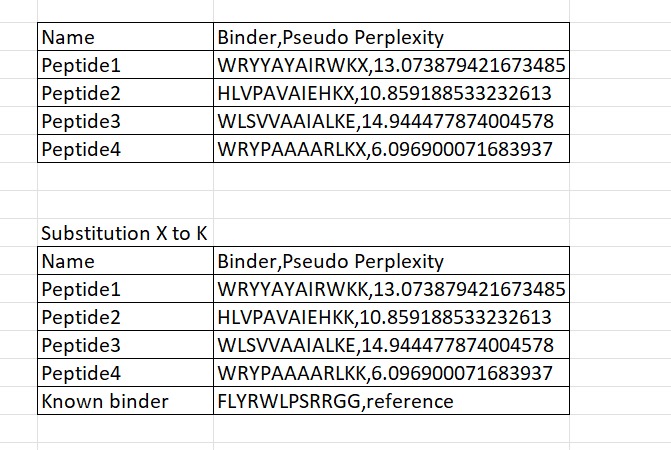
Four peptides of length 12 amino acids were generated using the PepMLM model conditioned on the mutant SOD1 A4V sequence. The generated peptides showed pseudo-perplexity scores ranging from 6.10 to 14.94, reflecting varying levels of model confidence. Among the generated candidates, peptide WRYPAAAARLKK displayed the lowest perplexity score (6.10), suggesting the highest likelihood of being a plausible binder according to the model. For comparison, the previously reported SOD1-binding peptide FLYRWLPSRRGG was included as a reference. These peptides were then used for further structural evaluation using AlphaFold3.
Part 2 Evaluate Binders with AlphaFold3
Peptide 1 : WRYYAYAIRWKK
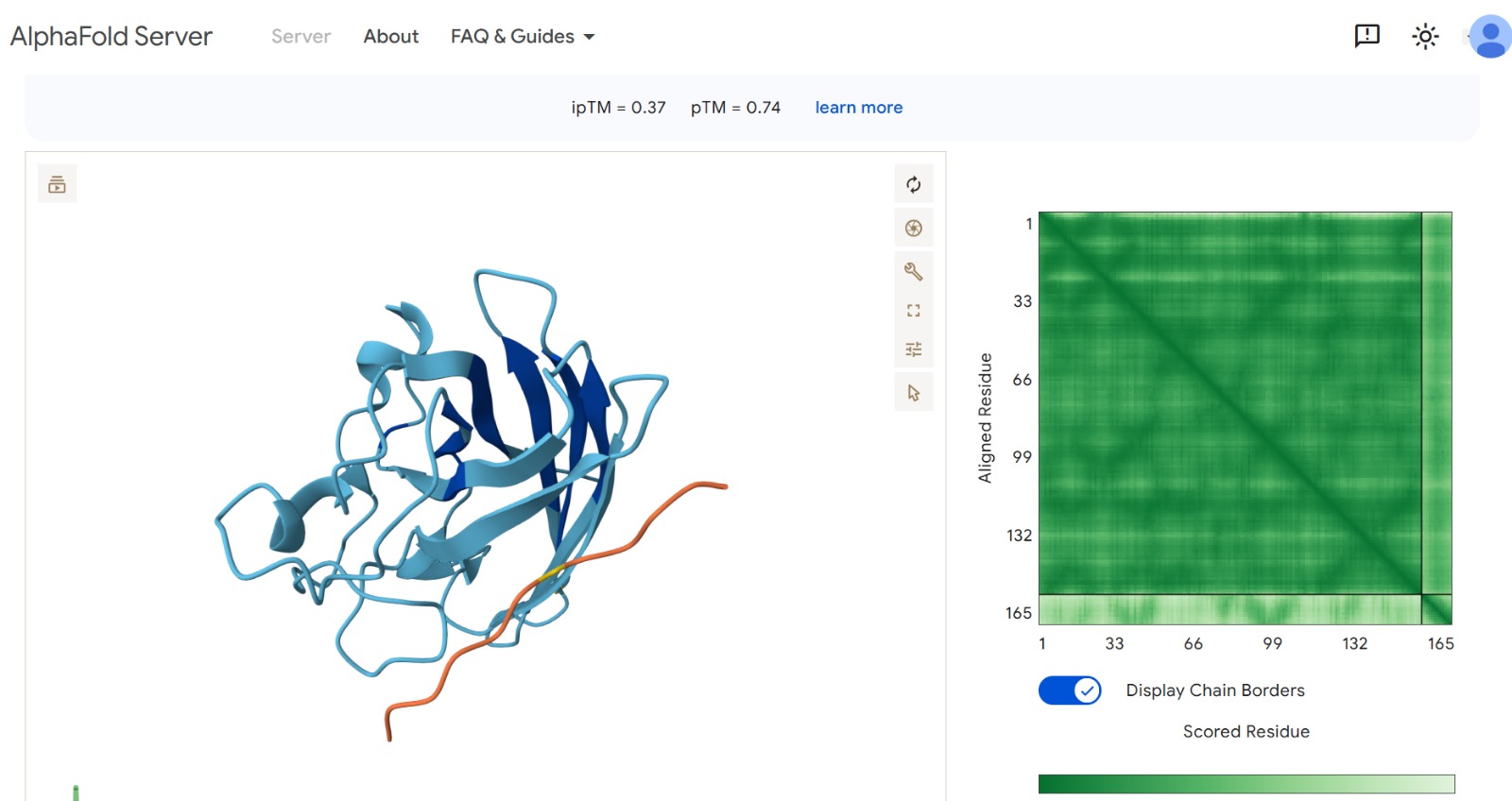
Peptide 2 : HLVPAVAIEHKK
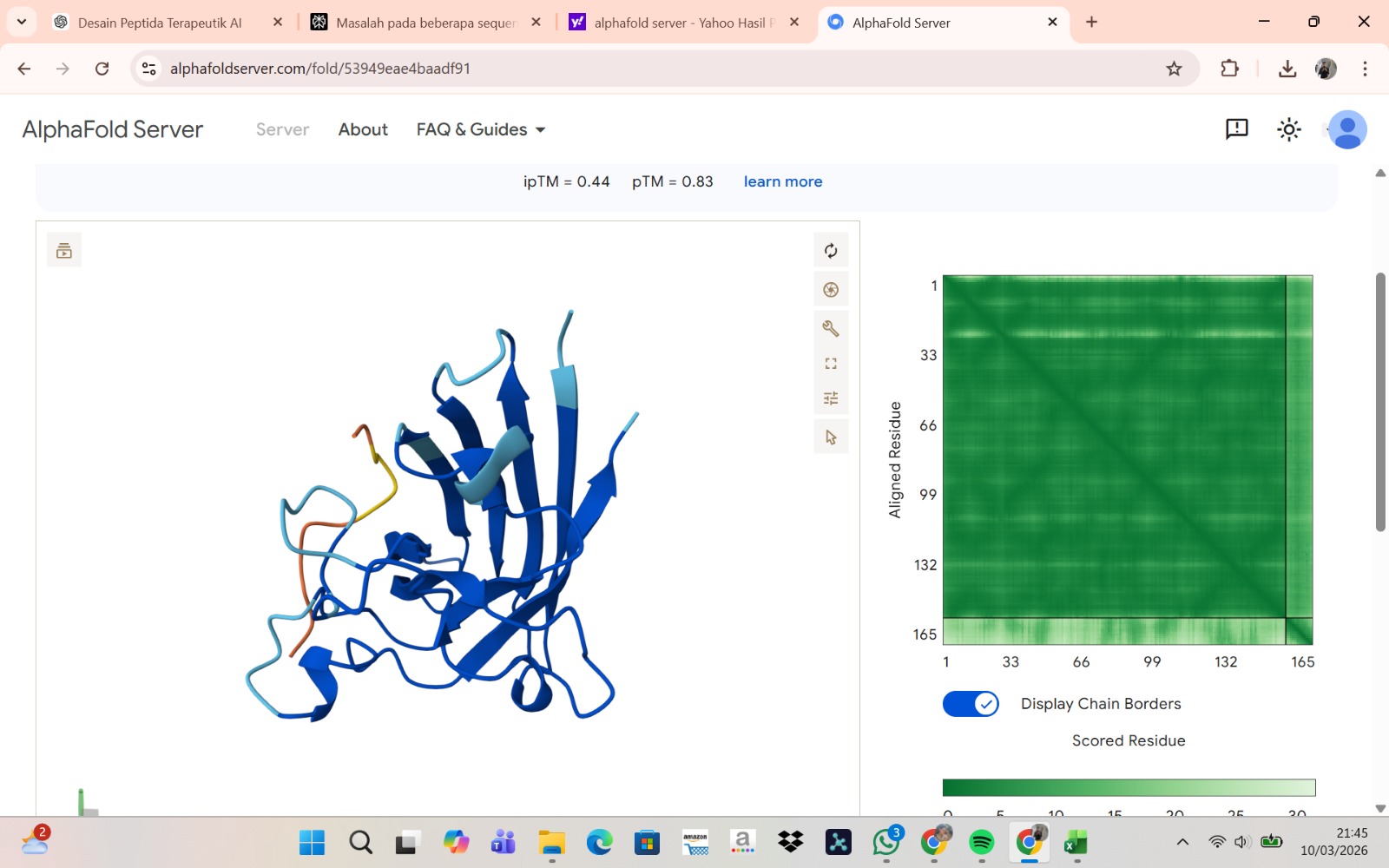
Peptide 3 : WLSVVAAIALKE
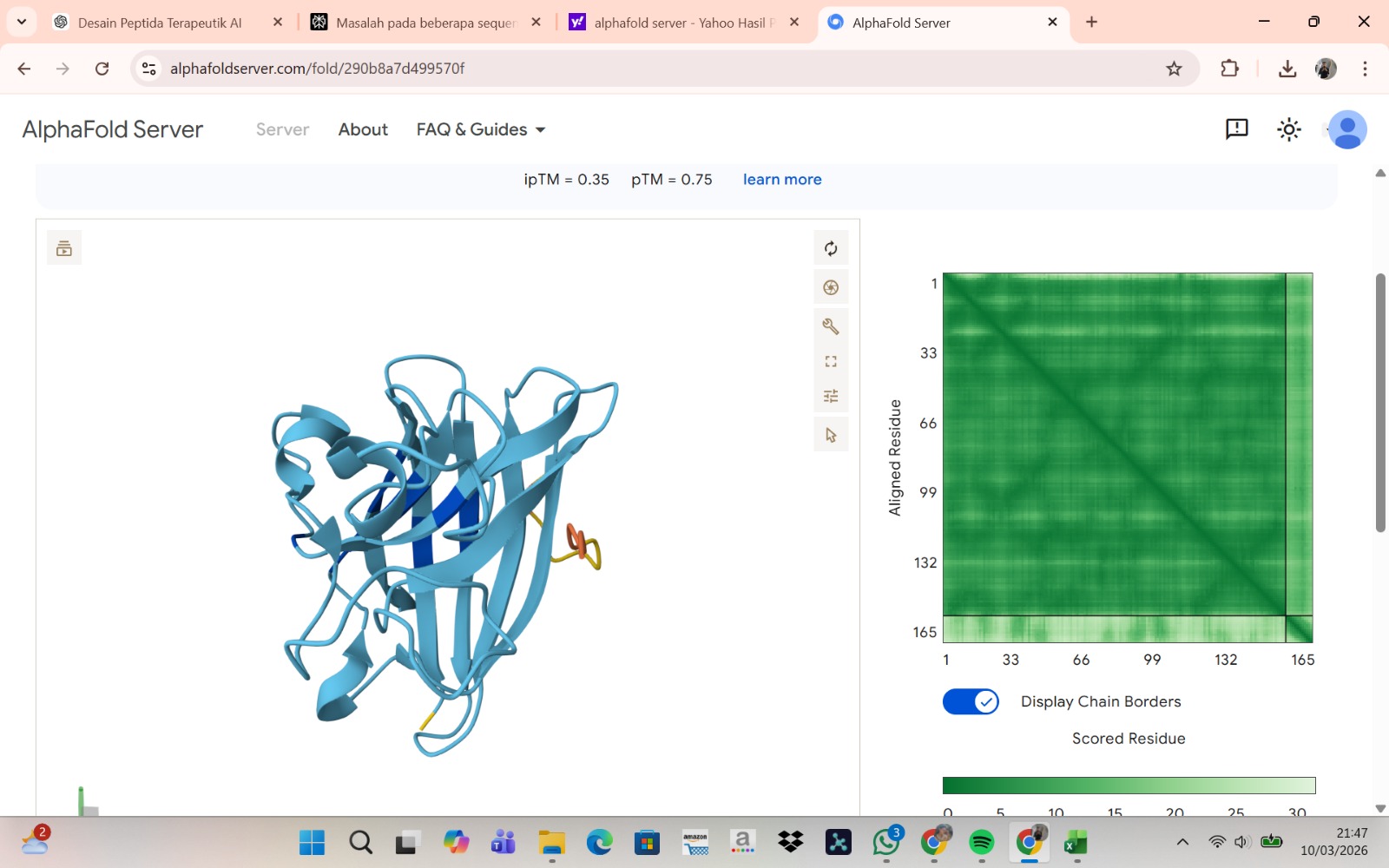
Peptide 4 : WRYPAAAARLKK
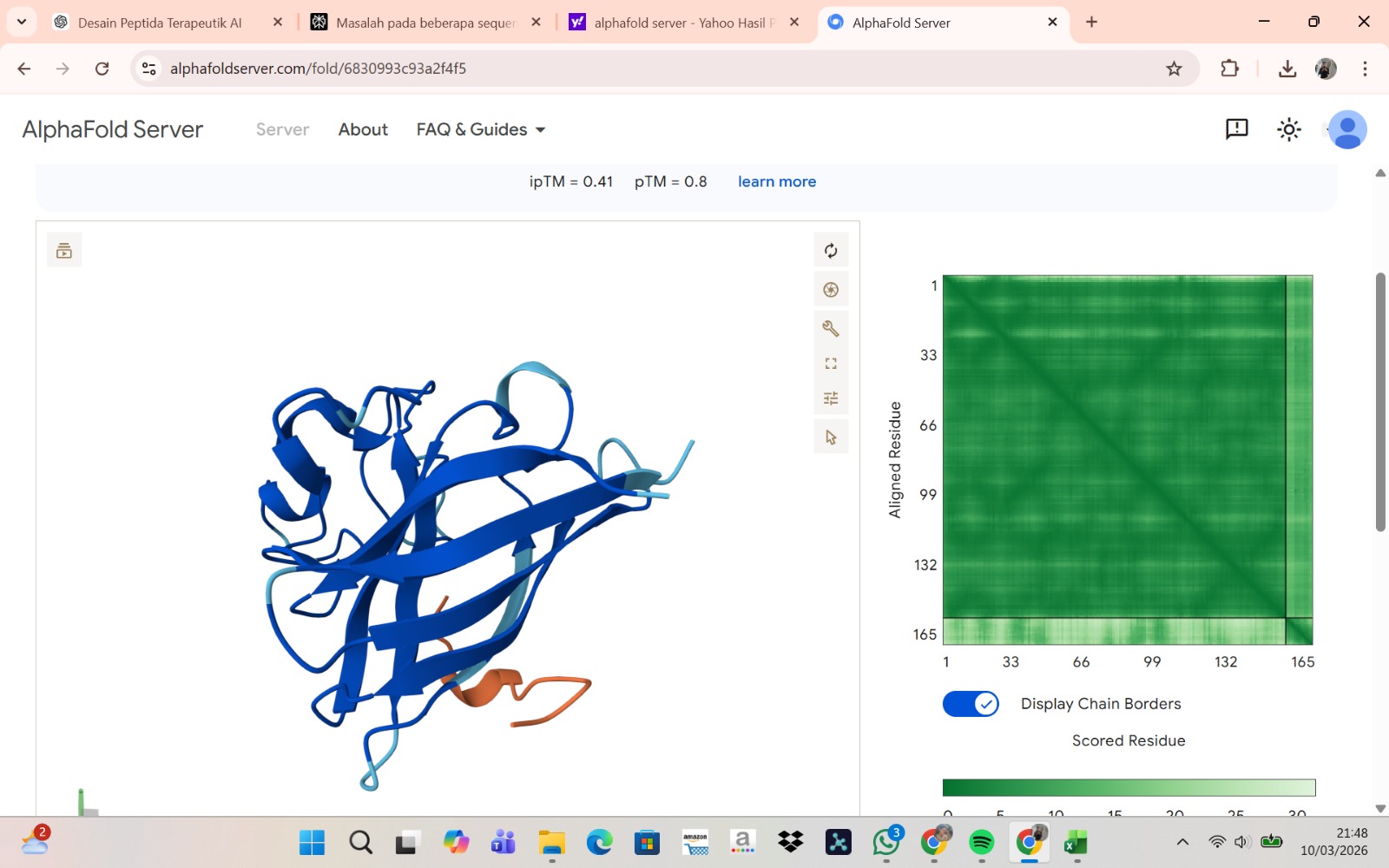
The peptide–protein complexes were modeled using AlphaFold3 by submitting the mutant SOD1 sequence as chain A and each peptide sequence as chain B. The predicted interface scores (ipTM) ranged from 0.35 to 0.44, indicating relatively weak but detectable interactions between the peptides and the SOD1 protein. Among the generated candidates, the peptide HLVPAVAIEHKK showed the highest ipTM value (0.44) and the highest pTM score (0.83), suggesting a more stable predicted complex compared to the other peptides. The remaining peptides displayed slightly lower ipTM scores, indicating weaker predicted interactions. Based on the predicted structures, the peptides appear to bind primarily to the surface of the SOD1 protein rather than being deeply buried within the structure. The interactions likely occur near exposed regions of the β-barrel or close to the N-terminal region where the A4V mutation is located. Although the binding confidence is modest, these results suggest that some of the PepMLM-generated peptides may interact with the mutant SOD1 surface and could serve as starting points for further optimization.
Part 3 Evaluate Properties with PeptiVerse
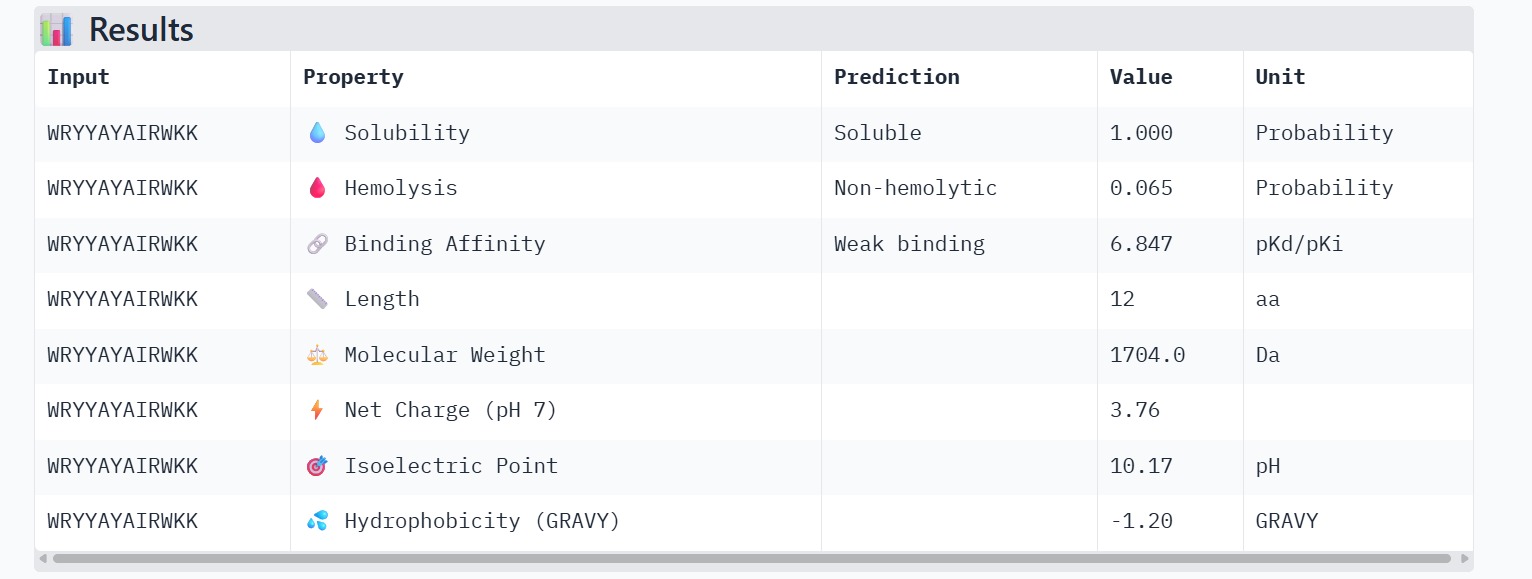
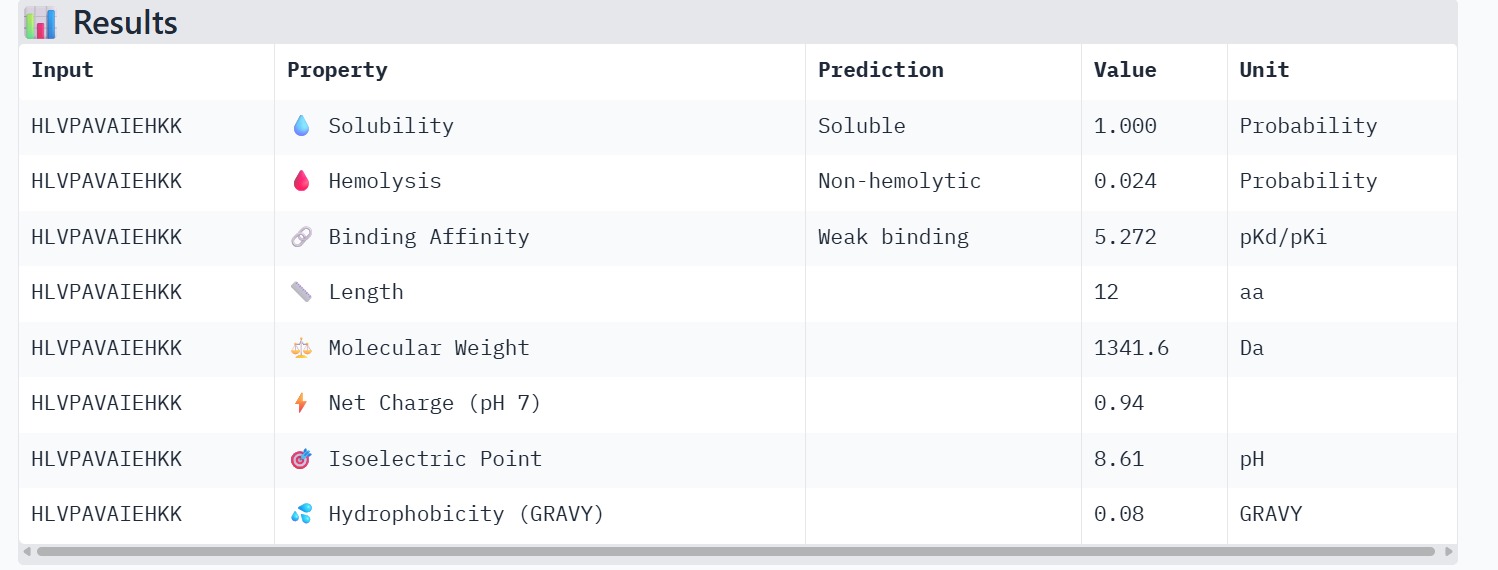
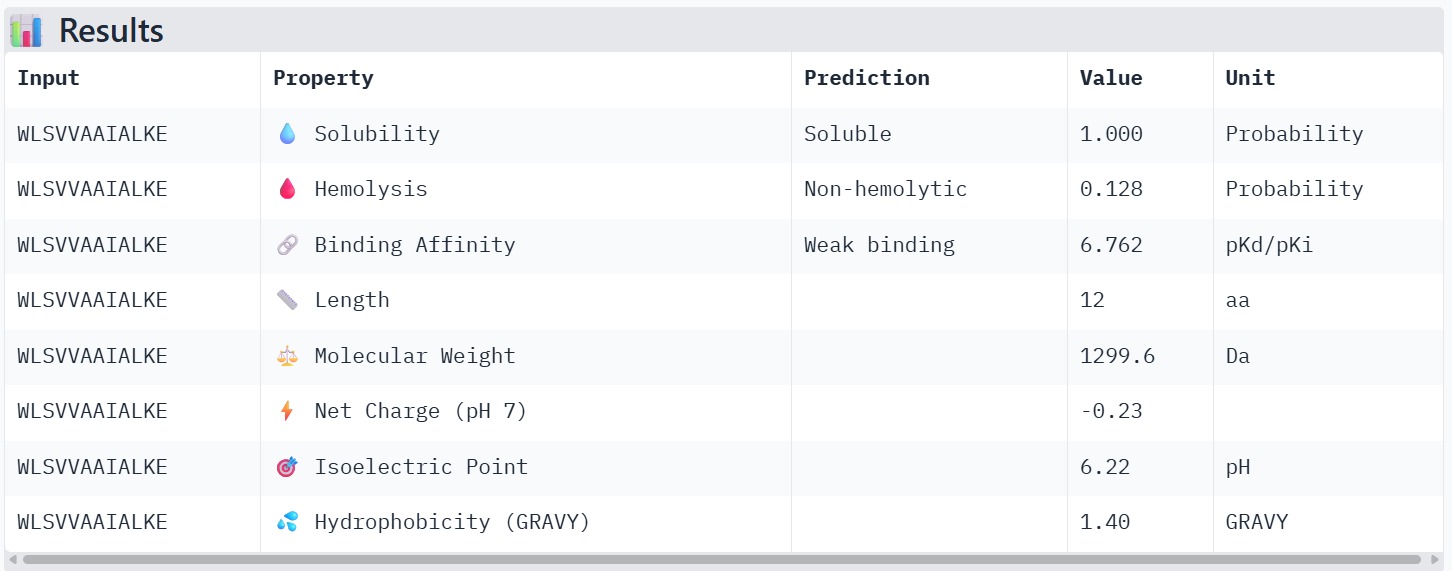
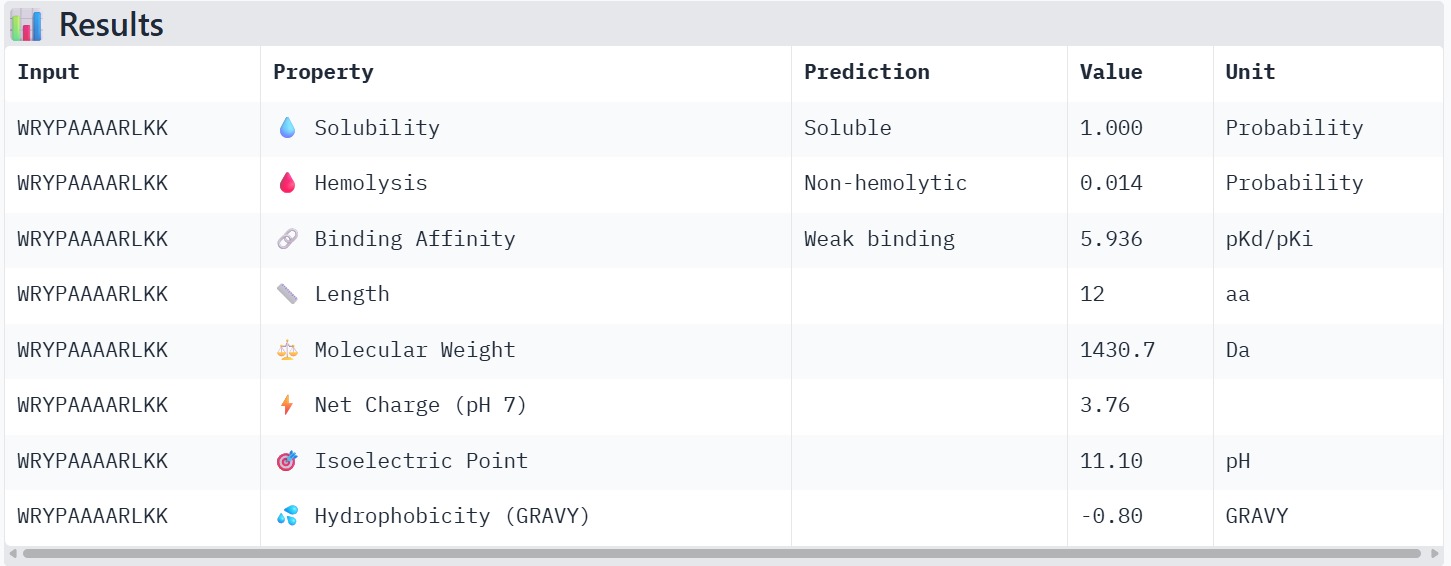
The therapeutic properties of the generated peptides were evaluated using PeptiVerse. All four peptides were predicted to be highly soluble with a probability of 1.000, indicating favorable physicochemical characteristics for biological applications. Hemolysis prediction suggested that all peptides are non-hemolytic, with WRYPAAAARLKK showing the lowest hemolysis probability (0.014). Binding affinity predictions ranged from pKd/pKi values of 5.27 to 6.85. Among the candidates, WRYYAYAIRWKK displayed the highest predicted binding affinity (6.847), suggesting stronger interaction with the A4V mutant SOD1 target. This peptide also exhibits a positive net charge (+3.76), which may enhance electrostatic interactions with the protein surface. Hydrophobicity analysis indicates that WRYYAYAIRWKK is moderately hydrophilic (GRAVY = −1.20), which supports solubility and structural stability. Overall, WRYYAYAIRWKK appears to provide the best balance between predicted binding affinity, solubility, and safety properties, making it the most promising candidate for further structural and experimental validation.
Part 4: Generate Optimized Peptides with moPPIt
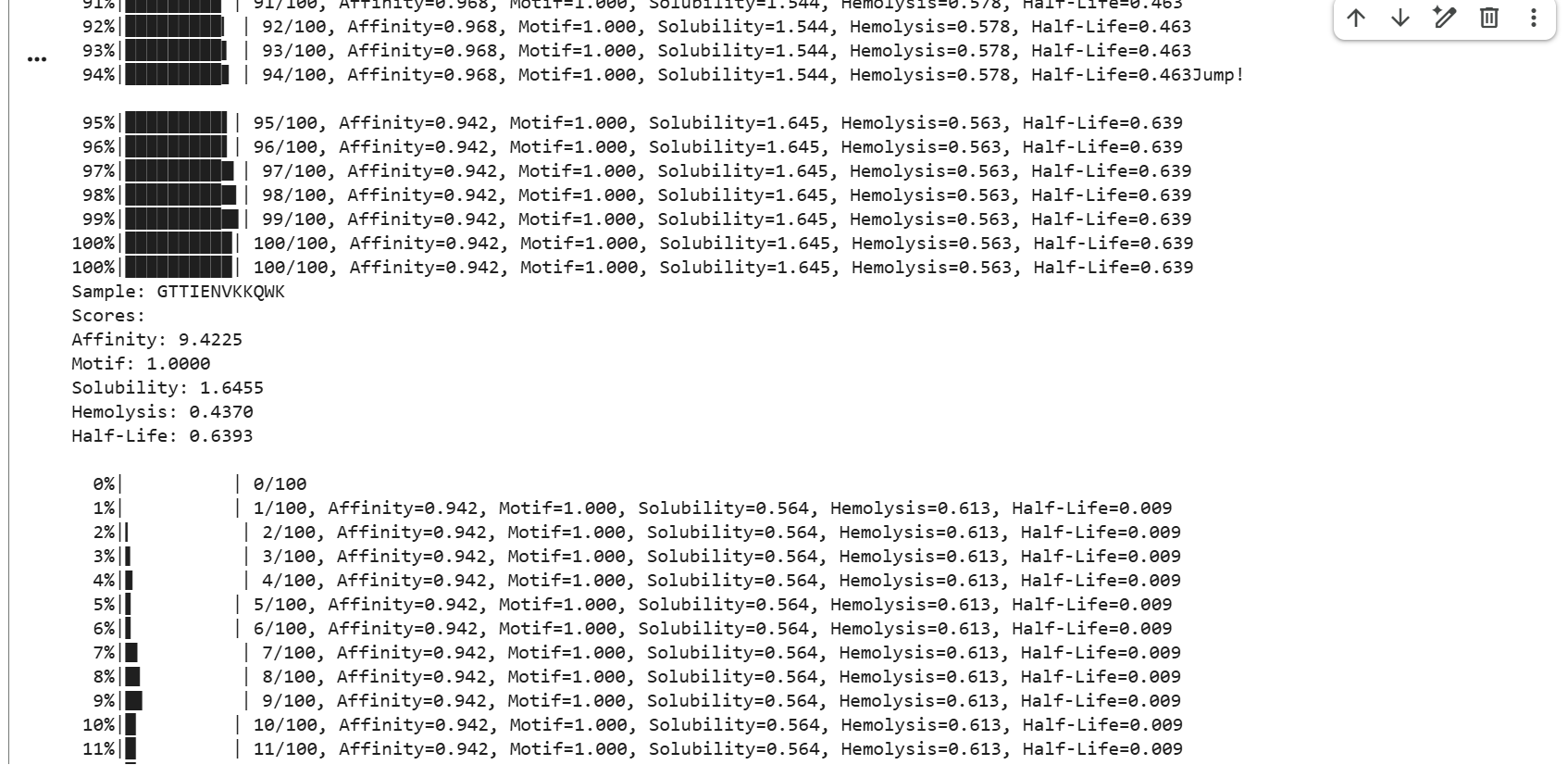
GTTIENVKKQWK showed strong affinity with a score of 9.4225, a perfect motif score of 1.0000, moderate solubility of 1.6455, hemolysis of 0.4370, and a half-life score of 0.6393.
ALWKWYRATAWQ showed strong affinity with a score of 9.4688, a perfect motif score of 1.0000, good solubility of 2.1135, low hemolysis of 0.3449, and a half-life score of 0.0134.
PSAAEWVEWLFK showed strong affinity with a score of 9.6628, a perfect motif score of 1.0000, good solubility of 2.0993, low hemolysis of 0.3619, and a half-life score of 0.0184.
LLAKIANPTQWK showed strong affinity with a score of 9.7345, a perfect motif score of 1.0000, moderate solubility of 1.5554, hemolysis of 0.4262, and a half-life score of 0.1850.
AWKPTALEFNWV showed strong affinity with a score of 9.5225, a perfect motif score of 1.0000, good solubility of 1.8301, hemolysis of 0.3823, and a half-life score of 0.0755.
ATETRFLPPWLW showed strong affinity with a score of 9.5070, a perfect motif score of 1.0000, moderate solubility of 1.4245, hemolysis of 0.3597, and a half-life score of 0.0264.
APTPEYEALFRF showed the highest affinity among these samples with a score of 9.8719, a perfect motif score of 1.0000, low solubility of 1.2404, hemolysis of 0.3469, and a half-life score of 0.0897.
TAKQFWDGWKWG showed strong affinity with a score of 9.6989, a perfect motif score of 1.0000, good solubility of 2.0194, hemolysis of 0.3731, and a half-life score of 0.2847.
NWKFAAWIHRPT showed strong affinity with a score of 9.6552, a near-perfect motif score of 0.9971, moderate solubility of 1.6367, hemolysis of 0.4038, and a very low half-life score of 0.0069.
FAGMFPLDAPTL showed the highest affinity in the set with a score of 9.8837, a near-perfect motif score of 0.9972, moderate solubility of 1.4887, hemolysis of 0.4193, and a half-life score of 0.0159.
The A4V mutant SOD1 sequence was used as the target protein for controlled peptide generation using moPPIt. Residues near the N-terminal region (positions 1–10), where the ALS-associated A4V mutation is located, were selected as the target binding region. Peptide length was set to 12 amino acids, and multiple optimization objectives including affinity, motif guidance, solubility, hemolysis, and half-life were enabled.
The generated peptides showed consistently high predicted affinity scores (9.42–9.88), indicating strong predicted interactions with mutant SOD1. Most peptides also achieved perfect motif scores, demonstrating successful targeting of the selected residue region near the mutation site. In addition, several peptides displayed favorable solubility and relatively low hemolysis probabilities, suggesting improved therapeutic properties. Among the generated candidates, GTTIENVKKQWK showed one of the best overall balances between affinity, solubility, safety, and predicted biological stability due to its relatively high half-life score.
Compared to the PepMLM-generated peptides, the moPPIt peptides were more optimized and therapeutically oriented. PepMLM primarily generated plausible binders based on sequence patterns, whereas moPPIt optimized multiple objectives simultaneously using guided generation. As a result, the moPPIt peptides displayed improved balance between binding affinity and drug-like properties such as solubility, reduced hemolysis, and stability. The peptide sequences generated by moPPIt therefore differed substantially from the PepMLM outputs, which is expected and reflects the different underlying design strategies of the two models.
Before advancing these peptides toward clinical studies, additional computational and experimental validation would be required. Molecular docking and molecular dynamics simulations should be performed to evaluate binding stability and interaction specificity. Experimental assays such as surface plasmon resonance or isothermal titration calorimetry would then be necessary to confirm binding affinity. Furthermore, cytotoxicity, hemolysis, serum stability, and cellular uptake assays would be required to assess safety and pharmacological potential before proceeding to animal studies and eventual clinical development.
Week 6 HW: Genetic Circuits I: Assembly Technologies
1. Components in Phusion High-Fidelity PCR Master Mix and Their Purpose
One of the main enzymes used is Phusion High-Fidelity DNA Polymerase, a DNA polymerase with an extremely low error rate (high fidelity), making it ideal for mutagenesis and cloning experiments.
The main components in Phusion High-Fidelity PCR Master Mix and their functions are:
| Components | Function |
|---|
| Phusion DNA Polymerase | Enzyme that synthesizes new DNA from primers during the extension phase. It has proofreading activity (3’→5’ exonuclease), resulting in very low replication errors. |
| dNTPs (dATP, dTTP, dGTP, dCTP) | Substrates or “building blocks” used by the polymerase to form the new DNA strand. |
| Reaction Buffer | Provides optimal chemical conditions (pH, salts, enzyme stability) for polymerase activity. |
| Mg²⁺ ions (MgCl₂) | Essential cofactor required by DNA polymerase to catalyze phosphodiester bond formation between nucleotides. |
| Stabilizing agents | Maintain enzyme stability during PCR. |
The master mix is typically at 2X concentration, so only primers, template DNA, and water need to be added.
2. Factors That Determine Primer Annealing Temperature
Annealing temperature determines how well primers bind to the DNA template. Factors influencing annealing temperature:
• Primer melting temperature (Tm): Annealing is usually 2–5°C lower than the primer Tm.
• Primer length: Longer primers typically have a higher Tm.
• GC content: G-C bonds have three hydrogen bonds, increasing stability and Tm.
• Primer-template complementarity: Mismatches reduce stability and Tm.
• Salt concentration in buffer: Ion concentration affects DNA duplex stability.
• Secondary structures in primers: Hairpins or dimers can disrupt proper annealing.
Too low an annealing temperature causes non-specific amplification, while too high prevents primer binding.
3. Comparison: PCR vs Restriction Enzyme Digestion
| Feature | PCR | Restriction Enzyme Digestion |
|---|
| Principle | DNA amplification using DNA polymerase | DNA cutting using restriction enzymes |
| Product | New DNA fragments from synthesis | DNA fragments from cutting existing DNA |
| Specificity | Determined by primers | Determined by enzyme recognition site |
| Time | Usually 1–2 hours | 30–60 minutes |
| Flexibility | Highly flexible (add mutations, overhangs, tags) | Limited to restriction site locations |
| When to use? | PCR preferred if: wanting to amplify DNA, introduce mutations, add new sequences, or no restriction sites available | Restriction digestion preferred if: precise plasmid cutting, traditional cloning with sticky ends, no mutations needed |
4. Ensuring DNA Fragments Are Suitable for Gibson Assembly
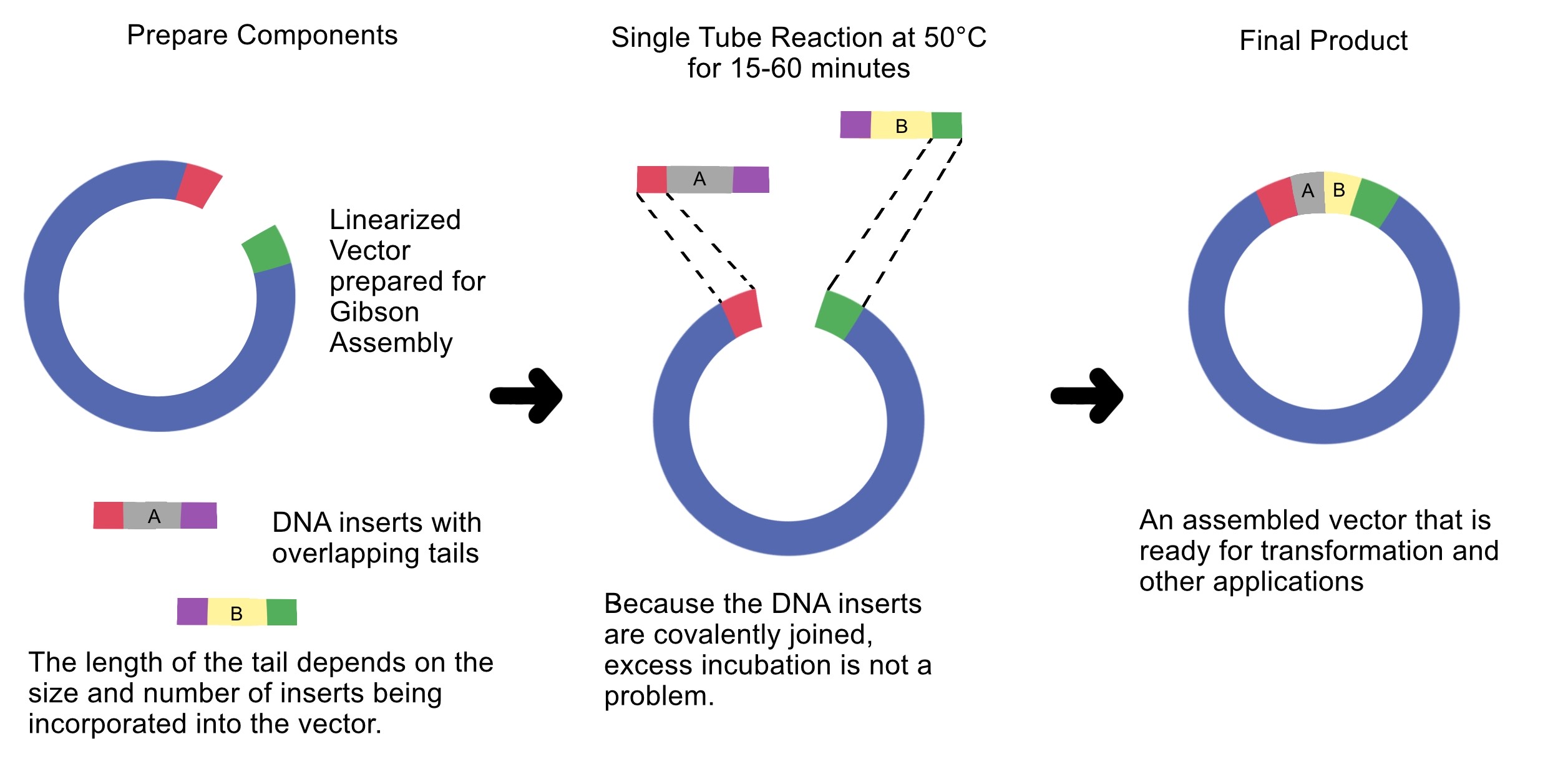
Gibson Assembly requires DNA fragments with homologous overlap sequences.
To ensure PCR or digested fragments are suitable for Gibson cloning:
• Design overlap sequences (20–40 bp) in primers.
• Ensure correct fragment orientation (5’ → 3’).
• Use design software like Benchling to verify overlaps.
• Confirm no unwanted mutations in overlaps.
• Verify fragment size via gel electrophoresis.
• Measure DNA concentration for proper insert:vector molar ratio (usually 2:1).
Overlap sequences enable the exonuclease in Gibson Assembly to generate single-stranded ends that can anneal to each other.
5. How Plasmid DNA Enters E. coli During Transformation
Plasmid DNA is introduced into Escherichia coli bacteria via heat-shock transformation. The process:
Bacterial cells are made competent, usually with CaCl₂ treatment.
Plasmid DNA is mixed with competent cells at cold temperature (0–4°C).
Heat shock at 42°C for ~45 seconds alters membrane permeability.
Temporary pores form in the cell membrane.
Plasmid DNA enters the cell via diffusion.
Cells recover in nutrient media (SOC) before antibiotic selection.
Only bacteria carrying the plasmid with antibiotic resistance genes will grow.
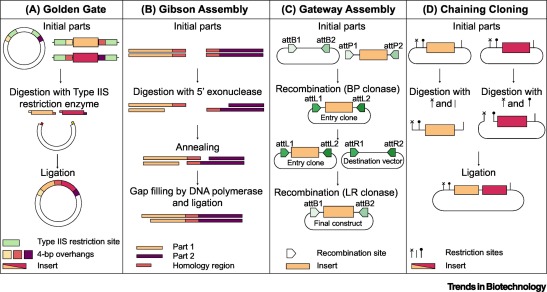
6. Another Assembly Method: Golden Gate Assembly
Golden Gate Assembly is a cloning technique that allows multiple DNA fragments to be joined in a single reaction. It uses Type IIS restriction enzymes that cut DNA outside their recognition sites, producing designer-specific overhangs. DNA fragments and vector plasmids are cut by these enzymes and then ligated by DNA ligase in a cyclic reaction alternating between cutting and ligation temperatures. Since the restriction sites are lost after ligation, assembled fragments are not recut. This enables directional assembly of multiple DNA fragments in one reaction. The method is highly popular in synthetic biology for efficiently constructing multigene assemblies. Golden Gate is often used for metabolic pathway construction or gene libraries.
Diagram of Golden Gate Assembly
Step 1: Restriction digestion
Fragment A Fragment B Fragment C
| BsaI | | BsaI | | BsaI |
Step 2: Custom overhangs created
A —-ATGC
B —-ATGC
C —-ATGC
Step 3: Ligation
A + B + C → assembled plasmid
simple concept:
DNA Fragment 1 + DNA Fragment 2
↓ BsaI digestion
Sticky overhangs created
↓ DNA ligase
Joined plasmid
7. Modeling Assembly with Benchling or Asimov Kernel
This method can be modeled using DNA design software like Benchling or Asimov Kernel.
Modeling steps:
Import plasmid and insert DNA sequences.
Add Type IIS restriction enzyme recognition sites (e.g., BsaI).
Design unique 4-bp overhangs for each fragment.
Simulate digestion using restriction analysis features.
Simulate ligation to ensure correct fragment orientation.
Verify the final plasmid sequence.
This software helps prevent frame shifts, orientation errors, or unwanted restriction sites before experiments.
Simulation
This construct models an inducible dual-reporter system commonly used in synthetic biology. The lac promoter allows controlled gene expression in the presence of IPTG. Upon induction, both GFP and RFP are expressed, enabling visualization of gene expression. The use of two reporter genes allows comparative analysis of expression levels. This system demonstrates how multiple genes can be co-expressed under a single regulatory element. Such designs are widely used in biosensors and gene circuit engineering.
Promotor
GGTCTCAATGTGTGAGTTAGCTCACTCATTAGGCACCCCAGGCTTTACACTTTATGCTTCCGGCTCGTATAATGTGTGGAGGTGAGACC
RBS1
GGTCTCAGGTAGGAGGGCTTGAGACC
GFP
GGTCTCGCTTATGGTGAGCAAGGGCGAGGAGCTGTTCACCGGGGTGGTGCCCATCCTGGTCGAGCTGGACCGCTGAGACC
Terminator1
GGTCTCCGCTGCCTCAGCGGTGGCGAACCTGCGCGTTGTTGCGGTTTTTTGCCGCCAGCGGTTATGAGACC
RBS2
GGTCTCTTATAGGAGGGCTTAATGGAGACC
RFP
GGTCTCAATGATGGTGAGCAAGGGCGAGGAGGATAACATGGCCATCATCAAGGAGTTCATGCGCTTCAAGCGCTGAGACC
Terminator2
GGTCTCCGCTGCCTCAGCGGTGGCGAACCTGCGCGTTGTTGCGGTTTTTTGCCGCCAGCGGACTAGAGACC
The promoter sequence was separated from the BsaI restriction sites to ensure correct biological annotation. The flanking BsaI sites are used only for Golden Gate assembly and are not part of the functional promoter region.
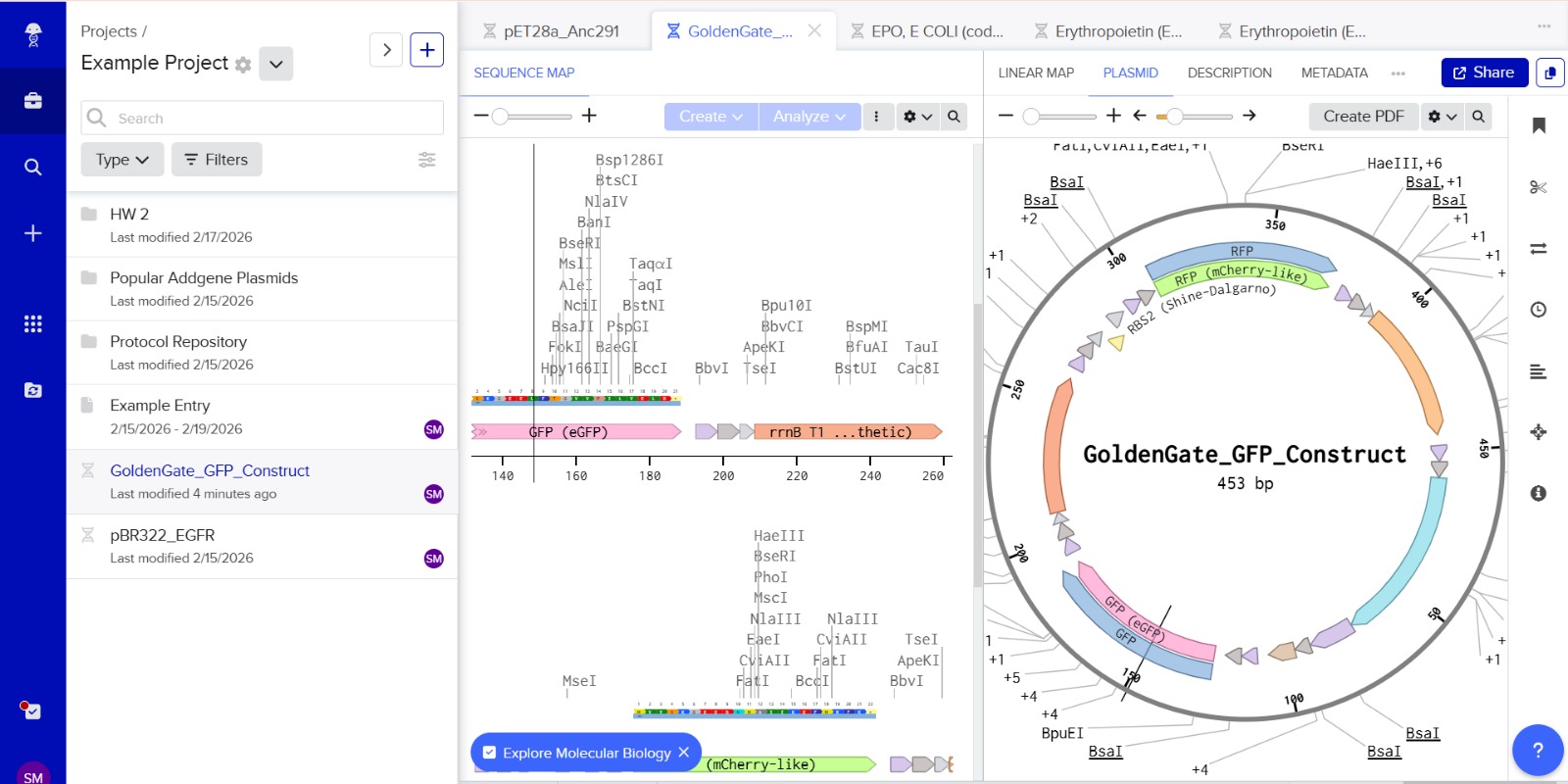
Stop codons were included at the end of each coding sequence (GFP and RFP) to ensure proper termination of translation. No additional stop codons were added downstream of terminators, as terminators function at the transcriptional level and do not affect translation termination.
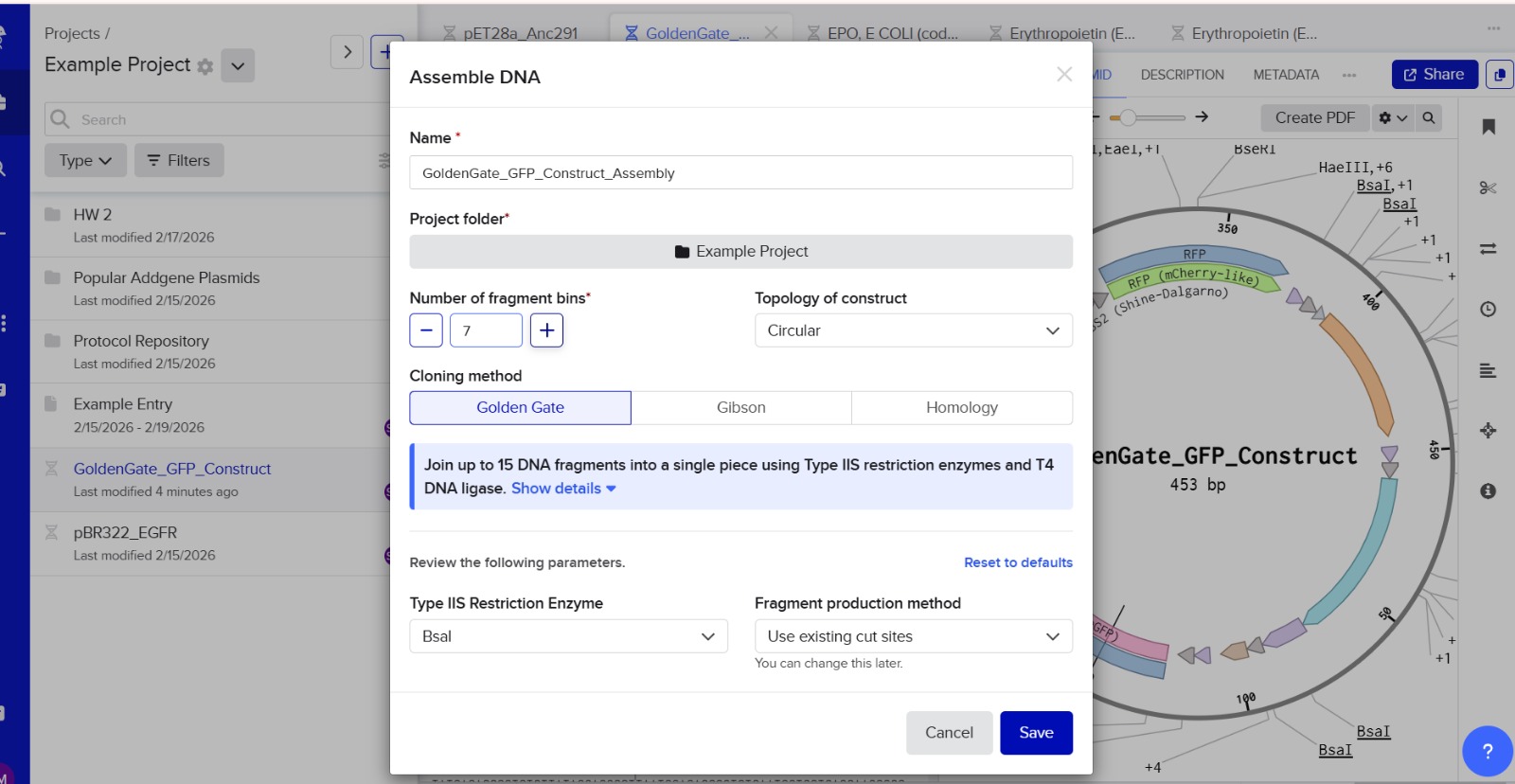
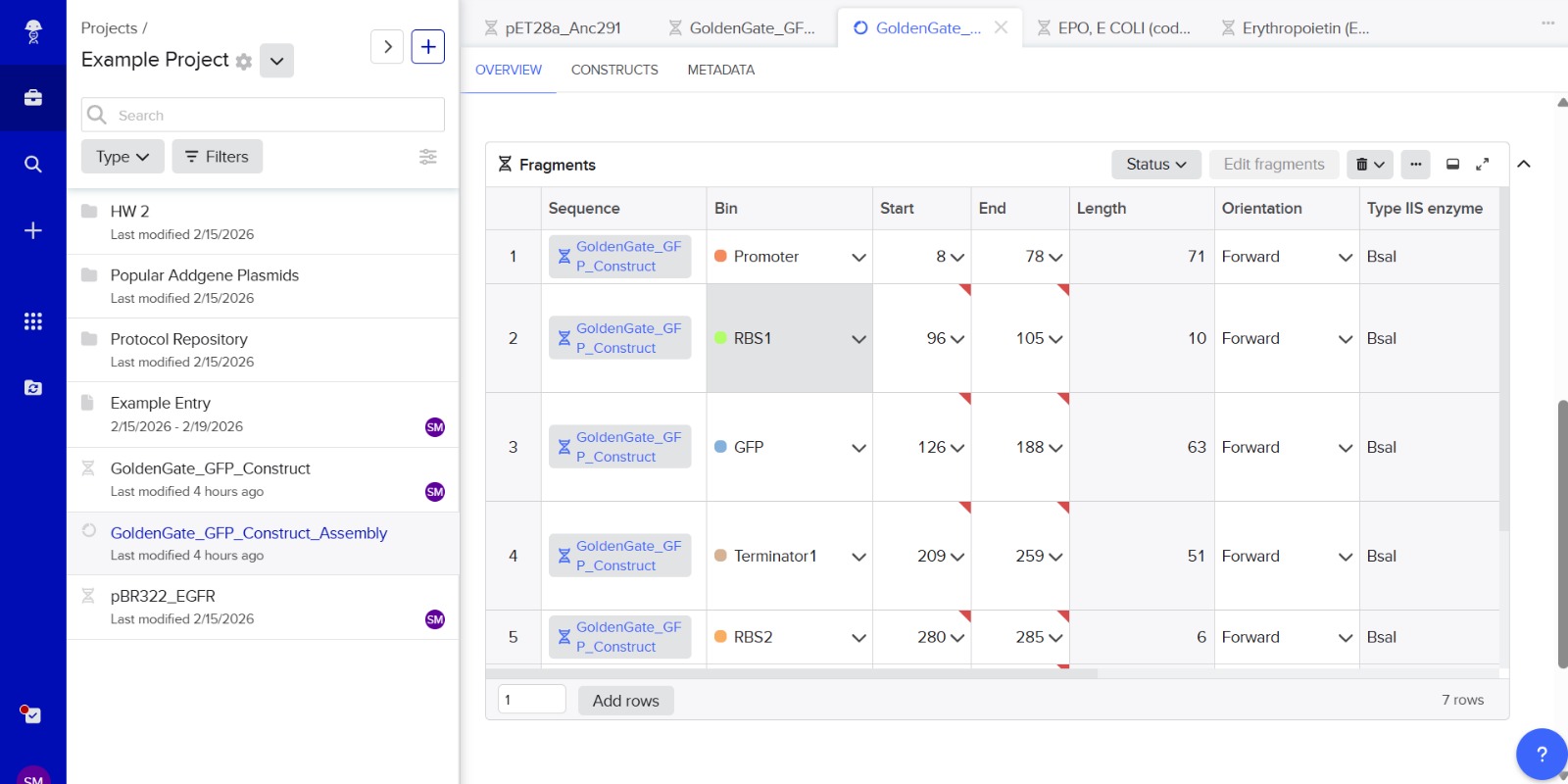
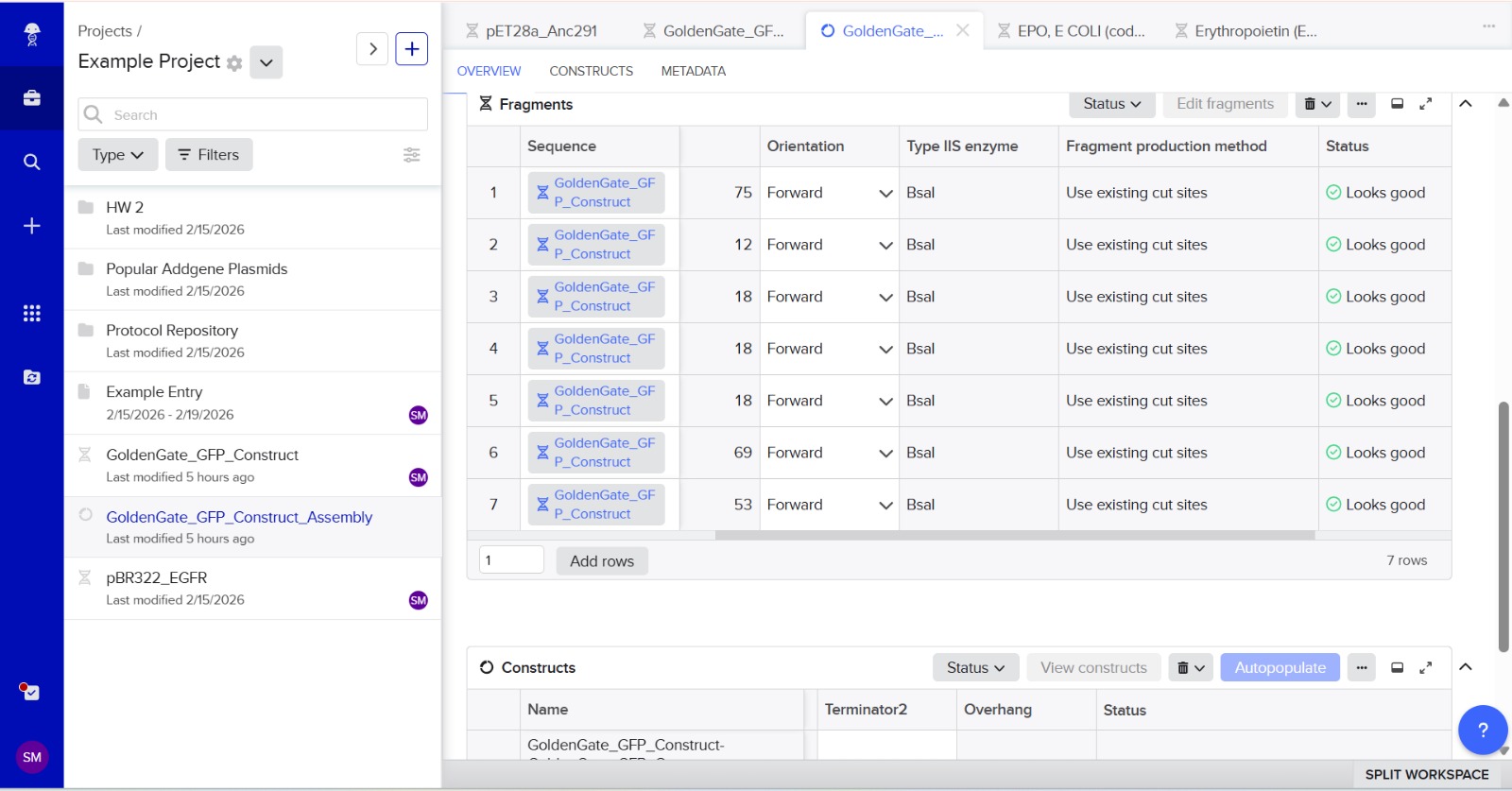
Assignment: Asimov Kernel
Bacterial Demo
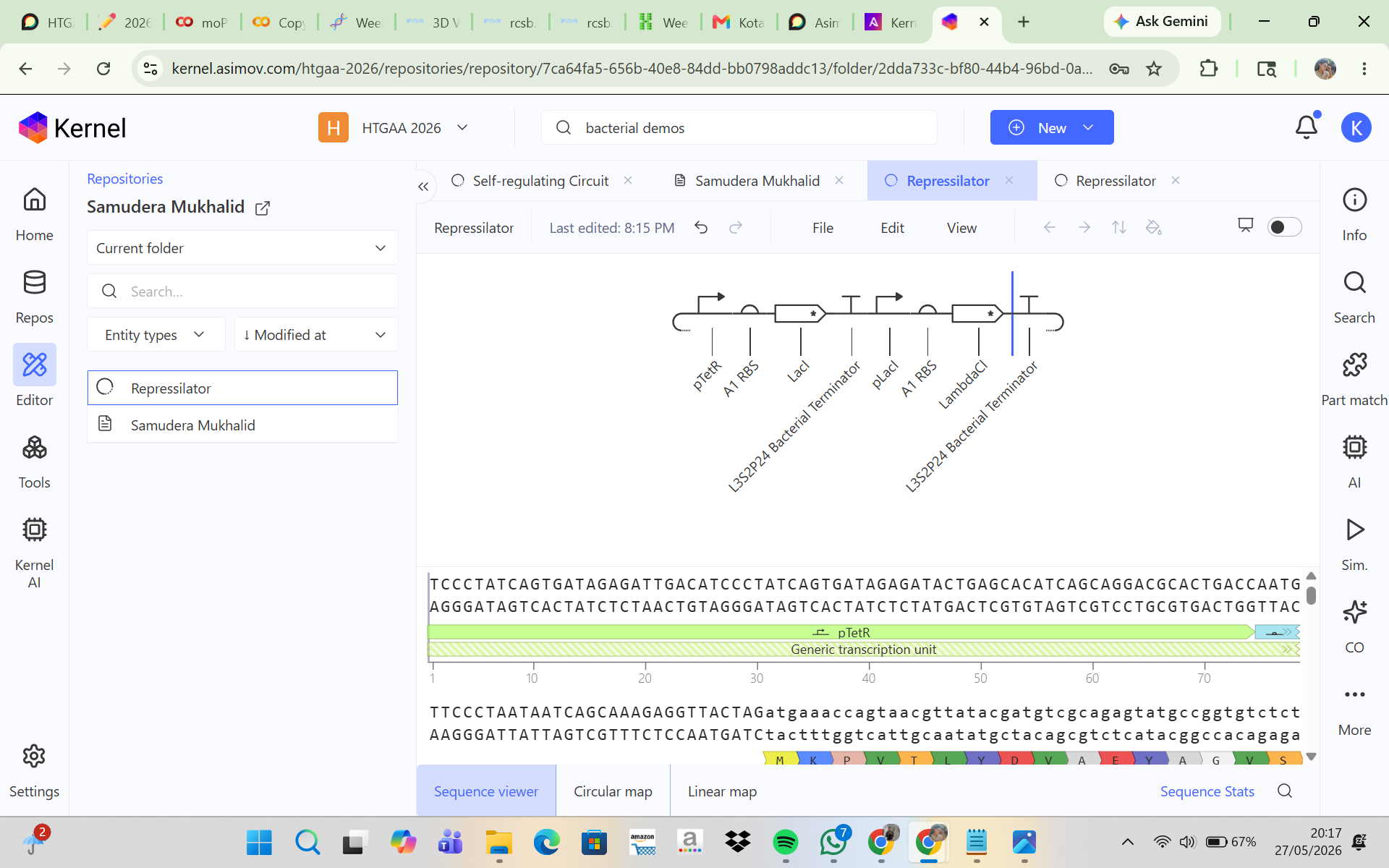
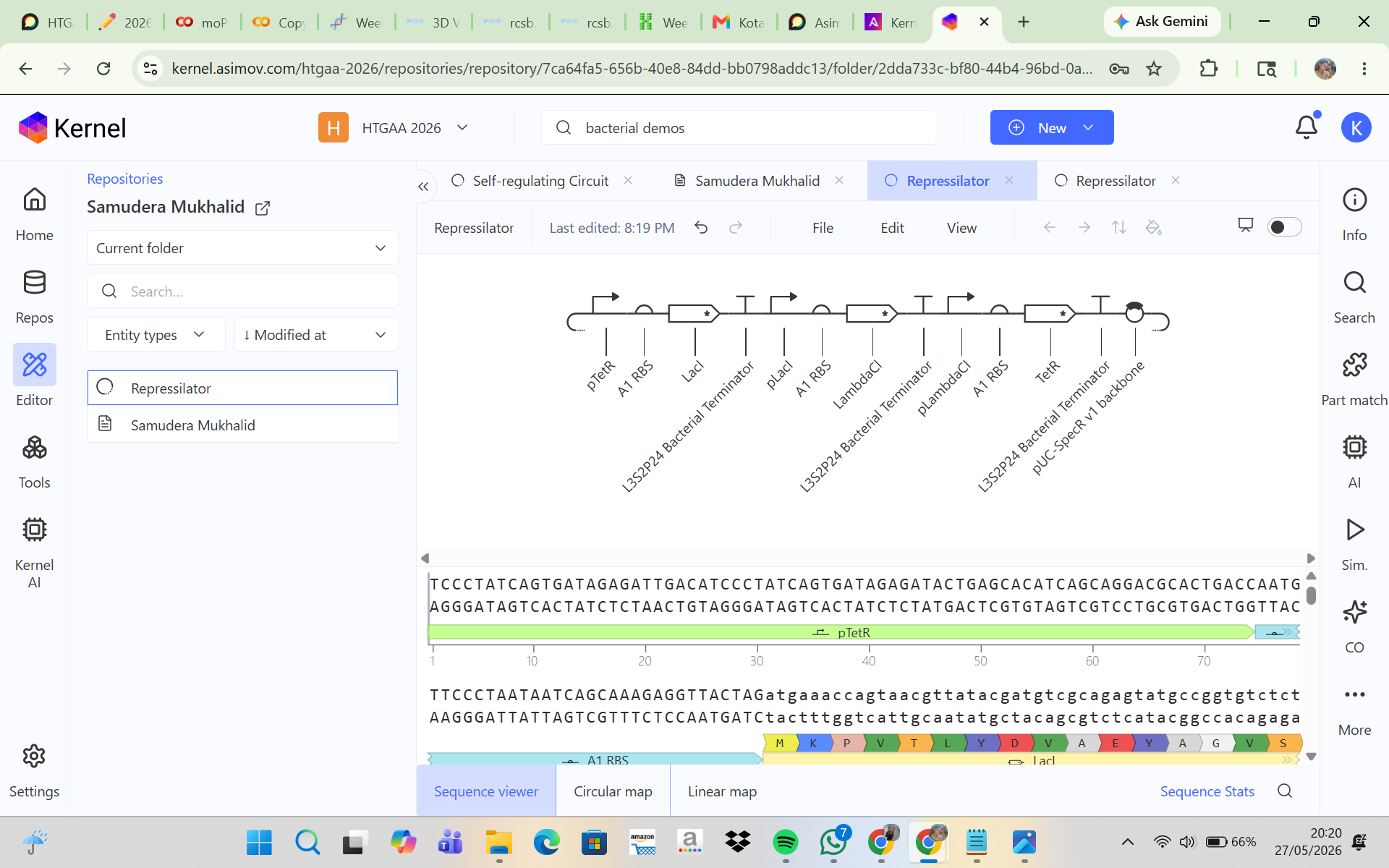
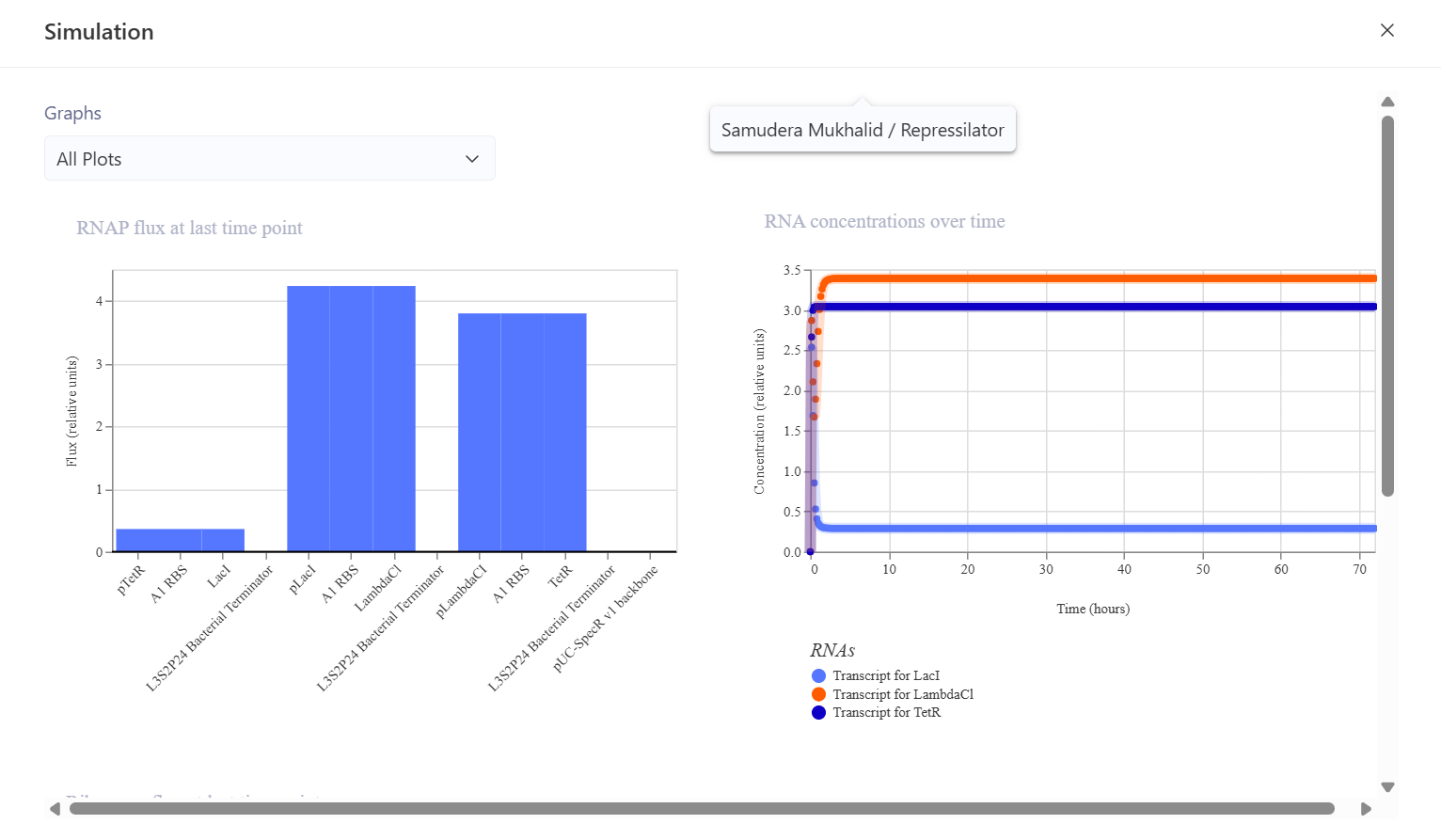
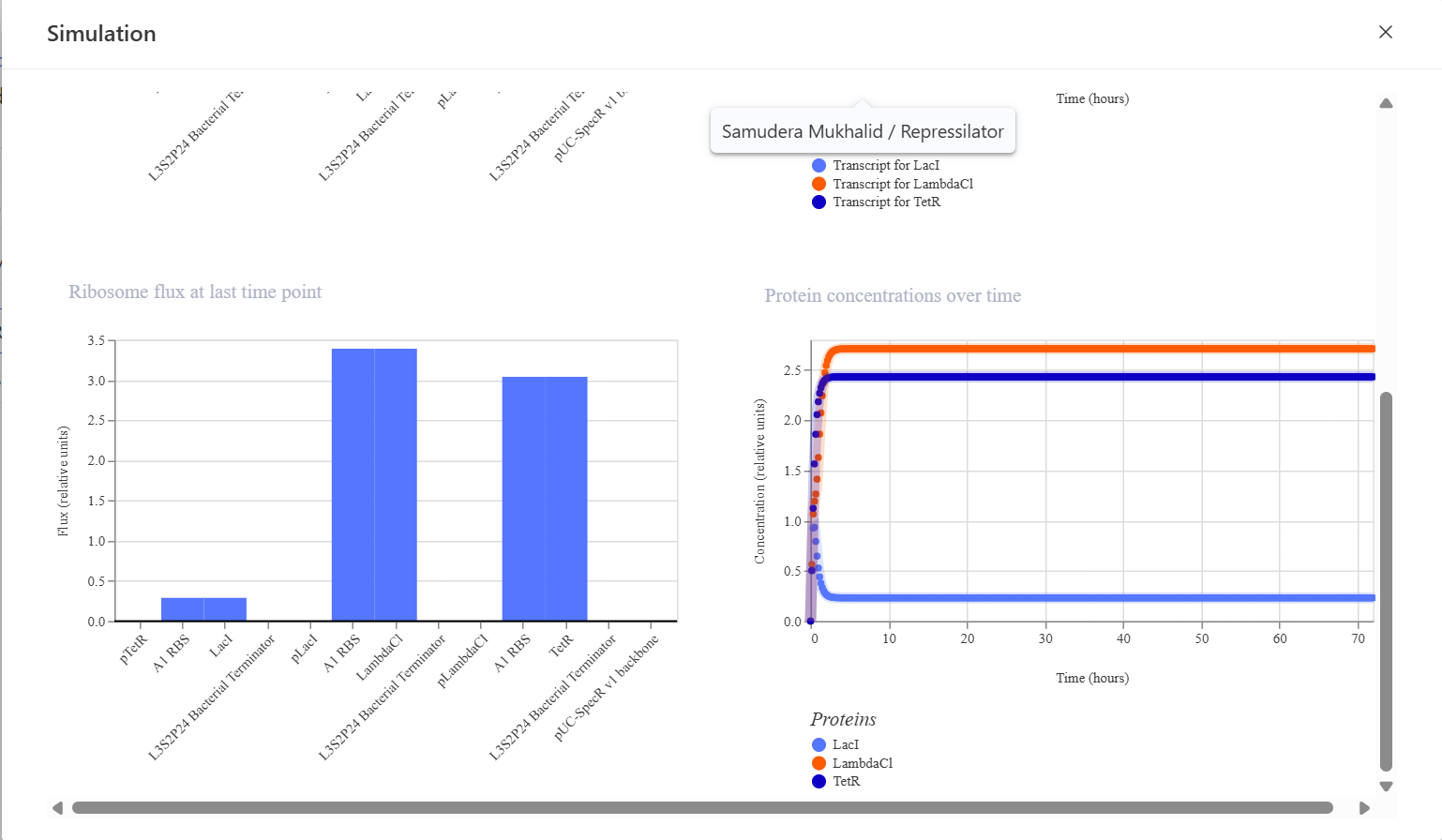
This is the simulation result I obtained. I think the simulation results are very different compared to the bacterial demos one.
CONSTRUCT 1. Auto-Repression GFP Circuit (Negative Feedback Circuit)
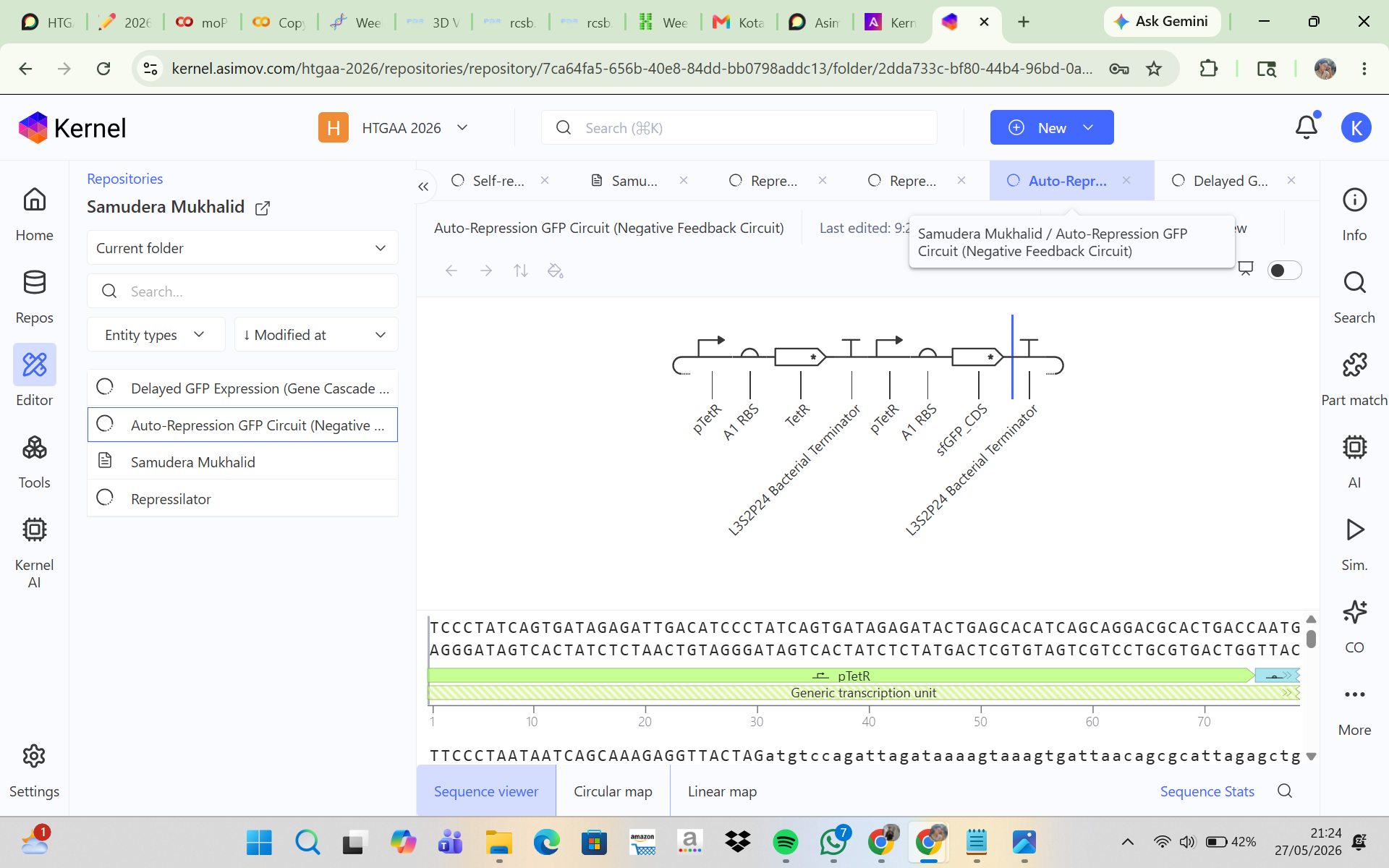
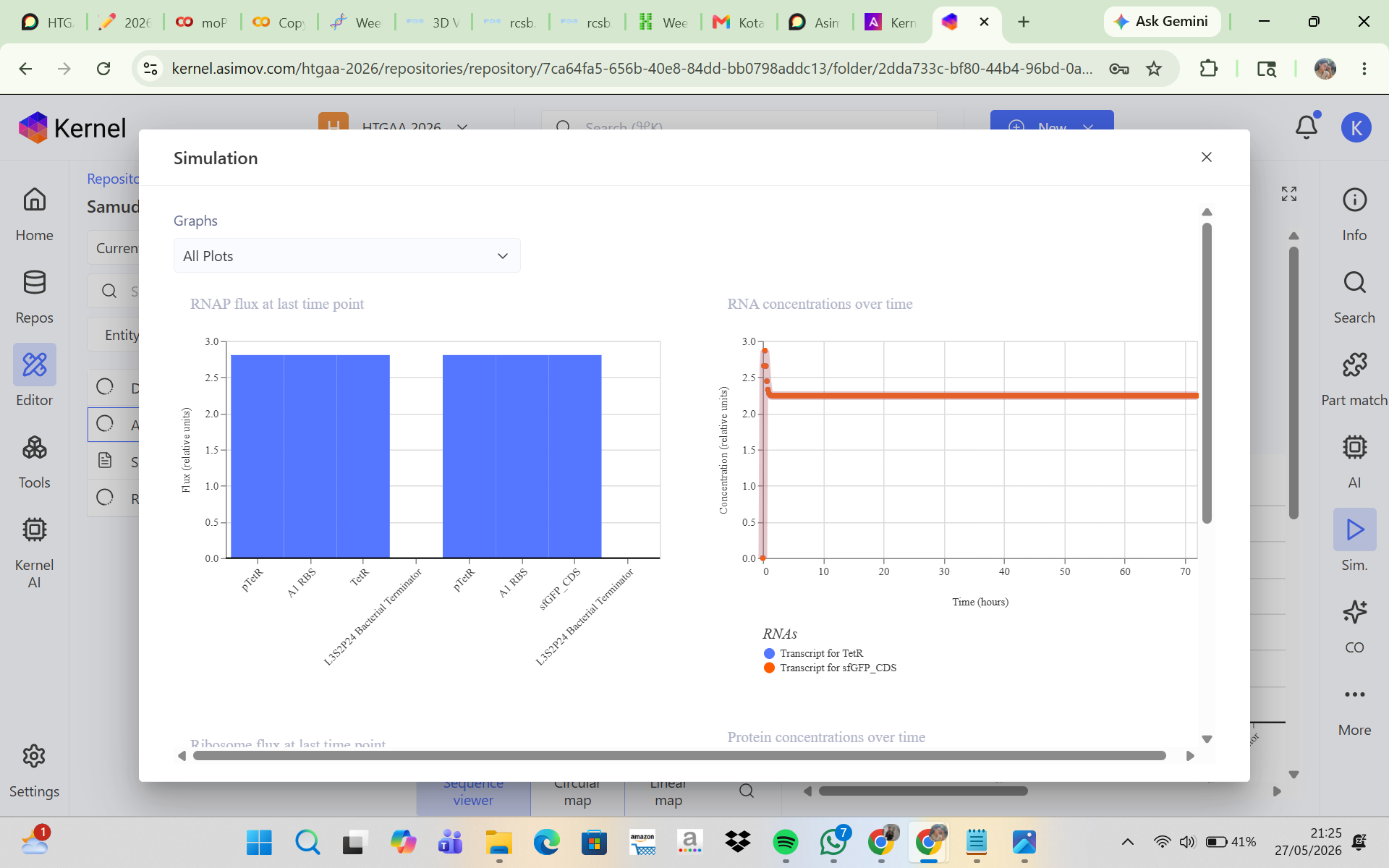
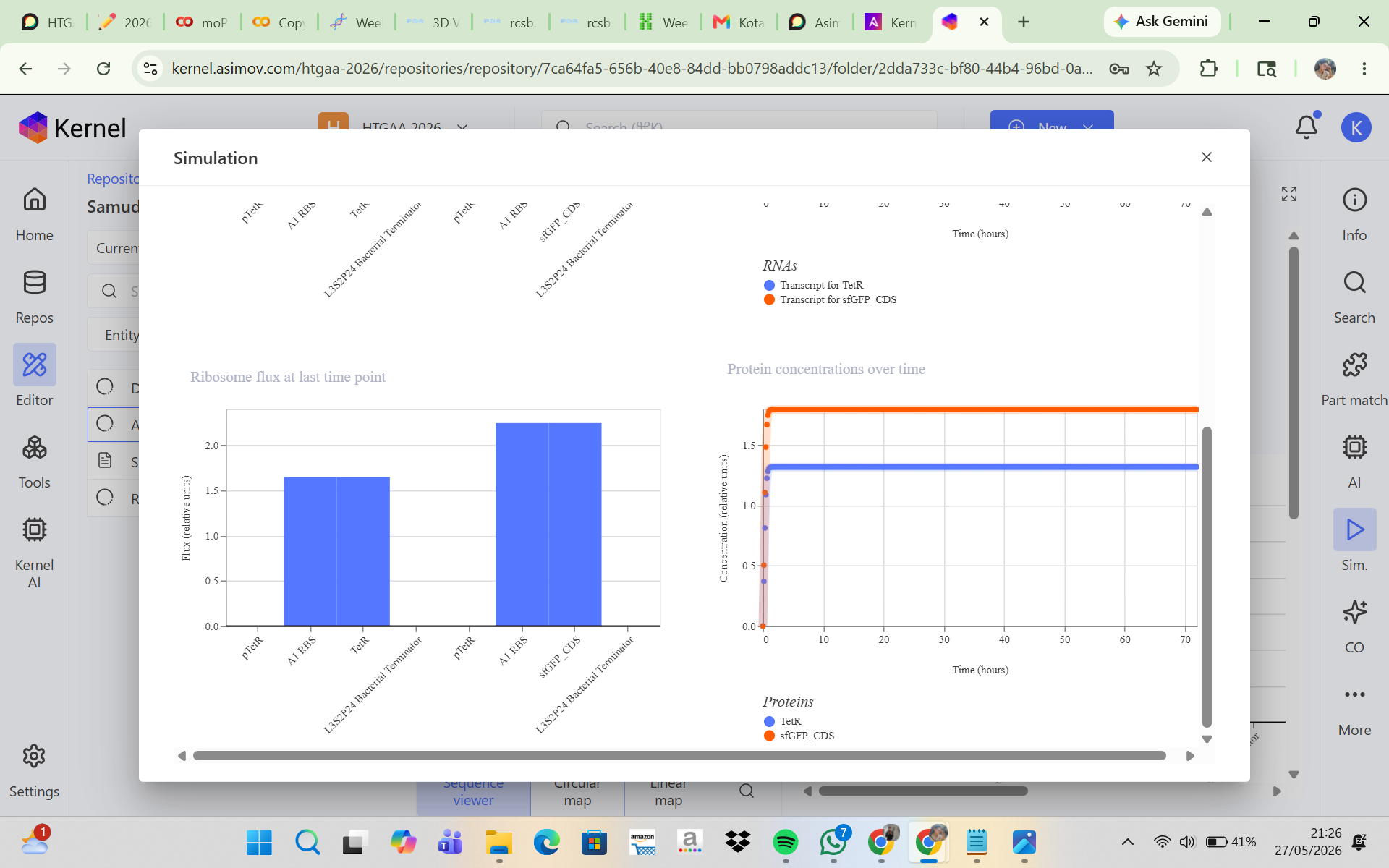
This construct was designed to produce GFP that initially turns on and is then self-repressed by a repressor protein. In this system, the pTetR promoter activates the expression of TetR and GFP, and the TetR protein then represses the pTetR promoter, which should cause GFP production to decrease over time. The initial hypothesis was that GFP would rise rapidly at the beginning of the simulation because the promoter is active, and then, as TetR accumulates, the promoter would become repressed so that GFP expression would decline and the system would eventually reach a low steady state. However, the simulation results and analysis showed that the construct successfully expressed TetR and sfGFP in a stable manner, but the negative feedback mechanism was not strong enough to produce the expected decrease in GFP expression. This may be due to a high promoter strength, a low protein degradation rate, or weak TetR repression. Adjusting the simulation parameters, such as increasing the degradation rate and reducing the promoter strength, may help produce a feedback behavior that is more consistent with the biological expectation.
CONSTRUCT 2. Delayed GFP Expression (Gene Cascade Circuit)
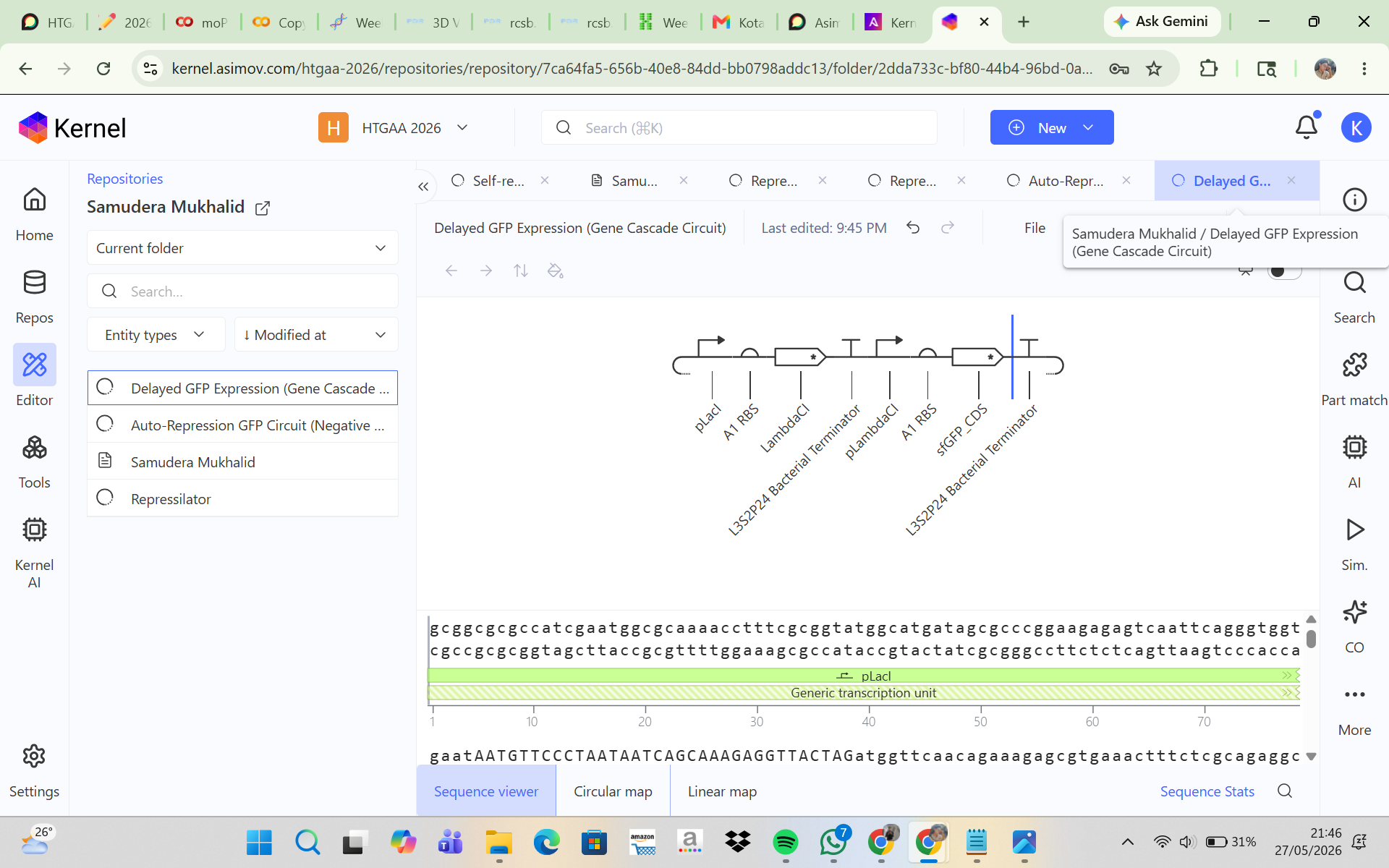
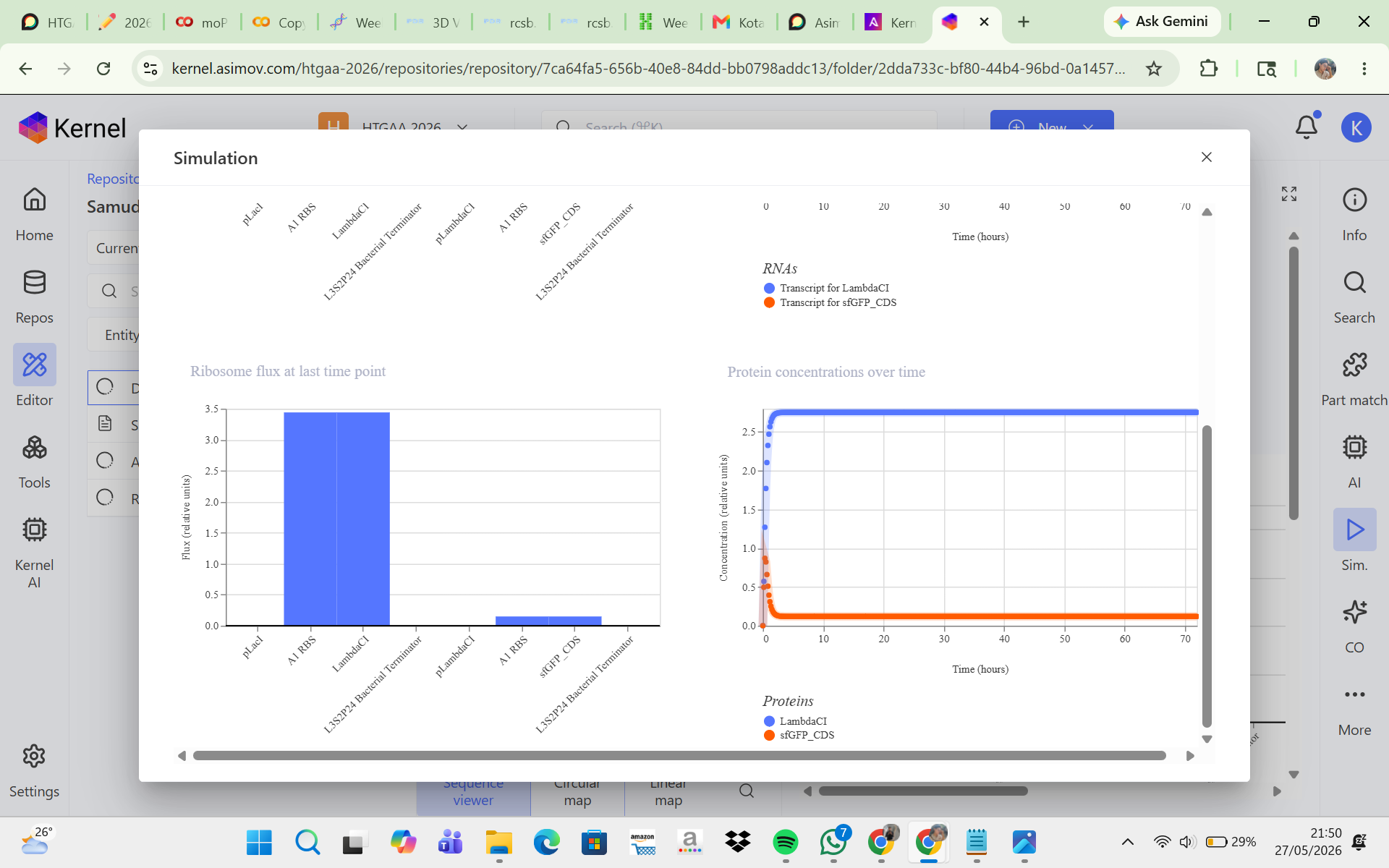
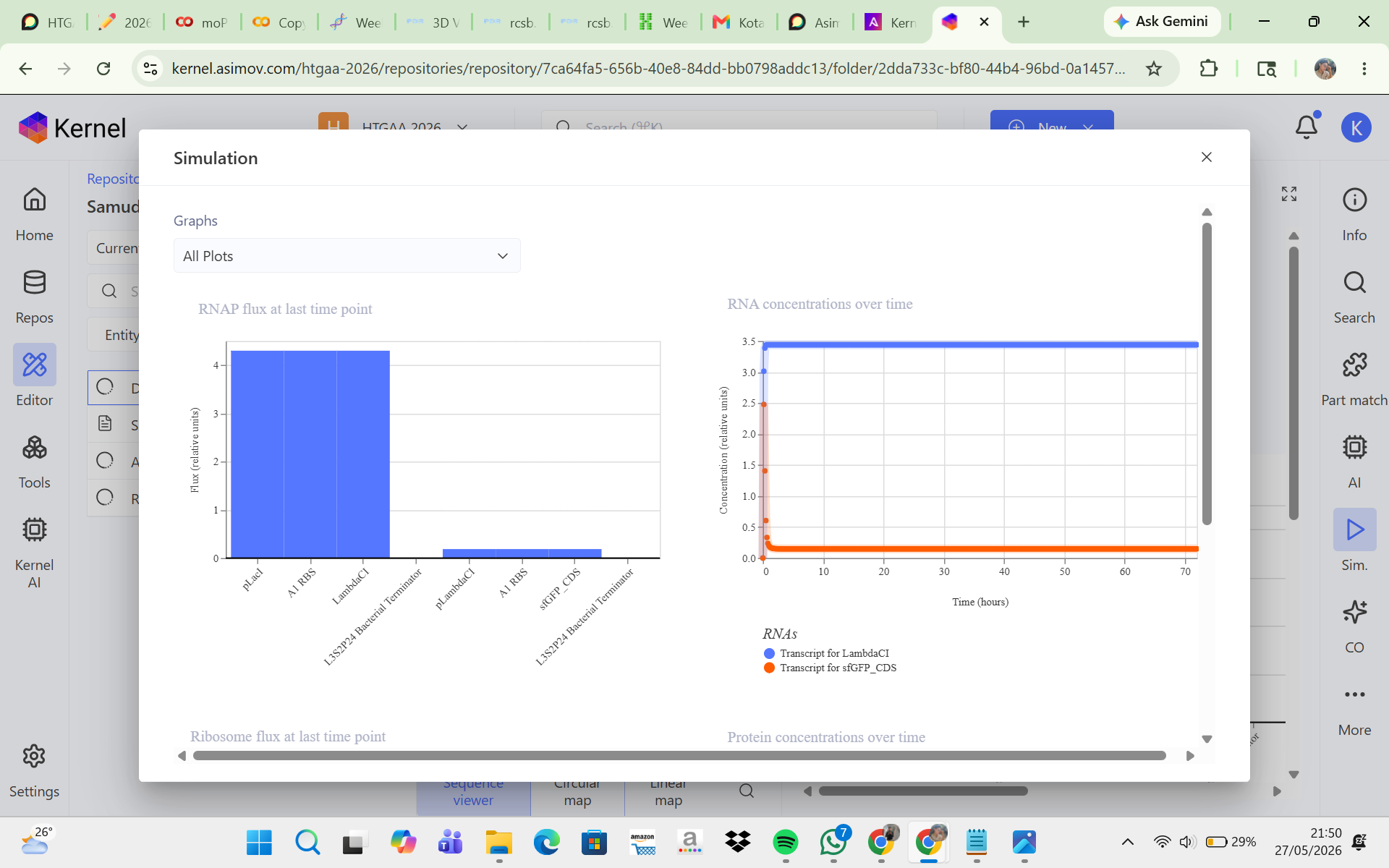
The goal of this construct was to create a delayed GFP expression system. In this design, the first promoter produces LambdaCI, and once LambdaCI accumulates to a sufficient level, the second promoter becomes active, allowing GFP to be expressed. The initial hypothesis was that GFP would not increase immediately at the beginning of the simulation because there would be a time delay while LambdaCI was first produced before it could activate GFP expression. However, the simulation results and analysis showed that LambdaCI was successfully expressed at a high and stable concentration, while sfGFP expression remained very low throughout the simulation. This suggests that the pLambdaCI promoter most likely functions as a promoter that is repressed by LambdaCI rather than activated by it. As a result, the circuit did not produce delayed GFP expression as expected. Adjustments such as replacing the second promoter, reducing repression strength, or increasing the sfGFP promoter strength may help generate a cascade behavior that is more consistent with the initial expectation.
CONSTRUCT 3. Simple Toggle Switch (Bistable Circuit)
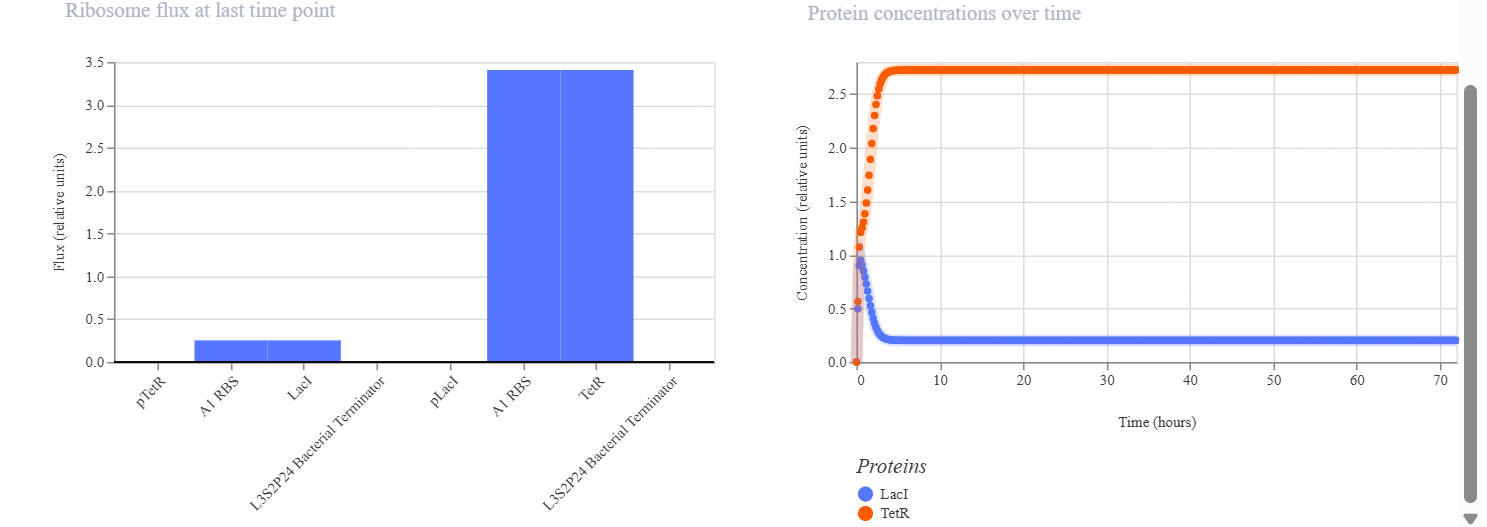
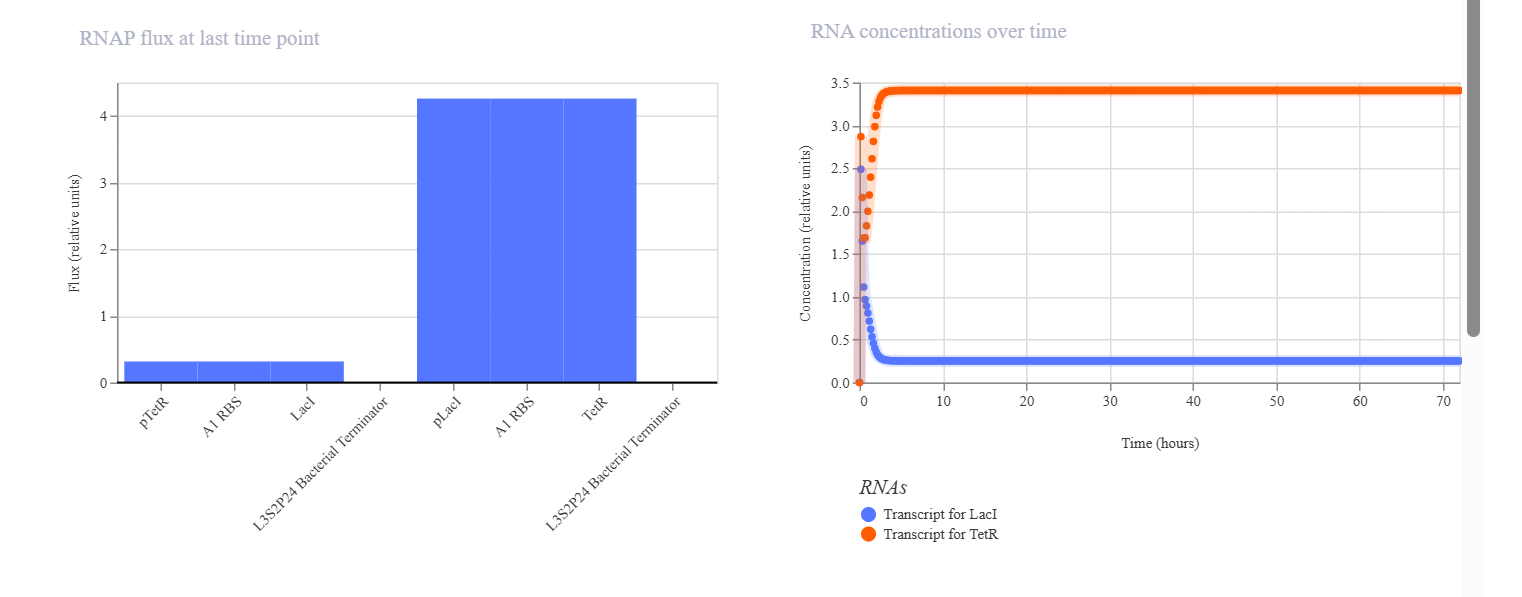
The goal of this construct was to make two proteins mutually inhibit each other so that only one would become dominant. In this system, LacI represses pLacI, while TetR represses pTetR, allowing the two proteins to compete with one another. The initial hypothesis was that the system would exhibit bistability, meaning that one protein would dominate while the other would be suppressed, and that the final state could depend on the initial concentration and simulation parameters. The simulation results and analysis showed that the toggle switch construct successfully produced bistable behavior. TetR became the dominant protein, maintaining a high and stable concentration, while LacI was suppressed to a low level. This indicates that the mutual repression between the two regulators worked effectively. TetR may have dominated because of stronger promoter activity, more effective repression, or an unbalanced initial condition. Adjusting simulation parameters such as promoter strength, degradation rate, and initial concentration may allow the alternative state, in which LacI dominates, to be observed.
Week 7 HW : Genetic Circuits Part II: Neuromorphic Circuits
PART 1: INTRACELLULAR ARTIFICIAL NEURAL NETWORKS
- Advantages of IANNs over Boolean Genetic Circuits
Intracellular Artificial Neural Networks (IANNs) provide key advantages over traditional Boolean logic-based genetic circuits. Conventional circuits produce discrete input-output relationships, limited to simple ON or OFF states, which restricts their information-processing power. In contrast, IANNs operate in an analog fashion, using molecular concentrations as continuous variables. This allows cells to generate nuanced, graded responses to fluctuating biological inputs.
These strengths make IANNs far more adaptable for handling the inherent complexity of biological systems, which rarely follow binary patterns. They also boost computational capacity inside cells by integrating multiple inputs at once, much like weighted summation in artificial neural networks. As a result, IANNs can support sophisticated functions, such as classifying biological signals or making multi-input decisions.
More specifically, IANNs offer:
• Continuous (analog) outputs, rather than just binary ON/OFF states
• Greater computational power through multi-input integration
• Robustness to biological noise, without relying on sharp thresholds
• Potential for multilayer network architectures
• More adaptive biological decision-making
Overall, IANNs offer a more realistic and advanced way to mimic how cells naturally process information.
- Applications of IANNs
One of the most promising uses for Intracellular Artificial Neural Networks (IANNs) is as smart biosensors for detecting complex diseases like cancer, which often involve multiple biomarkers. In such a system, engineered cells take in various biological inputs—for instance, two protein biomarkers (X1 and X2)—each weighted according to its relevance to the disease state. The cell then integrates these signals through molecular mechanisms resembling a perceptron: contributions from each input are summed and passed through a biological activation function. If the integrated result exceeds a threshold, the cell triggers an output, such as:
• Expression of a fluorescent protein for detection
• Activation of an apoptosis pathway to eliminate target cells
This approach outshines traditional biosensors, which detect only single biomarkers, by recognizing more specific, multifaceted disease patterns.
That said, deploying IANNs in biological systems comes with challenges, including:
• Biological noise that disrupts signal stability
• Limited dynamic range in gene expression
• Metabolic burden from extra components
• Cross-talk between regulatory pathways
• Difficulty in precisely tuning biological weights
- Diagram Intracellular Multilayer Perceptron
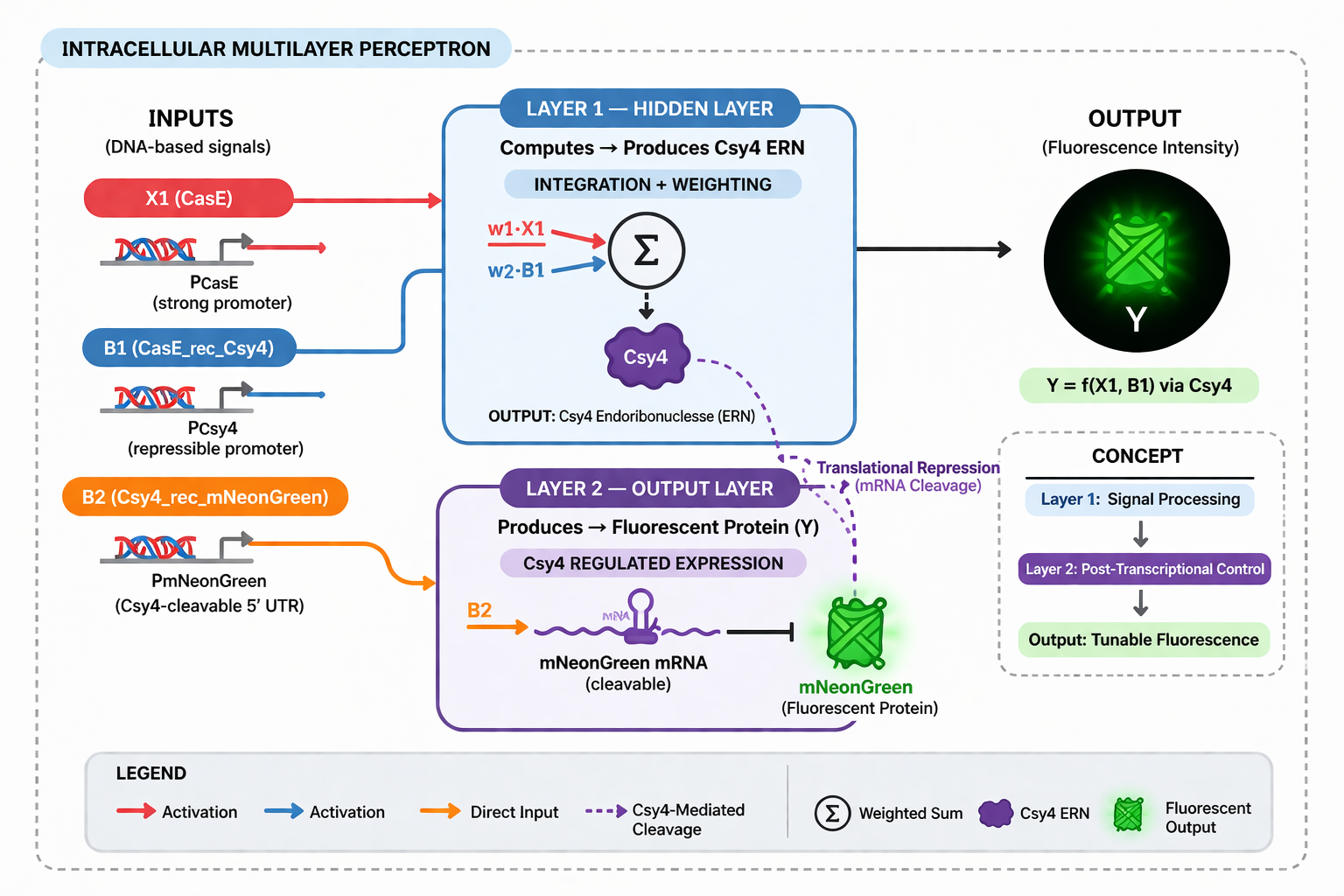
PART 2: FUNGAL MATERIALS
- Examples, Uses, Advantages, and Disadvantages of Fungal Materials
Fungal materials represent an innovative class of biomaterials made by growing fungal mycelium into sustainable, eco-friendly products. These have already found applications across industries, including:
• Mycelium-based packaging as a styrofoam alternative
• Synthetic mycelium leather for fashion
• Fungal bricks or construction panels
Producers cultivate mycelium on organic substrates like agricultural waste, forming dense, customizable structures. Compared to traditional materials, fungal options offer standout benefits. They’re fully biodegradable, avoiding environmental pollution, and require far less energy since they’re “grown” rather than manufactured through energy-intensive processes. Plus, they repurpose organic waste, aligning perfectly with circular economy principles.
Still, challenges remain:
• Lower mechanical strength than synthetic counterparts
• Sensitivity to humidity and environmental conditions
• Variability in outcomes due to the biological nature of fungi
• Longer production times versus conventional manufacturing
- Genetic Engineering in Fungi and Its Advantages
I am interested in developing environmentally responsive mycelium materials (smart responsive mycelium), living substances that sense changes around them and respond functionally. Using synthetic biology, we can engineer fungi to express specific genes in reaction to targeted stimuli, like pollutants, humidity shifts, or mechanical stress.
This engineering aims to produce active materials that don’t just sit passively but “sense” and “react,” opening doors to applications such as:
• Environmental biosensors (detecting pollutants or toxins)
• Adaptive construction materials (responding to humidity or cracks)
• Biomaterial-based monitoring systems (visual indicators of conditions)
Unlike conventional materials, these integrate biological functions directly into their structure, adding substantial value. Compared to bacteria, fungi offer distinct advantages for synthetic biology-driven biomaterials. As multicellular organisms, they naturally form three-dimensional mycelial networks, yielding solid, structured materials without extra manufacturing steps. Bacteria, being mostly unicellular, excel at molecule production but fall short for building bulk materials. Fungi also boast advanced protein secretion and thrive on cheap lignocellulosic waste substrates, making them more efficient and sustainable.
Challenges include slower growth rates and trickier genetic manipulation than bacteria. Yet for living, responsive, and adaptive materials, fungi shine by seamlessly blending biological functions with physical form.
Week 9 HW: Cell Free System
PART 1 — General Homework Questions
1. What are the main advantages of cell-free protein synthesis over in vivo methods, particularly in terms of flexibility and control of experimental variables? Provide at least two cases where CFPS is more beneficial.
Cell-free protein synthesis (CFPS) offers significantly greater flexibility compared to in vivo systems because it is not constrained by cellular viability. In this system, biological components are extracted from cells, allowing researchers to directly manipulate reaction conditions such as ion concentrations (Mg²⁺, K⁺), energy molecules (ATP), and enzymatic composition. This enables precise experimental control without interference from complex cellular regulatory networks. Furthermore, CFPS operates as an open system, meaning components can be added or removed dynamically during the reaction, enhancing experimental tunability.
CFPS is particularly advantageous in several scenarios, including:
• The production of toxic proteins (e.g., pore-forming proteins such as α-hemolysin), which would otherwise kill host cells.
• Rapid prototyping of genetic circuits in synthetic biology, as it eliminates the need for transformation and cell culture, thereby accelerating the design–build–test cycle.
2. What are the main components of a cell-free expression system and what are their respective roles?
A cell-free expression system consists of several essential components that collectively enable transcription and translation outside of living cells. The core component is the cell extract (cell lysate), typically derived from organisms such as E. coli, which contains ribosomes, transfer RNA (tRNA), transcription and translation enzymes, and other necessary factors. Additionally, a genetic template in the form of DNA or mRNA is required to encode the target protein. An energy system, including ATP and GTP, is crucial to drive biosynthetic reactions, often coupled with an energy regeneration mechanism to sustain the reaction over time.
Other important components include:
• Amino acids, which serve as building blocks for protein synthesis
• Ions (Mg²⁺, K⁺), which stabilize ribosomal structure and enzymatic activity
• Cofactors such as NAD⁺ and CoA, which support auxiliary metabolic reactions
3. Why is energy regeneration critical in cell-free systems, and how can continuous ATP supply be maintained?
Energy regeneration is critical in cell-free systems because protein translation is highly energy-intensive, requiring approximately four ATP molecules per peptide bond formation. Without an efficient regeneration system, ATP is rapidly depleted, leading to premature termination of protein synthesis. Therefore, maintaining a continuous ATP supply is essential for sustaining reaction efficiency and increasing protein yield.
One commonly used method is the phosphoenolpyruvate (PEP)-based system, where PEP is converted into pyruvate while generating ATP. Alternative approaches that offer improved stability include:
• Glucose-based energy systems that utilize metabolic pathways for ATP regeneration
• Creatine phosphate systems coupled with creatine kinase
These strategies help prolong reaction duration and enhance overall productivity.
4. Compare prokaryotic and eukaryotic cell-free systems. Provide one protein example for each and justify your choice.
Prokaryotic cell-free systems, such as those derived from E. coli, are known for their high speed, efficiency, and relatively low cost. However, they are limited in their ability to perform post-translational modifications such as glycosylation or complex disulfide bond formation. In contrast, eukaryotic systems, including wheat germ or mammalian extracts, are capable of more accurate protein folding and post-translational modifications, albeit at higher cost and lower production speed.
For example:
• Green Fluorescent Protein (GFP) is well-suited for prokaryotic systems because it does not require complex modifications.
• Antibodies (e.g., IgG) are better produced in eukaryotic systems due to their requirement for proper folding, disulfide bond formation, and glycosylation.
5. How would you design a cell-free experiment to optimize membrane protein expression? What challenges are involved and how can they be addressed?
Membrane protein expression in cell-free systems presents significant challenges because these proteins require a lipid environment to maintain proper structure and function. In the absence of such an environment, proteins are prone to misfolding and aggregation. Therefore, experimental design must incorporate strategies to mimic biological membranes.
Possible solutions include:
• Incorporating liposomes or nanodiscs to simulate membrane environments
• Using mild detergents to maintain protein solubility
• Enabling co-translational insertion, allowing proteins to integrate into membranes during synthesis
• Adding molecular chaperones such as GroEL/ES to assist in protein folding
These approaches can significantly improve protein stability and yield.
6. If low protein yield is observed, what are the possible causes and how would you troubleshoot them?
Low protein yield in cell-free systems can arise from several factors. One common cause is the degradation of DNA or mRNA by nucleases present in the extract. This issue can be mitigated by using more stable DNA templates or adding nuclease inhibitors. Another cause is insufficient energy supply due to ATP depletion, which can be addressed by optimizing the energy regeneration system, such as using PEP or glucose-based systems. Additionally, improper protein folding may result in aggregation and reduced yield. This can be improved by adding chaperones or optimizing reaction conditions such as temperature and ionic strength.
PART 2 — Synthetic Minimal Cell Design
7. Design a synthetic minimal cell: what is its function and how does it work?
The proposed synthetic minimal cell functions as a biosensor for detecting arsenic contamination in water. The system operates by sensing arsenic ions (As³⁺) through a regulatory protein such as ArsR. In the presence of arsenic, ArsR dissociates from the DNA, allowing transcription of a reporter gene such as GFP. The output is a measurable fluorescent signal that indicates the presence of arsenic in the environment.
8. Can this function be achieved without encapsulation or using natural cells?
This function can technically be achieved using a non-encapsulated cell-free system; however, it would lack the compartmentalization characteristic of a synthetic cell. It can also be implemented using genetically engineered natural cells such as E. coli, but this approach introduces limitations related to biosafety, regulatory complexity, and reduced modularity compared to synthetic vesicle-based systems.
9. What are the main components of the synthetic cell?
The synthetic cell consists of a lipid membrane composed of phospholipids such as POPC combined with cholesterol to enhance stability. Inside the vesicle, an E. coli-based cell-free system is encapsulated along with DNA encoding the arsR regulatory gene and the gfp reporter gene. This setup enables arsenic detection and signal generation within a controlled microenvironment.
10. How does the system communicate with the environment and how is the output measured?
Communication with the environment occurs through the diffusion of arsenic ions across the lipid membrane, as small molecules can permeate the vesicle. Upon detection, GFP is produced and can be measured using fluorescence spectroscopy or flow cytometry. This output provides a quantitative indication of arsenic concentration.
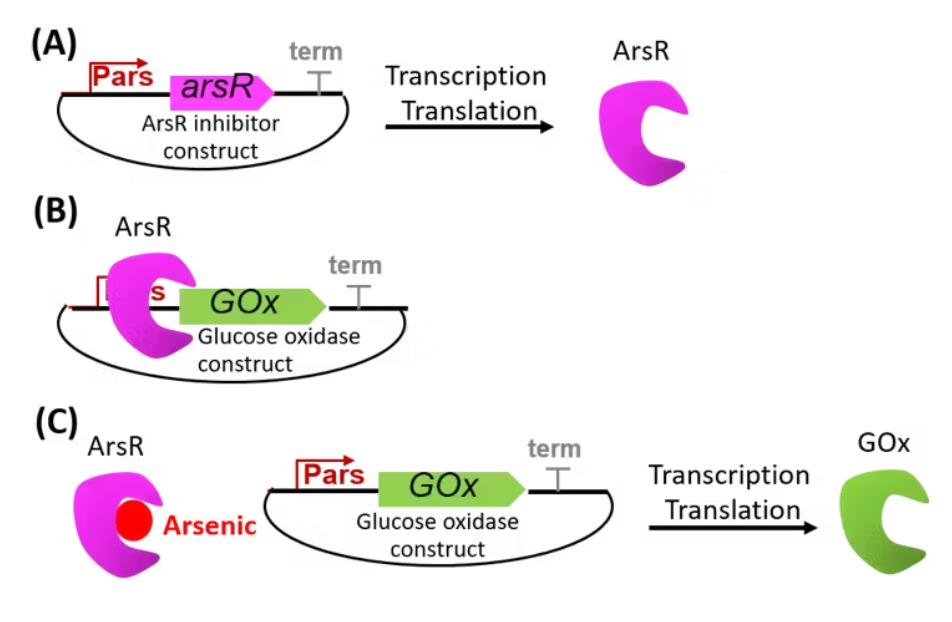
PART 3 — Freeze-Dried CFPS Application (Textiles)
11. What is the proposed application of cell-free systems in textiles?
The proposed concept involves smart textiles embedded with freeze-dried cell-free systems capable of detecting air pollutants. Upon exposure to environmental moisture, the system becomes activated and responds to pollutants such as nitrogen dioxide or heavy metals by producing a visible color change. This enables real-time monitoring of air quality through wearable materials.
12. How can the limitations of cell-free systems be addressed in this application?
Key limitations of cell-free systems include stability, activation requirements, and single-use constraints. These challenges can be addressed through:
• Enhancing stability via freeze-drying with protective agents such as trehalose
• Enabling activation through environmental moisture
• Designing modular and replaceable patches to mitigate single-use limitations
PART 4 — Genes in Space Proposal
13. What is the background and objective of this study?
Cosmic radiation in space poses a significant threat to DNA integrity and astronaut health, particularly through the induction of double-strand breaks. Understanding DNA repair mechanisms under microgravity conditions is essential for long-term space exploration and biotechnological advancements. This study aims to investigate the activity of the RecA protein in DNA repair using a cell-free system.
14. What is the molecular target and hypothesis?
The molecular target of this study is the recA gene and radiation-damaged DNA. The hypothesis is that microgravity and radiation conditions reduce the efficiency of RecA-mediated DNA repair. This effect is expected to be reflected in decreased fluorescence signals from a DNA repair reporter system.
15. What is the experimental plan?
The experiment will utilize the BioBits® cell-free protein expression system to produce RecA protein in vitro. Samples will include undamaged DNA as a control and DNA damaged by UV or radiation. DNA repair activity will be measured using a fluorescence reporter, with instruments such as the miniPCR thermal cycler and P51 Molecular Fluorescence Viewer. The resulting data will provide insights into DNA repair efficiency under space conditions.