Human chromosomes (and most eukaryotes) contain Linear DNA at the ends of which are telomeres which are long repititive DNA sequences. In linear chromosomes, DNA polymerase cannot fully replicate chromosome ends, leading to progressive shortening with each cell division. Telomeres help prevent loss of useful information during DNA replication and, in turn, are shortened with each cell division.
Hayflick Limit and Replicative Ageing
The shortening of telomeres is termed Telomere Attrition, and this is one of the 13 hallmarks of ageing (biological ageing at the cellular level). The maximum number of times a cell can replicate before cell division stops (due to the telomere region being exhausted) is known as the Hayflick limit (typically 40-60 times for a normal human somatic cell). Methods to increase this limit should positively impact this Ageing Hallmark, while ensuring that excessive telomere length does not lead to unwanted effects (long telomeres are often observed in multiple types of cancer cells).
Circular DNA without telomeres performing rolling-circle replication[1]
In this individual project (to be formally proposed in the ongoing HTGAA 2026 cohort), I mainly dwell upon two potential approaches that could help remove this nature-imposed cellular replication limit (discovered by Leonard Hayflick) thereby increasing organismal lifespan; the other bottlenecks of lifespan or how whether this will have impact of healthspan are considered out of scope for this project ideation (although I wish to perform experiments on single and multi-cellular organisms regarding the after effects of such synthetic tweaks which could also provide some understanding on the effects on cellular/tissue healthspans).
References
Sakatani, Y., Yomo, T. & Ichihashi, N., Self-replication of circular DNA by a self-encoded DNA polymerase through rolling-circle replication and recombination.,
Scientific Reports, 2018.
https://www.nature.com/articles/s41598-018-31585-1
About me
Born in Kolkata, India, I have spent around two-thirds of my life in my hometown (also known as the City of Joy)
Zealous to strategize on colonizing planets in our Solar System (favourite: Saturn) with a multi-pronged approach of surviving the Solar Supernovae (1. Moving away from the Sun in search of other stars, as well as 2. Developing heat-resistant materials and organisms to survive the Solar Supernovae event by shifting Earth)
I like playing Chess and Football, and solving Problems…
Power Engineer (renewable energy systems); Operational Researcher (mathematical modelling and heuristic design for large scale optimization)
First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
Answer 1
All living cells perform cell division; however, every cell division causes telomere shortening (Telomeres are protective caps at the ends of chromosomes). The limit of the number of cell divisions till a safe limit, such that no useful information is lost (directly from the DNA; telomeres still get shortened in the process), is known as the Hayflick Limit (discovered by Leonard Hayflick in the 1960s). Telomerase elongating a telomere[1] The process of telomere shortening/attrition is one of the (currently 13) Hallmarks of Ageing; therefore, understanding how to increase this limit will be a game-changer.
Scientists & Researchers have been trying to do this using different techniques; the purpose of this HTGAA Individual Project is to suggest a few novel methods and try to understand how implementing them fares wrt. to other methods, as well as understand the bio-technical nuances/problems which might occur due to these changes in the DNA, subsequently, during cell division stages… The methods to be explored include:
Telomerase activity control (to increase Telomere length)
Developing a circular DNA (cDNA, from the linear DNA, by joining the two ends)
Testing the above (cDNA) approach along with a torsion release mechanism (more on this later)
Developing a new protein which can bind at the end of the T and D-Loop of a Telomere and allow the last few nucleotides to be copied (preventing any loss of the DNA during copying)
Question 2
Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals. Below is one example framework (developed in the context of synthetic genomics) you can choose to use or adapt, or you can develop your own. The example was developed to consider policy goals of ensuring safety and security, alongside other goals, like promoting constructive uses, but you could propose other goals for example, those relating to equity or autonomy.
Answer 2
To ensure the technology does not cause disruption in the evolutionary process of species, biosystems, etc. or allow the development of bioweapons, a few governance or policy goals are suggested.
Extending Deep Understanding of Potential (malicious) Use-cases: Understand (new) possible pathways that can arise from the technology itself or as an application of the underlying technology; identify potentially promising pathways which may lead to unintended outcomes and leverage mechanisms to halt them, thereby ensuring biosecurity.
Increasing Traceability and Improving Transparency: It is necessary to understand (holistically) the current (government/private research) labs that have mastered the technology and keep track of their proliferation intent.
Approving appropriate Biosafety levels[2] for eliminating environmental contamination possibilities, and ensuring the safety standards are upheld throughout: Initially classifying necessary biosafety standards that may be appropriate for this kind of experimentation (with a considerable factor of safety), followed by, (a-)periodic lab checks to ensure all lab facilities are up to the mark (and even red-teaming efforts to understand internal sabotage potential) should be performed (maybe by independent organisations or an overseeing body).
Question 3
Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”). Try to outline a mix of actions (e.g. a new requirement/rule, incentive, or technical strategy) pursued by different “actors” (e.g. academic researchers, companies, federal regulators, law enforcement, etc). Draw upon your existing knowledge and a little additional digging, and feel free to use analogies to other domains (e.g. 3D printing, drones, financial systems, etc.). • Purpose: What is done now and what changes are you proposing? • Design: What is needed to make it “work”? (including the actor(s) involved - who must opt-in, fund, approve, or implement, etc) • Assumptions: What could you have wrong (incorrect assumptions, uncertainties)? • Risks of Failure & “Success”: How might this fail, including any unintended consequences of the “success” of your proposed actions?
Answer 3
Action Plan A: • Purpose: Build a network of organizations/institutions that possess the technology or are working to develop the same, and allow cautious expansion of the network, while continually assessing the "(state) intent to proliferate"[3]. Develop a Knowledge Graph/Tree of research labs and individuals who have technical know-how about the scientific technology and are pursuing active research in the same topic. • Design: Incentivise collaboration within the network and regularly educate (through conferences and seminars) about the necessity to have strict access controls for proliferation prevention. • Assumptions: Organizations/Institutions are assumed to not themselves be bad nodes (in a decentralized system) entirely. Periodically reach out to other labs (that may be able to pivot to the same domain) regarding information of whether they are actively pursuing to develop the same scientific tool (either independently or via collaborations). • Risks of Failure & “Success”: Possible failure modes include splitting up of a single collaborative structure into two or more frameworks (may be due to ideological differences)...
Action Plan B: • Purpose: Build an Oversight Body which will request reports from the individuals and research labs (from the dynamically expanding knowledge tree) regarding their concerns about proliferation, and especially to understand whether consensus about halting research in the domain (similar to Mirror life[4,5]) needs to be developed immediately or communicated more effectively. • Design: The oversight body would need to develop partnerships with the national research frameworks of various countries and the United Nations (WHO, etc), allowing a swift trigger of national-level investigations or request international scrutiny in case unchecked proliferation of the technology (either developed independently or through collaboration/technology transfer) is detected from any part of the knowledge tree. • Assumptions: The recommendations of the Oversight Body are taken seriously by all members, and effective execution of the same is followed swiftly. • Risks of Failure & “Success”: There is a chance that such a system could become powerless when the individual members have less intent to prosecute or break ties with another member found indulging in questionable practices.
Action Plan C: • Purpose: Leverage biological agent detection kits[6] to continually monitor surrounding areas of each lab. • Design: Provide capability of risk assessment to discriminate harmful (and harmless) environmental biologics. • Assumptions: Detection systems are highly effective, set up and monitored by a third party or the overseeing body, and cannot be tampered with by individuals or the surrounding organisation(s). • Risks of Failure & “Success”: This is the last stage and any contamination detection would mean lapse in some of the previous stages. Essentially, detection of such harmful substances would be code-red for the area and the surrounding regions!
Question 4
Next, score (from 1–3, with 1 as best, or n/a) each of your governance
actions against your rubric of policy goals:
Answer 4
Does the option:
Action Plan A
Action Plan B
Action Plan C
Identify Malicious Use-cases and Enhance Biosecurity
• By preventing incidents
2
1
2
• By helping respond
2
2
1
Increase Traceability, Improve Transparency and Accountability, while Fostering Lab Safety
• By preventing incidents
1
1
2
• By helping respond
3
2
1
Ensure Biosafety Levels to prevent contamination and also protect the environment
• By preventing incidents
3
2
3
• By helping respond
3
3
1
Other considerations
• Not impede research
1
2
1
• Promote constructive applications
1
1
3
Question 5
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties. For this, you can choose one or more relevant audiences for your recommendation, which could range from the very local (e.g. to MIT leadership or Cambridge Mayoral Office) to the national (e.g. to President Trump or the head of a Federal Agency) to the international (e.g. to the United Nations Office of the Secretary-General, or the leadership of a multinational firm or industry consortia). These could also be one of the “actor” groups in your matrix.
Answer 5
Many of the governance opinions suggested hereinabove are already practised in some or the other form (in varying intensities)[7] to prevent biowarfare. However, with the advent of powerful AI infrastructure allowing real-time decision-making, integration of the proposed Knowledge Tree with a continuous data stream from detection units is a promising future (although enhancing cybersecurity risks), allowing immediate detection of environmental contamination at a wider level than previously possible. Furthermore, an international decentralised governing framework of the technology development direction by the scientific community itself is suggested to prevent misuse and/or proliferation. In this regard, a combination of the Technology-Knowledge Graph of participating members, the establishment of a decentralised Oversight Body, and the leveraging of state-of-the-art biologics detection systems, along with real-time data analysis for immediate threat perception through autonomous (AI-enabled, with human in the loop) decision-making, is key towards the development of a tight-knit, trustworthy and unbiased ecosystem.
Question 6
Reflecting on what you learned and did in class this week, outline any ethical concerns that arose, especially any that were new to you. Then propose any governance actions you think might be appropriate to address those issues. This should be included on your class page for this week.
Answer 6
Philosophically speaking, the class focused on why D/Acc [8] (i.e. cautiously moving towards technological progress, ensuring existing or in-research technologies cannot cause near-doomsday events or something even close) is more important than E/Acc[9] (a techno-optimistic utopian idea of allowing unrestricted technological progress). For my individual project idea, the ultimate goal is to test the suggested methods on embryos of smaller organisms (such as worms, flies, and mice). The final implementation in larger organisms and humans needs to be handled extremely carefully. Governance mechanisms must ensure that this does not cascade to humans until the holistic, deep after-effects of such methods are well understood; these mechanisms should intend to extend our current understanding of ripple/butterfly effects across massive timescales, e.g. how much of the chromatic/DNA/genetic edits are actually inherited (if at all) and what could be the evolutionary impact of the same.
References
Udroiu, I., Marinaccio, J., & Sgura, A., Many Functions of Telomerase Components: Certainties, Doubts, and Inconsistencies,
International Journal of Molecular Sciences, 2022.
https://doi.org/10.3390/ijms232315189
1. Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome? How does biology deal with that discrepancy?
DNA polymerase has a raw error rate of approximately 10-4-10-5 errors per nucleotide added; this can cause high errors when compared to the ~3 × 109 base pairs of the human genome, as this can introduce thousands of mutations per cell division. This discrepancy is tackled through multiple layers of error control, including polymerase proofreading, post-replication mismatch repair, and cell-cycle checkpoints or apoptosis that eliminate heavily damaged cells, reducing the effective mutation rate to ~10-9–10-10 per base per division.
2. How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
An average human protein is ~300 amino acids, and each amino acid is encoded by 1–6 synonymous codons (let's take an average of ~3 codons per amino acid). This makes the number of possible DNA sequences encoding the same protein roughly ≈ 3300 ≈ 10143 possible nucleotide sequences. In practice, synonymous codons can affect translation dynamics and mRNA stability; rare codons affect translation speed & tRNA bias, slowing ribosomes (waiting for low-abundance tRNAs).
~from Dr. LeProust:
1. What’s the most commonly used method for oligo synthesis currently?
Phosphoramidite solid-phase synthesis seems the most commonly used method for oligonucleotide (oligo) synthesis currently, as it is an automated chemical process that builds oligonucleotides nucleotide-by-nucleotide on a solid support.
2. Why is it difficult to make oligos longer than 200nt via direct synthesis?
Direct chemical synthesis of oligonucleotides longer than 200nt is extremely difficult due to cumulative errors; even a 1% failure per step eliminates >90% of the desired product by 200nt due to error accumulation.
3. Why can’t you make a 2000bp gene via direct oligo synthesis?
- Per step error increases exponentially over thousands of cycles, making such a long synthesis impossible - Longer chains on solid supports block reagent diffusion, dropping coupling efficiency - Additionally, extremely large quantities of chemicals will be required for the steps (which are performed in batches)
~from George Church:
1. What are the 10 essential amino acids in all animals, and how does this affect your view of the “Lysine Contingency”?
10 essential amino acids required by most animals are: - Histidine - Isoleucine - Leucine - Lysine - Methionine - Phenylalanine - Threonine - Tryptophan - Valine - Arginine The "Lysine Contingency" from Jurassic Park (1993) was related to genetically engineered dinosaurs unable to synthesise lysine, making them dependent on other lysine sources (thereby making them dependent on humans to feed them Lysine). This is actually not possible as Lysine is already available in meat/fish/grains, etc and even in many single-celled organisms. Thus the dinosaurs could actually still get Lysine from their prey; herbivorous dinosaurs can also obtain Lysine through microbial gut fermentation (through micro-organisms within their guts; it would be impossible for no microbiota to exists as then the digestive system would collapse; it would be another interesting project to understand the consequences of removing all microbiota from a healthy gut of a mouse and seeing the consequences, both computationally via metabolic pathway analysis as well as experimentally).
My understanding of Gl. Ep. is that it simply pulls DNA through a maze, which has channels and pores; each DNA fragment experiences the same force per unit length (so essentially the intended forward acceleration for all DNA fragments would have been the same if there was no friction), but the maze structure creates more resistance/friction to the longer fragments due to which they slow down. Thus we get different bands; unless the pore formation is deterministic and can be atomistically replicated, this is essentially a heuristic which works in real life (as pores in different lanes can also be different; I think there should be a metric to measure if the lanes should have the same weight; this can be achieved by puting the test DNA in the lanes, but also placing a much longer and a much shorter DNA-fragment in all the lane-wells; ideally there should be a straight line formed at the top and bottom (imagine the first and last step of the ladder stretched out across all lanes) and the metric should be how straight the line is! Straighter the line the more holistically equal all the DNA-racing lanes are!).
The answer to the question on this website (under “2. DNA Gel Ladders”) “Because DNA has the same charge per mass for any number of nucleotides, gel electrophoresis separates DNA purely based on length (can you think why?)” is:
~ because the force per unit mass is same, and the only differentiating factor becomes the DNA-travel/movement resistance due to the gel which is proportional to the length (more the length there are more contact points with the gel and longer DNA cant pass through those pores easily, essentially creating a length-specific bottleneck…
Part 1:
1. a.1. b.Image 1 (a, b) reminds us of the shifting temperatures across the globe due to global warming and motivates us to work to prevent climate change.
(Due to lack of time and the problem being a fathomable combinatorial problem, I chose to use my imagination and get the best of whatever sequence was generated; in case any1 is interested in getting hold of a mathematical formulation which can directly output the enzymes required for each lane of a specific design pattern, I can develop such an MILP formulation…)
2. a.2. b.Image 2 (a, b) reminds us of an anomalous intelligence drop in the Gen-Z population (the Reverse Flynn effect), which is possibly an effect of high smartphone/social media/internet usage (and more research may be necessary to understand the actual causes and develop policies to combat them).
Part 2: Not applicable for Global Committed Listeners without Lab access (therefore I am skipping this)
The organism selected was human, mentioning this allows further optimization of the codon, for this case of the human nuclear protein.
Codon optimization is generally necessary for improving translation efficiency and protein yield (reducing ribosomal pausing and improving folding); the safeguard against specific restriction enzymes is to ensure that the DNA does not get cut in case any of the subsequent future workflows requires usage of such an enzyme…
3.4. You have a sequence! Now what?
To synthesize this protein from within my DNA (assuming that it is not already present), we can use some technique to insert the obtained (codon optimized) DNA sequence into a viral vector to insert it inside the human DNA. After successful DNA insertion, the Central Dogma will take care of the rest of the protein synthesis process…
3.5. How does it work in nature/biological systems?
A single gene can code for multiple proteins at the transcriptional level, as there can be multiple start sites, alternate splicing of exons, and the 3-nucleotide reading frame, which can essentially pack three times the information.
Part 4: Prepare a Twist DNA Synthesis Order
4.1. Twist account and a Benchling account created
(i) What DNA would you want to sequence (e.g., read) and why?
This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I am interested in developing a sample of mammalian circular DNA (mice, monkeys, etc.), and understand the complications during such DNA replication (to prevent cancers during cell division). Therefore, I am interested in developing a circular DNA (cDNA; clipping away the telomeres) and merging the two ends and then sequencing that entire cDNA. My motive behind this is to disprove my hunch that cDNA is a viable path to human lifespan/healthspan extension!?!
(ii) In lecture, a variety of sequencing technologies were mentioned.What technology or technologies would you use to perform sequencing on your DNA and why?
I will use the latest Oxford Nanopore sequencing technique because:
- it can read entire circular DNA molecules without any need for fragmentation
- it can sequence the DNA using rolling-circle–amplified replication
- it uses natural motor enzymes
Is your method first-, second- or third-generation or other? How so?
The Oxford Nanopore third-generation sequencing performs single-molecule sequencing without DNA amplification, producing long reads, and preserves the circular DNA topology.
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
The input will be the (purified) circular mammalian DNA; no fragmentation and no PCR is required.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
A motor enzyme feeds DNA through a biological nanopore; each nucleotide creates a change in ionic current, which can be decoded uniquely.
What is the output of your chosen sequencing technology?
From the raw electrical signal data (FAST5), long-read nucleotide sequences (FASTQ/FASTA) are derived, which preserve structural information like junctions, repeats, and circular continuity
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
I wish to synthesise a mammalian cDNA and probe its self-replicating properties during cellular division (ensuring that the DNA copy generated does not exhibit features of cancer or other abnormalities).
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I will use oligonucleotide synthesis followed by enzymatic DNA assembly to construct the full mammalian cDNA, because this approach allows precise sequence control, modular assembly, and is compatible with commercial gene synthesis pipelines (e.g., Twist).
What are the essential steps of your chosen sequencing methods?
- the approx 1842 nt long DNA is split into 12 overlapping oligos (175 bp each)
- Chemical synthesis of each short DNA oligonucleotides
- Enzymatic assembly of oligos into the full-length cDNA
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
- Possibly high error rates as sequence length nears the limit of 200 bp for oligonucleotide synthesis; this can be bypassed by splitting DNA further
- Whole-genome or very large circular constructs may require multi-step assembly
5.3 DNA Edit
(i) What DNA would you want to edit and why?
In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
I am interested to edit DNA of most test aminals (ncluding but not limited to, C elegans, fruitflies, mice, monkeys, etc.) starting from the smaller organisms to large mammals. However this is not gene editing; simply clipping off the telomeres and joining the ends of each linear chromosome to make it circular. Next I wish to observe cell-division processes and how such cDNA fares in large animals (mapping out the Hayflick limit changes due to this alternation).
Additionally, I am interested in looking into telomere maintenance and end-protection, especially if their controlled modulation can extend cellular lifespan without inducing genomic instability or cancer.
(ii) What technology or technologies would you use to perform these DNA edits and why?
How does your technology of choice edit DNA? What are the essential steps? What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing? What are the limitations of your editing methods (if any) in terms of efficiency or precision?
There seem to be enzymes that are already used to develop circular DNA from linear DNA by clipping telomeres and joining both ends:
- Restriction Endonucleases make staggered cuts in linear DNA
- DNA Ligase joins the ends of linear DNA fragments together
- Protelomerase specifically resolves telomeres, converting linear DNA into circular form
I am also interested in looking at CRISPR-based editors for targeted modifications in case it is necessary to arrest certain telomere(-ase) or related pathways.
The main challenges are in delivering edits uniformly across all chromosomes, and the high risk of genomic instability (especially during cell divisions). Therefore, all experiments will be restricted to somatic cells in model organisms, and will need extensive validation steps.
Additionally, smaller circular DNA exhibits torsion; in case this might also be a concern when large mammalian DNA is made circular, mechanisms to periodically release the torsion need to be developed (whether such a system may be necessary also needs to be ideated -- requires expert guidance and advice).
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
The Million Molecule Challenge by Ora Biomedical aims to find the best combination-drugs for enhancing C. Elegans longevity. They developed an autonomous lab that can capture this data continuously throughout the worm’s lifespan (and the worm with the longest healthspan makes its combinatorial drug intervention win). This can be extended to larger lifeforms with novel automations.
(Post this point, I have had to use AI help extensively for my homeworks, as almost everything is brand new to me.)
Final Project Automation Strategy
For my final project investigating co-translational folding differences between wild-type and retro-proteins (GB1/Ubiquitin), precision and reproducibility are critical. Because I am testing how environmental factors (like temperature gradients and microgravity) affect folding pathways, doing this manually would introduce high pipetting variance and human error. I plan to automate the sample preparation and expression phases using the following three approaches:
1. Ginkgo Nebula (Cloud Lab) for Thermal Gradient CFPS
To rigorously test my hypothesis regarding temperature-dependent final states (and simulated global warming effects), I will use the cloud lab to run a high-throughput cell-free protein synthesis (CFPS) array.
Echo Liquid Handler: Transfer specific molarities of wild-type and retro-DNA templates into a 384-well plate.
Bravo/Multiflo: Dispense the CFPS lysate and energy master mix into all wells to initiate translation.
PlateLoc: Seal the plate to prevent evaporation.
Inheco Thermocyclers: Incubate different zones of the plate at precise, distinct temperature brackets (e.g., 25°C, 30°C, 37°C, 42°C) simultaneously.
PHERAstar: Read initial baseline fluorescence/absorbance if tagged, before routing the plates to downstream purification.
2. Automated Tryptic Digestion for LC-MS/MS (Opentrons OT-2)
To confirm the primary structure and sequence inversion, the proteins must be digested into peptide fragments for tandem mass spectrometry. Tryptic digestion is highly sensitive to enzyme ratios and timing.
I will script an Opentrons OT-2 protocol to automate the addition of denaturation buffers, DTT (reduction), IAA (alkylation), and Trypsin.
The robot will handle the precise micro-volume washing and desalting steps on a magnetic module before LC-MS/MS injection, ensuring my bottom-up peptide mapping is perfectly standardized.
3. Custom 3D Printed Microfluidic Holders for Microgravity
For the space-based cell-free expression chambers, standard well plates cannot be used due to fluid behavior in zero gravity. I will design and 3D-print a custom hardware holder (compatible with automated liquid handler decks on Earth) that securely locks down sealed, microfluidic cell-free chips. This allows the robots to prepare the space-bound assays perfectly before they are shipped to the ISS.
I am also interested in understanding how climate change-induced changes within the internal (homeostatic; thermal) energy landscape can increase the probability of protein folding errors (or even change the protein structures), which can lead to a host of other biological problems due to global warming!
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Considering the average meat contains 25% of proteins by weight[1], and proteins are approximately 100% composed of amino acids, we need to find the number of amino acids present in 125 grams. As it is already provided that an average amino acid is about 100 Daltons, the estimated number of amino acids is equal to:
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
The human digestive system breaks down all raw materials into its basic forms (for example proteins are broken down into the amino acids) and then these are used for the body's own processes. If the proteins are somehow magically ingested as is, in their same form in the original organism and those proteins somehow get inside a living human cell, then there could be some other issues related to creation of new pathways which are similar to what the protein was used for in the original organism and this could have unintended consequences ultimately killing the carnivore. Nucleic Acids (constituents of DNA/RNA) are also broken down in our digestive system preventing any possibility of incorporating the external DNA fragments within our cells; in the case of a leaky-gut when some protein or DNA fragments might enter the body, the immune system responds appropriately. Thus, the digestive system acts as an information shredder, passing on raw memory-less ingredients to the host.
3. Why are there only 20 natural amino acids?
There are >500 amino acids that occur naturally, but only 22 of them are expressed through living genes.[2] Why only 22 seems a tough question for evolutionists, my hunch is that even if life started with more than 500 types of animo acids being expressed, there could be eating preferences which could have led to the specific pathways (which we cuurently have) being reinforced while the other pathways disappeared slowly; or, the origin of life that posibly evolved in a pond-cluster started with these 22 essentials while the other pond-clusters didn't make it to large scale organisms. Solid proof can only be found upon trying to replicate a fast-tracked evolutionary process using all pathways consisting of 500 AAs and then observing if the evolution converges to the same 22 AAs.
4. Can you make other non-natural amino acids? Design some new amino acids.
As per the definition of Amino Acids, they must contain Amino groups and Carboxylic Acids (and an alpha carbon connecting both groups; research about beta and gamma AAs are also worthwhile). So it should be pretty simple to design a new AA as per this definition. An easy way to generate new AA's (so that they are unique) is to find the heaviest AA designed till date and add another carbon atom somewhere (or at the other end); this is a general trick to keep on extending artificial AAs, but instability issues should be kept in mind. Another important reason for the limit of 22 AAs could be their size, which allows them to pass through the cellular membranes via AA-transporter proteins (larger/heavier AAs may have higher resistance to do that). Further just having a new AA wont matter much unless a new Aminoacyl-tRNA synthetase (aaRS, essential enzymes that attach specific amino acids to their corresponding tRNAs) is designed; humans have 20 different types of aaRS for attaching the 20 standard-essential AAs to their respective tRNAs.
Worth Mentioning: Simply adding single carbons can be often ignored by ribosomes, and the translation machinery, better to add larger groups; in this regard adding Phosphoserine analogs with non-hydrolyzable bonds are used to study signaling.
5. Where did amino acids come from before enzymes that make them, and before life started?
Before enzymes and life existed, amino acids formed spontaneously through natural geochemical and atmospheric processes. They were synthesised on primitive Earth and supposedly also delivered from space. The water reservoirs that they accumulated into form the raw chemical building blocks that sparked life are also known as The Primordial Soup.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
D-amino acids are exact mirror images of natural L-amino acids and thus they form exact mirror-image structures. Natural proteins made of L-amino acids form right-handed helices; therefore, reversing the chirality of the building blocks reverses the handedness of the helix. The answer is therefore LEFT.
7. Can you discover additional helices in proteins?
Proteins possess several other structures apart from the well-known alpha helices and beta sheets because the amino acid backbone allows for various bending angles. While alpha helices and beta sheets are the most common repeating patterns of secondary structure, others exist and serve critical functions:
– Beta Turns and Loops: Short, non-repetitive segments connecting alpha helices and beta sheets. They allow the protein chain to fold back on itself, giving the protein its compact, 3D shape.
– Random Coils: Irregular, unordered stretches of the polypeptide chain. Unlike helices or sheets, they don't have a stable, repeating geometry and are highly flexible.
– Other Helices: Less common structures like the 310-helix or π-helix, which are tighter and sometimes found at the ends of alpha helices
8. Why are most molecular helices right-handed?
In nature, all amino acids found are of the chirality type L, and therefore, they form right-handed curls. A possibility regarding this is that the first life from the primordial soup was of the L-type and that subsequent superstructures (i.e., proteins) took away much of the intermediate raw materials that could have been used to form D-type amino acids. Thus, D-type structures died in the primordial evolutiona nd we don't see them anymore, except when artificially created in labs. However, this does not directly imply that the L-amino acids physically cannot form left-handed helices. Left-handed L-helices are physically possible and L-amino acids can theoretically fold into left-handed alpha helices; but the issue is steric hindrance: In a left-handed helix made of L-amino acids, the bulky side chains (Rgroups) are forced too close to the backbone atoms. This creates severe physical crowding (steric clash). Right-handed is energetically cheaper: In a right-handed helix made of L-amino acids, the side chains point outward and away from the backbone, minimizing crowding and maximizing stability.
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets tend to aggregate because they possess exposed, unsatisfied hydrogen bonds along their outer edges and highly hydrophobic flat surfaces. This architectural vulnerability drives them to stack or extend infinitely to achieve thermodynamic stability (frequently leading to the formation of pathological amyloid fibrils).
The Primary Driving Forces are:
– Enthalphy gain via backbone-to-backbone Unsatisfied Edge Hydrogen Bonding
– The Hydrophobic Effect and Solvation Entropy: When flat, hydrophobic side chains (e.g., Leu, Ile, Val, Phe) stack face-to-face, trapped, highly ordered water molecules are released back into the bulk solvent. This creates large, concentrated patches of hydrophobic residues (such as Leucine, Isoleucine, and Valine); to escape the surrounding water, these greasy, flat surfaces crash together, driving face-to-face stacking of multiple β-sheets. This massive increase in solvent entropy is the primary thermodynamic engine driving aggregation.
10. Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Amyloid diseases (like Alzheimer's, Parkinson's, and Type 2 Diabetes) do not happen because β-sheets are inherently toxic, but because the cross-β sheet architecture is the lowest global thermodynamic energy minimum for almost any polypeptide chain. Almost any protein, if unfolded or destabilized long enough, can convert into a β-sheet-rich amyloid.
Amyloid β-sheets make extraordinary, high-performance nanomaterials. While biology views them as pathological, material scientists exploit their steel-like mechanical properties, chemical resilience, and self-assembling nature, to create Protective Biofilm Matrices, Conductive Nanowire, Injectable Hydrogels, High-Strength Composites, etc.
11. Design a β-sheet motif that forms a well-ordered structure.
General steps to design a β-sheet motif:
– [Step 1: Length] ──> Choose 8 to 16 amino acids (peptide needs to be long enough to provide sufficient surface area for self-assembly, but short enough to remain highly soluble)
– [Step 3: Charge] ──> Engineer Electrostatic Complementarity: Alternate + and - charges on the Hydrophilic face to prevent the strands from shifting randomly. The hydrophilic residues/sheets then line up with these opposing (alternating positive and negative) charges locking into a precise, pristine grid of salt bridges.
– [Step 4: Termini] ──> Cap the ends (Acetylation / Amidation) to remove raw charges: Natural peptide ends carry a positive charge at the N-terminus and a negative charge at the C-terminus. These raw, terminal charges can disrupt the sheet geometry; they can be neutralised by adding an Acetyl group (Ac-) to the front and an Amide group (-NH₂) to the back.
The following template of RAD16 is supposedly the Gold-Standard Design Template widely used in biomedical engineering to create stable biomaterials and scaffolds: Ac-R A D A R A D A R A D A R A D A-NH₂ (Arginine - Alanine - Aspartic Acid - Alanine... repeated)
Part B: Protein Analysis and Visualization
Questions
Answers
1. Briefly describe the protein you selected and why you selected it.
I selected the protein Ubiquitin for this assignment. Ubiquitin is a small regulatory protein found in eukaryotic cells that plays an important role in protein degradation, signalling, and cellular regulation. It has a well-characterised three-dimensional structure, and many experimentally determined structures are available in the Protein Data Bank (PDB). I initially considered using GB1, but found that its initiator methionine is retained in the mature protein. Since my broader project involves studying retro (reverse) protein sequences, I wanted a protein in which the initial methionine is naturally removed during post-translational processing. In many proteins, the initiator methionine is cleaved by methionine aminopeptidase when the second amino acid has a small side chain. Ubiquitin is therefore a suitable choice because its mature form does not retain the starting methionine, allowing cleaner comparison between the natural sequence and a retro/reverse sequence without introducing an artificial terminal methionine residue. Additionally, ubiquitin is small, structurally stable, and extensively studied, making it convenient for computational and structural analysis.
2. Identify the amino acid sequence of your protein. How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids. How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs. Does your protein belong to any protein family?
The protein I selected is Ubiquitin from humans, a highly conserved regulatory protein involved in protein degradation and cellular signalling. Ubiquitin. The mature amino acid sequence of UBIQUITIN is QIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRGG; it contains 75 amino acids with the most frequent AA being Leucine (L appears 9 times, frequency 11.84%). Lysine (K) is also highly abundant and biologically important because ubiquitin forms polyubiquitin chains through lysine residues. Ubiquitin has thousands of homologs across eukaryotes because it is one of the most evolutionarily conserved proteins known. Human and yeast ubiquitin, for example, share about 96% sequence identity, differing in only 3 out of 76 amino acids; (these were validated using UniProt BLAST). Ubiquitin belongs to the Ubiquitin protein family (Pfam accession: PF00240), a family of small regulatory proteins involved in ubiquitination and intracellular protein turnover. Members of this family are small proteins or protein domains that adopt the characteristic ubiquitin fold. Proteins in this family are involved in post-translational modification pathways and intracellular protein turnover.
3. Identify the structure page of your protein in RCSB. When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å). Are there any other molecules in the solved structure apart from protein?
Does your protein belong to any structure classification family?
The structure page of my protein is available in the Protein Data Bank (RCSB PDB) under the entry 1UBQ titled “Structure of ubiquitin refined at 1.8 Å resolution.” The structure was solved in 1987 using X-ray diffraction, with the reported resolution indicating a very high-quality protein structure. In structural biology, lower resolution values correspond to more accurate atomic positions, and structures below about 2.0 Å are generally considered excellent quality. Apart from the protein itself, the solved structure also contains water molecules that were resolved in the crystal structure. No additional ligands or cofactors are present in the basic ubiquitin structure. Ubiquitin belongs to the ubiquitin fold structural family, a highly conserved protein fold characterized by a compact globular structure containing β-sheets and an α-helix. It is also classified within the Ubiquitin-like superfamily in structural classification databases such as SCOP and CATH. Members of this structural family are involved in protein regulation, signalling, and intracellular protein turnover.
4. Open the structure of your protein in any 3D molecule visualization software: PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands). Visualize the protein as “cartoon”, “ribbon” and “ball and stick”. Colour the protein by secondary structure. Does it have more helices or sheets? Colour the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues? Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
I used RCSB's 3D Viewer to visualize ubiquitin in cartoon, ribbon, molecular surface, ball-and-stick, and spacefill representations.
Cartoon with Ball-and-stick representationMoleculesMolecular surface representationSecondary structure viewColored by residue chargeColored by residue typeColored by HydrophobicityColoured by Accessible Surface AreaColoured by Geometry Quality
Ubiquitin contains more β-sheets than α-helices (consistent with the characteristic ubiquitin β-grasp fold).
Hydrophobic residues are mainly buried in the interior core, while charged and hydrophilic residues are exposed on the surface.
It has one hole (binding pocket). Surface visualization shows a compact globular structure with shallow interaction grooves rather than deep catalytic binding pockets.
Part C. Using ML-Based Protein Design Tools
Questions
Answers
C1.1. Deep Mutational Scans. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
Single Mutation Scan (purple indicates strongly unfavorable mutations relative to the wild-type residue) (download to view interactive HTML)The first Amino acid mutation is highly unfavourable to anything. However, peculiarly, the second AA mutation to Methionine (M) is extremely favourable. Does that mean that after the second place mutation to M, the first place AA is removed through post-translational modifications; otherwise in this case there will be an AA before the methionine (M) which generally does not happen as M is the start codon. For the retro protein sequence, the first place's mutation is similarly highly unfavourable but a specific position 40 seems to have many favourable mutations...Mutation Scan of the Retro-Protein Sequence (download to view interactive HTML)
C1.2. Latent Space Analysis: Use the provided sequence dataset to embed proteins in reduced dimensionality. Analyze the different formed neighborhoods: do they approximate similar proteins? Place your protein in the resulting map and explain its position and similarity to its neighbours.
Protein sequuence similarity in latent space representation (download to view interactive HTML)Protein sequences were first converted into 320-dimensional latent embeddings using the ESM2 protein language model. These embeddings are numerical representations learned from large-scale evolutionary protein sequence data and encode biochemical, structural, and evolutionary features of proteins. Similar proteins tend to occupy nearby regions in this high-dimensional embedding space. To visualize these embeddings, t-distributed Stochastic Neighbor Embedding (t-SNE) was used to reduce the 320-dimensional vectors into three dimensions. Since only five protein sequences were analyzed, the perplexity parameter was reduced from the default value of 30 to 3 (because t-SNE requires the perplexity value to be smaller than the number of samples). Perplexity approximately controls the effective number of neighboring points considered during dimensionality reduction. Lower perplexity values emphasize local neighborhood relationships between nearby proteins. The three plotted axes (TSNE1, TSNE2, and TSNE3) do not correspond to specific physical or biochemical quantities such as molecular weight, hydrophobicity, or sequence length. Instead, they are abstract latent coordinates generated by t-SNE to preserve local similarity relationships between the original high-dimensional embeddings. Proteins that appear closer together in the plot are interpreted as having more similar learned sequence representations, while proteins farther apart are considered more dissimilar in the latent embedding space. In the resulting embedding map, Ubiquitin and the retro-Ubiquitin sequence occupied different positions despite containing the same amino acid composition in reversed order, suggesting that sequence order strongly influences the learned representation of the protein language model and that the retro sequence is interpreted as biologically distinct from native ubiquitin. Other proteins, such as haemoglobin, insulin, and albumin fragments, also occupied separate regions, reflecting their differing sequence patterns and biological functions. However, t-SNE visualizations should be interpreted qualitatively rather than quantitatively. The distances and cluster sizes in t-SNE plots can vary depending on initialization and parameter choices such as perplexity, and therefore do not represent exact evolutionary or structural distances between proteins.
C2. Protein Folding: Fold your protein with ESMFold. Do the predicted coordinates match your original structure? Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
ESMFold was used to predict the structure of both native ubiquitin and a retro-ubiquitin sequence generated by reversing the amino acid order. The model directly predicted atomic coordinates from sequence without requiring multiple sequence alignments. The native ubiquitin prediction showed high confidence, with most residues having pLDDT values above 85–90. The PAE heatmap also showed low predicted aligned error across most residue pairs, indicating that the model was confident about the global fold and residue positioning. The predicted structure retained the compact ubiquitin-like fold with well-defined β-sheets and α-helices. pLDDT is a per-residue confidence score ranging from 0–100, where higher values indicate more reliable structural predictions.
In contrast, the retro-ubiquitin model showed much lower pLDDT values (~30–55 across most residues), suggesting low confidence and reduced structural stability. The PAE heatmap also displayed much larger predicted errors across residue pairs, indicating uncertainty in the relative arrangement of structural regions.
Even though retro-ubiquitin contains the same residues as native ubiquitin, reversing the sequence disrupts the learned structural and evolutionary patterns required for forming the exact original folds.
C3. Protein Generation: Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one. Input this sequence into ESMFold and compare the predicted structure to your original.
ProteinMPNN was used to perform inverse folding on the Ubiquitin backbone structure. Instead of predicting structure from sequence, ProteinMPNN predicts amino acid sequences that are compatible with a given protein backbone geometry. The generated heatmap represents the probability of each amino acid occurring at every residue position based on the backbone structure. Bright regions indicate amino acids strongly preferred by the model at specific positions, while diffuse regions represent positions that tolerate greater sequence variability.ProteinMPNN HeatMap indicating model predictions for a given Backbone StructureThe designed sequence differed substantially from native Ubiquitin while still remaining compatible with the same backbone fold. The sequence recovery value was ~54%, indicating that roughly half of the residues were conserved relative to the original sequence.
Structurally important positions showed stronger conservation, whereas surface or flexible positions tolerated more substitutions. Interestingly, the generated sequence achieved a lower ProteinMPNN score than the native sequence, suggesting that the designed sequence may fit the backbone geometry more favorably (according to the model!). This demonstrates that many different amino acid sequences can potentially encode similar protein folds, highlighting the robustness and degeneracy of protein structural organization.
ESMFold structure of protein predicted by MPNN based on Ubiquitin-BackBone
Part D. Group Brainstorm on Bacteriophage Engineering
Since I am solo, I am proposing Recursive Protein Optimization using beam-search over Mutation Space: considering ESM2 likelihoods, conditional mutational exploration, ESMFold structural validation, recursive branching search, and local + global optimization.
– The pipeline starts with athe MS2 L protein sequence as input. First, an ESM2 mutational scan is performed to estimate which single-residue mutations are evolutionarily plausible based on protein language model likelihoods.
– Instead of selecting only one mutation, the algorithm performs a beam-search style exploration of the top candidate mutations. A parameter termed beam_width controls how many of the highest-scoring mutations are retained at each stage.
– For every candidate mutation, ESMFold is used to predict the resulting structure and estimate metrics such as pLDDT confidence, pTM score, and structural similarity relative to both the original protein and the locally mutated parent sequence.
– Mutations that improve or preserve structural confidence are retained, while destabilizing mutations are discarded. The retained sequences are then recursively reintroduced into the same pipeline, allowing conditional mutations to accumulate over multiple generations.
– This creates a branching mutation graph where each node corresponds to a protein variant and edges correspond to accepted mutations. The search terminates when no further stabilizing or plausible mutations can be identified.
– Finally, all terminal branches are compared to identify candidate proteins with improved stability while preserving the original fold topology.
This pipeline takes into consideration condition-dependent mutations and evaluates then to choose the next pathways, and ultimately reach the best stable form. This recursive strategy attempts to approximate epistatic interactions between mutations, where the effect of one mutation depends on the presence of earlier mutations.
– Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
– Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
– To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
– Record the perplexity scores that indicate PepMLM’s confidence in the binders.
The human SOD1 sequence is: MATVAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Post-translation, the starting Methionine gets removed. After A4V Mutation, it becomes: ATVVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Generated four candidate binder peptides (length = 12 aa) using PepMLM conditioned on the mutant SOD1 (A4V) sequence. Lower pseudo-perplexity values indicate higher model confidence in the generated binder sequence.
The following abbreviations ae used in the table: PeptiVerse (PV); AlphaFold (AF);
Among the generated candidates, WSYPAVAARLKX achieved the lowest pseudo-perplexity (6.81), suggesting the highest confidence prediction by PepMLM among the generated sequences.
Important Note: X denotes a masked/unspecified amino acid token generated by the model pipeline and may require post-processing or substitution depending on downstream analysis. The “X” causes problems in Alphafold and so the first four were generated by editing that Google Collab…
Part 2: Evaluate Binders with AlphaFold3
– Navigate to the AlphaFold Server: alphafoldserver.com
– For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
– Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
– In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
Most information is consolidated in the above table.
Among the peptides, I will be going forward with WRSYAVAIGHKK as its parameters (in the above table) seem to be the closest to the known binding peptide. The peptide is localised near the C-terminus; but both the C and N terminus are also near each other…
For FLYRWLPSRRGG, the peptide seems to be closer to the beta-sheets and more near to the N-terminus than the C-terminus.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, we evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
– Paste the peptide sequence..
– Check the boxes: Predicted binding affinity, Solubility, Hemolysis probability, Net charge (pH 7), Molecular weight
– Paste the A4V mutant SOD1 sequence in the target field.
– Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
– Choose one peptide you would advance and justify your decision briefly.
(Above table is to be referred…)
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
– Open the moPPit Colab linked from the HuggingFace moPPIt model card (Make a copy and switch to a GPU runtime.)
– In the notebook: - Paste your A4V mutant SOD1 sequence. - Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch). - Set peptide length to 12 amino acids. - Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
– After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
The below peptides were generated using moPPIt:
Binder
Hemolysis
Solubility
Affinity
Motif
CATGCNVWPGVI
0.047789514
1
6.598640442
0.667649984
ADSEFTAPSEAH
0.057091117
1
5.709741116
0.711770117
ESEKYGVQCHIT
0.064232945
1
6.120092869
0.721853793
CFAGIYKQKEQT
0.048995256
1
6.007740021
0.786077559
QAQCGQFQFNVE
0.04008162
1
6.198196411
0.903030217
SQCTRVLVPTIC
0.114275873
1
6.355286598
0.730438828
ARKPCFAALQSA
0.026957572
1
6.220830917
0.638868332
EKPDYHDGPCWI
0.046631336
0.99999994
6.483778477
0.73085916
These peptides have similar ranges for the parameters mentioned in the above table when compared to the first table on this page; further analysis on multiple other dimensions need to be performed for a thorough comparison.
Part C: Final Project: L-Protein Mutants
L-Protein Engineering | Option 3: Random Mutagenesis
Based on the Table Information provided, and filtering to only the mutations where Lysis is happenning and protein levels are assigned as 1, I identify the following mutations should take place:
Position of the mutation in L
Base Pair Changed (RNA nucleotide coordinate of the MS2 phage genome)
Amino Acid Position
Amino Acid Change
Lysis
Protein Levels (ND=Not determined)
38
C->T
13
P->L
1
1
43
T->G
15
S->A
1
1
52
A->G
18
R->G
1
1
53
G->T
18
R->I
1
1
89
G->A
30
R->Q
1
1
89
G->T
30
R->L
1
1
92
G->T
31
R->I
1
1
131
T->C
44
L->P
1
1
131
T->C
44
L->P
1
1
133
G->C
45
A->P
1
1
136
A->T
46
I->F
1
1
We use an L-protein with the AA mutations at the following positions 13, 30 and 46(from the above table) thereby leading to this L-protein sequence: METRFPQQSQQTLASTNRRRPFKHEDYPCLRQQRSSTLYVLIFLAFFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
DnaJ is completely wild-type (unmutated): MAKQDYYEILGVSKTAEEREIRKAYKRLAMKYHPDRNQGDKEAEAKFKEIKEAYEVLTDSQKRAAYDQYGHAAFEQGGMGGGGFGGGADFSDIFGDVFGDIFGGGRGRQRAARGADLRYNMELTLEEAVRGVTKEIRIPTLEECDVCHGSGAKPGTQPQTCPTCHGSGQVQMRQGFFAVQQTCPHCQGRGTLIKDPCNKCHGHGRVERSKTLSVKIPAGVDTGDRIRLAGEGEAGEHGAPAGDLYVQVQVKQHPIFEREGNNLYCEVPINFAMAALGGEIEVPTLDGRVKLKVPGETQTGKLFRMRGKGVKSVRGGAQGDLLCRVVVETPVGLNERQKQLLQELQESFGGPTGEHNSPRSKSFFDGVKKFFDDLTR
Alphofold3 co-folding result of 3-mutated L-protein with DnaJ
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion High-Fidelity PCR Master Mix contains all the necessary components for robust, high-fidelity DNA amplification. Its primary components include: Phusion DNA Polymerase (for high processivity and proofreading activity, resulting in an error rate roughly 50-fold lower than traditional Taq polymerase), Deoxynucleotide Triphosphates (dNTPs; serve as the fundamental molecular building blocks that the polymerase polymerizes to synthesize the complementary nascent DNA strand), Reaction Buffer & Proprietary Stabilizers (to maintain pH stability during thermal cycling), Magnesium Ions (acts as an essential cofactor for DNA polymerase activity by coordinating with the phosphate groups of the dNTPs and the DNA backbone, facilitating the nucleophilic attack required for phosphodiester bond formation).
What are some factors that determine primer annealing temperature during PCR?
The primer annealing temperature is critical for balancing reaction specificity and yield, and is dictated by the following interconnected factors:
Primer Melting Temperature: The temperature at which 50% of the primer-template duplex is dissociated.
GC Content: G-C base pairs share three hydrogen bonds, whereas A-T pairs share only two. Primers with a higher percentage of GC pairs require higher temperatures to denature and thus exhibit a higher melting temp.
Primer Length: Longer primers have more total base-pairing interactions, increasing the total thermal energy required to disrupt the hybrid structure, which raises the melting temp.
Salt and Buffer Concentration: Monovalent and divalent cations mask the negative charges on the phosphodiester backbone of DNA. Higher salt concentrations stabilize the duplex, reducing electrostatic repulsion and raising the effective melting temp.
Primer Mispriming/Secondary Structure: The presence of internal hairpins, self-dimers, or cross-dimers lowers the concentration of free, accessible primer, occasionally requiring temperature adjustments to avoid off-target amplifications.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests (REDs). Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Both methods are foundational molecular biology techniques used to generate linear DNA fragments, but they differ fundamentally in:
Mechanism: PCR occurs through enzymatic synthesis and exponential amplification of a specific target region using flanking oligonucleotides; in RED, the chemical cleavage of pre-existing phosphodiester bonds at specific sequence-defined palindromic restriction sites occur.
Protocol Requirements: PCR requires sequence-specific primers, dNTPs, a thermostable polymerase, and a thermal cycler. RED requires a sequence containing the target restriction site, specific endonuclease enzymes, and an isothermal incubation block.
Fidelity & Modifications: PCR can introduce unwanted point mutations (minimized by high-fidelity enzymes); allows easy addition of custom flanking sequences (e.g., Gibson overhangs) via primer tails. RED is high fidelity because it cuts exact biological DNA; limited strictly to the locations of existing or engineered restriction sites.
Yield & Scalability: PCR amplifies fragments exponentially from trace amounts of template (nanogram scale). REDs yield is strictly limited by the starting mass of the source plasmid/DNA (microgram scale).
PCR is preferred when a gene needs to be isolated from a genomic or plasmid source while simultaneously adding flanking homology arms (overhangs) for seamless cloning methods like Gibson Assembly, or when working with tiny amounts of template DNA. Restriction Digests are preferred when cutting open a large recipient vector backbone to minimize the risk of mutations across a large sequence, or when performing quick diagnostic analytical checks (restriction mapping) to confirm if a plasmid contains the correct insert.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
- Homology Arms: Ensure adjacent fragments share 20–40 bp of identical sequence at their tips. - Blunt Ends: Use a proofreading polymerase (like Phusion) so there are no non-templated $3'$ A-overhangs. - Clean Fragments: Run a gel to verify sizes, then column-purify to strip out background dNTPs and enzymes.
How does the plasmid DNA enter the E. coli cells during transformation?
- Chemical Competence: Ca++ neutralizes negative charges so DNA sits on the cell wall. Heat shock (42°C) creates a rapid thermal draft that physically opens transient membrane pores. - Electrocompetence: Cells are washed to remove salts. A high-voltage shock induces localized dielectric breakdown, creating hydrophilic pores that pull DNA inward via electrophoresis.
Describe another assembly method in detail (such as Golden Gate Assembly). Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate Assembly relies on Type IIS restriction enzymes, such as BsaI, which cleave DNA at a precise distance outside of their non-palindromic recognition sequences to generate custom 4-base-pair sticky overhangs. Because these cuts are offset from the binding site, the recognition sequences are completely discarded from the insert fragments during the digestion process. This strategic removal ensures that the final assembled product no longer contains active restriction sites, rendering it entirely immune to subsequent cleavage. Due to this one-way directionality, the donor fragments, Type IIS enzyme, and T4 DNA Ligase can all be mixed simultaneously within a single tube. By cycling the temperature between optimal digestion (37°C) and ligation (16°C) conditions, the reaction equilibrium is driven relentlessly toward the final assembled construct. This brilliant mechanism enables seamless, scarless, and highly efficient multiplexed cloning of dozens of unique DNA fragments in a single reaction step.
Donor Fragment Architecture:
5'-- [BsaI Site] -> (4-bp Overhang A) -> [ Promoter/Gene/Terminator (Functional Genetic Material) ] -> (4-bp Overhang B) -> [BsaI Site] --3'
|
| + BsaI Enzyme Cleavage
v
(4-bp Overhang A) -> [ Target DNA Insert ] -> (4-bp Overhang B) <-- Ready for Scarless Ligation
Model this assembly method with Benchling or Asimov Kernel!
Benchling Simulation & Strategy Documentation: To simulate a functional Golden Gate Assembly, I modelled the insertion of a Green Fluorescent Protein (GFP) reporter gene into a modified pUC19 expression vector backbone using the Type IIS restriction enzyme BsaI.
Finally, I opened Benchling's Assembly Wizard toolbar on the right side of the screen and selected Golden Gate Assembly. I designated the modified pUC19 file as the "Backbone" (selecting the LacZ region) and the GFP file as the "Insert", setting the enzyme parameter to BsaI. The software automatically scanned the sequences, aligned the perfectly matched 4-bp sticky overhang junctions (ATGC and TAAA), and simulated the one-pot digestion-ligation reaction. The tool successfully generated a final, circularized, fully annotated recombinant plasmid map where the GFP gene is seamlessly and scarlessly integrated into the vector frame, ready to drive green fluorescence expression in competent bacterial cells.
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
IANNs are prediction systems trained to ensure that the active genetic circuits have higher outputs and inactive ones have lower outputs (continuous domain outputs); they can be trained also to have boolean output incase the penultimate layer exceeds a specific prediction threshold (for specific reactions), as well as can be trained to give quantitative reaction flux as outputs. Ideally, they should be paired with traditional genetic circuits for enhanced results (quantitative reactions can also be found using traditional Flux Balance Analysis - FBA/FVA/FPA, etc.).
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
IANNs can be also used when the input data is scarce or not fully available. They can also be favoured or act as an additional pipeline within large computational biological pipelines where timing is of the essence and algorithmic suggestions would anyways be validated using wetlab experimentations.The inputs can be continuous molecular signals, such as varying extracellular metabolite concentrations or precise intracellular temperature readings. The network integrates these signals to output suggested genetic interventions (e.g., specific promoter strength combinations) to maximize a target metabolic flux.
A diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4 (Tx: transcription; Tl: translation) is provided. Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
This diagram is retained, and at the X1 position (which is the first input of the original diagram), the output of another perceptron is sent:
Intracellular 2-layer perceptronWe can similarly connect the original X2 input with another perceptron making it a 2-layer 3 perceptron model.
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Mushrooms are fungi; probably some cakes can also be made out of them. There are upcoming climate startups which are designing fungi-based materials that can fix carbon on tiles, the tiles will be fixed on public places or inside/outside of buildings and the genetically modified fungi would do the trick. References:
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Genetically engineer fungal mycelium to express self-healing proteins or secrete natural hydrophobic resins directly into their chitin-glucan cell wall matrix. This would allow the creation of living, water-resistant bio-composites that can autonomously repair structural micro-cracks when exposed to moisture or specific nutrients. Fungi could also be engineered to secrete heavy-metal-binding peptides for the in-situ bioremediation of contaminated soil while simultaneously forming structural blocks.
The advantages of performing synthetic biology in fungi as opposed to bacteria include:
Eukaryotic Expression Machinery: Fungi possess advanced post-translational modification capabilities (like complex glycosylation and proper disulfide bond folding) necessary to express intricate eukaryotic proteins that form inclusion bodies or fail in bacteria.
Hyphal Network Growth: Fungi grow via three-dimensional, interconnected hyphal networks (mycelium) that physically bind loose agricultural byproducts into solid macro-structures, whereas bacteria typically form amorphous biofilms or liquid cultures. (A fungi network is supposed to be the largest living organism in the world).
Extracellular Secretion: Fungi naturally secrete massive quantities of enzymes and proteins directly into their surroundings, significantly simplifying downstream harvesting and processing pipelines compared to lysing bacterial cells.
Assignment Part 3: First DNA Twist Order
Review Part 3: DNA Design Challenge of the week 2 homework. Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above. Document the backbone vector it will be synthesized in on your website.
I will continue this on the benchling link already posted within the week 2 homework section (see the end of my week 2 homework page).
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free systems offer direct access to the reaction environment without a protective, living cell membrane barrier. This allows precise manipulation of chemical variables—such as adjusting pH, tweaking redox potentials, and adding non-canonical amino acids or toxic chemical inhibitors—without killing a host organism.
Case 1: Production of highly toxic proteins (e.g., antimicrobial peptides or cytotoxic enzymes) that would kill living E. coli hosts.
Case 2: Rapid, high-throughput screening of massive variant libraries where transforming and culturing live cells takes too much time.
Case 3: When experimenting on systems that are not all found within a single cell, like testing plant chloroplasts on human skin cells for biohackers (or other ethical sci-fi topics allowing human sensory enhancements, including quantum electromagnetic sensings, which many global presenters talked about).
Describe the main components of a cell-free expression system and explain the role of each component.
Cellular Extract: Crude cytoplasmic lysate (usually from E. coli, wheat germ, or CHO cells) containing ribosomes, aminoacyl-tRNA synthetases, and endogenous translation factors.
DNA Template: Plasmids or linear PCR products encoding the target gene under an appropriate promoter (e.g., T7).
Energy Mix & Substrates: A mixture of dNTPs/NTPs, amino acids, and vital salts (Mg++, K+) to feed transcription and translation.
Energy Regeneration System: High-energy secondary substrates (like phosphoenolpyruvate or creatine phosphate) used to continually replenish ATP supplies.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Protein synthesis is thermodynamically expensive; each peptide bond consumes multiple high-energy phosphate bonds. Without a living metabolic network to generate power, free ATP pools deplete in minutes due to rapid hydrolysis and accumulation of inhibitory inorganic phosphate. To ensure a continuous supply, you can use a PEP-pyruvate kinase system where phosphoenolpyruvate (PEP) acts as a high-energy donor, allowing a recycling enzyme to continuously re-phosphorylate spent ADP back into active ATP.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic systems (like E. coli TX-TL) are high-yield, cheap, and fast, but they lack complex post-translational modifications (PTMs) like glycosylation or proper disulfide bonding. Eukaryotic systems (like HeLa or CHO lysates) are slower and yield less total protein, but they have native endoplasmic reticulum/Golgi machinery for complex folding.
Prokaryotic Choice: Green Fluorescent Protein (GFP), because it requires no glycosylation or complex mammalian chaperone folding, making it ideal for cheap, massive yields in E. coli lysates.
Eukaryotic Choice: Human Erythropoietin (EPO), because it requires intricate sialic acid glycosylation to function biologically, which prokaryotic machinery cannot perform.
Photosynthesis pathway may be mimicked within cell-free systems to allow novel space structures for future exoplanet exploration by evolved humans.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
The Challenge: Membrane proteins are highly hydrophobic and will rapidly misfold, aggregate, and precipitate out of solution when translated into a watery, open extract without a structural lipid bilayer to shield them.
The Solution: Supplement the cell-free reaction matrix with synthetic nanodiscs or detergent micelles (like Triton X-100 or DDM). As the ribosome synthesizes the hydrophobic domains, the proteins can spontaneously insert directly into these soluble lipid structures, maintaining proper folding and activity.
Actually, membranes can also be designed (or generated) such that certain specific nutrients or other solutions are encapsulated within spherical membranes and put inside the cell-free systems. Later, after some time, these spherical membranes can be removed to observe how the cell interacts with the internal components of the membrane, which are designed to mimic the external world. This system may actually invert the topology such that the cell-free system acts like the interior of the cell and the region within the spherical membrane-bound structures is actually made to represent the outside of he cell. (This inverts the inside-out cellular topology!)
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Reason: Rapid depletion of the energy source. Fix: Supplement the reaction with an improved, higher-capacity secondary energy system like creatine phosphate/creatine kinase.
Reason: High endogenous nuclease/protease activity degrading your DNA template or target protein. Fix: Add an RNase inhibitor cocktail or switch to a commercial extract optimized with protease gene knockouts (like a $\Delta rne$ or $\Delta lon/\Delta ompT$ strain).
Reason: Inefficient codon usage slowing down translation. Fix: Perform codon optimization on your template DNA sequence to match the specific tRNA abundance profile of the extract’s origin organism.
Reason: The protein is generated in high quantities but is also utilized very quickly because of high demand from downstream pathways. Fix: Perform pathway analysis using Computational and Systems Biology to block downstream mechanisms allowing the target protein to accumulate…
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
Pick a function and describe it.
a. What would your synthetic cell do? What is the input and what is the output?
b. Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
c. Could this function be realized by genetically modified natural cell?
d. Describe the desired outcome of your synthetic cell operation.
Design all components that would need to be part of your synthetic cell.
a. What would be the membrane made of?
b. What would you encapsulate inside? Enzymes, small molecules.
c. Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promoters, like Tet-ON, you need mammalian)
d. How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
Experimental details
a. List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
b. How will you measure the function of your system?
Design of a Synthetic Minimal Cell (SMC) for Environmental Contaminant Sensing
1. Function and System Logic
Concept: A synthetic minimal cell designed as an environmental diagnostic tool to detect localized contamination of the agricultural pollutant atrazine.
Input/Output: The input is extracellular atrazine (which freely diffuses across the lipid membrane). The output is the molecular release of IPTG into the surrounding environment.
Realization without encapsulation: No, this cannot be accomplished using open cell-free TX-TL alone. Without the physical barrier of a lipid membrane, the signaling molecule (IPTG) would diffuse immediately into the media and trigger downstream reporter systems blindly, eliminating the conditional sensor gating.
Realization via natural cells: Yes, an atrazine-responsive riboswitch could be engineered into live bacteria. However, using an SMC eliminates the risk of genetic drift, mutation, or cellular death caused by environmental selection pressure or toxicity.
Desired Outcome: In the presence of atrazine, the SMC synthesizes a pore-forming protein, allowing internal IPTG to escape and activate nearby engineered E. coli biosensors, transforming them into a bright, visible fluorescent green indicator line.
2. Component Architecture and Communication
Membrane Composition: A structural lipid bilayer composed of POPC (Palmitoyl-oleoyl-phosphatidylcholine) supplemented with cholesterol to control membrane fluidity and minimize unmediated small-molecule leakage.
Encapsulated Material: Crude E. coli TX-TL extract, a baseline pool of free internal IPTG molecules, and an expression plasmid encoding a pore-forming channel controlled by an upstream atrazine-binding RNA riboswitch.
Extract Strain Origin: A bacterial transcription/translation system (E. coli extract) is chosen because prokaryotic riboswitches and standard RNA aptamers integrate seamlessly with bacterial translation machinery.
Environmental Communication: The input molecule (atrazine) is small and hydrophobic enough to pass through the lipid membrane naturally. The output molecule (IPTG) is membrane-impermeable and remains trapped inside until the atrazine triggers the transcription and translation of the membrane channels.
3. Experimental Details and Validation
Lipids and Specific Genes: • Lipids: POPC and Cholesterol. • Genes: The Alpha-hemolysin (aHL) pore gene, fused directly downstream of an engineered atrazine riboswitch aptamer loop sequence. • Reporter Cells: Non-pathogenic E. coli transformed with a simple GFP reporter gene driven by a standard T7 promoter and a Lac operator.
Functional Measurement: The SMCs and reporter bacteria are co-cultured in solution. Following exposure to varying concentrations of atrazine, the activation profile is evaluated by tracking bulk green fluorescence output over time using a P51 Molecular Fluorescence Viewer or a standard laboratory plate reader.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
An interactive smart jacket woven with freeze-dried, cell-free biosensors that changes colour when it detects airborne toxic organophosphate pesticides in agricultural fields.
How will the idea work, in more detail? Write 3-4 sentences or more.
The fabric of the jacket is embedded with freeze-dried E. coli extract, a constitutive transcriptional setup, and a specific butyrylcholinesterase enzyme logic circuit. When an agricultural worker is exposed to pesticide overspray, the ambient moisture or sweat rehydrates the cell-free reagents woven into the fibres. The target organophosphates immediately inhibit the enzyme circuit, stopping a background reaction and causing a visible, safe enzymatic colour change across the sleeve.
What societal challenge or market need will this address?
It addresses acute chemical exposure and long-term poisoning risks faced daily by agricultural labourers worldwide, providing an immediate, low-cost wearable warning system without requiring complex electronic displays.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
The cell-free system is stabilized inside porous paper matrices stitched as modular, disposable patches into the jacket’s shoulders and sleeves. It remains completely inert while dry, activates instantly upon absorbing atmospheric droplets or fluid, and can be cleanly unclipped and replaced with a fresh dry patch after a positive exposure event.
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/.
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Long-duration space travel exposes astronauts to chronic cosmic radiation, driving high rates of DNA double-strand breaks and cellular oxidative stress. Monitoring this progressive molecular damage in real time is a critical challenge for crew safety. Current medical diagnostic platforms require heavy, power-hungry equipment that is infeasible for long missions. Developing zero-footprint, lightweight biological monitoring solutions is essential for tracking astronaut health and testing the effectiveness of spacecraft radiation shielding materials over time.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
The target is the expression of the human p53 protein, a universal cellular biomarker that activates exponentially in response to direct DNA damage and radiation stress.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
The p53 pathway serves as the primary master regulator for DNA damage responses in human cells. By monitoring the real-time transcription and translation of this specific molecular target under microgravity conditions, we can directly gauge the precise kinetic rate and cellular intensity of radiation-induced mutations. This provides an immediate, quantifiable readout of the current onboard radiological threat level to human tissue.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
Hypothesis: A freeze-dried cell-free expression system can be successfully rehydrated aboard the International Space Station (ISS) to accurately quantify radiation damage in blood samples without active cell culture maintenance. Because cell-free systems bypass the metabolic requirements of living organisms, they remain structurally stable during long-term storage in microgravity, providing a reliable, low-resource diagnostic platform.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
Astronaut blood samples will be collected, and their isolated DNA will be mixed with BioBits® freeze-dried extracts containing a customised p53-activated fluorescent aptamer plasmid. Samples will be incubated inside the miniPCR® thermal cycler, and visual output will be captured using the P51 Molecular Fluorescence Viewer. Control reactions will include a pre-flight radiation-damaged positive control and a shielded ground-control setup to isolate space-specific baseline drift.
Homework Part B: Individual Final Project
I submitted my Twist order late, but then realised a few changes were needed! I request that a few twist orders be allowed to process in the next semester for Global Committed Listeners. This will help us a lot as we have refined our individual projects together…
Identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
For my final project investigating how protein synthesis direction changes a protein's structure for identical amino acid sequences, the core physical metrics I need to measure are:
1) Primary Structure Verification: Confirming that both the forward-synthesized protein (Ubiquitin/GB1) and the reverse-synthesized retro-protein contain the identical amino acid sequence but in inverted directions.
2) Tertiary Fold and Structural Topography: Measuring the actual 3D conformation of the retro-GB1 protein to see if it adopts a different stable fold than the highly structured wild-types.
3) Protein Purity and Mass: Verifying the molecular weight of the expressed variants to ensure full-length translation without premature truncation.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
Sequence Orientation & Intact Mass: Measure the exact molecular weight of the intact, full-length retro-protein chain. To confirm the directional sequence, the protein will be broken down into specific peptide fragments.
Higher-Order Structure (Secondary/Tertiary Fold): Measure the atomic-scale spatial coordinates, secondary structures (beta-sheets, alpha-helices), and the overall 3D folded geometry of the crystallized retro-protein.
Environmental/Gravitational Co-translational Dynamics: Monitor structural differences when the proteins are synthesized under normal Earth gravity versus a microgravity environment to isolate convective or gravitational confounding variables during co-translational folding.
Temperature-dependent final state: Monitor the final protein structure if the stable temperature is changed across multiple experimental setups. This will also help understand how the temperature inside bodies can affect protein folding and whether climate change-induced global warming can affect final protein structures (temperature definitely affects the protein structure, but it is necessary to find when this effect starts to cause problems). Temperature-dependent protein structure differences for the same/retro sequence can be different for different proteins and for different homeostatic environments, and therefore, some specific proteins could be worse affected by global warming than others. This will also help identify those proteins which are more thermally stable for humans (and ultimately allow synthesis of the other proteins to evolve into more thermally stable stages) ~ heat-resistant humans!
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS): Use intact mass spectrometry to confirm the exact total mass of the expressed protein variants. Following this, I will perform bottom-up peptide mapping via tryptic digestion and MS/MS sequencing to read the exact primary sequence directionally, verifying the complete inversion of amino acid coordinates.
X-ray Crystallography or Cryo-EM: To rigorously evaluate my core hypothesis that retro-variants fold into different structural topologies, I will use X-ray crystallography (or high-resolution Cryo-EM) to map out the explicit atomic-scale tertiary fold of retro-Ubiquitin. This provides the physical ground truth to compare directly against my low-pLDDT AlphaFold structural predictions.
Space-based Cell-Free Expression Chambers: To test whether gravitational fields act as a confounding force during co-translational synthesis, I will use automated microfluidic cell-free expression systems running in microgravity environments (such as on the ISS) contrasted directly against identical Earth-bound control reactions.
Waters Part III — Peptide Mapping - primary structure
Waters Part IV — Oligomers
Waters Part V — Did I make GFP?
raw lab spectrum data sheets, peak lists, or observed m/z values generated from the Waters Immerse Lab equipment is necessary to calculate exact experimental molecular weights, isotopic charge states, and parts-per-million (ppm) mass errors! I did not attend any labs being a Global Committed Listener! Imp. Note to self: Check all the related videos and see if you can actually ask for a grant to perform these experiements within IIT Kharagpur’s Central Research facility, or other places near Kolkata. Also check YoutTube tutorials and pages of fellow students who were able to complete this…
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
I made a long-necked tall green dinosaur with its bottom half in the fourth quadrant and its long neck and head stretching into the first quadrant; someone else (should be nicknamed Asteroid) changed those pixels and made the chaotic drawing more symmetric and distinguishing quadrants (not allowing any common information flow across the different drawing quadrants).
Instead of allowing everyone to change all pixels with a time-gated manner, I think everyone should be allowed to have their own full canvas to make the full design. Then others will be able to vote on which of those drawings could be integrated into the main canvas. Like say my dino could be 10*10 pixels and if selected it could be placed somewhere in the main canvas; or everyone’s canvas could be superimposed, or just integrate individual tiles for a mega picture!!!
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Component Roles in the Cell-Free Reaction
E. coli Lysate / BL21 (DE3) Star Lysate (includes T7 RNA Polymerase)
This is the core biological engine of the reaction. It provides the heavy machinery (like ribosomes) to read the genetic code and build proteins, while the T7 polymerase specifically transcribes the DNA template into RNA.
This acts as the environmental stabilizer. It maintains a steady pH so the machinery doesn’t degrade, while the salt ions (especially magnesium) serve as essential chemical helpers that allow the ribosomes to clip together correctly.
Energy / Nucleotide System (Ribose, Glucose, AMP, CMP, GMP, UMP, Guanine)
This functions as the fuel station and letter pool. The nucleotides (AMP, CMP, GMP, UMP) are the raw alphabet letters used to build RNA, while sugars like glucose and ribose are broken down to keep charging up the system’s battery.
These are the physical construction bricks. They represent the full set of 20 standard amino acids that the ribosome strings together to assemble the final physical protein chain.
Additives (Nicotinamide)
This is a metabolic booster. It helps protect and stabilize key energy-carrying helper molecules in the mix, ensuring the reaction keeps running smoothly without stalling out.
Backfill (Nuclease Free Water)
This is the clean liquid filler. It brings the entire mixture up to the exact required operational volume without introducing any dirty enzymes that might accidentally chew up the DNA or RNA.
PEP-NTP vs. NMP-Ribose-Glucose Master Mixes
The 1-hour optimized PEP-NTP mix uses a high-energy “rocket fuel” molecule (PEP) and pre-made premium batteries (NTPs) to create an explosive burst of protein production that burns out very quickly. In contrast, the 20-hour NMP-Ribose-Glucose mix relies on cheap, raw starter ingredients (NMPs and sugars) that force the lysate to slowly build its own energy network from scratch, resulting in a much longer, sustained marathon reaction.
Bonus Question: Transcription without GMP but with Guanine
Even though the pre-made nucleotide letter GMP is missing, the biological engine in the lysate can use a built-in salvage pathway to grab the raw base Guanine and chemically stick a ribose sugar and phosphate onto it. This transforms the plain Guanine into a fully functional nucleotide building block, allowing transcription to proceed normally.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Biophysical and Functional Properties of the 6 Fluorescent Proteins
sfGFP (Superfolder GFP)
This protein has incredibly robust, fast-folding kinetics. It folds so efficiently that it resists misfolding and aggregates much less than standard proteins, giving a very bright and reliable readout quickly.
mRFP1 (Monomeric Red Fluorescent Protein 1)
This protein suffers from relatively slow chemical maturation times and a lower quantum yield (innate brightness). Because it takes a long time for the internal chemical “lightbulb” to finish forming, its red signal can be quite faint early on.
mKO2 (Monomeric Kusabira Orange 2)
This orange protein is highly sensitive to the pH of its environment. If the reaction mixture becomes even slightly too acidic during incubation, its fluorescence output drops off sharply.
mTurquoise2
This cyan reporter has a phenomenal quantum yield (it shines very brightly once folded), but it has complex maturation kinetics. It requires a steady supply of molecular oxygen to successfully run the internal chemical reaction that activates its fluorescence.
mScarlet_I
This is a cutting-edge red protein with superior brightness, but it is notoriously prone to photobleaching (fading under continuous light exposure). It needs to be kept in a stable, well-supported environment so its delicate structure doesn’t wear out over time.
Electra2
This variant is explicitly engineered for rapid translation and immediate readout. However, its fast-paced assembly makes it highly dependent on a massive, uninterrupted initial pool of raw resources, so it doesn’t stall out.
Master Mix Optimization Hypothesis
Hypothesis: To maximize mRFP1 red fluorescence over a long 36-hour marathon incubation, we should supplement the custom reagent slots with an enhanced Energy Regeneration System consisting of Creatine Phosphate and Creatine Kinase (alongside a mild increase in HEPES-KOH buffer concentration).
Expected Effect: Because mRFP1 is a slow-maturing protein, a standard system will run out of batteries before the protein has time to chemically ignite its lightbulb. The custom energy supplement will continuously replenish spent ADP with ATP over the long haul, while the extra buffer prevents the system from becoming acidic, providing the sluggish mRFP1 chains with a stable, high-energy environment they need to fully mature and reach maximum brightness.
Initially my idea was to try and connect two-ends of a chromosome (the telomeres) so that the phenomenon of telomere attrition does not happen during cell-divisions; I felt this was a good way to tackle one of the hallmarks of ageing. However, since humans have linear DNA, the problem of catenane formation is generally not encountered; due to this humans dont have the DNA topoisomerase IV (Topo IV) which helps separate the catenated chromosomes, by breaking DNA strands of one chromosome and allowing the other chromosome to pass through the break. (Thus, the replication of multiple circular DNA will have this issue of one circle passing through the other and if these circular chromosomes are being pulled to different daughter cells, then the Topo IV activity will be a must.)
Initially my idea was to try and connect two-ends of a chromosome (the telomeres) so that the phenomenon of telomere attrition does not happen during cell-divisions; I felt this was a good way to tackle one of the hallmarks of ageing. However, since humans have linear DNA, the problem of catenane formation is generally not encountered; due to this humans dont have the DNA topoisomerase IV (Topo IV) which helps separate the catenated chromosomes, by breaking DNA strands of one chromosome and allowing the other chromosome to pass through the break. (Thus, the replication of multiple circular DNA will have this issue of one circle passing through the other and if these circular chromosomes are being pulled to different daughter cells, then the Topo IV activity will be a must.)
The general rule [1] seems that circular chromosome organisms need dedicated decatenation machinery per chromosome: Human cells don’t have this machinery at scale, which is the fundamental problem for the circularization-idea (alongwith the additional problems of what other mutations creep in due to the Topo IV activity of breaking and making the double-stranded DNA).
This idea is thus discarded!
Protein Folding Direction for the same AA Sequence
I am therefore pivoting to another idea related to protein foldings and improving AlphaFold accuracy, where I wish to understand: “How does the protein synthesis direction change the folded protein’s structure and function for the same AA-sequence?”
I have a hunch that the same AminoAcid Sequence when synthesized in the opposite direction can have drastically different folds. This is due to the fact that the rate of protein synthesis is slower than the rate of protein folding, and so the protein does not wait to be folded after it has been synthesized entirely but keeps folding on the go. Some pionees in the field suggested that these could be mirror images of the protein structure that is generated by the forward directional synthesis (Claude also thinks this is possible for some proteins, although it says that the opposite coils are not found in nature; hope I am not getting into mirror life intricacies and safety issues through this). Thus, for this experiment, I wish to find the structure of a well-known GB1 protein and its retro (reversed version), and compare these folding predictions with Alphafold’s predictions.
Final UPDATE (before website freeze): Please find my final presented slides here
Do connect if you are interested to collaborate on retro-protein foldings or global-warming induced changes in internal homeostatic/thermal landscapes leading to changes in the protein folds and potentially in the final structure.
The question to ask here is whats the maximum temperature exposure when internally there will be a lot of protein folding errors within the organism for it to sustain itself…
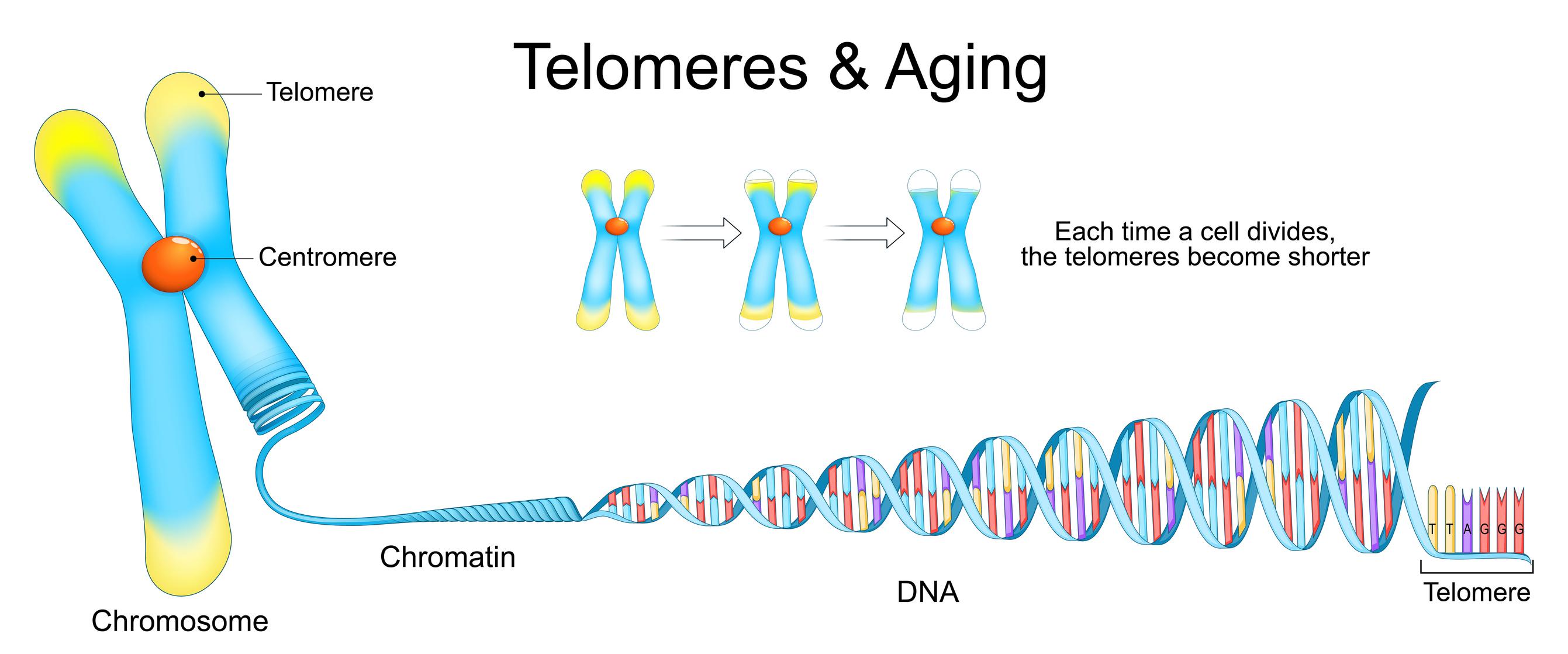
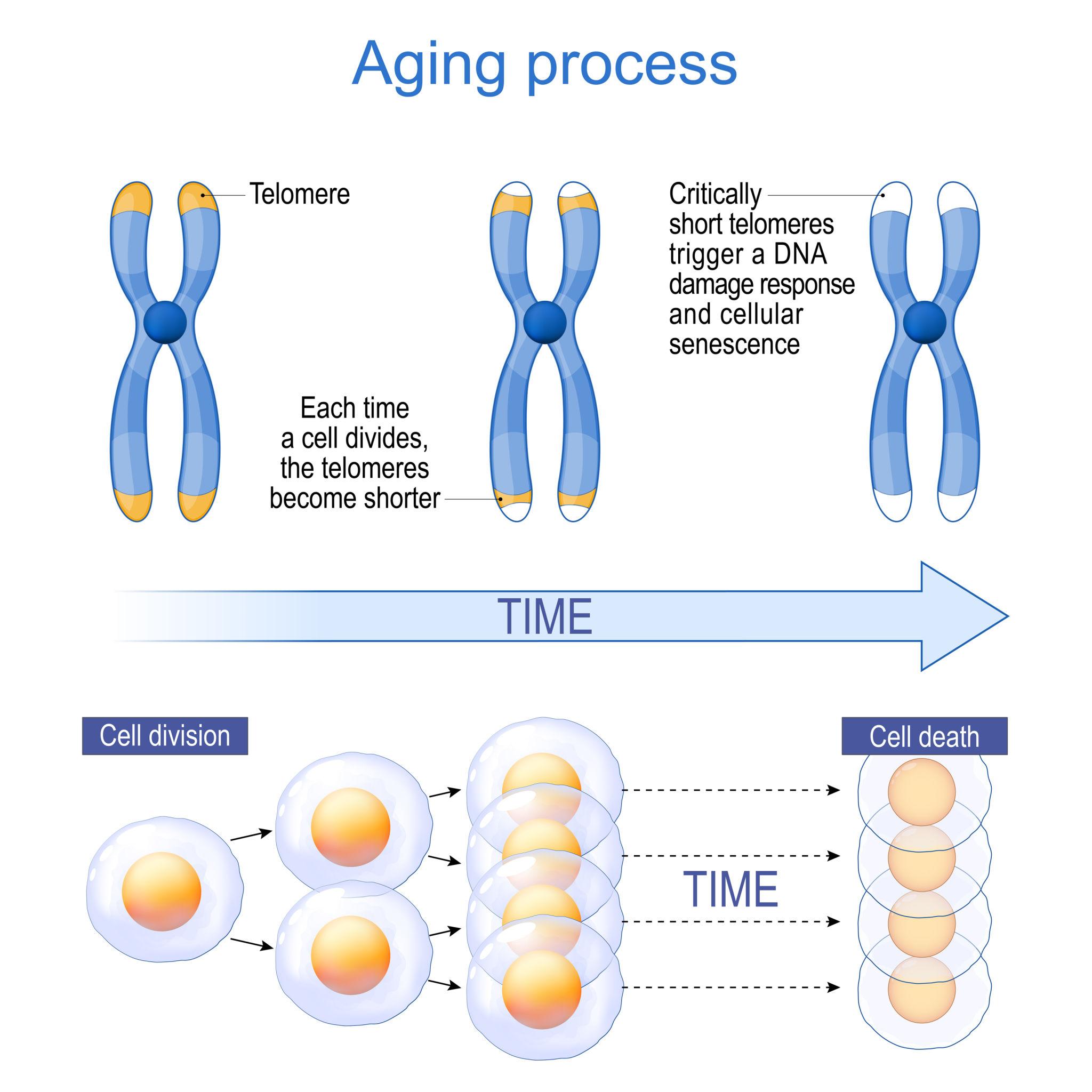
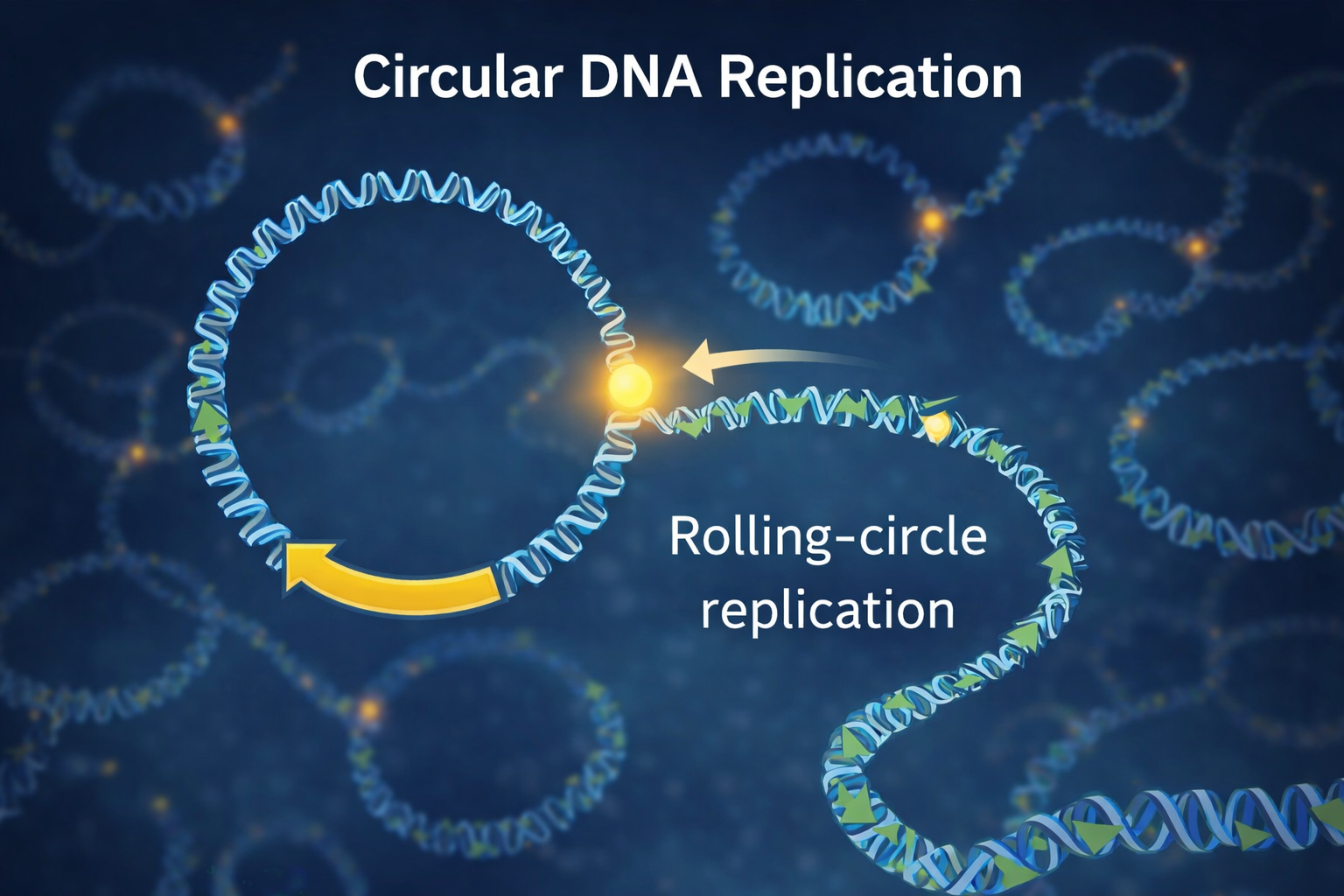


































{kind=link}