Week 4 HW: Protein Design Part I
This page tackles all homeworks of week 4.
Part A. Conceptual Questions
| Questions | Answers |
|---|---|
| 1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) | Considering the average meat contains 25% of proteins by weight[1], and proteins are approximately 100% composed of amino acids, we need to find the number of amino acids present in 125 grams. As it is already provided that an average amino acid is about 100 Daltons, the estimated number of amino acids is equal to: 125 g 100 Dalton = 125 g 100 g/mol = 1.25 mol × 6.022 × 1023 mol-1 ≈ 7.53e23 molecules |
| 2. Why do humans eat beef but do not become a cow, eat fish but do not become fish? | The human digestive system breaks down all raw materials into its basic forms (for example proteins are broken down into the amino acids) and then these are used for the body's own processes. If the proteins are somehow magically ingested as is, in their same form in the original organism and those proteins somehow get inside a living human cell, then there could be some other issues related to creation of new pathways which are similar to what the protein was used for in the original organism and this could have unintended consequences ultimately killing the carnivore. Nucleic Acids (constituents of DNA/RNA) are also broken down in our digestive system preventing any possibility of incorporating the external DNA fragments within our cells; in the case of a leaky-gut when some protein or DNA fragments might enter the body, the immune system responds appropriately. Thus, the digestive system acts as an information shredder, passing on raw memory-less ingredients to the host. |
| 3. Why are there only 20 natural amino acids? | There are >500 amino acids that occur naturally, but only 22 of them are expressed through living genes.[2] Why only 22 seems a tough question for evolutionists, my hunch is that even if life started with more than 500 types of animo acids being expressed, there could be eating preferences which could have led to the specific pathways (which we cuurently have) being reinforced while the other pathways disappeared slowly; or, the origin of life that posibly evolved in a pond-cluster started with these 22 essentials while the other pond-clusters didn't make it to large scale organisms. Solid proof can only be found upon trying to replicate a fast-tracked evolutionary process using all pathways consisting of 500 AAs and then observing if the evolution converges to the same 22 AAs. |
| 4. Can you make other non-natural amino acids? Design some new amino acids. | As per the definition of Amino Acids, they must contain Amino groups and Carboxylic Acids (and an alpha carbon connecting both groups; research about beta and gamma AAs are also worthwhile). So it should be pretty simple to design a new AA as per this definition. An easy way to generate new AA's (so that they are unique) is to find the heaviest AA designed till date and add another carbon atom somewhere (or at the other end); this is a general trick to keep on extending artificial AAs, but instability issues should be kept in mind. Another important reason for the limit of 22 AAs could be their size, which allows them to pass through the cellular membranes via AA-transporter proteins (larger/heavier AAs may have higher resistance to do that). Further just having a new AA wont matter much unless a new Aminoacyl-tRNA synthetase (aaRS, essential enzymes that attach specific amino acids to their corresponding tRNAs) is designed; humans have 20 different types of aaRS for attaching the 20 standard-essential AAs to their respective tRNAs. Worth Mentioning: Simply adding single carbons can be often ignored by ribosomes, and the translation machinery, better to add larger groups; in this regard adding Phosphoserine analogs with non-hydrolyzable bonds are used to study signaling. |
| 5. Where did amino acids come from before enzymes that make them, and before life started? | Before enzymes and life existed, amino acids formed spontaneously through natural geochemical and atmospheric processes. They were synthesised on primitive Earth and supposedly also delivered from space. The water reservoirs that they accumulated into form the raw chemical building blocks that sparked life are also known as The Primordial Soup. |
| 6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect? | D-amino acids are exact mirror images of natural L-amino acids and thus they form exact mirror-image structures. Natural proteins made of L-amino acids form right-handed helices; therefore, reversing the chirality of the building blocks reverses the handedness of the helix. The answer is therefore LEFT. |
| 7. Can you discover additional helices in proteins? | Proteins possess several other structures apart from the well-known alpha helices and beta sheets because the amino acid backbone allows for various bending angles. While alpha helices and beta sheets are the most common repeating patterns of secondary structure, others exist and serve critical functions:
|
| 8. Why are most molecular helices right-handed? | In nature, all amino acids found are of the chirality type L, and therefore, they form right-handed curls. A possibility regarding this is that the first life from the primordial soup was of the L-type and that subsequent superstructures (i.e., proteins) took away much of the intermediate raw materials that could have been used to form D-type amino acids. Thus, D-type structures died in the primordial evolutiona nd we don't see them anymore, except when artificially created in labs. However, this does not directly imply that the L-amino acids physically cannot form left-handed helices. Left-handed L-helices are physically possible and L-amino acids can theoretically fold into left-handed alpha helices; but the issue is steric hindrance: In a left-handed helix made of L-amino acids, the bulky side chains (Rgroups) are forced too close to the backbone atoms. This creates severe physical crowding (steric clash). Right-handed is energetically cheaper: In a right-handed helix made of L-amino acids, the side chains point outward and away from the backbone, minimizing crowding and maximizing stability. |
| 9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation? | β-sheets tend to aggregate because they possess exposed, unsatisfied hydrogen bonds along their outer edges and highly hydrophobic flat surfaces. This architectural vulnerability drives them to stack or extend infinitely to achieve thermodynamic stability (frequently leading to the formation of pathological amyloid fibrils).
The Primary Driving Forces are:
|
| 10. Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials? | Amyloid diseases (like Alzheimer's, Parkinson's, and Type 2 Diabetes) do not happen because β-sheets are inherently toxic, but because the cross-β sheet architecture is the lowest global thermodynamic energy minimum for almost any polypeptide chain. Almost any protein, if unfolded or destabilized long enough, can convert into a β-sheet-rich amyloid. Amyloid β-sheets make extraordinary, high-performance nanomaterials. While biology views them as pathological, material scientists exploit their steel-like mechanical properties, chemical resilience, and self-assembling nature, to create Protective Biofilm Matrices, Conductive Nanowire, Injectable Hydrogels, High-Strength Composites, etc. |
| 11. Design a β-sheet motif that forms a well-ordered structure. | General steps to design a β-sheet motif:
|
Part B: Protein Analysis and Visualization
| Questions | Answers |
|---|---|
| 1. Briefly describe the protein you selected and why you selected it. | I selected the protein Ubiquitin for this assignment. Ubiquitin is a small regulatory protein found in eukaryotic cells that plays an important role in protein degradation, signalling, and cellular regulation. It has a well-characterised three-dimensional structure, and many experimentally determined structures are available in the Protein Data Bank (PDB). I initially considered using GB1, but found that its initiator methionine is retained in the mature protein. Since my broader project involves studying retro (reverse) protein sequences, I wanted a protein in which the initial methionine is naturally removed during post-translational processing. In many proteins, the initiator methionine is cleaved by methionine aminopeptidase when the second amino acid has a small side chain. Ubiquitin is therefore a suitable choice because its mature form does not retain the starting methionine, allowing cleaner comparison between the natural sequence and a retro/reverse sequence without introducing an artificial terminal methionine residue. Additionally, ubiquitin is small, structurally stable, and extensively studied, making it convenient for computational and structural analysis. |
| 2. Identify the amino acid sequence of your protein. How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids. How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs. Does your protein belong to any protein family? | The protein I selected is Ubiquitin from humans, a highly conserved regulatory protein involved in protein degradation and cellular signalling. Ubiquitin. The mature amino acid sequence of UBIQUITIN is QIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRGG; it contains 75 amino acids with the most frequent AA being Leucine (L appears 9 times, frequency 11.84%). Lysine (K) is also highly abundant and biologically important because ubiquitin forms polyubiquitin chains through lysine residues. Ubiquitin has thousands of homologs across eukaryotes because it is one of the most evolutionarily conserved proteins known. Human and yeast ubiquitin, for example, share about 96% sequence identity, differing in only 3 out of 76 amino acids; (these were validated using UniProt BLAST). Ubiquitin belongs to the Ubiquitin protein family (Pfam accession: PF00240), a family of small regulatory proteins involved in ubiquitination and intracellular protein turnover. Members of this family are small proteins or protein domains that adopt the characteristic ubiquitin fold. Proteins in this family are involved in post-translational modification pathways and intracellular protein turnover. |
| 3. Identify the structure page of your protein in RCSB. When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å). Are there any other molecules in the solved structure apart from protein? Does your protein belong to any structure classification family? | The structure page of my protein is available in the Protein Data Bank (RCSB PDB) under the entry 1UBQ titled “Structure of ubiquitin refined at 1.8 Å resolution.” The structure was solved in 1987 using X-ray diffraction, with the reported resolution indicating a very high-quality protein structure. In structural biology, lower resolution values correspond to more accurate atomic positions, and structures below about 2.0 Å are generally considered excellent quality. Apart from the protein itself, the solved structure also contains water molecules that were resolved in the crystal structure. No additional ligands or cofactors are present in the basic ubiquitin structure. Ubiquitin belongs to the ubiquitin fold structural family, a highly conserved protein fold characterized by a compact globular structure containing β-sheets and an α-helix. It is also classified within the Ubiquitin-like superfamily in structural classification databases such as SCOP and CATH. Members of this structural family are involved in protein regulation, signalling, and intracellular protein turnover. |
| 4. Open the structure of your protein in any 3D molecule visualization software: PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands). Visualize the protein as “cartoon”, “ribbon” and “ball and stick”. Colour the protein by secondary structure. Does it have more helices or sheets? Colour the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues? Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)? | I used RCSB's 3D Viewer to visualize ubiquitin in cartoon, ribbon, molecular surface, ball-and-stick, and spacefill representations.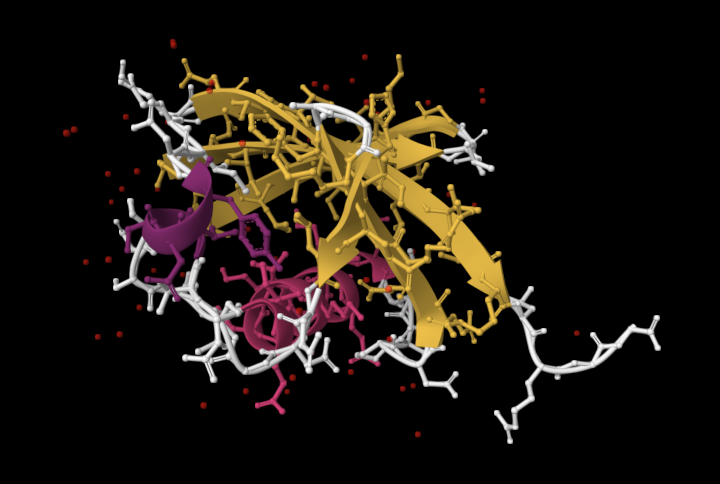 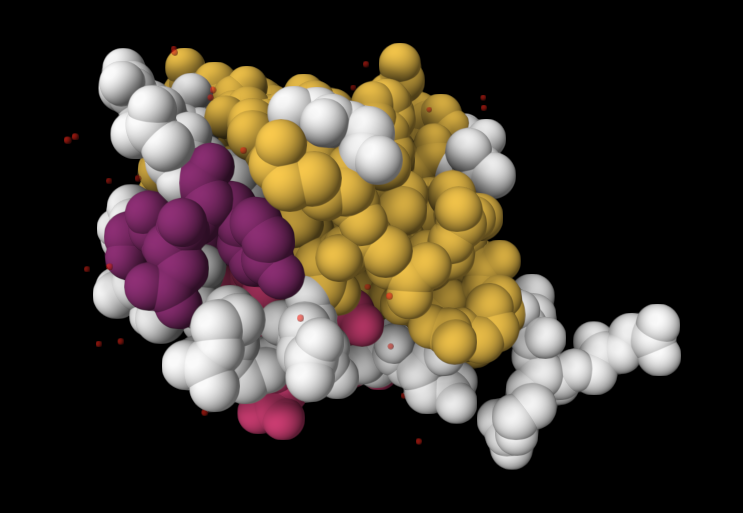 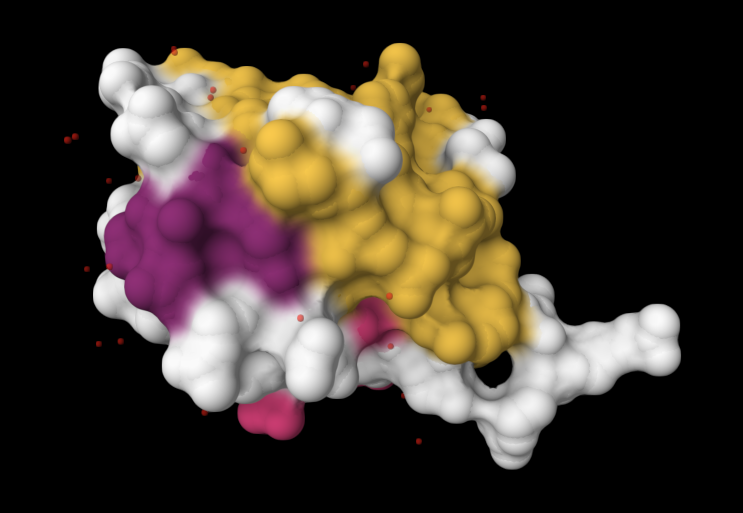 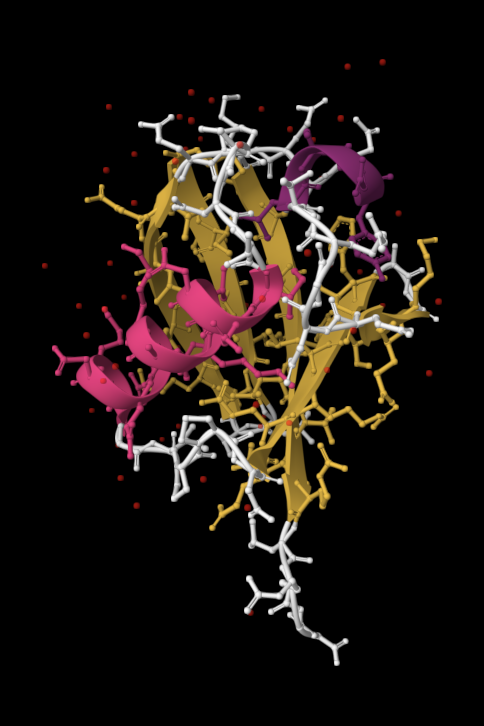 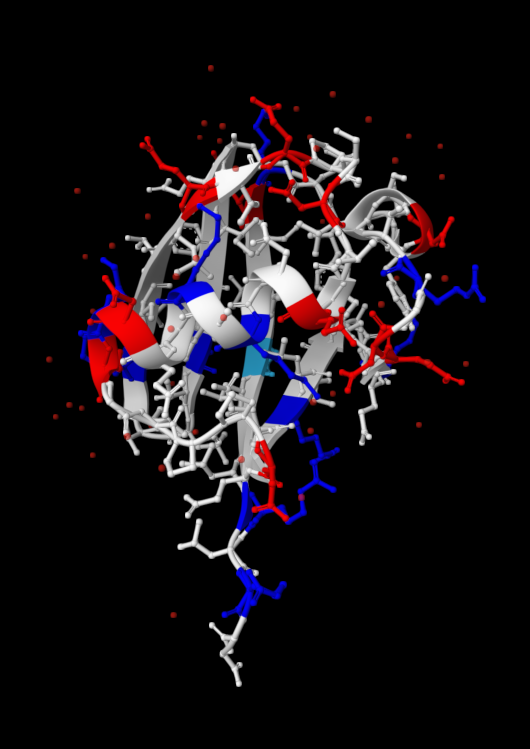 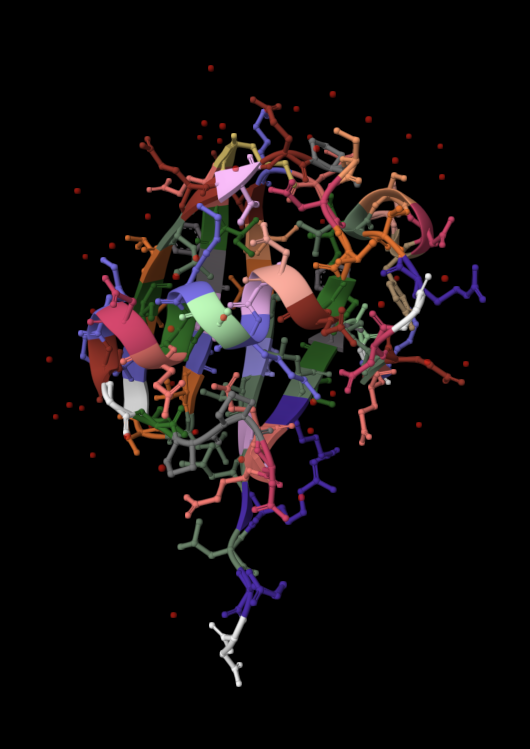 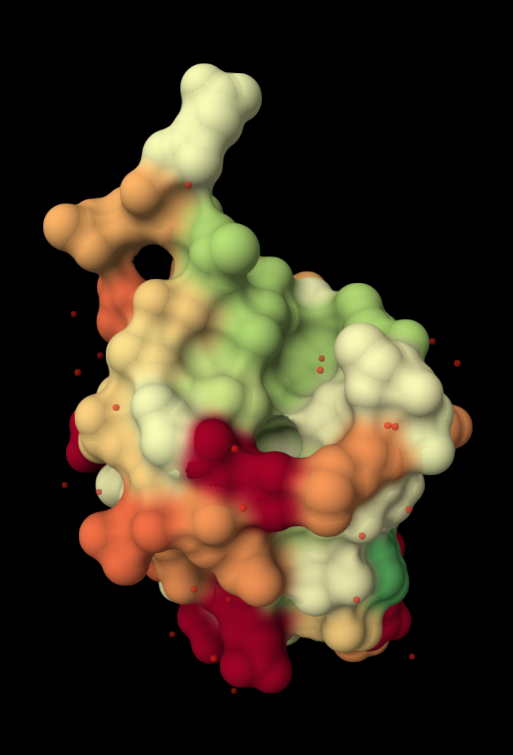 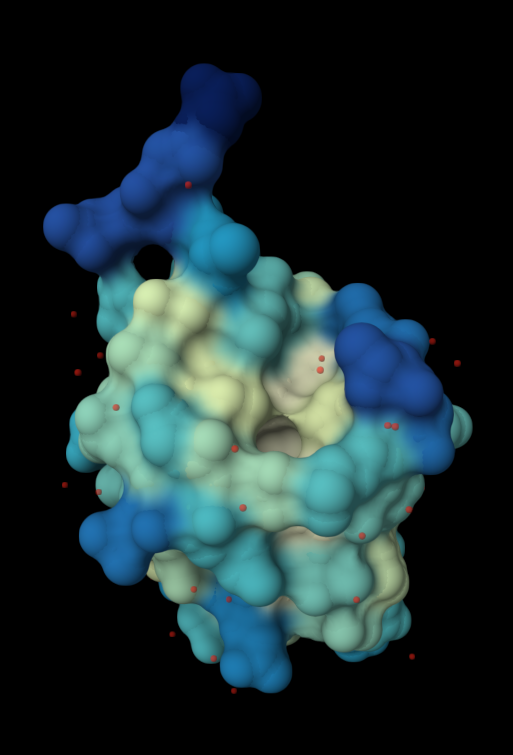 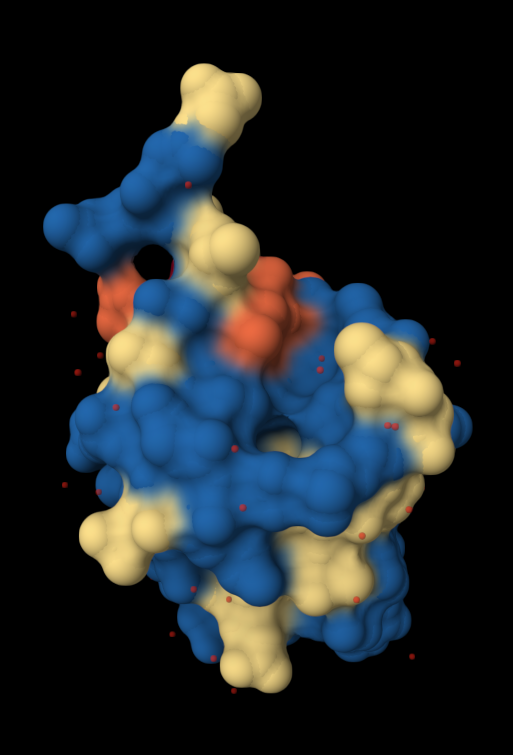 Ubiquitin contains more β-sheets than α-helices (consistent with the characteristic ubiquitin β-grasp fold). Hydrophobic residues are mainly buried in the interior core, while charged and hydrophilic residues are exposed on the surface. It has one hole (binding pocket). Surface visualization shows a compact globular structure with shallow interaction grooves rather than deep catalytic binding pockets. |
Part C. Using ML-Based Protein Design Tools
| Questions | Answers |
|---|---|
| C1.1. Deep Mutational Scans. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods. Can you explain any particular pattern? (choose a residue and a mutation that stands out) | 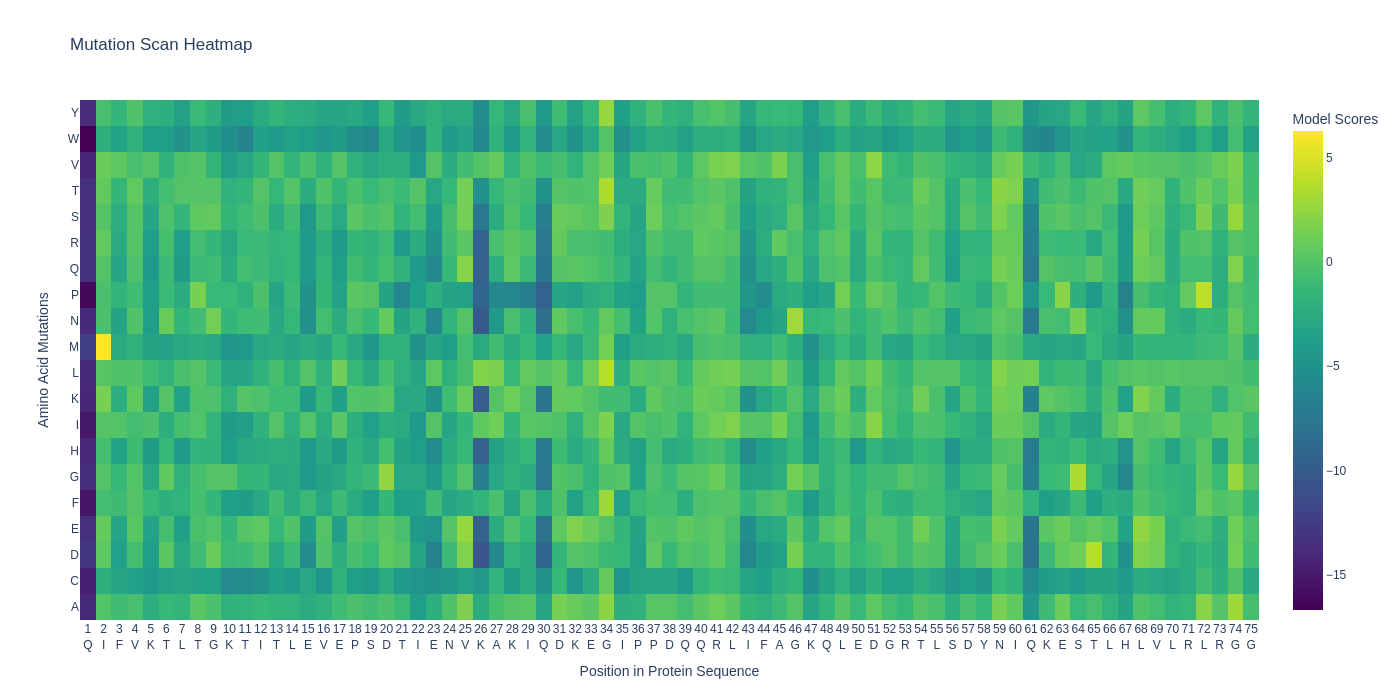 For the retro protein sequence, the first place's mutation is similarly highly unfavourable but a specific position 40 seems to have many favourable mutations... 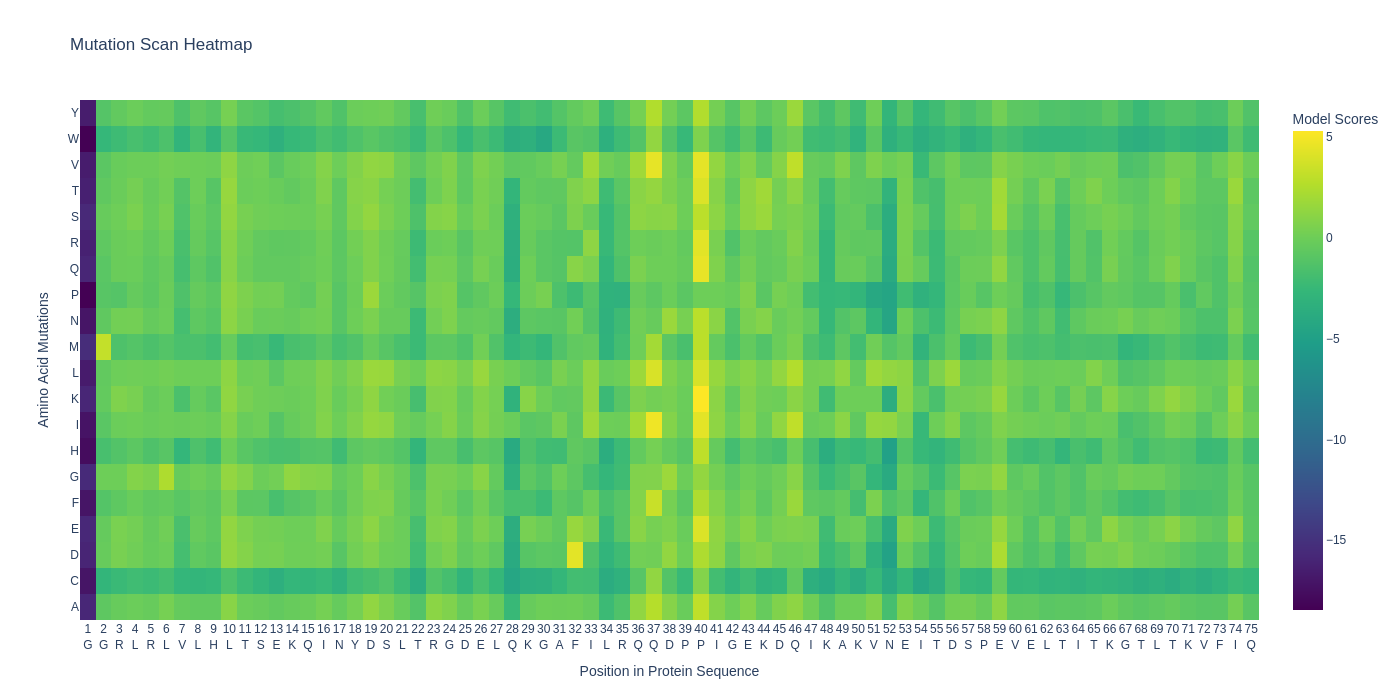 |
| C1.2. Latent Space Analysis: Use the provided sequence dataset to embed proteins in reduced dimensionality. Analyze the different formed neighborhoods: do they approximate similar proteins? Place your protein in the resulting map and explain its position and similarity to its neighbours. | 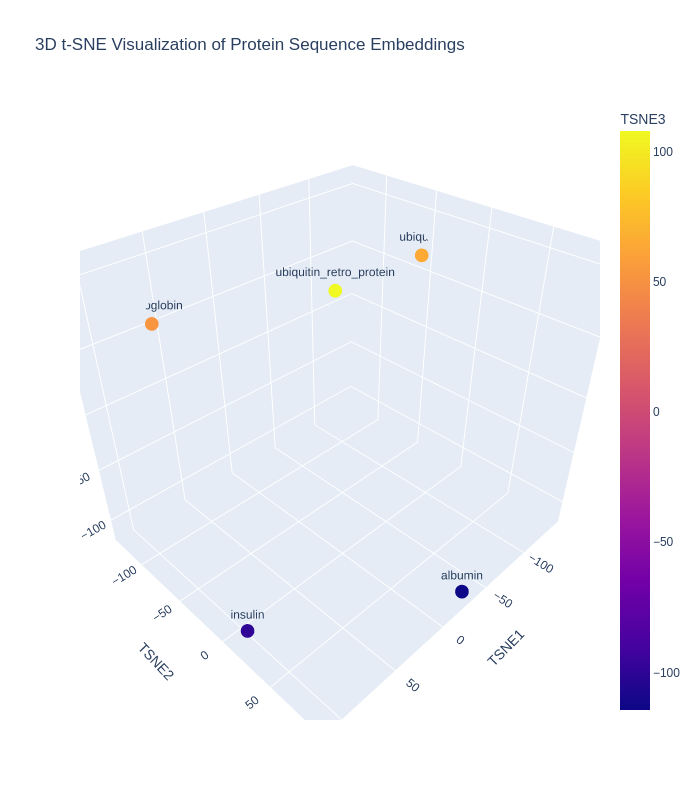 To visualize these embeddings, t-distributed Stochastic Neighbor Embedding (t-SNE) was used to reduce the 320-dimensional vectors into three dimensions. Since only five protein sequences were analyzed, the perplexity parameter was reduced from the default value of 30 to 3 (because t-SNE requires the perplexity value to be smaller than the number of samples). Perplexity approximately controls the effective number of neighboring points considered during dimensionality reduction. Lower perplexity values emphasize local neighborhood relationships between nearby proteins. The three plotted axes (TSNE1, TSNE2, and TSNE3) do not correspond to specific physical or biochemical quantities such as molecular weight, hydrophobicity, or sequence length. Instead, they are abstract latent coordinates generated by t-SNE to preserve local similarity relationships between the original high-dimensional embeddings. Proteins that appear closer together in the plot are interpreted as having more similar learned sequence representations, while proteins farther apart are considered more dissimilar in the latent embedding space. In the resulting embedding map, Ubiquitin and the retro-Ubiquitin sequence occupied different positions despite containing the same amino acid composition in reversed order, suggesting that sequence order strongly influences the learned representation of the protein language model and that the retro sequence is interpreted as biologically distinct from native ubiquitin. Other proteins, such as haemoglobin, insulin, and albumin fragments, also occupied separate regions, reflecting their differing sequence patterns and biological functions. However, t-SNE visualizations should be interpreted qualitatively rather than quantitatively. The distances and cluster sizes in t-SNE plots can vary depending on initialization and parameter choices such as perplexity, and therefore do not represent exact evolutionary or structural distances between proteins. |
| C2. Protein Folding: Fold your protein with ESMFold. Do the predicted coordinates match your original structure? Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations? | ESMFold was used to predict the structure of both native ubiquitin and a retro-ubiquitin sequence generated by reversing the amino acid order. The model directly predicted atomic coordinates from sequence without requiring multiple sequence alignments. The native ubiquitin prediction showed high confidence, with most residues having pLDDT values above 85–90. The PAE heatmap also showed low predicted aligned error across most residue pairs, indicating that the model was confident about the global fold and residue positioning. The predicted structure retained the compact ubiquitin-like fold with well-defined β-sheets and α-helices. pLDDT is a per-residue confidence score ranging from 0–100, where higher values indicate more reliable structural predictions. 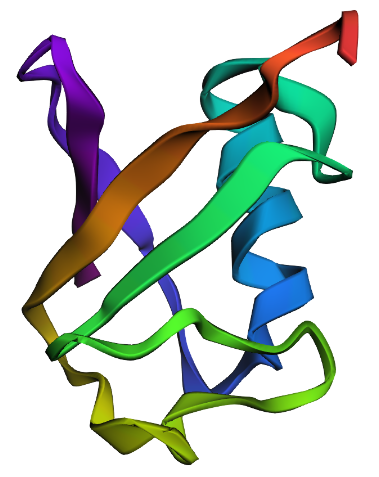 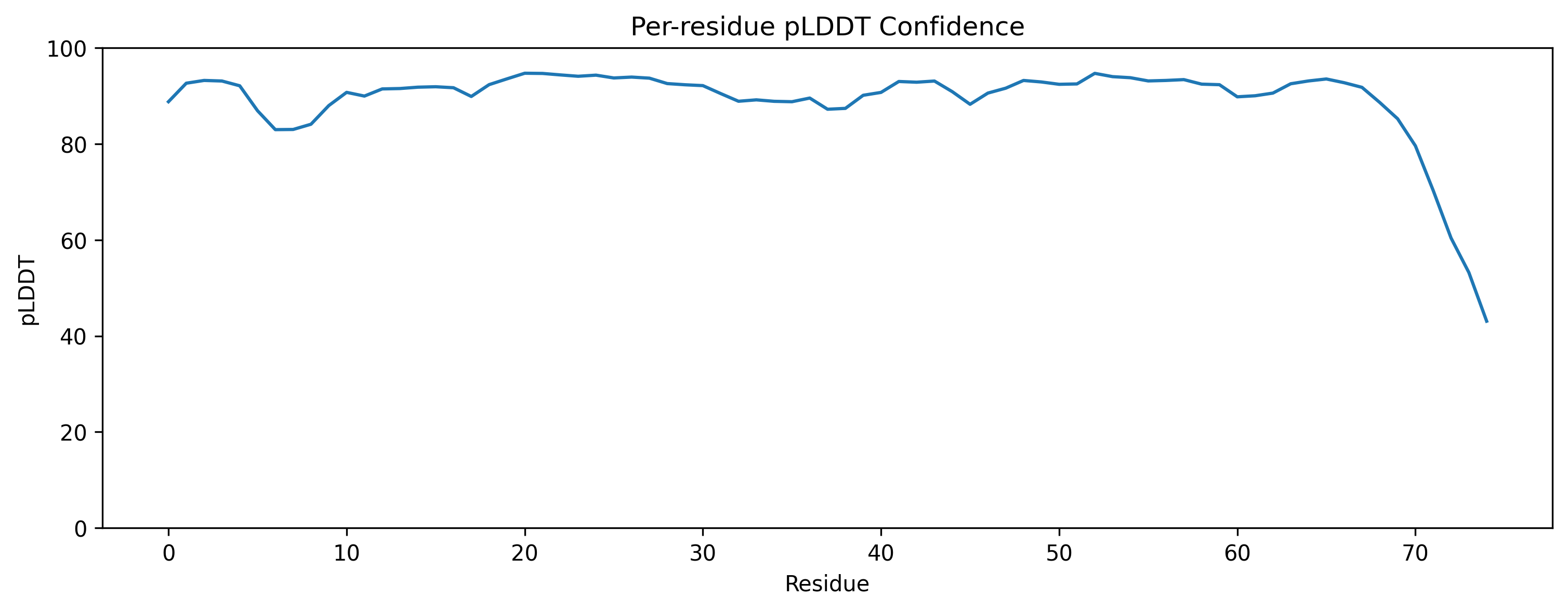 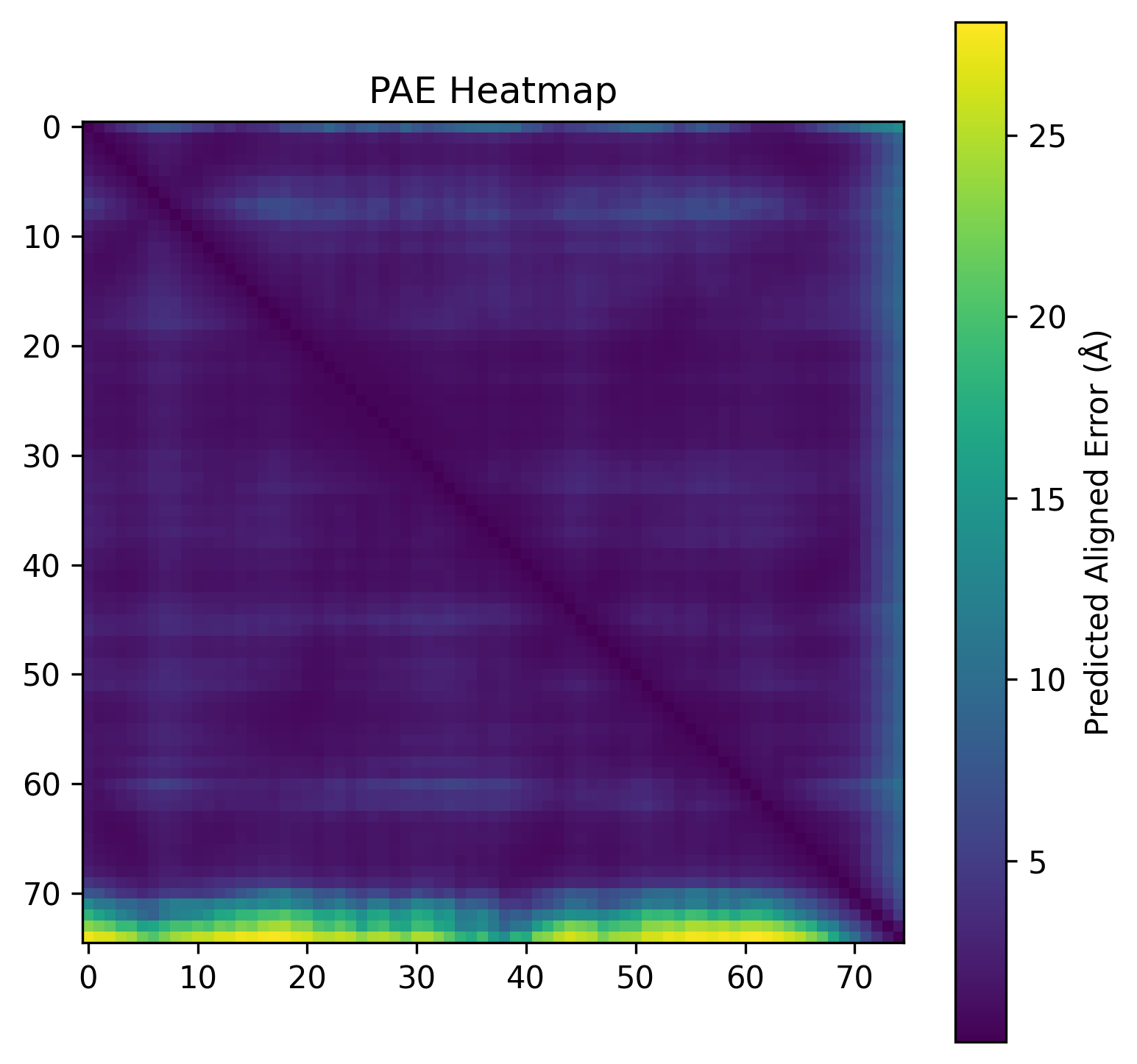 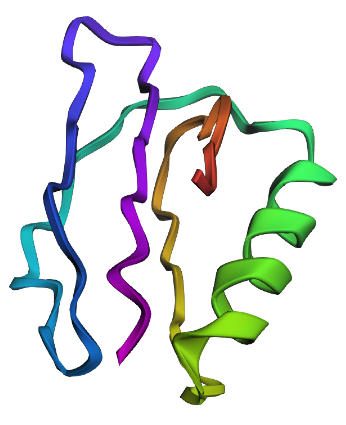 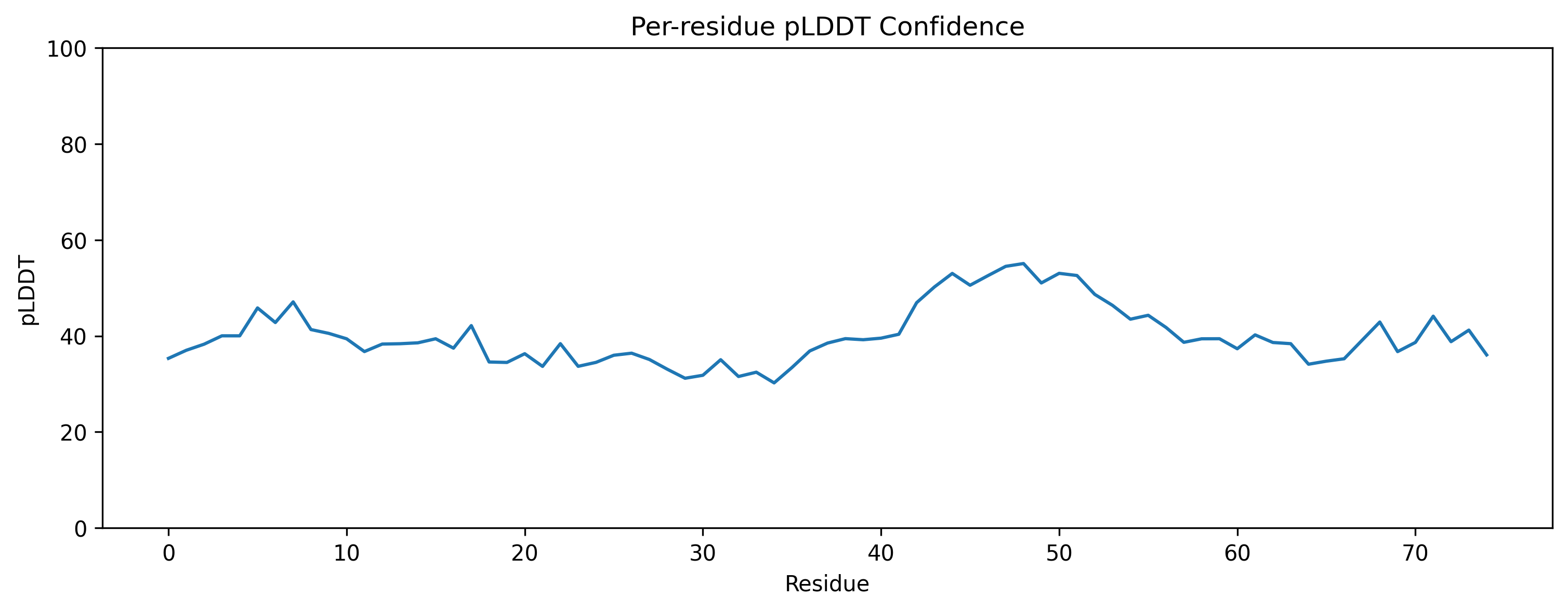 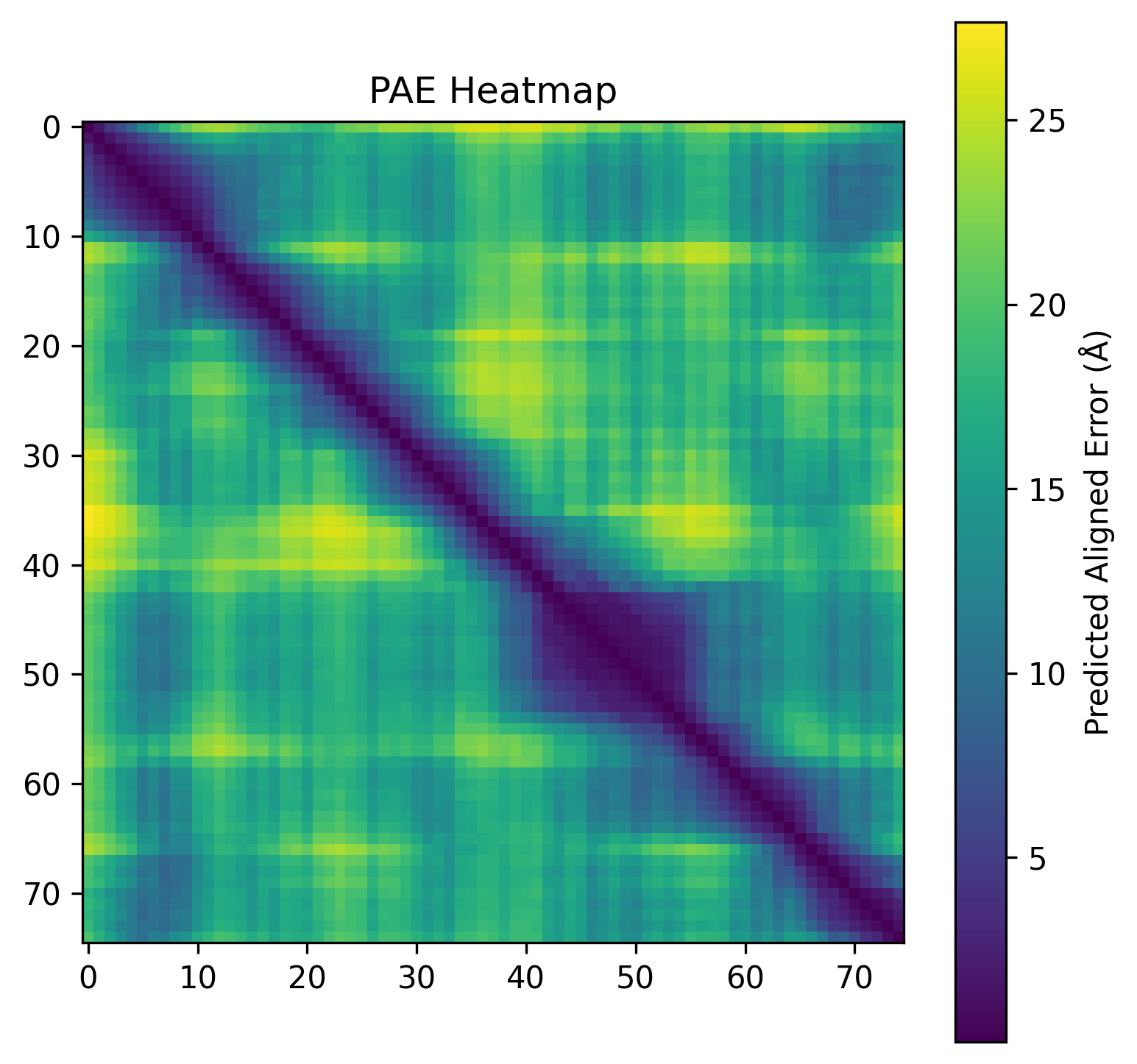 |
| C3. Protein Generation: Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one. Input this sequence into ESMFold and compare the predicted structure to your original. | ProteinMPNN was used to perform inverse folding on the Ubiquitin backbone structure. Instead of predicting structure from sequence, ProteinMPNN predicts amino acid sequences that are compatible with a given protein backbone geometry. The generated heatmap represents the probability of each amino acid occurring at every residue position based on the backbone structure. Bright regions indicate amino acids strongly preferred by the model at specific positions, while diffuse regions represent positions that tolerate greater sequence variability.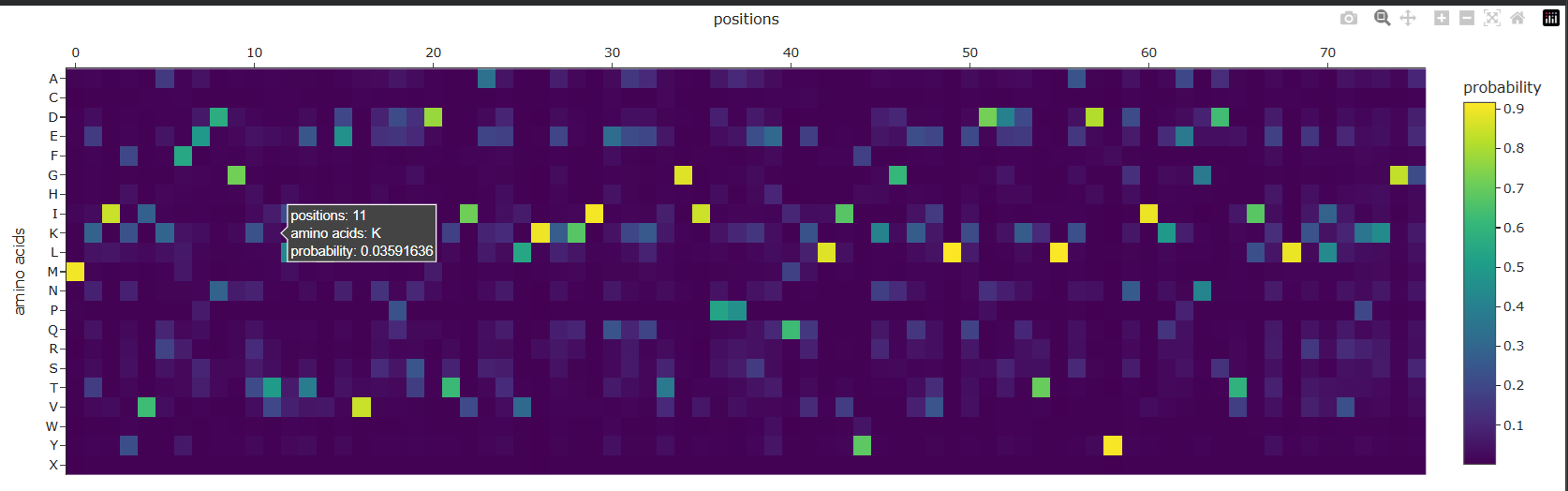 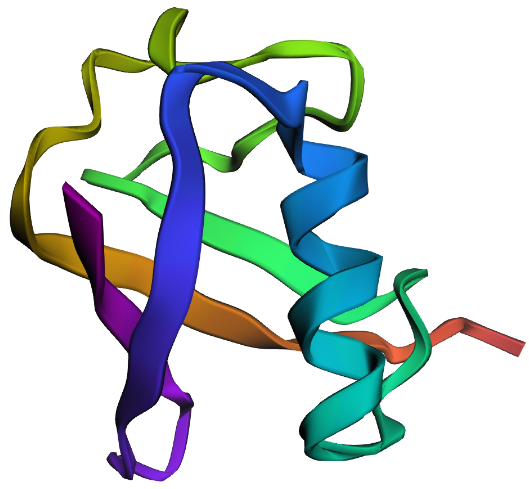 |
Part D. Group Brainstorm on Bacteriophage Engineering
Since I am solo, I am proposing Recursive Protein Optimization using beam-search over Mutation Space: considering ESM2 likelihoods, conditional mutational exploration, ESMFold structural validation, recursive branching search, and local + global optimization.
- – The pipeline starts with athe MS2 L protein sequence as input. First, an ESM2 mutational scan is performed to estimate which single-residue mutations are evolutionarily plausible based on protein language model likelihoods.
- – Instead of selecting only one mutation, the algorithm performs a beam-search style exploration of the top candidate mutations. A parameter termed beam_width controls how many of the highest-scoring mutations are retained at each stage.
- – For every candidate mutation, ESMFold is used to predict the resulting structure and estimate metrics such as pLDDT confidence, pTM score, and structural similarity relative to both the original protein and the locally mutated parent sequence.
- – Mutations that improve or preserve structural confidence are retained, while destabilizing mutations are discarded. The retained sequences are then recursively reintroduced into the same pipeline, allowing conditional mutations to accumulate over multiple generations.
- – This creates a branching mutation graph where each node corresponds to a protein variant and edges correspond to accepted mutations. The search terminates when no further stabilizing or plausible mutations can be identified.
- – Finally, all terminal branches are compared to identify candidate proteins with improved stability while preserving the original fold topology.
Process Flowchart:
Protein Sequence
│
▼
ESM2 Mutational Scan
│
▼
Rank Mutations by Score
│
▼
Select Top-k Mutations
(beam_width = 1–3)
│
┌─────────┴─────────┐
▼ ▼
Mutation 1 Mutation 2
│ │
▼ ▼
ESMFold Prediction ESMFold Prediction
│ │
▼ ▼
Evaluate pLDDT / pTM / RMSD / PAE
│ │
└─────────┬─────────┘
▼
Keep Improved Variants
│
▼
Recursive Mutational Search
│
▼
Build Mutation Graph
│
▼
Select Best Final Variant
An Example mutation tree:
Original Protein Sequence
│
├── V12A
│ ├── V12A + K18R
│ │ └── V12A + K18R + L27I
│ │
│ └── V12A + S22T
│
├── K18R
│ ├── K18R + L27I
│ └── K18R + E31D
│
└── L27I
└── L27I + S22T
This pipeline takes into consideration condition-dependent mutations and evaluates then to choose the next pathways, and ultimately reach the best stable form. This recursive strategy attempts to approximate epistatic interactions between mutations, where the effect of one mutation depends on the presence of earlier mutations.