Week 6 HW: Genetic Circuits Part I
This page tackles all homeworks of week 6.
- What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
- What are some factors that determine primer annealing temperature during PCR?
- Primer Melting Temperature: The temperature at which 50% of the primer-template duplex is dissociated.
- GC Content: G-C base pairs share three hydrogen bonds, whereas A-T pairs share only two. Primers with a higher percentage of GC pairs require higher temperatures to denature and thus exhibit a higher melting temp.
- Primer Length: Longer primers have more total base-pairing interactions, increasing the total thermal energy required to disrupt the hybrid structure, which raises the melting temp.
- Salt and Buffer Concentration: Monovalent and divalent cations mask the negative charges on the phosphodiester backbone of DNA. Higher salt concentrations stabilize the duplex, reducing electrostatic repulsion and raising the effective melting temp.
- Primer Mispriming/Secondary Structure: The presence of internal hairpins, self-dimers, or cross-dimers lowers the concentration of free, accessible primer, occasionally requiring temperature adjustments to avoid off-target amplifications.
- There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests (REDs). Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
- Mechanism: PCR occurs through enzymatic synthesis and exponential amplification of a specific target region using flanking oligonucleotides; in RED, the chemical cleavage of pre-existing phosphodiester bonds at specific sequence-defined palindromic restriction sites occur.
- Protocol Requirements: PCR requires sequence-specific primers, dNTPs, a thermostable polymerase, and a thermal cycler. RED requires a sequence containing the target restriction site, specific endonuclease enzymes, and an isothermal incubation block.
- Fidelity & Modifications: PCR can introduce unwanted point mutations (minimized by high-fidelity enzymes); allows easy addition of custom flanking sequences (e.g., Gibson overhangs) via primer tails. RED is high fidelity because it cuts exact biological DNA; limited strictly to the locations of existing or engineered restriction sites.
- Yield & Scalability: PCR amplifies fragments exponentially from trace amounts of template (nanogram scale). REDs yield is strictly limited by the starting mass of the source plasmid/DNA (microgram scale).
- How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
- How does the plasmid DNA enter the E. coli cells during transformation?
- Describe another assembly method in detail (such as Golden Gate Assembly). Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
The Phusion High-Fidelity PCR Master Mix contains all the necessary components for robust, high-fidelity DNA amplification. Its primary components include: Phusion DNA Polymerase (for high processivity and proofreading activity, resulting in an error rate roughly 50-fold lower than traditional Taq polymerase), Deoxynucleotide Triphosphates (dNTPs; serve as the fundamental molecular building blocks that the polymerase polymerizes to synthesize the complementary nascent DNA strand), Reaction Buffer & Proprietary Stabilizers (to maintain pH stability during thermal cycling), Magnesium Ions (acts as an essential cofactor for DNA polymerase activity by coordinating with the phosphate groups of the dNTPs and the DNA backbone, facilitating the nucleophilic attack required for phosphodiester bond formation).
The primer annealing temperature is critical for balancing reaction specificity and yield, and is dictated by the following interconnected factors:
Both methods are foundational molecular biology techniques used to generate linear DNA fragments, but they differ fundamentally in:
PCR is preferred when a gene needs to be isolated from a genomic or plasmid source while simultaneously adding flanking homology arms (overhangs) for seamless cloning methods like Gibson Assembly, or when working with tiny amounts of template DNA. Restriction Digests are preferred when cutting open a large recipient vector backbone to minimize the risk of mutations across a large sequence, or when performing quick diagnostic analytical checks (restriction mapping) to confirm if a plasmid contains the correct insert.
- Homology Arms: Ensure adjacent fragments share 20–40 bp of identical sequence at their tips.
- Blunt Ends: Use a proofreading polymerase (like Phusion) so there are no non-templated $3'$ A-overhangs.
- Clean Fragments: Run a gel to verify sizes, then column-purify to strip out background dNTPs and enzymes.
- Chemical Competence: Ca++ neutralizes negative charges so DNA sits on the cell wall. Heat shock (42°C) creates a rapid thermal draft that physically opens transient membrane pores.
- Electrocompetence: Cells are washed to remove salts. A high-voltage shock induces localized dielectric breakdown, creating hydrophilic pores that pull DNA inward via electrophoresis.
Golden Gate Assembly relies on Type IIS restriction enzymes, such as BsaI, which cleave DNA at a precise distance outside of their non-palindromic recognition sequences to generate custom 4-base-pair sticky overhangs. Because these cuts are offset from the binding site, the recognition sequences are completely discarded from the insert fragments during the digestion process. This strategic removal ensures that the final assembled product no longer contains active restriction sites, rendering it entirely immune to subsequent cleavage. Due to this one-way directionality, the donor fragments, Type IIS enzyme, and T4 DNA Ligase can all be mixed simultaneously within a single tube. By cycling the temperature between optimal digestion (37°C) and ligation (16°C) conditions, the reaction equilibrium is driven relentlessly toward the final assembled construct. This brilliant mechanism enables seamless, scarless, and highly efficient multiplexed cloning of dozens of unique DNA fragments in a single reaction step.
Donor Fragment Architecture:
5'-- [BsaI Site] -> (4-bp Overhang A) -> [ Promoter/Gene/Terminator (Functional Genetic Material) ] -> (4-bp Overhang B) -> [BsaI Site] --3'
|
| + BsaI Enzyme Cleavage
v
(4-bp Overhang A) -> [ Target DNA Insert ] -> (4-bp Overhang B) <-- Ready for Scarless Ligation
Benchling Simulation & Strategy Documentation:
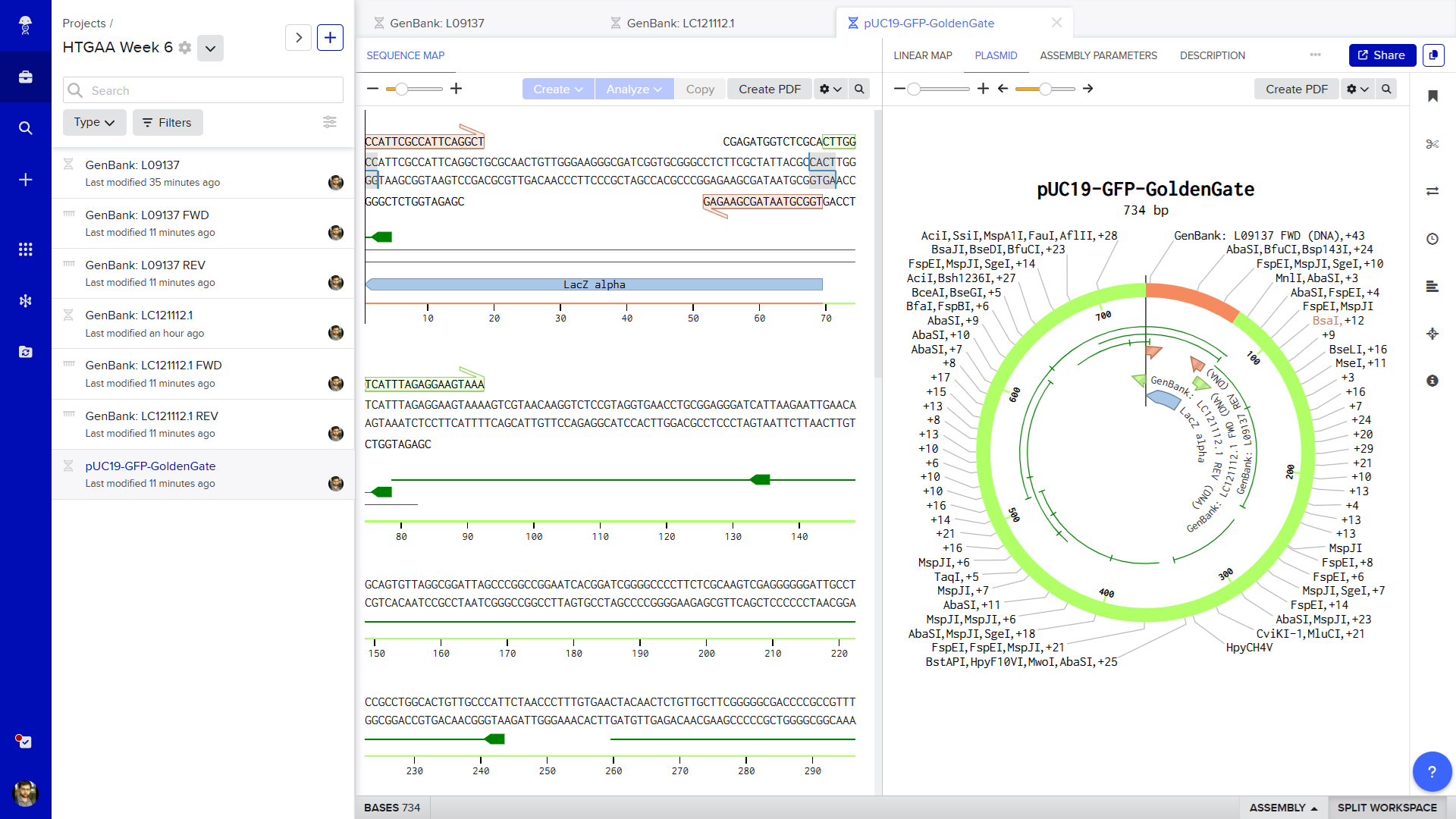
To simulate a functional Golden Gate Assembly, I modelled the insertion of a Green Fluorescent Protein (GFP) reporter gene into a modified pUC19 expression vector backbone using the Type IIS restriction enzyme BsaI.
First, I imported the standard circular pUC19 cloning vector plasmid sequence (GenBank: L09137) into Benchling. I auto-annotated this and identified the LacZ Alpha region (as this is used during the Assembly)
Next, I created a separate linear DNA sequence file for the GFP insert using the coding sequence found in the Aequorea victoria GFP expression vector (GenBank: LC121112.1).
Finally, I opened Benchling's Assembly Wizard toolbar on the right side of the screen and selected Golden Gate Assembly. I designated the modified pUC19 file as the "Backbone" (selecting the LacZ region) and the GFP file as the "Insert", setting the enzyme parameter to BsaI. The software automatically scanned the sequences, aligned the perfectly matched 4-bp sticky overhang junctions (ATGC and TAAA), and simulated the one-pot digestion-ligation reaction. The tool successfully generated a final, circularized, fully annotated recombinant plasmid map where the GFP gene is seamlessly and scarlessly integrated into the vector frame, ready to drive green fluorescence expression in competent bacterial cells.
The full Benchling Project is make publickly accessible (click this link).