Week 2 HW: How DNA read, write and edit
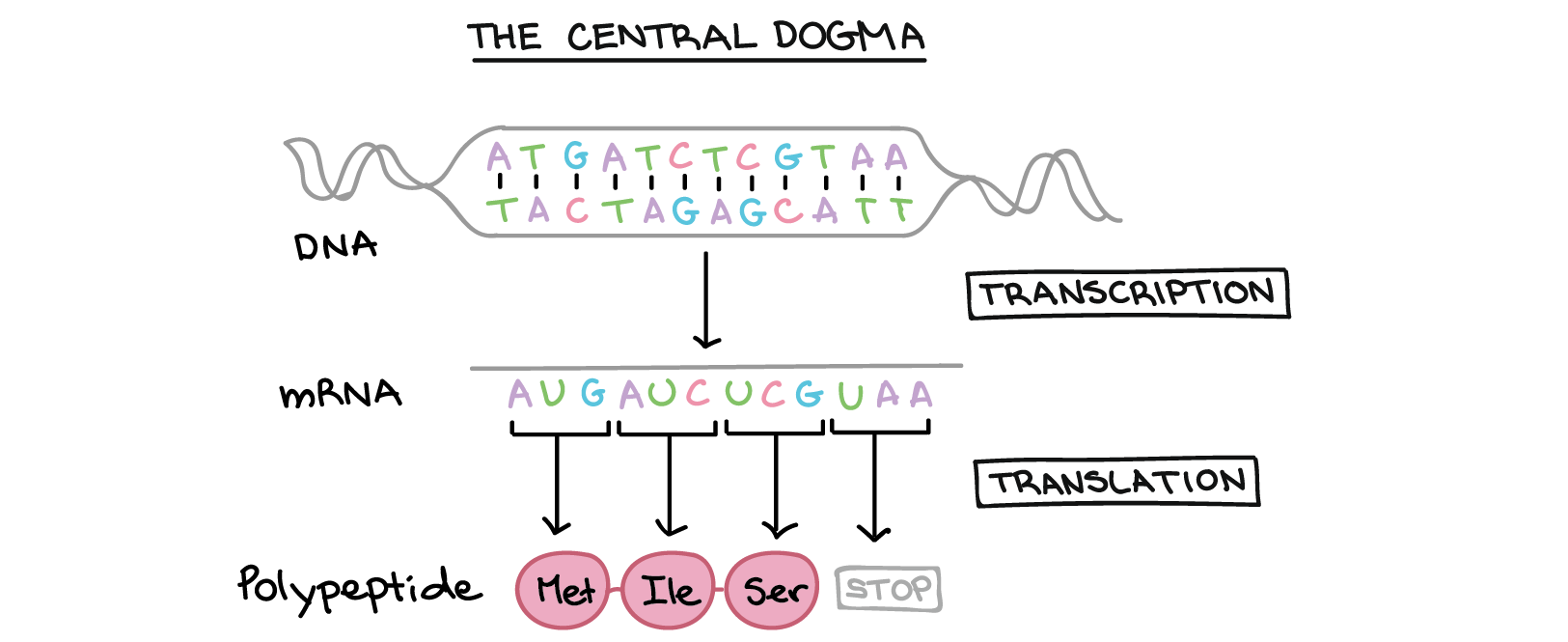
Part 1: Benchling & In-silico Gel Art
1.1 Restriction digestion simulation
Using Benchling, I simulated λ DNA digests with the following restriction enzymes: EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, and SalI.

Digest reaction setup
| Condition | M | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Water | - | - | 13 µL | 13 µL | 13 µL | 13 µL | 13 µL | 13 µL | 14 µL | - | - |
| CutSmart | - | - | 2 µL | 2 µL | 2 µL | 2 µL | 2 µL | 2 µL | 2 µL | - | - |
| λ DNA | - | - | 3 µL | 3 µL | 3 µL | 3 µL | 3 µL | 3 µL | 3 µL | - | - |
| Enzymes | - | - | NdeI + SacI | EcoRI + SacI | EcoRI | EcoRI + EcoRV | EcoRI + SacI | NdeI + XhoI | NdeI | - | - |
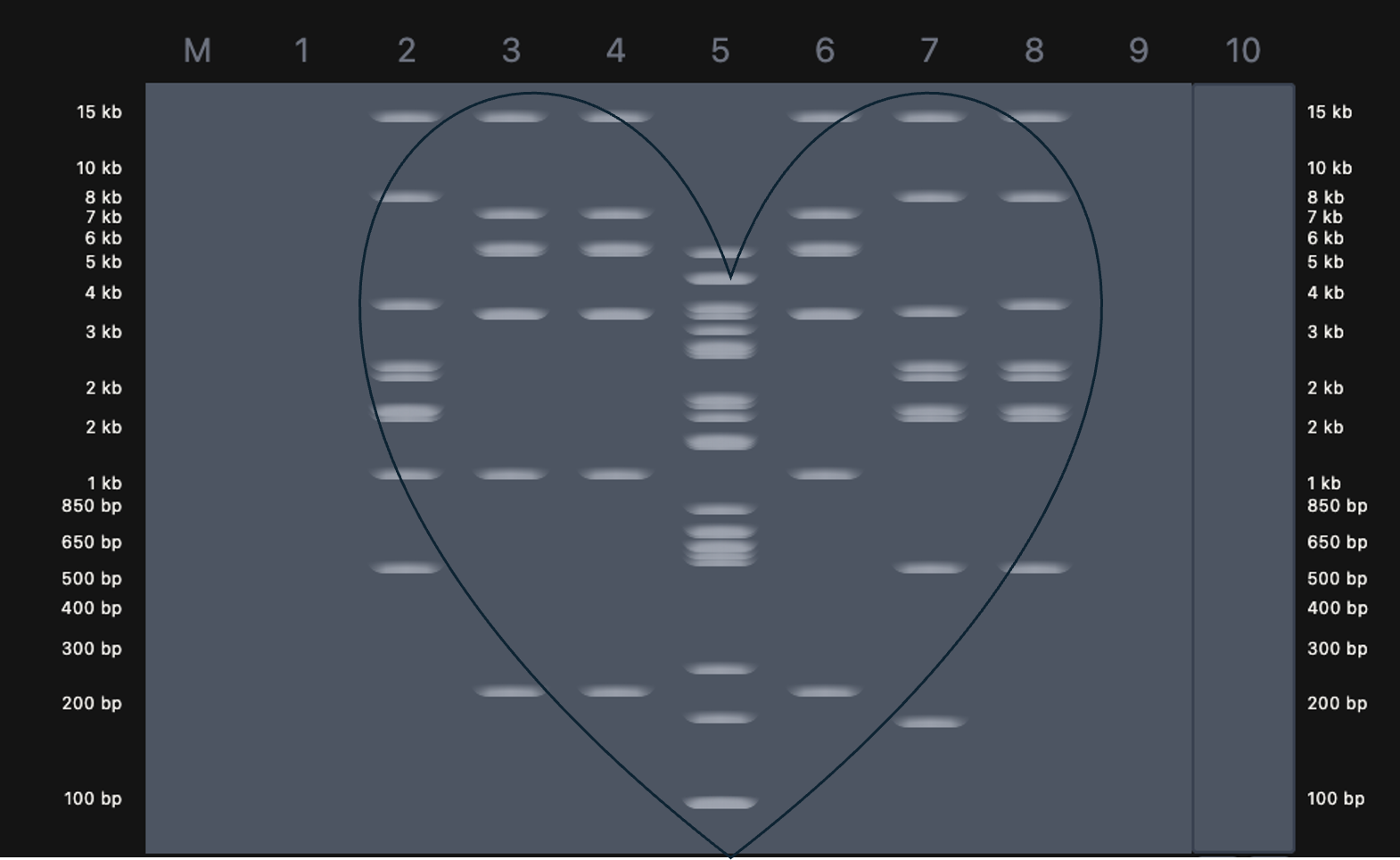
1.2 Latent Figure–style gel art
My pattern is a heart, in the spirit of Paul Vanouse’s Latent Figure Protocol artworks and in honor of Valentine’s Day.
Part 2: DNA Design Challenge
2.1 Chosen protein: NF1
My chosen protein is NF1 (neurofibromin), a tumor suppressor that negatively regulates the RAS pathway and helps prevent uncontrolled cell growth.
- Germline NF1 mutations cause Neurofibromatosis Type 1, which increases risk of neurofibromas and malignant peripheral nerve sheath tumors.
- Somatic NF1 mutations also appear in ~5–10% of sporadic cancers, including lung, breast, and melanoma.
UniProt entry: NF1_HUMAN (P21359)
Example N‑terminal sequence:MAAHRPVEWVQAVVSRFDEQLPIKTGQQNTHTKVSTEHNKECLINISKYKFSLVISGLTT
2.2 Reverse translation to DNA
Using an online protein-to-DNA tool, I reverse translated the NF1 amino‑acid sequence to obtain a candidate coding DNA sequence (CDS).
I then cross‑checked the resulting sequence against the NF1 gene in the NCBI Gene database to confirm that the codons match an authentic NF1 coding region.
Example tool used: CUSABIO Protein-to-DNA Sequence Generator.
2.3 Codon optimisation
After I determined the nucleotide sequence encoding human NF1, I codon-optimised it for expression in yeast. Even though the amino‑acid sequence stays the same, different organisms do not use all synonymous codons equally. Yeast has its own pattern of preferred codons, linked to which tRNAs are abundant. If I kept the original human codon usage, the yeast translation machinery might stall on rare codons, leading to slower translation, ribosome pausing, and lower NF1 yield. Codon optimisation rewrites the DNA sequence using codons common in yeast, without altering the protein sequence, thereby improving translation efficiency and protein expression levels.I optimised the NF1 coding sequence for Saccharomyces cerevisiae (strain S288C) because this is the baker’s yeast strain I plan to use as the expression host. Matching the codon usage to S. cerevisiae should maximise NF1 expression in this yeast background while maintaining the correct human NF1 protein sequence.
I would place the codon-optimised NF1 sequence under the control of a yeast promoter (for example, GAL1) on an expression plasmid and transform it into S. cerevisiae. When I grow the yeast in inducing conditions, the GAL1 promoter is activated, and yeast RNA polymerase recognises the promoter region and transcribes the NF1 coding sequence into messenger RNA. This NF1 mRNA is processed and exported to the cytoplasm, where ribosomes read the codons and, using yeast tRNAs, assemble the NF1 polypeptide chain. Because the sequence was codon-optimised for yeast, the ribosomes mostly encounter codons that match abundant tRNAs, which should make translation more efficient and increase NF1 yield.
Some extra notes and readings: The organism I have chosen is S. cerevisiae. That plasmid is set up for Gateway cloning, so I don’t add NF1 with restriction enzymes into a random MCS; I will just swap NF1 into the ccdB Gateway cassette under the GAL1 promoter. blog.addgene
In this vector:
- Promoter: GAL1 (yeast‑inducible).
- Destination cassette: attR1–ccdB–CmR–attR2 between GAL1 and the terminator. This is where NF1 gene goes.
- I first clone NF1 into a Gateway entry vector (attL1–NF1–attL2), then do an LR reaction with pAG423GAL‑ccdB. The LR reaction recombines attL/attR sites and replaces the ccdB cassette with your NF1 coding sequence, so the final plasmid has:
What is the Gateway LR assembly?
Gateway LR is a recombination cloning step that moves your gene from an entry vector into a destination vector (like pAG423GAL‑ccdB) using site‑specific recombination instead of restriction enzymes. blog.addgene
Basic idea
- In the LR reaction, a mix of Gateway recombinase enzymes recombines attL with attR sites, swapping NF1 in and ccdB out.
- After LR, the destination plasmid now has:
GAL1 promoter → NF1 ORF → terminator, plus new attB sites at the junctions.
Because ccdB is toxic to standard E. coli strains, only plasmids that successfully replace ccdB with my gene survive, which gives strong selection for correct recombinants.
What you actually do in the lab
- Mix:
- Entry plasmid (attL1‑NF1‑attL2)
- Destination plasmid (pAG423GAL‑ccdB)
- LR Clonase enzyme mix + buffer
- Incubate (often 1 hour / room temperature).
- Transform E. coli and plate on the antibiotic for the destination plasmid.
- Surviving colonies carry pAG423GAL‑NF1.
Part 3: Prepare a Twist DNA Synthesis Order
I attempted to create my NF1 gene construct and optimize it; however, the gene is too large for a plasmid construct. As a result, I couldn’t proceed further. Below, I am outlining the steps I took to create a plasmid that expresses GFP, as mentioned on the webpage. Check the files below:
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would I sequence and why?
I would sequence synthetic DNA strands used for DNA-based digital data storage, where arbitrary binary data (e.g., images, text, archives) are encoded into nucleotide sequences and written as pools of oligonucleotides. [
DNA-based storage is attractive because it offers extremely high information density, long-term stability (centuries under proper conditions), and technology-agnostic decoding (any future sequencer that can read A/T/C/G can, in principle, recover the data).
Sequencing these DNA pools accurately is critical because even small base-calling errors can corrupt the decoded file, so this is an ideal testbed for thinking about tradeoffs between read length, error profile, and redundancy in the encoding scheme.
(ii) What technology would I use and why?
I would primarily use Illumina sequencing-by-synthesis (SBS), complemented by Oxford Nanopore Technologies (ONT) if I wanted rapid, on-demand reads.
Illumina SBS:
- Mature, high-throughput platform with very low per-base error rates, widely used as the gold standard for reading DNA data storage constructs.
- Excellent for large pools of short oligos (e.g., 100–300 bp) which are typical in DNA data storage systems.
Oxford Nanopore:
- Single-molecule, real-time, third-generation sequencing that can read DNA without amplification and is increasingly used for rapid DNA data retrieval, despite higher raw error rates.
Given that DNA data storage typically tolerates some raw error through redundancy and error-correcting codes, combining Illumina for “archival, highly accurate” reads and ONT for “quick access” reads is attractive.
5.1: Method generation and classification
- Illumina SBS is considered second-generation (NGS):
- Characteristics: massively parallel sequencing of clonal clusters on a flow cell, short reads (typically 50–300 bp), imaging of fluorescence during synthesis.
- Oxford Nanopore is third-generation (single-molecule):
- Characteristics: direct sequencing of single DNA molecules as they translocate through a nanopore; no PCR is strictly required, long reads (kb–Mb), higher raw error but random.
5.1: Input and sample preparation
For DNA data storage, the input is a pool of synthetic DNA oligonucleotides (each encoding a chunk of digital data plus indices and error-correction). For Illumina SBS, essential prep steps:
DNA input
- Synthetic DNA pool (e.g., freeze-dried or in aqueous solution) containing thousands to millions of unique oligos (typically 100–300 nt).
Fragmentation (if needed)
- For short oligos used in data storage, additional fragmentation is usually unnecessary; for longer constructs, mechanical or enzymatic fragmentation can be used.
End repair and A-tailing
- Blunt-ending and addition of a single A overhang to make DNA compatible with T-tailed adapters.
Adapter ligation
- Ligate platform-specific adapters containing:
- Flow cell binding sequences
- Sequencing primer binding sites
- Optional indices/barcodes.
- Ligate platform-specific adapters containing:
PCR amplification (library enrichment)
- Limited-cycle PCR to enrich adapter-ligated molecules and add full adapter sequences; in data storage, PCR cycles are minimized to reduce bias.
Library QC and normalization
- Check size distribution and concentration; normalize and pool libraries as needed. [pmc.ncbi.nlm.nih] For Oxford Nanopore, prep differs:
Input DNA
- Ideally longer DNA fragments if using data storage schemes that encode longer blocks, but short-oligo protocols exist. [academic.oup]
End-repair / dA-tailing
- Prepare DNA ends compatible with nanopore adapters.
Adapter ligation
- Ligate ONT’s motor-protein–containing adapters so that a motor controls DNA translocation through the pore.
(Optional) Amplification
- Some workflows (e.g., PCR-based or rolling-circle amplification) are used when input quantity is low, but direct sequencing of native DNA is also common.
Load library onto flow cell
- Introduce library onto the nanopore device for sequencing.
5.1: Essential steps and base calling
Illumina sequencing-by-synthesis (second-generation)
Cluster generation
- Adapter-ligated DNA binds to oligos on the flow cell surface and undergoes bridge amplification to form clonal clusters, each cluster representing many copies of one original molecule. pmc.ncbi.nlm.nih
Sequencing-by-synthesis cycles
- Reversible terminator nucleotides (A, C, G, T), each with a distinct fluorescent label, are added.
- DNA polymerase incorporates a single nucleotide at each cluster; imaging detects fluorescence at every cycle. pmc.ncbi.nlm.nih
Base calling
- For each cycle, the color at each cluster is recorded.
- Image data are converted into intensity traces, then into base calls with quality scores (e.g., Phred Q-scores). pmc.ncbi.nlm.nih
Output
- Short reads (FASTQ files) with per-base qualities, typically accompanied by indices to map reads back to specific oligos. pmc.ncbi.nlm.nih
- In data storage, these reads are de-multiplexed and decoded to reconstruct the original digital file. academic.oup
Oxford Nanopore sequencing (third-generation)
DNA translocation through nanopores
- DNA–adapter complexes are captured by nanopores in a membrane.
- A motor protein ratchets DNA through the pore one or a few nucleotides at a time. chemistryworld
Signal measurement
- Ionic current across the pore fluctuates according to the sequence of nucleotides occupying the pore; this generates a continuous electrical trace. chemistryworld
Base calling
- Machine-learning models (e.g., RNNs, CNNs) translate current patterns into base sequences, assigning probabilities to each base at each position. chemistryworld
Output
- Long reads in FASTQ or FAST5 format with per-base quality scores. chemistryworld
- For data storage, reads are aligned to expected oligo designs, consensus is built, then digital bits are reconstructed via the encoding scheme. academic.oup
5.2 DNA Write
(i) What DNA would I synthesize and why?
I would synthesize a small DNA origami scaffold plus staple set encoding a recognizable 2D pattern (e.g., a smiley face or logo) that also embeds a short DNA data storage message in some staple sequences. academic.oup
This merges structural DNA nanotechnology (origami) with information-encoding DNA, demonstrating dual use: nanoscale art or devices and simultaneous archival of metadata (e.g., author, date, or a URL encoded in staples). academic.oup
Concretely, this could involve:
- A ~7 kb scaffold (e.g., M13mp18 derived) with designed folding path.
- 200–250 staple oligos (20–60 nt) whose sequences are chosen both to fold the structure and to encode a short binary payload in some of their variable regions via an ATCG coding scheme. academic.oup
(ii) What technology would I use and why?
To synthesize these DNA sequences, I would use a commercial array-based phosphoramidite solid-phase synthesis platform such as that offered by Twist Bioscience or similar vendors. pmc.ncbi.nlm.nih
Reasons:
- Array-based synthesis on silicon supports massively parallel synthesis of thousands of oligos (all the origami staples, plus any data-encoding variants) in one run at relatively low cost. healthandwealth.substack
- Phosphoramidite chemistry is the current workhorse for high-throughput, custom oligo synthesis and is well supported by industrial pipelines. pmc.ncbi.nlm.nih
For longer constructs (e.g., custom scaffolds beyond single-oligo length), I would rely on gene synthesis workflows where shorter oligos are synthesized and then enzymatically assembled into longer double-stranded fragments. healthandwealth.substack
5.2: Essential steps of the synthesis method
(A) Chemical oligonucleotide synthesis (phosphoramidite)
Design sequences
- Use DNA origami design software (e.g., caDNAno) to design scaffold routing and staple sequences; optionally overlay a digital data encoding scheme onto selected staples. academic.oup
Solid-phase phosphoramidite cycles
- Oligos are synthesized 3’→5’ on a solid support (e.g., controlled-pore glass or silicon chip).
- Each nucleotide addition involves:
- De-protection of the terminal 5’ hydroxyl
- Coupling of activated phosphoramidite nucleotide
- Capping of unreacted 5’ OH
- Oxidation to stabilize the phosphodiester bond. pmc.ncbi.nlm.nih
Cleavage and deprotection
- Oligos are cleaved from the solid support and base-protecting groups are removed. pmc.ncbi.nlm.nih
Purification
- Depending on quality needs, crude oligos may be desalted, HPLC-purified, or PAGE-purified. healthandwealth.substack
Assembly (for longer DNA)
- For long scaffolds or genes:
- Overlapping oligos are combined and assembled by PCR, Gibson assembly, or other enzymatic methods to yield kilobase-scale dsDNA. pmc.ncbi.nlm.nih
- For long scaffolds or genes:
Quality control
- Verify oligo pools or assembled genes by mass spectrometry, capillary electrophoresis, and/or test sequencing. healthandwealth.substack
(B) DNA origami folding
Mix scaffold and staples
- Combine scaffold strand with large excess of staple oligos in appropriate buffer (e.g., Mg²⁺-containing folding buffer).
Thermal annealing
- Heat to denature, then slowly cool to allow staples to hybridize and fold scaffold into target shape.
Validation
- Observe structures by AFM or TEM (in principle for the project, though not strictly required for the “writing” step).
5.2: Limitations of the synthesis method
Length limitations
- Phosphoramidite synthesis suffers from cumulative error; typical high-fidelity oligos are reliable up to ~200 bp, beyond which error rate and synthesis failures increase. healthandwealth.substack
- Platforms like Twist can assemble longer gene fragments (up to ~1.8 kb) by combining shorter oligos, but this adds complexity and cost. healthandwealth.substack
Error profile
- Errors include substitutions, deletions, and truncated products; these require purification or downstream error-correction (e.g., cloning and sequencing, or redundancy for data storage). pmc.ncbi.nlm.nih
Speed and throughput
- Parallel synthesis arrays are fast for thousands of oligos, but turnaround time is still on the order of days to weeks from design to delivery. healthandwealth.substack
Scalability and cost
- Cost per base remains higher than ideal for very large-scale DNA data storage or genome-scale synthesis; enzymatic synthesis methods (e.g., TdT-based) are being developed to address this but have their own biases and limitations. pmc.ncbi.nlm.nih
5.3 DNA Edit
(i) What DNA would I edit and why?
I would edit the genomes of closely related extant species to introduce traits from extinct animals, in line with de-extinction efforts such as Colossal Biosciences’ woolly mammoth–Asian elephant and dodo–Nicobar pigeon projects.
Specifically, edits would:
- Install cold-adaptation traits (e.g., thick fur, fat metabolism adaptations) into Asian elephant genomes to create mammoth-like hybrid elephants that could help restore Arctic grassland ecosystems.
- Introduce morphological and ecological traits of the dodo into the genome of the Nicobar pigeon to re-establish a functional analog of the extinct bird in its native habitat.
These edits have both conservation (ecosystem restoration, genetic rescue) and scientific (understanding genotype-phenotype relationships) motives.
(ii) What technology would I use and why?
I would use CRISPR–Cas9 genome editing, combined with cell culture and reproductive technologies (e.g., iPSCs and cloning), because CRISPR allows programmable, multiplexable edits at specific genomic loci with relatively high efficiency. CRISPR is already being applied in de-extinction pipelines: researchers extract and sequence ancient DNA, compare genomes between extinct and extant species, then program CRISPR guides to install key variants in cells of the living relative.
5.3: How CRISPR edits DNA – essential steps
Ancient DNA reconstruction and target selection
- Extract DNA from well-preserved remains of the extinct species (e.g., mammoth tusk, dodo bone).
- Use high-throughput sequencing and assembly methods to reconstruct as much of the extinct genome as possible.
- Align extinct and extant genomes to identify candidate genes and variants associated with desired traits (e.g., fur density, fat storage, beak shape).
CRISPR design
- For each target gene:
- Design guide RNA (gRNA) sequences that match the extant species’ genomic sequence near the intended edit (adjacent to appropriate PAM sites).
- Design donor DNA templates (ssODN or plasmid donors) encoding the extinct-species variant(s) if precise knock-ins are required.
- For each target gene:
Editing in cells
- Choose a suitable cell type from the extant species (e.g., fibroblasts, endothelial progenitor cells, or iPSCs).
- Deliver the CRISPR components:
- Cas9 protein or mRNA
- gRNA(s)
- Donor DNA (for homology-directed repair)
- Delivery by electroporation, nucleofection, or viral vectors.
Mechanism of edit
- Cas9–gRNA complexes bind the target DNA sequence and introduce a double-strand break.
- Cellular repair pathways act:
- Non-homologous end joining (NHEJ) introduces insertions/deletions (useful for knockouts).
- Homology-directed repair (HDR) uses supplied donor DNA to introduce precise nucleotide substitutions or insertions (needed for dodo/mammoth variants).
Clone selection and validation
- Screen edited cells by PCR and sequencing to confirm correct edits, check for off-target changes, and isolate clones carrying multiple desired edits.
Embryo generation and development
- Reprogram edited cells into iPSCs (if not already), differentiate where needed, or use nuclear transfer to create embryos.
- Implant embryos into surrogate mothers or use ex utero gestation platforms if developed.
Phenotypic assessment
- Assess whether edited animals show target traits (e.g., cold resistance, morphology) and evaluate ecological impacts before any reintroduction.
5.3: Inputs and preparation
Design-stage inputs
- Reconstructed extinct-species genome sequence.
- High-quality reference genome of the extant species.
- Computational pipelines for variant calling, functional annotation, and gRNA design.
Wet-lab inputs
- Living cells from the extant species (e.g., gray wolf, Asian elephant, Nicobar pigeon).
- Cas9 (protein or mRNA).
- Synthetic gRNAs for each targeted locus.
- Donor DNA templates for precise edits.
- Plasmids or RNP complexes for delivering the CRISPR machinery.
5.3: Limitations of the editing method
Efficiency and multiplex editing
- Editing many loci simultaneously (required to approximate an extinct genome’s phenotype) lowers efficiency per site and complicates clone selection.
Precision and off-target effects
- gRNAs can sometimes bind partially mismatched sites, causing off-target cuts and unintended mutations, which is a concern in both animal welfare and ecological safety.
Context dependence
- Introducing a few key genes from an extinct species into a modern genome may not fully recapitulate the original phenotype due to epistasis and regulatory differences.
Developmental and reproductive challenges
- Edited embryos may fail to develop properly; gestation in surrogate species presents immunological, anatomical, and ethical hurdles.
Ethical and ecological limits (conceptual, not technical)
- Even with powerful CRISPR tools, decisions about which traits to edit, how many individuals to release, and how to manage ecological consequences are non-trivial and may limit real-world deployments.