Individual Final Project
In Silico Design of a Humanized Chimeric Arginine Deiminase
ABSTRACT
Arginine deiminase (ADI) is a bacterial enzyme with promising therapeutic potential for the treatment of arginine-auxotrophic cancers such as hepatocellular carcinoma, melanoma, and mesothelioma. ADI exerts its antitumor effect by depleting extracellular arginine, an amino acid essential for the survival of certain tumor cells lacking argininosuccinate synthetase expression. Despite its therapeutic promise, the bacterial origin of ADI leads to high immunogenicity, which may trigger immune responses, reduce enzyme efficacy, and limit repeated administration. This project aims to design a humanized chimeric ADI in silico by identifying immunogenic epitopes in bacterial ADI and substituting them with structurally analogous regions from the human arginine-metabolizing enzyme while preserving catalytic function. Structural modeling, immunogenic epitope prediction, molecular docking, and protein validation analyses will be performed to evaluate the stability and substrate-binding ability of the engineered chimera. The resulting humanized ADI is expected to exhibit reduced immunogenicity while maintaining therapeutic activity, providing a potential strategy for improving enzyme-based cancer therapies.
PROJECT AIMS
To identify immunogenic epitopes in bacterial arginine deiminase using in silico epitope prediction tools.
To identify structurally compatible regions in the human arginine-metabolizing enzyme for epitope substitution.
To construct a chimeric ADI model by replacing immunogenic bacterial epitopes with human peptide fragments.
BACKGROUND
Cancer cells often undergo metabolic reprogramming to sustain rapid proliferation. Some tumors, including melanoma, hepatocellular carcinoma, and mesothelioma, are unable to synthesize sufficient arginine due to low expression of argininosuccinate synthetase (ASS1). As a result, these cancers become dependent on extracellular arginine for growth and survival. This metabolic vulnerability makes arginine deprivation an attractive therapeutic strategy.
Arginine deiminase (ADI) is a microbial enzyme that catalyzes the hydrolysis of arginine into citrulline and ammonia, thereby depleting extracellular arginine and suppressing tumor growth. ADI derived from bacterial sources has shown significant antitumor activity, making it a promising candidate for enzyme therapy. However, as a foreign bacterial protein, ADI can induce immune responses in patients, resulting in antibody production, rapid clearance, hypersensitivity, and decreased therapeutic efficacy.
One promising approach to overcome this limitation is protein humanization, in which immunogenic regions of a bacterial therapeutic enzyme are replaced with structurally similar fragments from human proteins. This strategy has been successfully demonstrated in previous in silico studies on bacterial therapeutic enzymes to reduce antigenicity while maintaining enzymatic activity.
Therefore, this project proposes the in silico design of a humanized chimeric arginine deiminase, where immunogenic epitopes of bacterial ADI are replaced with homologous regions from a human arginine-metabolizing protein. This design aims to reduce immunogenicity while preserving the enzyme’s ability to bind arginine, thereby improving its therapeutic potential for cancer treatment.
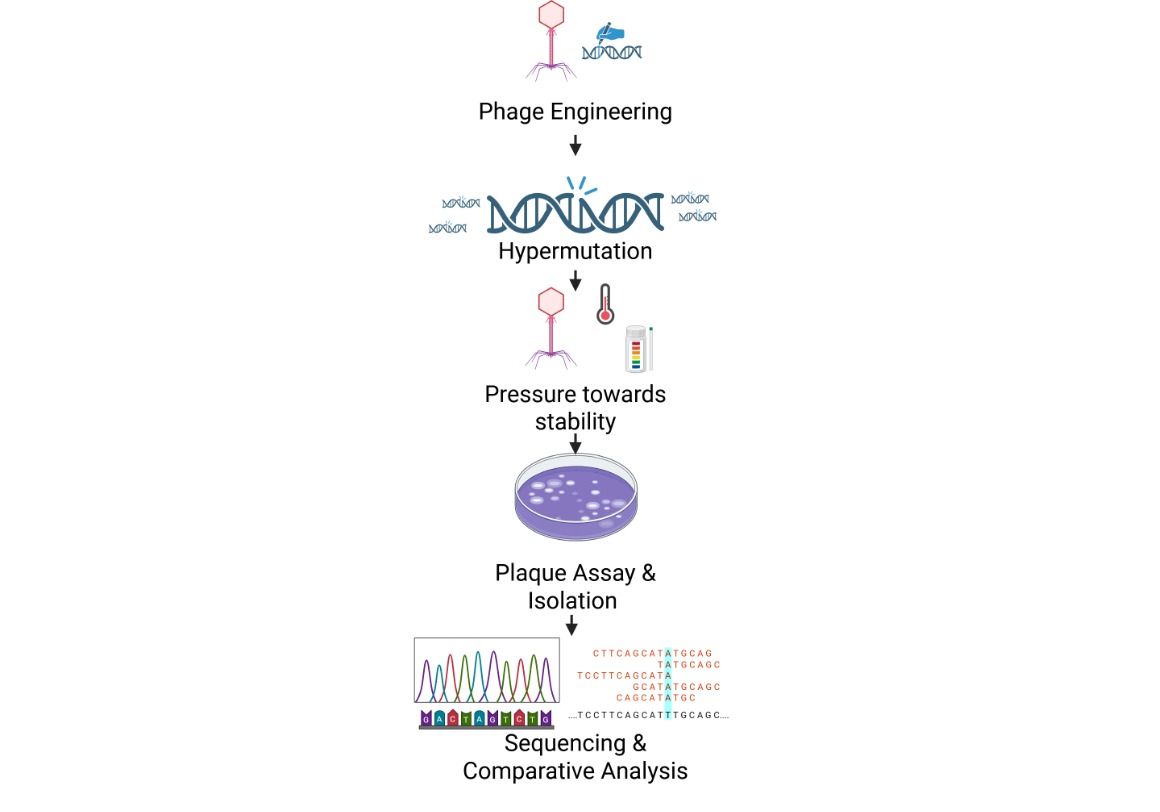
STEP-BY-STEP METHODS (IN SILICO WORKFLOW)
1. Retrieval of Protein Structures and Sequences
The amino acid sequences and three-dimensional structures of arginine deiminase (ADI) from Mycoplasma arginini (PDB ID: 1LXY) and human arginase I (PDB ID: 2AEB) will be retrieved from the Protein Data Bank (PDB).
2. Prediction of Immunogenic T-cell Epitopes in Bacterial ADI
To predict immunogenic T-cell epitopes in bacterial arginine deiminase (ADI; PDB ID: 1LXY), the protein’s amino acid sequence will be submitted to the NetMHCIIpan server for MHC class II binding prediction, focusing on regions likely to elicit T-cell immune responses. The sequence will be fragmented into overlapping peptides of 9–15 amino acids, with binding affinities evaluated against common HLA-DR alleles—HLA-DRB101:01, HLA-DRB103:01, HLA-DRB104:01, HLA-DRB107:01, HLA-DRB108:01, HLA-DRB111:01, HLA-DRB113:01, and HLA-DRB115:01—which serve as reference alleles with broad global frequency in the literature. Epitope density will be determined for each protein, selecting peptides classified as strong binders (SB, IC50 ≤ 1%) or weak binders (WB, IC50 ≤ 5%) as candidate immunogenic epitopes for targeted substitution.
3. Allergenicity Prediction of Candidate Epitopes
Candidate immunogenic epitopes identified from bacterial ADI will undergo allergenicity prediction using AllerTOP v.2.0 to assess their potential to provoke allergic responses. Epitopes classified as probable allergens will be prioritized for substitution, ensuring targeted modification of regions most likely to elicit adverse immune reactions
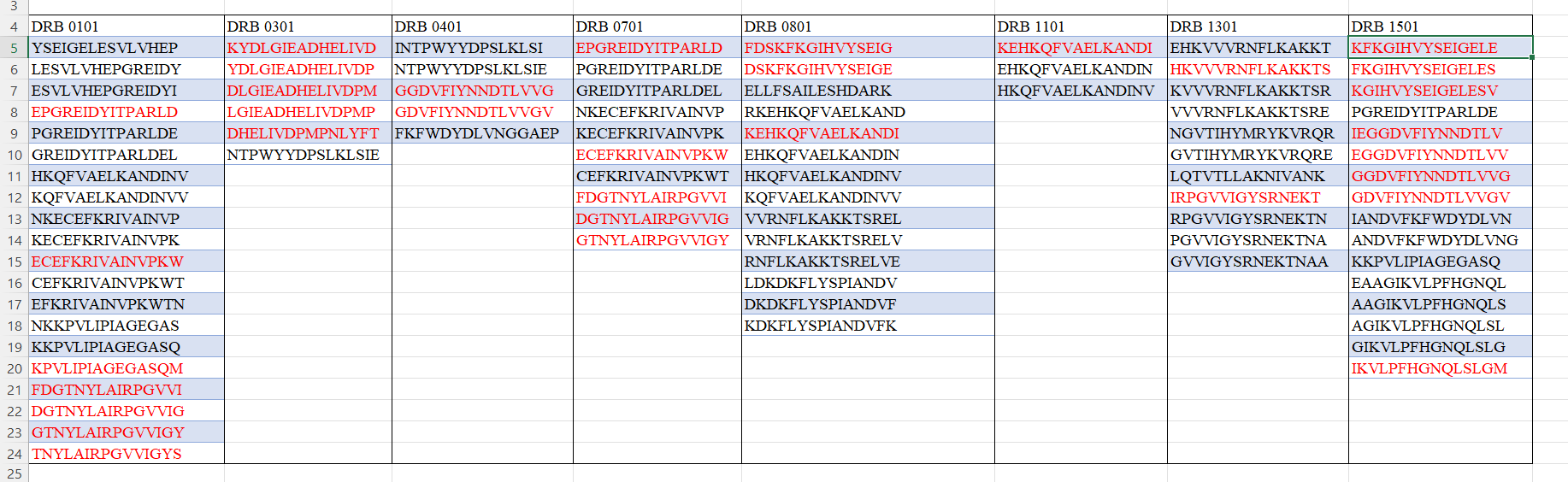
4. Epitope Alignment and Mapping
Not all predicted allergenic epitopes will be substituted; selection will prioritize those that are surface-exposed (solvent-accessible), readily recognizable by the immune system, located outside the active site, and non-essential for structural stability. This focuses substitution on the “most exposed” residues—defined as surface-accessible epitopes—rather than solely those with the highest immunogenicity scores. Active site residues were identified by detecting amino acid residues located within 4 Å of the native ligand in the crystal structure using BioPython. These residues were excluded from epitope substitution to preserve catalytic activity.
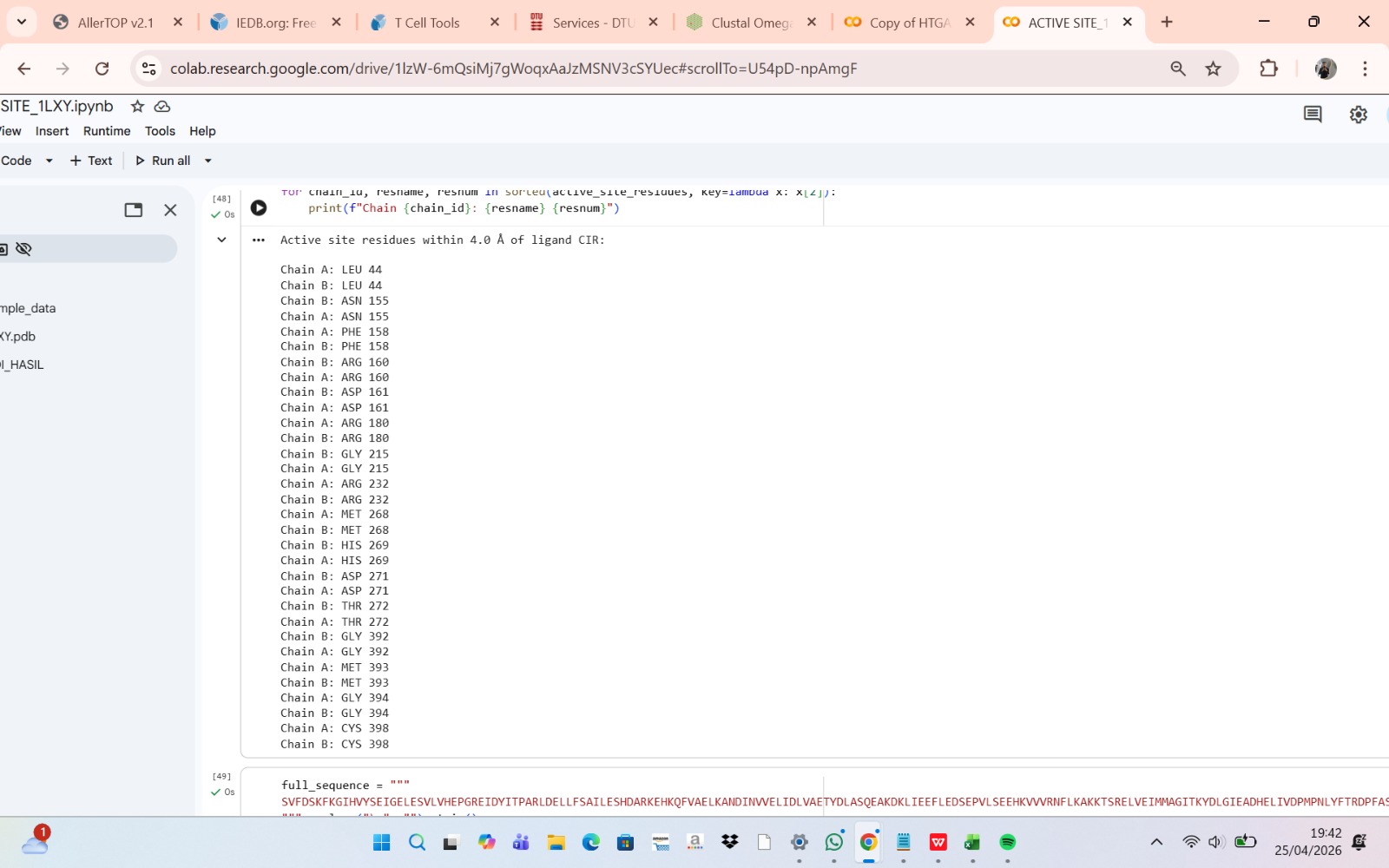
The linear positions of each epitope within the target protein sequence are mapped using Biopython (SeqIO module). The full protein sequence is read from a FASTA file, and each epitope peptide is located via substring matching to determine the start and end residues. This positional information facilitates overlap analysis with active site residues and three-dimensional structural mapping.
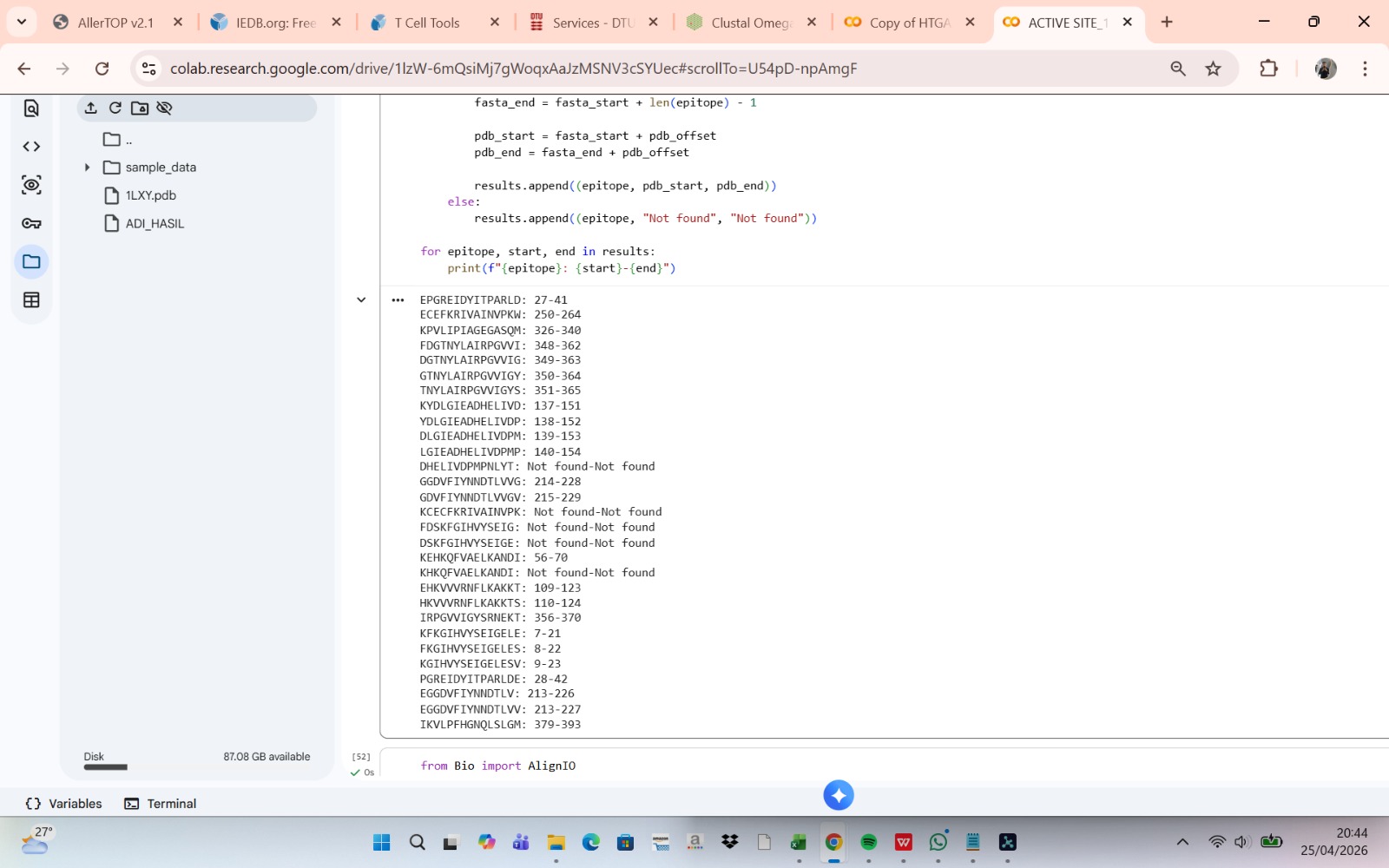
After identifying the active site location, the epitope sequence order, and the RSA value per residue for each epitope calculated using the NetSurfp 3.0 web tool. The selected epitope for the next step meets the criteria of not overlapping with the active site and having a large RSA value of at least 25%. The RSA value is determined by averaging the RSA values of each residue in the epitope divided by the number of residues in the epitope.
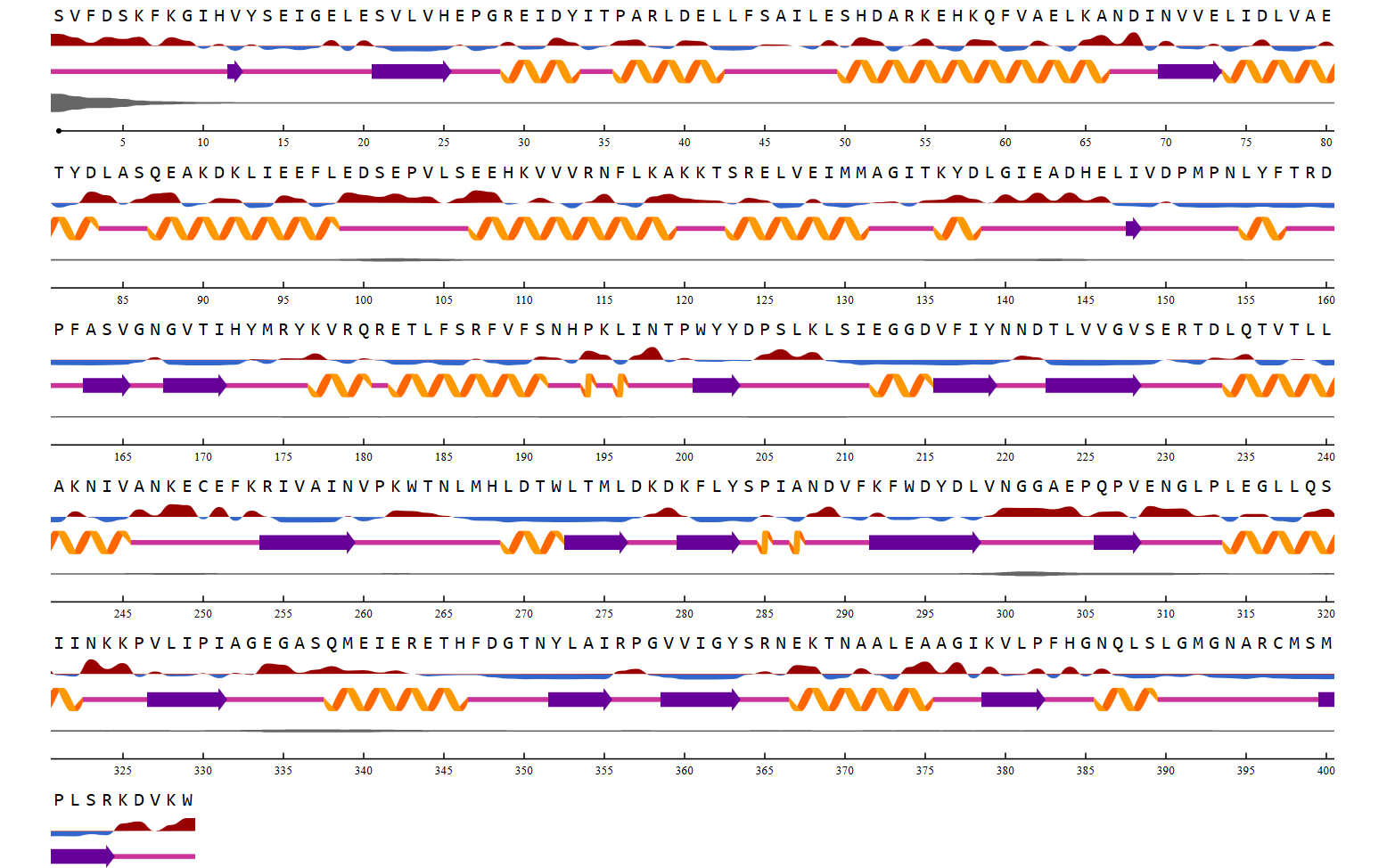
5. Epitope Conservation Analysis and Candidate Selection
Sequence epitopes were aligned against the human protein (2AEB) to evaluate sequence conservation. The alignment was performed using a pairwise alignment approach, and conservation scores were calculated as the proportion of identical or similar residues within each epitope region.
The conservation score was defined as:
Highly conserved: >0.8
Moderately conserved: 0.4–0.8
Low conserved: <0.4
Epitopes with high conservation were excluded from mutation due to their potential structural or functional importance. Moderately conserved epitopes were considered suitable candidates for conservative mutation strategies, aiming to reduce immunogenicity while preserving protein stability.
After identifying low-conserved epitopes, the next step was to evaluate their identity and similarity scores. Identity is defined as the percentage of residues that are exactly identical between the bacterial epitope and the homologous region in the human protein, reflecting direct sequence equivalence. A higher identity indicates greater similarity to the human protein, fewer required substitutions, and a lower risk of structural disruption. Similarity, on the other hand, represents the percentage of residues that are either identical or share similar physicochemical properties. Higher similarity suggests that substitutions are more conservative and that the overall biochemical characteristics of the epitope are preserved, making it a key parameter for rational humanization.
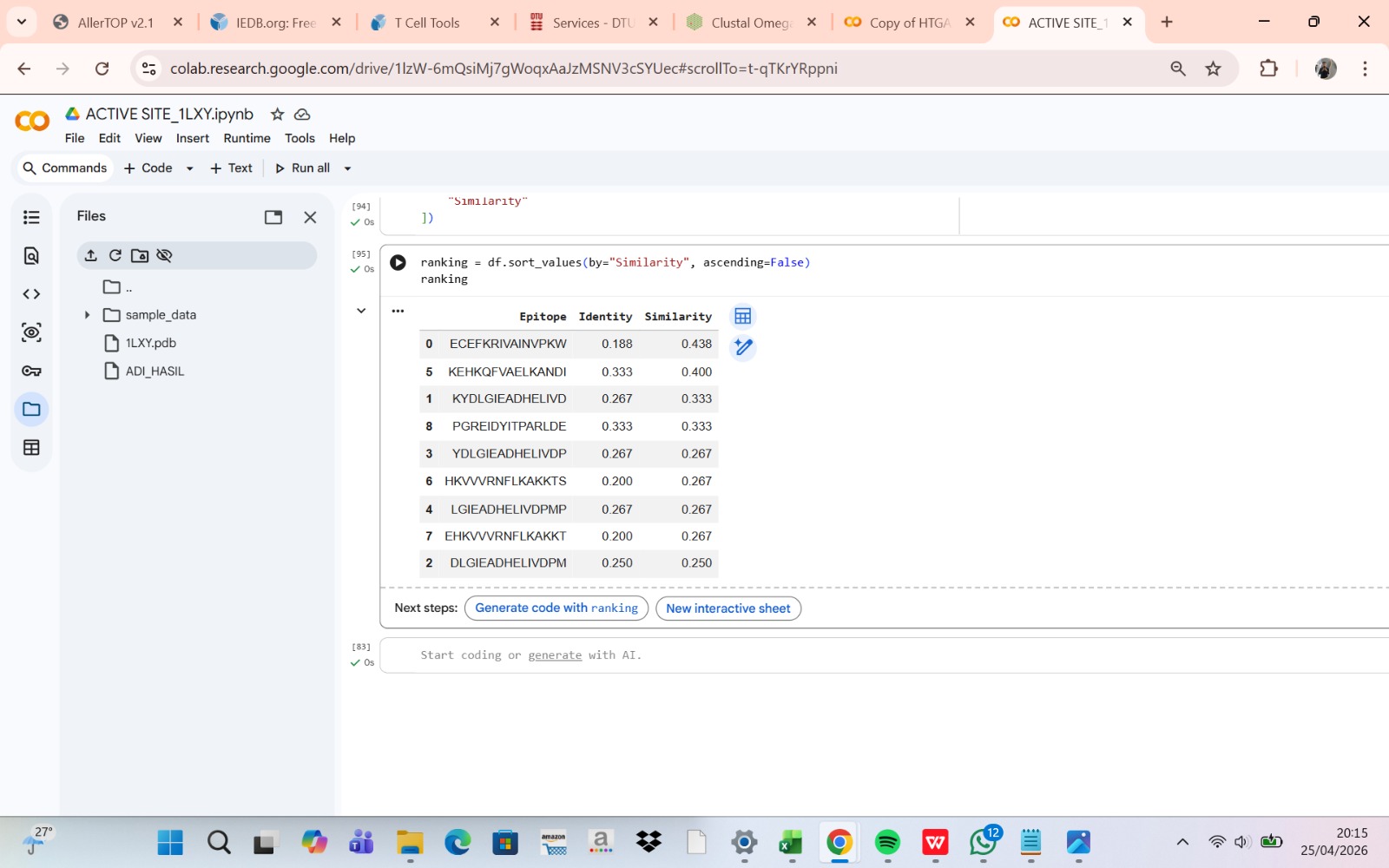
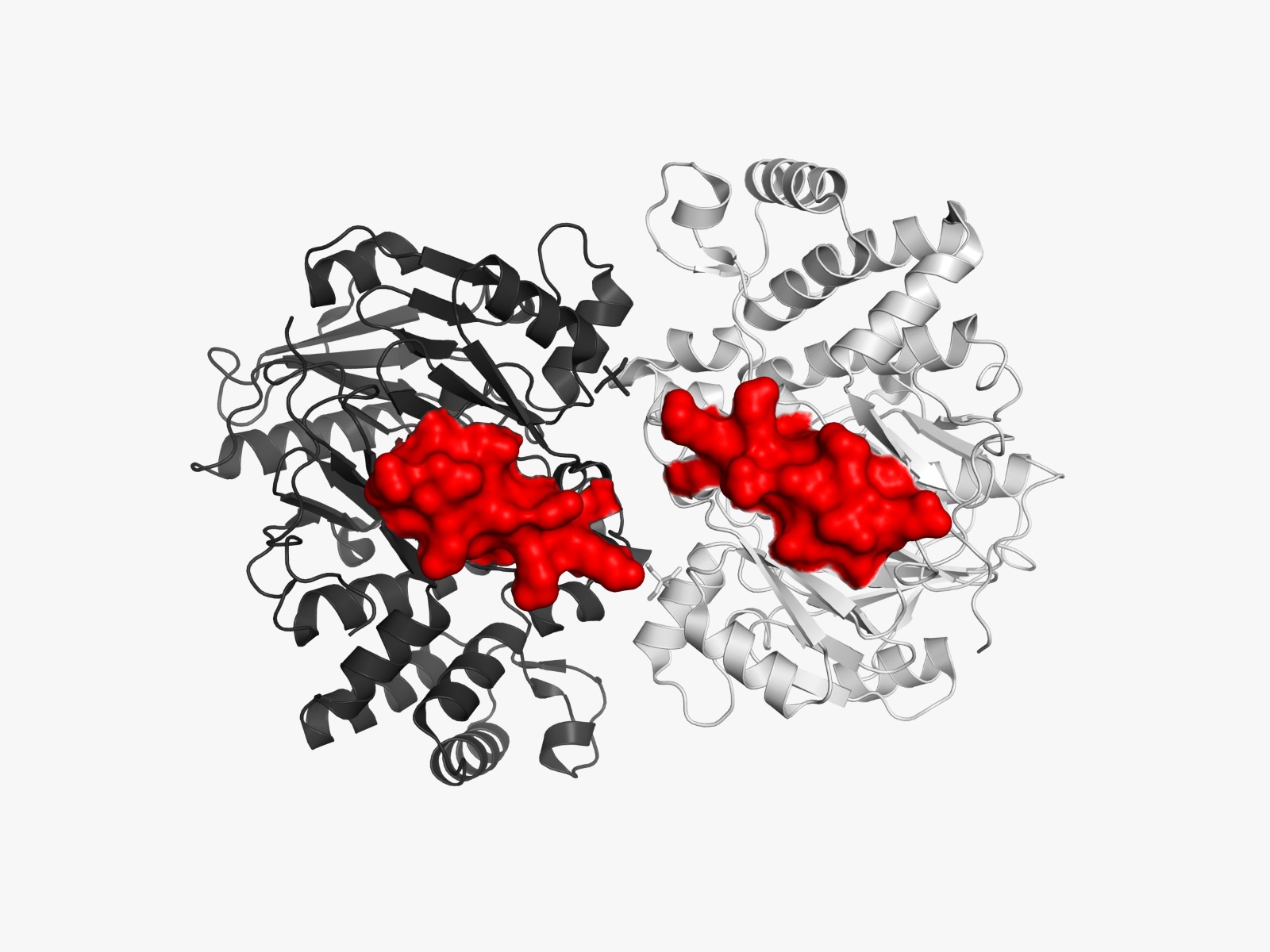
Among the evaluated epitopes, KEHKQFVAELKANDI exhibited the highest identity (0.33) and relatively high similarity (0.40) compared to the corresponding human arginase region. This indicates that minimal and conservative substitutions would be required to humanize this epitope while maintaining its structural and functional integrity. Therefore, this epitope was selected as the most suitable candidate for rational humanization.
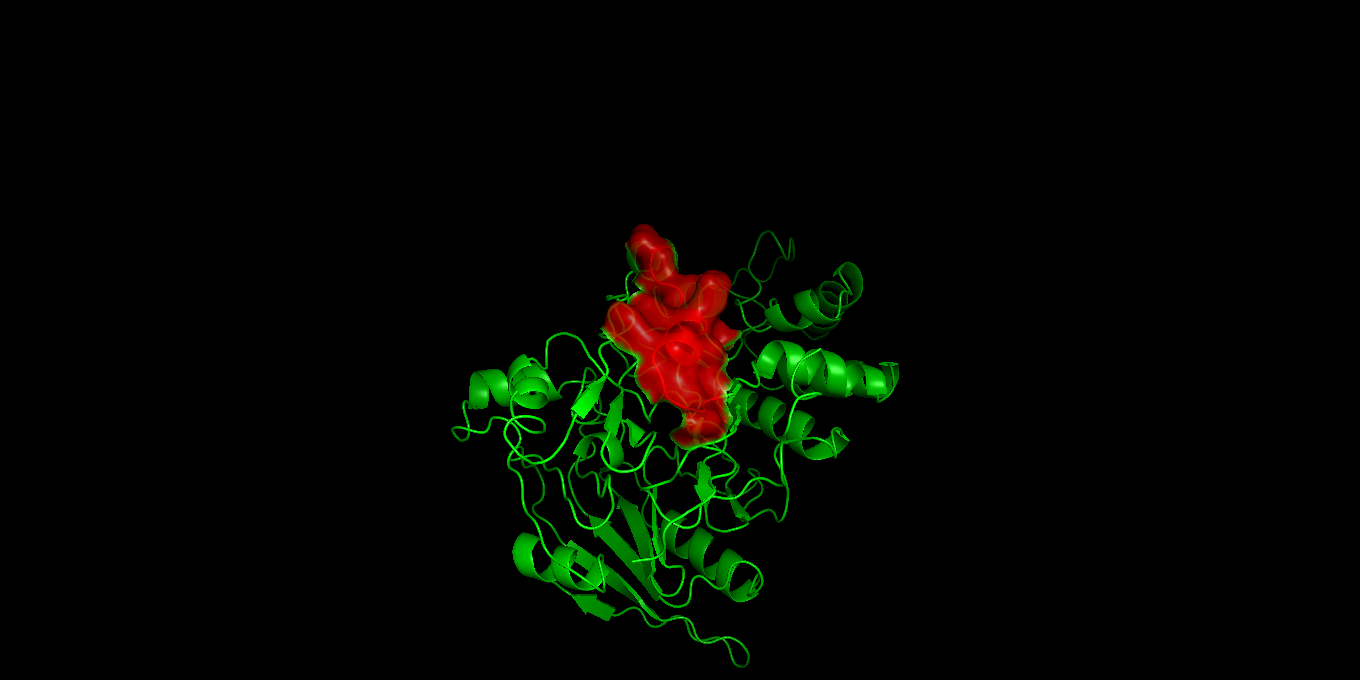
Visualization of the epitope KEHKQFVAELKANDI on its native protein structure (PDB: 1LXY).
6. Identification of Local Homologous Segments Using 15-mer Smith–Waterman Alignment
To identify the human protein region that is locally homologous to the bacterial epitope, a 15-mer peptide-based local alignment approach was applied. The bacterial epitope sequence (15 amino acids) was used as a query and aligned against the full-length human arginase protein (PDB: 2AEB) using the Smith–Waterman algorithm, which is designed to detect optimal local similarities between sequences. This method scans the entire human sequence and calculates alignment scores for all possible regions, allowing the identification of the segment with the highest local alignment score. The region with the best score—reflecting the highest combination of identity and similarity—was defined as the local homologous segment.
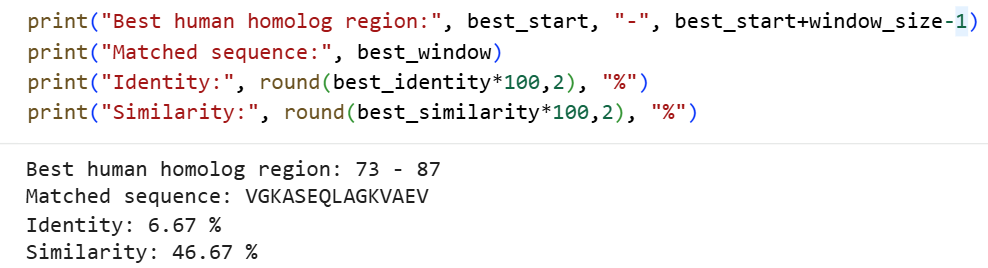
The resulting best-matching human segment VGKASEQLAGKVAEV (residues 73–87) exhibited moderate similarity (46.67%) but very low identity (6.67%) compared to the bacterial epitope. This indicates that, although there is minimal exact sequence overlap, several residues share similar physicochemical properties, suggesting the presence of conservative substitutions. Therefore, this region provides a rational basis for epitope humanization, where targeted substitutions can be designed to increase similarity to the human sequence without significantly disrupting protein stability or function.
7. Homology Modeling and Structural Validation of the Humanized Chimeric Protein
The human homologous sequence VGKASEQLAGKVAEV was inserted into the protein sequence of 1LXY to replace the bacterial epitope KEHKQFVAELKANDI, and the resulting structure was modeled using SWISS-MODEL. The purpose of this visualization was to evaluate the three-dimensional structure formed and to assess whether the humanized chimeric Arginine Deiminase affects overall structural stability.
SWISS sturcture visualization
PyMOL visualization
Rachmacandram plot
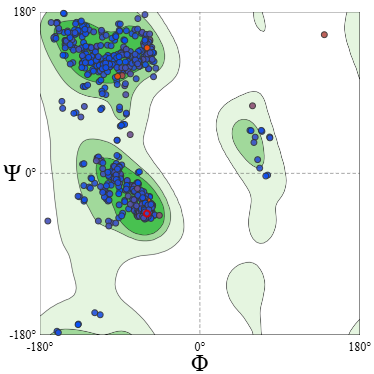
Based on the evaluation of the Ramachandran plot, the modeled structure shows good stereochemical quality, with the majority of residues located in favored regions and only a very small proportion classified as outliers. This indicates that the backbone conformation is largely acceptable and that the substitution of the epitope does not significantly disrupt the overall protein structure. Therefore, the humanized modification can be considered structurally stable and suitable for further analysis.
MolProbity Results
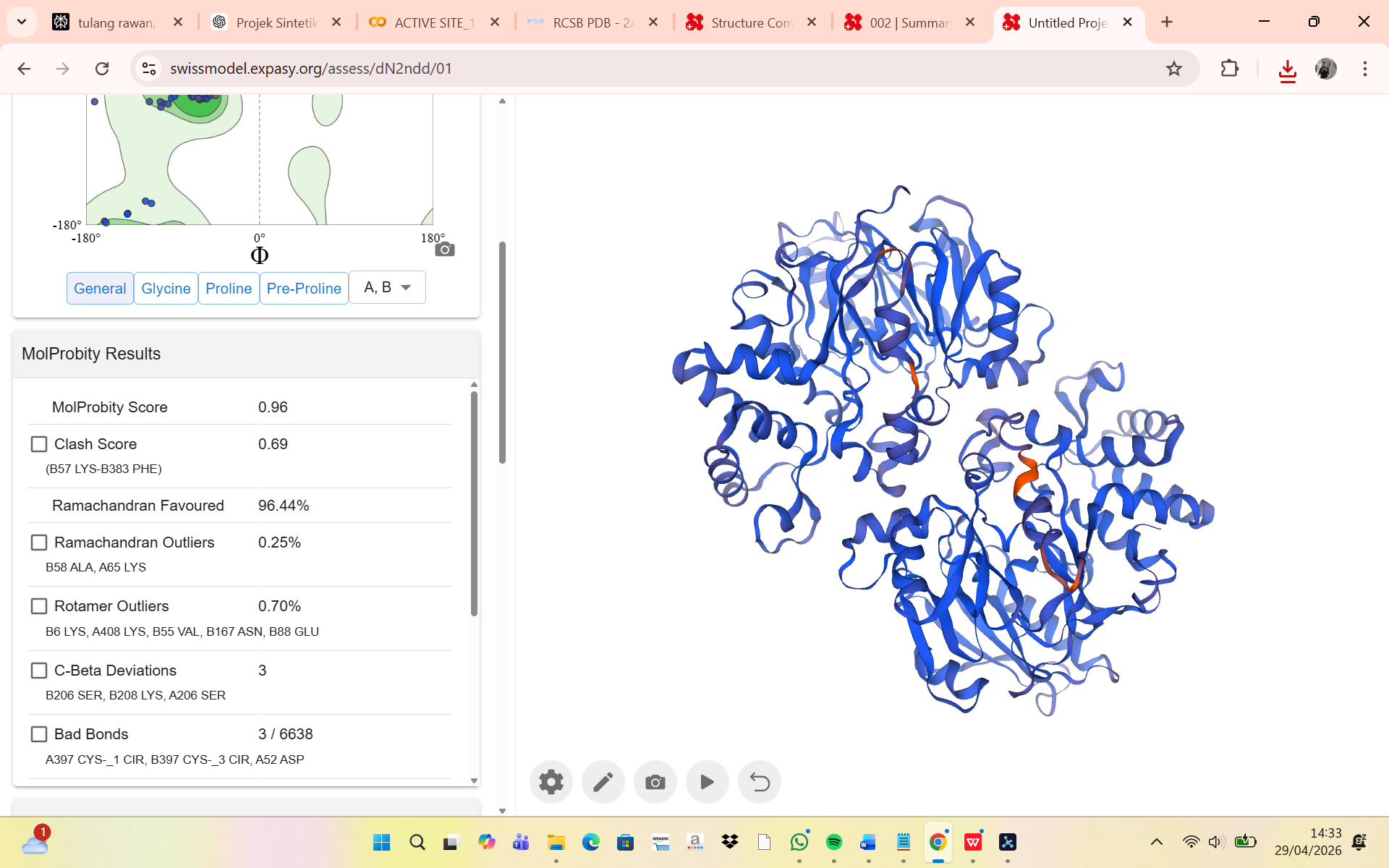
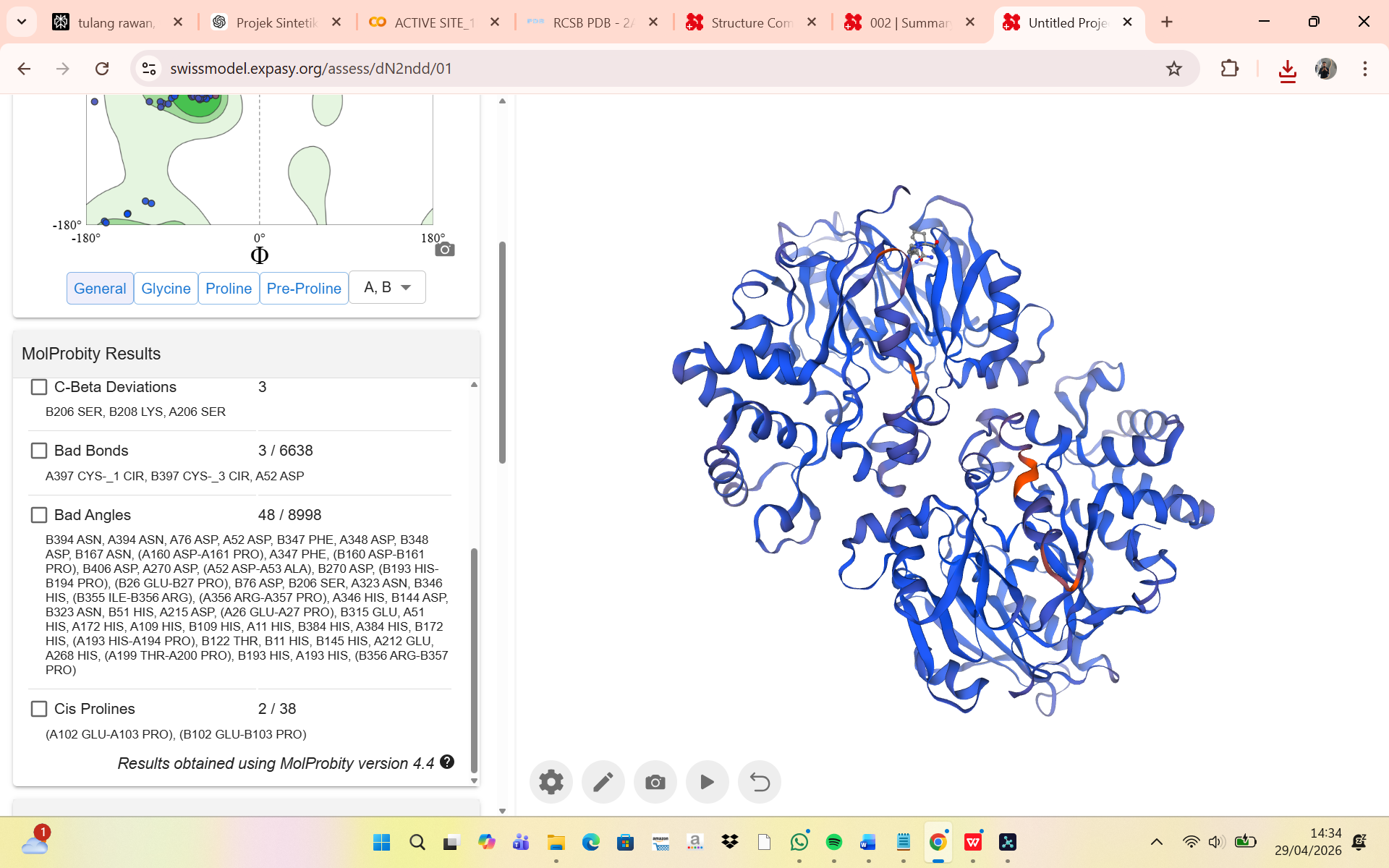
The structural quality of the humanized chimeric protein model was evaluated using the MolProbity validation server, which assesses stereochemical accuracy through several parameters, including MolProbity score, clashscore, Ramachandran analysis, rotamer quality, bond geometry, and angle deviations. The obtained MolProbity score was 0.96, indicating an excellent overall structural quality comparable to high-resolution experimental crystal structures. The clashscore value of 0.69 suggests that the model contains very few steric clashes between atoms, demonstrating that the introduced mutations did not generate significant spatial conflicts within the protein structure.
Ramachandran plot analysis showed that 96.44% of residues were located in favored regions, while only 0.25% were classified as outliers. Based on standard structural validation criteria, this result indicates that the protein backbone conformation is highly stable and that the humanization process did not introduce major structural distortions. Similarly, the rotamer outlier percentage was only 0.70%, showing that nearly all side chains adopted energetically favorable conformations. Minor deviations were observed in several flexible residues such as lysine, glutamate, and asparagine, which are commonly associated with local conformational variability.
Additional geometric validation showed only 3 C-beta deviations, 3 abnormal bond lengths out of 6638 total bonds, and 48 abnormal bond angles out of 8998 total angles, all of which remain within acceptable ranges for computationally modeled proteins. The presence of two cis-proline residues was also considered tolerable, as cis conformations may naturally occur in loop or flexible regions. Overall, these results demonstrate that the humanized chimeric Arginine Deiminase maintains good stereochemical quality and structural stability, indicating that the epitope substitution strategy did not significantly compromise the integrity of the protein model and that the structure is suitable for further computational and functional analyses.
RMSD Value
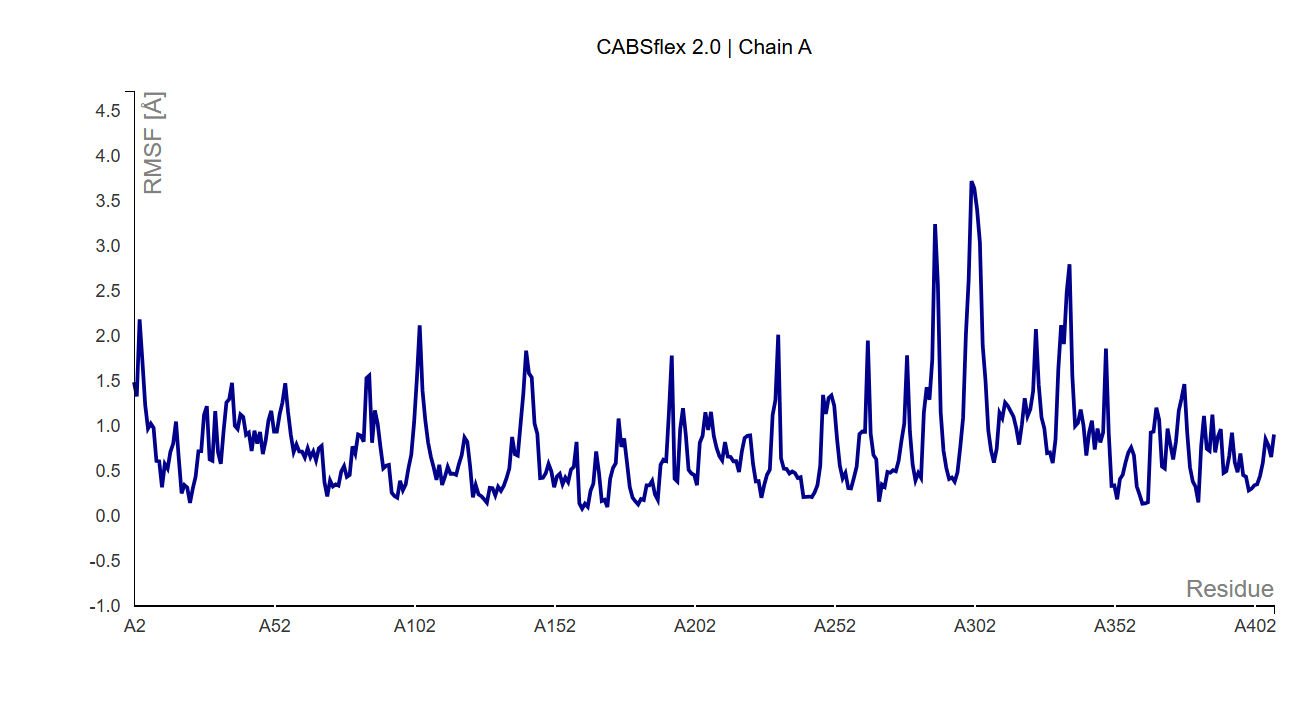
The structural flexibility of the humanized chimeric protein was evaluated using CABS-flex simulation by analyzing the Root Mean Square Fluctuation (RMSF/RMSD-like flexibility profile) of each residue. RMSD and RMSF values describe the magnitude of atomic movement during structural simulation, where lower values indicate higher structural stability and lower conformational fluctuation, while higher values indicate flexible or mobile regions within the protein structure. In general, protein regions with RMSD or RMSF values below approximately 1–2 Å are considered relatively stable and structurally well maintained.
Based on the simulation results, the chimeric protein showed an average RMSD value of 0.791 Å across 409 residues, indicating that the overall structure remained highly stable throughout the simulation. Most residues exhibited fluctuations below 1.5 Å, suggesting that the majority of the protein backbone experienced only minor movements and maintained its structural integrity after epitope substitution.
Importantly, the overall low mean RMSD value demonstrates that the humanization process did not significantly destabilize the protein structure. The fluctuations observed were mainly localized rather than global, indicating that the chimeric Arginine Deiminase retained a stable overall fold while allowing limited flexibility in certain surface regions.
8. Molecular Docking Analysis of the Humanized Chimeric Arginine Deiminase
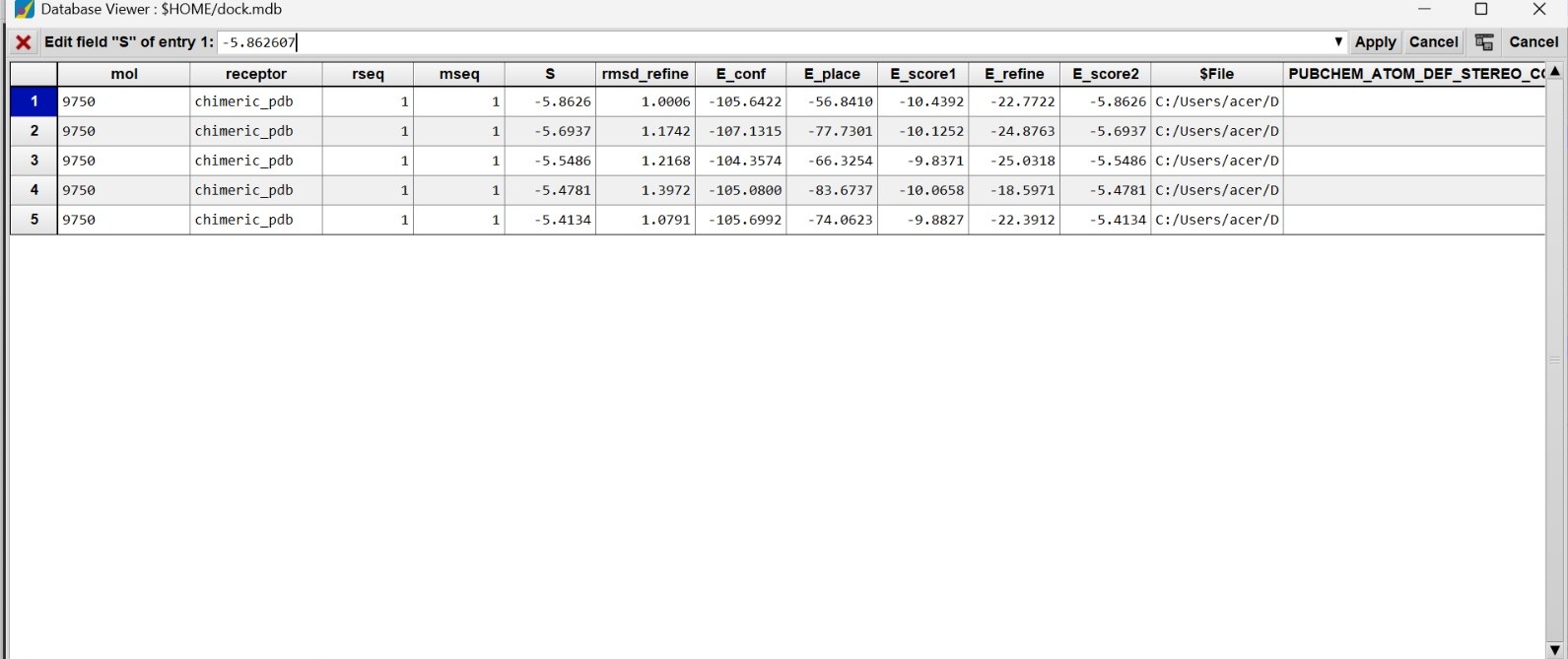
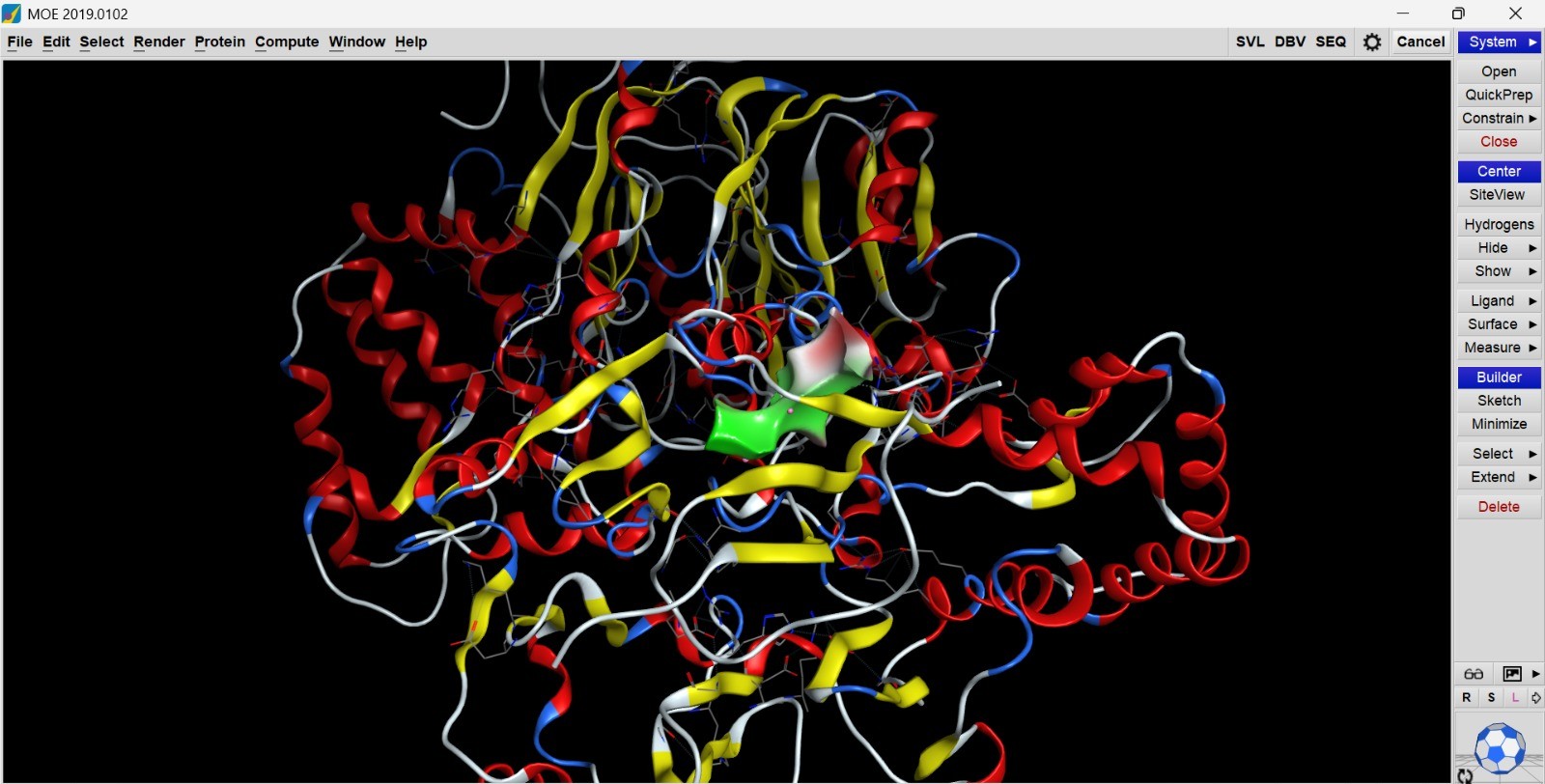
Molecular docking analysis was performed using the MOE 2019 Software to evaluate the interaction between the humanized chimeric Arginine Deiminase protein and its ligand at the predicted active site. Docking interpretation was primarily based on the docking score (S score), RMSD refinement value, binding energy components, and intermolecular interaction profiles such as hydrogen bonding and electrostatic interactions. In molecular docking studies, a more negative docking score indicates stronger predicted binding affinity and more favorable interaction stability between the protein and ligand. Meanwhile, RMSD values close to or below 2.0 Å generally indicate that the ligand binding pose is stable and structurally reliable.
The docking results showed that the best docking pose produced an S score of −5.86 kcal/mol with an RMSD refine value of approximately 1.00 Å. This result indicates that the ligand was able to bind stably within the active site pocket of the chimeric protein. All five docking poses displayed relatively similar docking scores (−5.41 to −5.86 kcal/mol), suggesting consistent ligand accommodation and stable binding conformations within the catalytic region. The low RMSD values (approximately 1.0–1.4 Å) further support the structural consistency of the docking poses and indicate minimal conformational deviation during refinement.
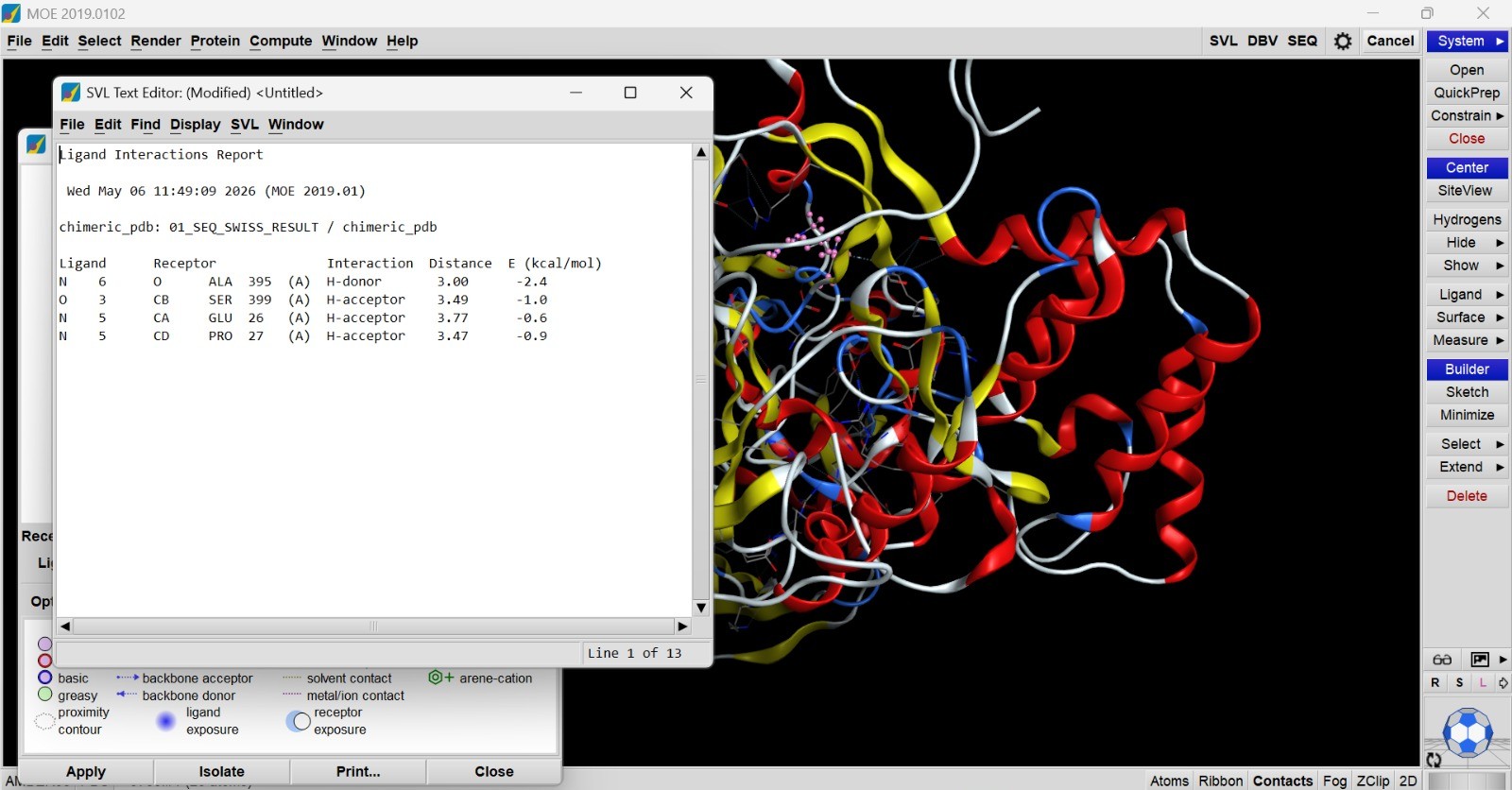
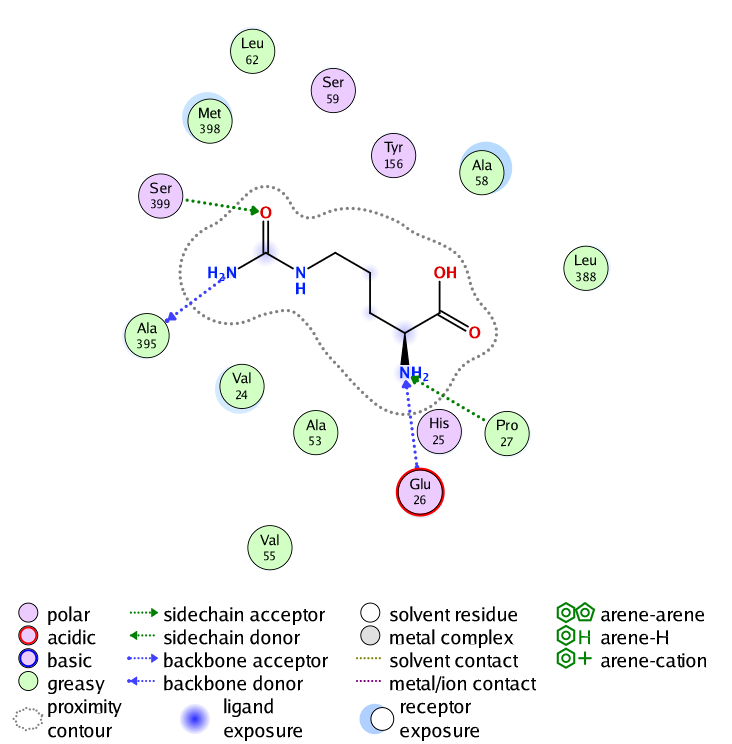
Interaction analysis demonstrated that the ligand formed several stabilizing hydrogen bond interactions with nearby amino acid residues, including Alanine 395, Serine 399, Glutamate 26, and Proline 27. Hydrogen bond distances ranged from approximately 3.00 to 3.77 Å, which falls within the acceptable range for biologically relevant intermolecular interactions. The strongest interaction was observed with Ala395, showing a binding energy contribution of −2.4 kcal/mol, indicating a favorable stabilizing effect on ligand binding. These interactions suggest that the active site architecture of the chimeric protein remained functionally accessible after epitope humanization.
Visualization of the docking complex further demonstrated that the ligand occupied the central binding pocket without causing major structural distortion to the surrounding secondary structure elements. The overall protein fold remained compact and well organized, indicating that substitution of the bacterial epitope with the human homologous segment did not significantly disrupt active-site geometry or ligand accessibility.
Overall, the docking analysis suggests that the humanized chimeric Arginine Deiminase retained favorable ligand-binding capability and structural integrity after epitope substitution. The combination of negative docking scores, low RMSD values, and stable hydrogen bond interactions indicates that the engineered protein is structurally stable and potentially capable of maintaining its catalytic function following rational humanization.
BIBLIOGRAPHY REFERENCE
Pedroso, A., Herrera Belén, L., Beltrán, J. F., Castillo, R. L., Pessoa, A., Pedroso, E., & Farias, J. G. (2023). In silico design of a chimeric humanized L-asparaginase. International Journal of Molecular Sciences, 24(8), Article 7550. https://doi.org/10.3390/ijms24087550