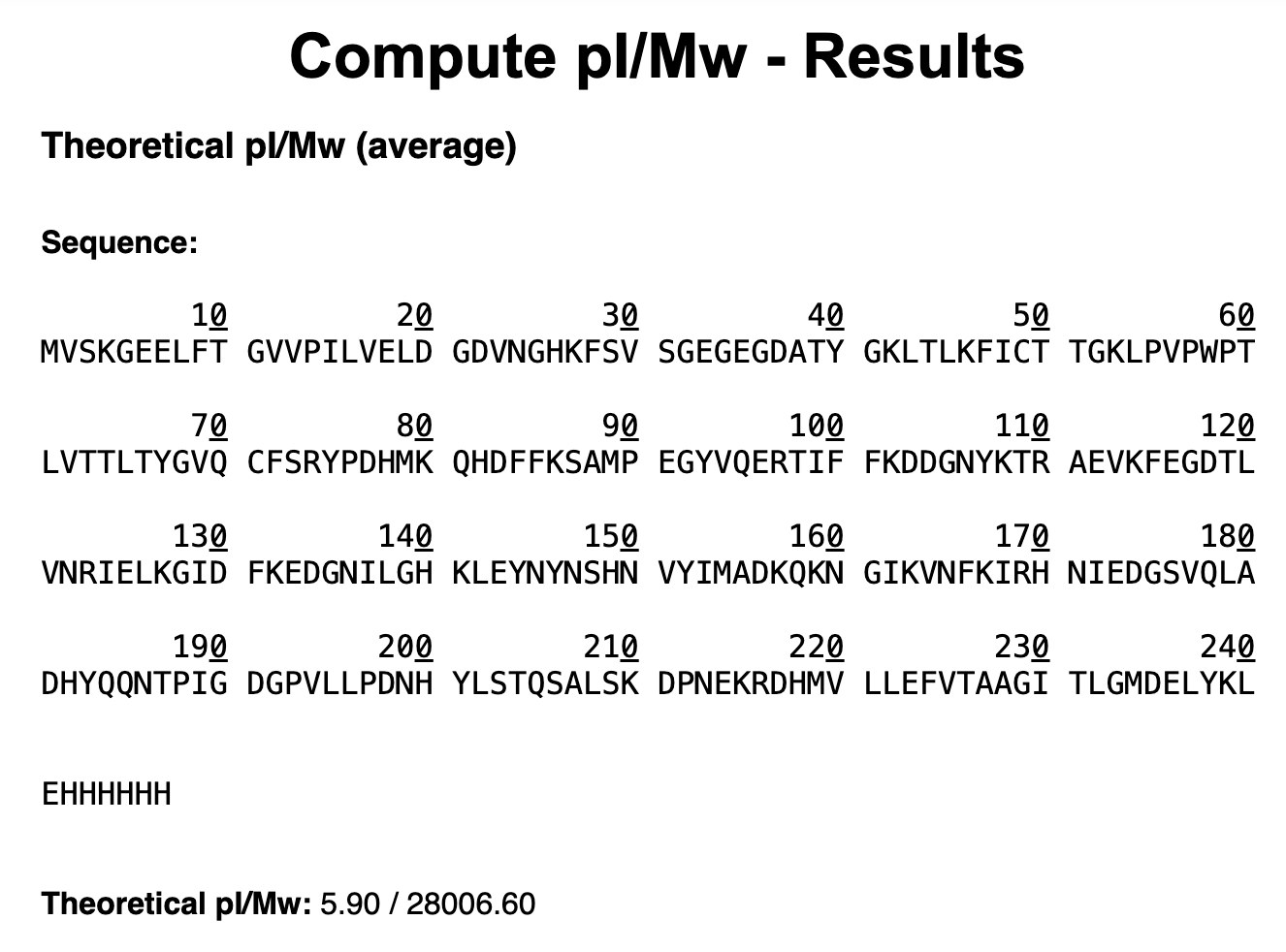
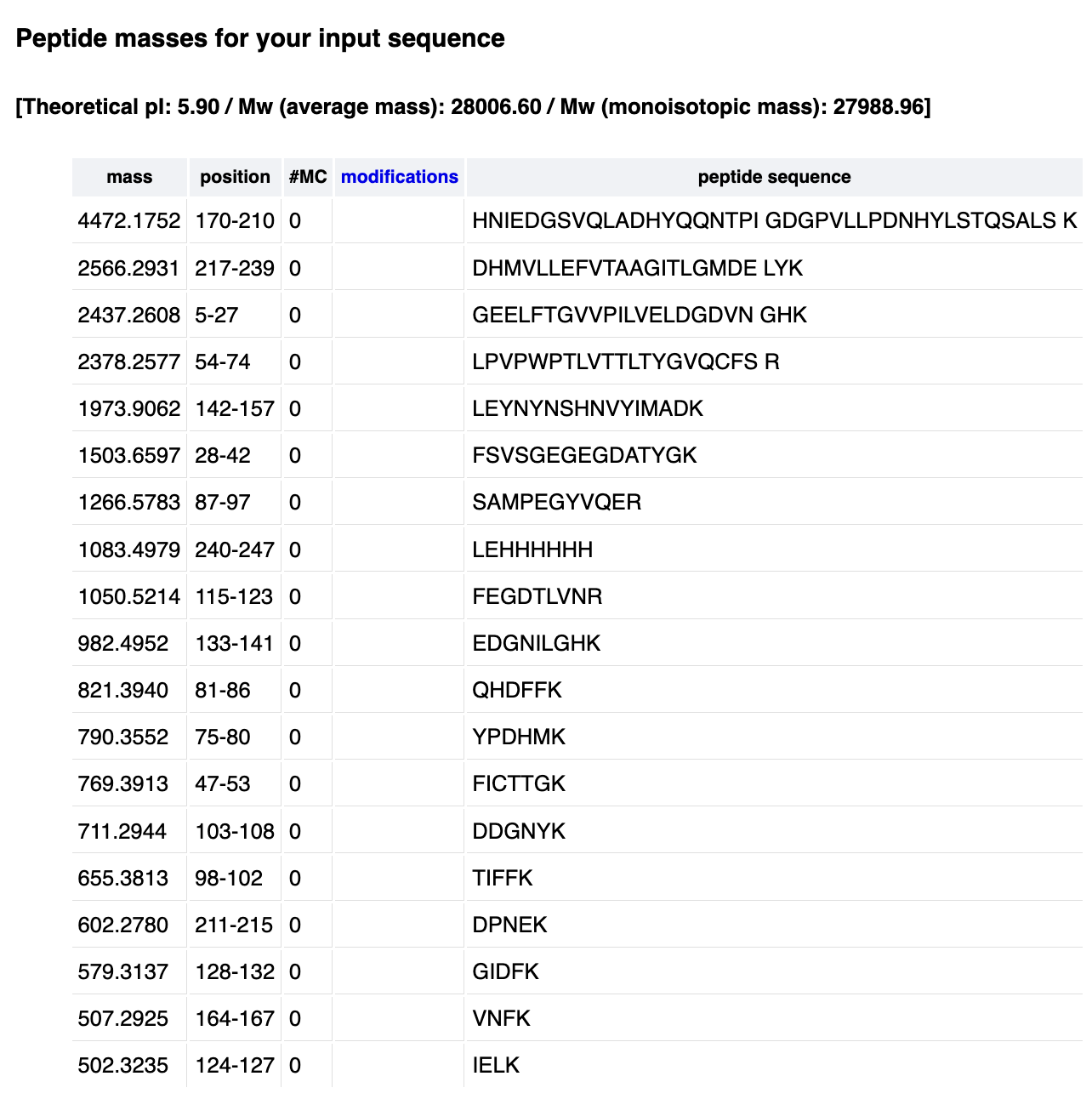
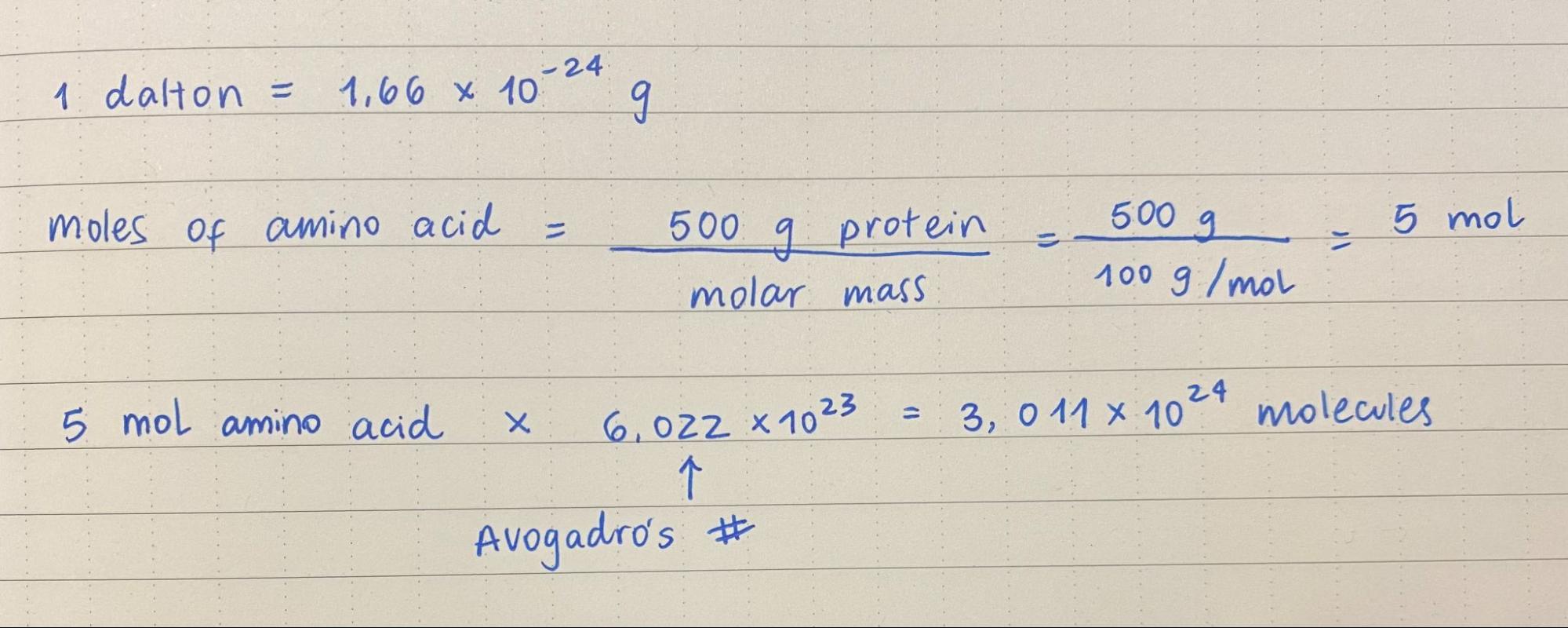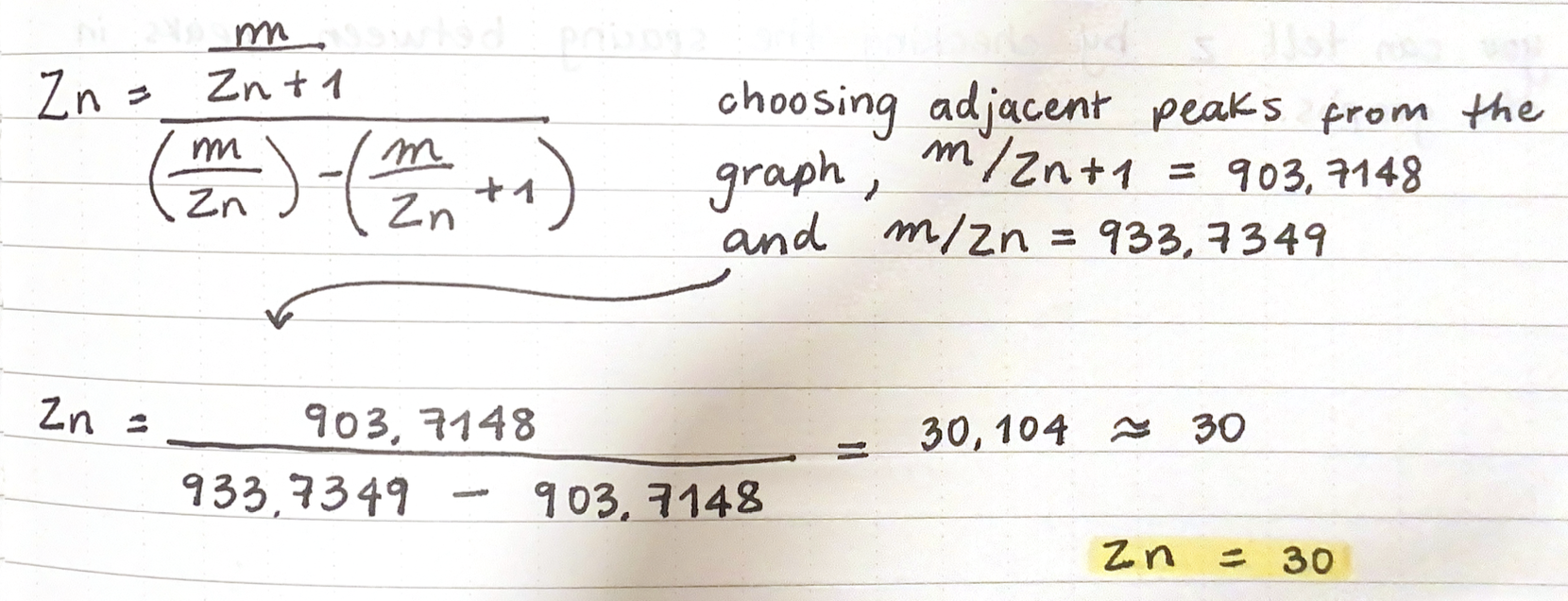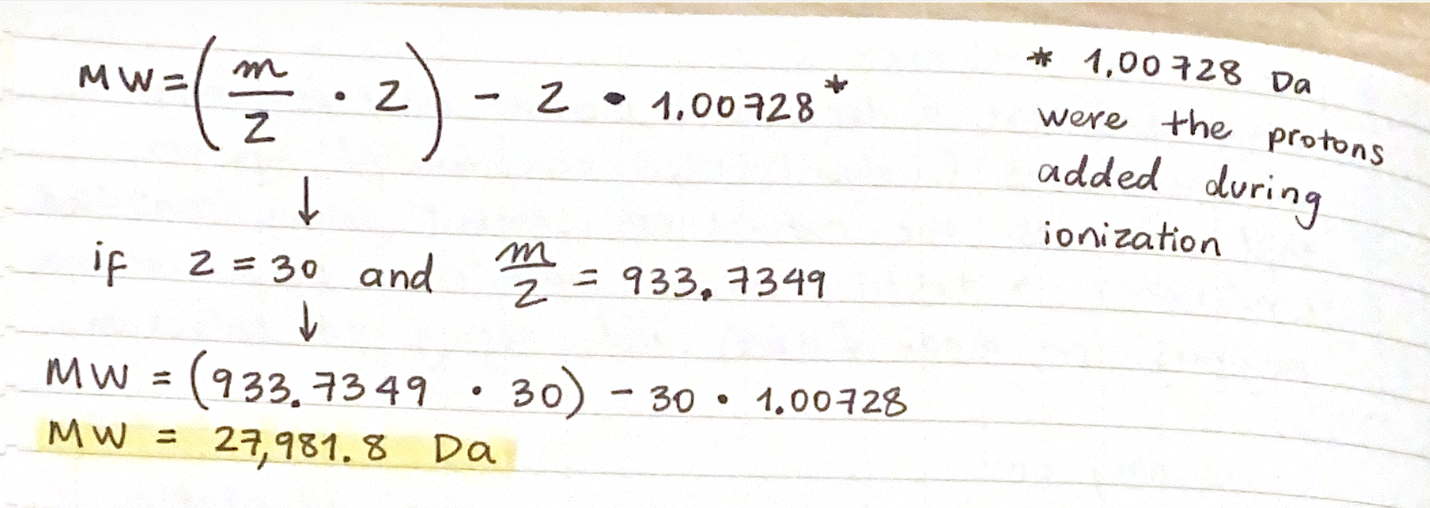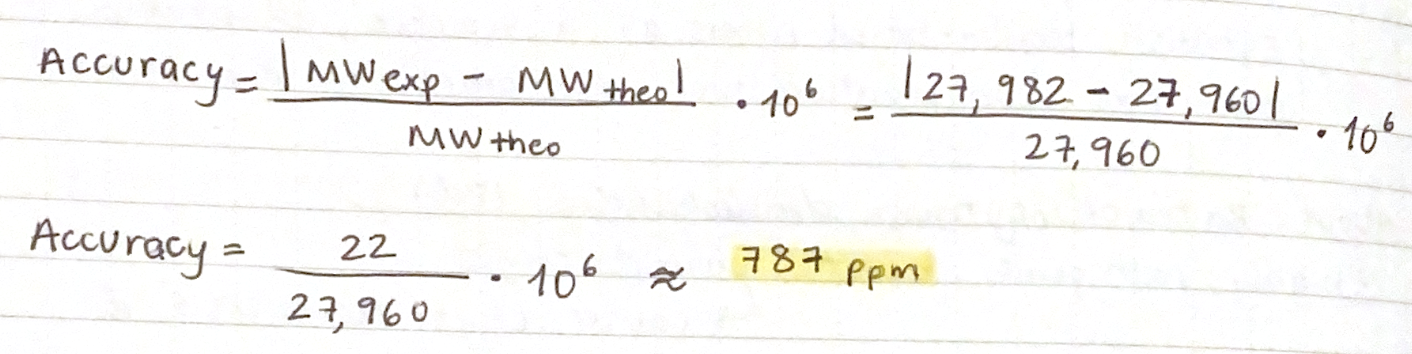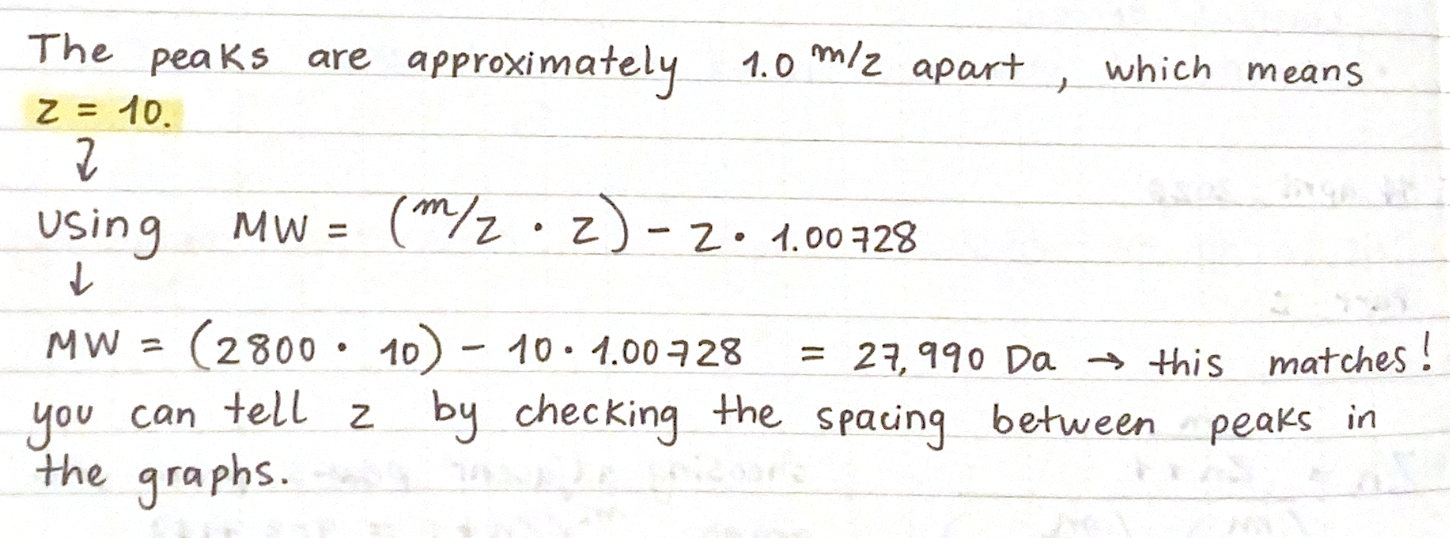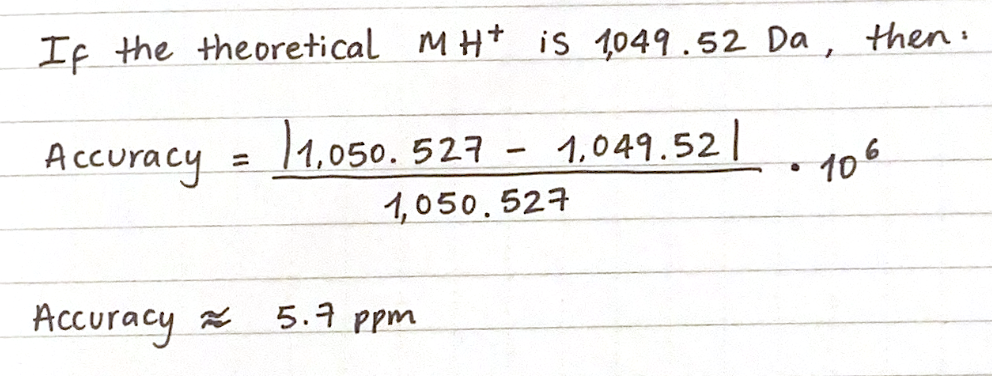First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
As a Biologist, I have a macro-scale perspective on life, from organisms to ecosystems to planetary systems, and have always been drawn to technological innovations. However, I am now curious about the fundamental question of what constitutes life at a micro-scale, and what does engineering its core principles entail. Still interested in biocomputational methods, I want to learn more about the intersection of bio-artificial intelligence and synthetic biology.
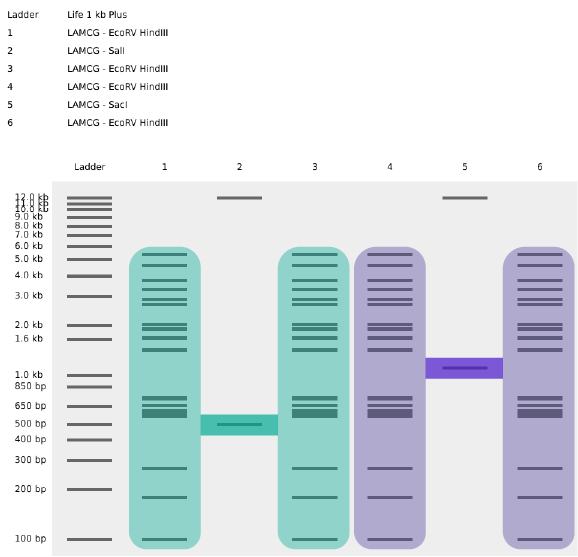
Part 1: Benchling & In-silico Gel Art My original idea was to create two sister chromatids, since most of the patterns from the Enzymes were scattered vertical lines, and they kind of looked like alleles inside a chromosome. I had some trouble creating the centromere of the chromosome because none of the enzymes alone created just one line in the middle of the ladder (so around 800 bp), so I picked SacI and SalI and ignored the top line at 12.0 kb.
Part A. Conceptual Questions 1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Part A: SOD1 Binder Peptide Design Part 1: Generate Binders with PepMLM To get the human SOD1 sequence, I went to UniProt. The ID for this protein is P00441 and the sequence is the following:
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Now, if the mutation is A4V, that means that in position 4 there’s a change from alanine to valine. The mutated sequence is then the following:
Assignment: DNA Assembly 1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion High-Fidelity PCR Master Mix contains all the core reagents necessary for accurate and efficient DNA amplification:
Phusion DNA Polymerase: A high-fidelity enzyme with 3′→5′ exonuclease proofreading activity that minimizes errors during DNA synthesis, especially important for mutation-based cloning (NEB, 2023). dNTPs (deoxynucleotide triphosphates): Provide the nucleotide building blocks (A, T, G, C) for DNA strand elongation. Reaction Buffer (with Mg²⁺): Maintains the ionic strength and conditions needed for optimal enzyme activity and DNA strand stability. Stabilizers & enhancers: Help maintain enzyme performance across temperature ranges and buffer pH changes during thermocycling. (New England Biolabs (NEB), 2023).
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) 1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Characteristic Intracellular Artificial Neural Networks Traditional Genetic Circuits (that use Boolean functions) Input-output mapping Continuous logic that can sum multiple inputs with determined importance or “weights”. This allows for classification of complex patterns. Discrete simple logic (AND, OR, NAND) with ON/OFF behaviors. Vulnerability to noise Since they rely on graded responses, they can average across inputs. This makes them less vulnerable to change output when exposed to noise. Sensitive to noise around thresholds. If there are small fluctuations the ON/OFF gate can be flipped. Decision-making They classify inputs into categories at once and produce signals to different “effector modules” (also called “winner-take-all decisions” in mammalian cells, as mentioned in Chen et al., 2024). This also allows for higher adaptive behavior. They often produce a single binary output per circuit. This makes them less adaptable. Table created using information taken from:
Homework Part A: General and Lecturer-Specific Questions General homework questions 1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Final Project Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc. Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements. What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork 1) Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST. 2) Make a note on your HTGAA webpages including: what you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”); what you liked about the project; and what about this collaborative art experiment could be made better for next year.
Subsections of Homework
Week 1 HW: Principles and Practices
First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
As a Biologist, I have a macro-scale perspective on life, from organisms to ecosystems to planetary systems, and have always been drawn to technological innovations. However, I am now curious about the fundamental question of what constitutes life at a micro-scale, and what does engineering its core principles entail. Still interested in biocomputational methods, I want to learn more about the intersection of bio-artificial intelligence and synthetic biology.
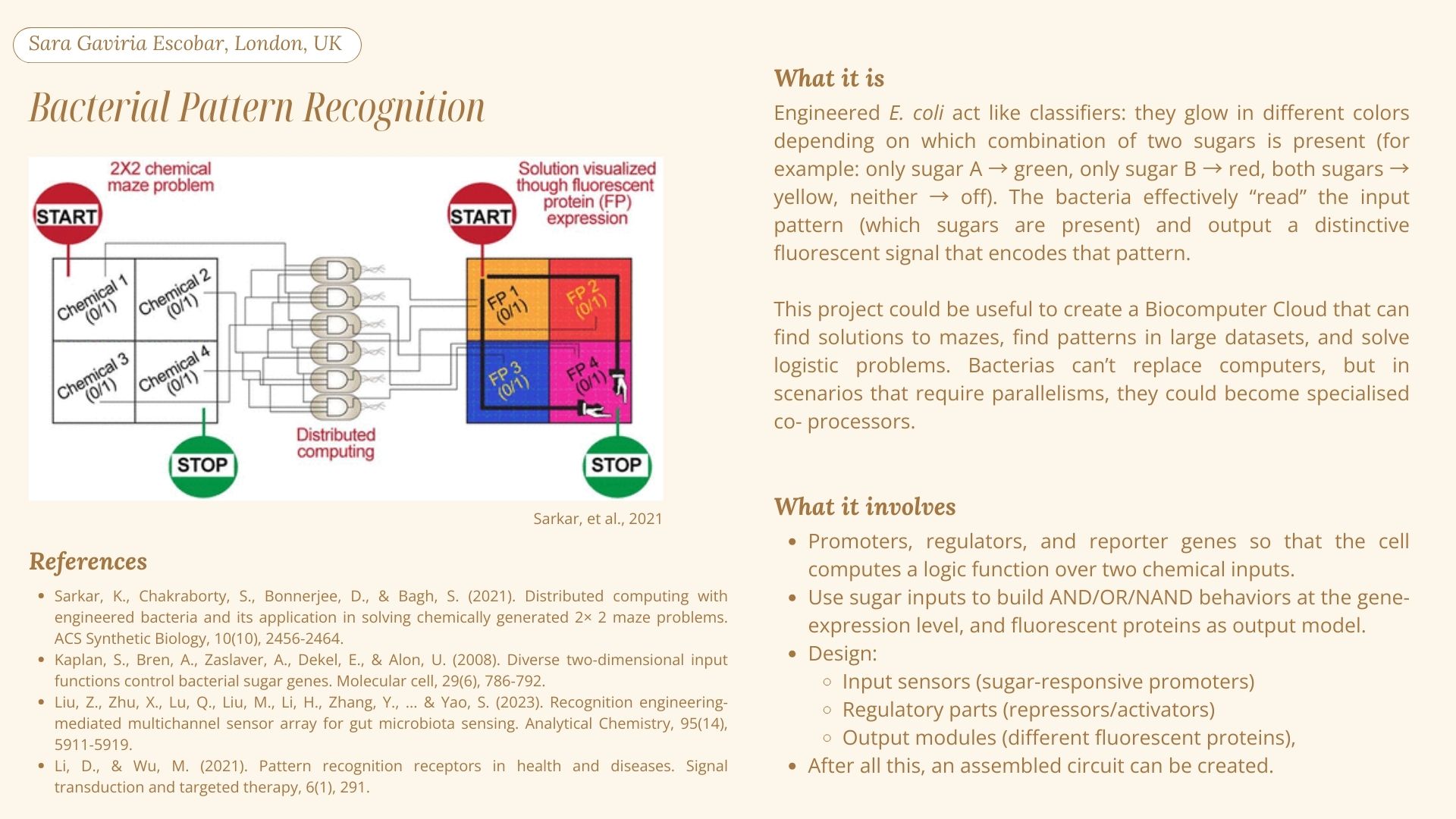
My initial research led me to concepts such as distributed computing, logic gates and perceptron-based learning algorithms. Then, I first encountered the term “biocomputer”, which I understand is analysing how living systems perform computation functions, and in some cases the living systems are used to perform those functions as well. In the research paper by Sarkar et al. (2021) titled “Engineered Bacteria Computationally Solve Chemically Generated 2X2 Maze Problems”, the authors programmed E.coli with genetic circuits to solve maze problems within a chemical mixture introduced inside the tubes where the bacterias were incubated in (Siobhan Roberts, 2021). They observed that the bacteria were able to solve the maze problems by analysing different maze configurations.
Inspired by this and other similar research, I would like to further explore the problem-solving capacities of other microorganisms. I am curious to see if similar genetic programming can be applied to other microbial species to solve maze problems and hopefully translate these results in a way that helps us understand new ways to optimize human-made machines.
I am excited to learn more about this in my HTGAA journey, especially knowing that Neuromorphic circuits/computing is part of the course’s curriculum. If I find new topics that spark my interest, I will add them to the list below:
Biocomputers, logic gates, learning algorithms
Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
For an “ethical” future in relation to biocomputer and bio-artificial intelligence research, I propose three main principles:
Non-malfeasance ✮
Safety ⚘
Respect ☀
Transparency ✿
I propose the following goals, encompassed within one or more of the main principles mentioned previously:
A. Prevent creation/release of harmful organisms ⚘:
When collaborating or working with living microorganisms, researchers should always avoid creating and/or releasing pathogenic organisms. This involves a thorough previous investigation on the particular species’ characteristics and potential risks of it being engineered and exposed to different lab procedures.
B. Minimize harm and resource use in experimentation ✮ ☀:
Firstly, researchers should aspire to always minimize harm to all living organisms when working with them inside and out of the lab. Additionally, they should also avoid using more resources than they need, this requires a well thought out initial plan and constant readjustments of materials, time and procedures throughout the experimental portion of the research.
C. Ensure accurate public and scientific understanding ✿:
Science has to be more democratized, especially when it is cutting-edge innovations like synthetic biology. I believe a way of doing so is by open communication with the general public using accessible friendly language.
D. Promote constructive applications of the technology ✮ ✿:
True innovation should inspire applications that are ethical, fair, and beneficial for both human and more-than-human life. Achieving this requires active collaboration among diverse groups and expertise. By integrating diverse perspectives, we can better study expectations and needs, hopefully creating shared, mutualistic goals for our collective future.
Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”). Try to outline a mix of actions (e.g. a new requirement/rule, incentive, or technical strategy) pursued by different “actors” (e.g. academic researchers, companies, federal regulators, law enforcement, etc). Draw upon your existing knowledge and a little additional digging, and feel free to use analogies to other domains (e.g. 3D printing, drones, financial systems, etc.).
Purpose: What is done now and what changes are you proposing?
Design: What is needed to make it “work”? (including the actor(s) involved - who must opt-in, fund, approve, or implement, etc)
Assumptions: What could you have wrong (incorrect assumptions, uncertainties)?
Risks of Failure & “Success”: How might this fail, including any unintended consequences of the “success” of your proposed actions?
Previous risk analysis: the projects should be reviewed and approved by an Institutional Biosafety Committee (IBC) (Institutional Biosafety Committee, n.d.) and/or an established Ethics Review Board after presenting a thorough risk assessment of the chosen organism and genetic programming for biocomputational research.
Establishing welfare margins: there could be an international guideline created by a wide community of academics from science to ethics where there is an established welfare margin for microbial stress in experimental designs to minimize demonstrable harm without scientific necessity. These guidelines would be based on known and measurable physiological indicators, and would help promote a duty of care for all living systems, including microorganisms.
(I recognize this can be considered unnecessary as it is ambitious and could involve an almost philosophic discussion on the care for microorganisms in scientific research. However, I feel that as researchers we should prioritize not generating stress and/or pain to any living organism.)
Bioethics compliance: as a condition for publication, scientific journals should require a statement/certificate of ethical review by the researcher team and an established Ethics Review Board. This certificate states that the research methods are compliant with international bioethical laws and guidelines (such as the Universal Declaration on Bioethics and Human Rights or Oviedo Convention in Europe) (Fondation Brocher, 2023). Peer reviewers are also encouraged to revise and comment on the bioethical approaches of the experimental procedures.
Research efficiency and sustainability standards: Synthetic biology labs (and all research institutions in general) should focus on research efficiency and establishing sustainability standards. I propose a series of documents that would provide a skeleton for periodic resource efficiency check-ins during lab meetings. To motivate research teams to adhere to this strategy, institutions could create an annual recognition for research teams that demonstrate a responsible use of resources and waste while maintaining rigorous science. Also, being awarded previously could increase the chances of acquiring further funding for the research.
Public engagement and education: A portion of research funding must be used for the researchers to actively engage with the public using (or teaming up with) scientific communication initiatives (public forums, workshops, interactive talks, etc.), explaining the key takeaways from their research and the limits of biocomputation to avoid sensationalism or misinterpretation.
Key actors summary:
Research team
Institutional Biosafety Committee (IBC)
Ethics Review Board
Scientific journal
Peer reviewers
Funding agencies
The general public
Scientific communicators
Institutions
Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Governance actions are scored 1 (least effective) to 3 (most effective).
Governance Action
Prevent creation/release of harmful organisms ⚘
Minimize harm and resource use in experimentation ✮ ☀
Ensure accurate public and scientific understanding ✿
Promote constructive applications of the technology ✮ ✿
Previous risk analysis
3
1
2
3
Establishing welfare margins
1
3
1
2
Bioethics compliance
1
3
1
3
Research efficiency and sustainability standards
2
3
1
2
Public engagement and education
1
1
3
3
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties. For this, you can choose one or more relevant audiences for your recommendation, which could range from the very local (e.g. to MIT leadership or Cambridge Mayoral Office) to the national (e.g. to President Biden or the head of a Federal Agency) to the international (e.g. to the United Nations Office of the Secretary-General, or the leadership of a multinational firm or industry consortia). These could also be one of the “actor” groups in your matrix.
Based on the scoring matrix, my top priorities are: the previous risk analysis, the bioethics compliance and the public engagement and education. I believe these three address the most critical breaking points. Risk analysis is non-negotiable because it prevents harmful microorganisms from spreading and endangering other living forms; bioethics compliance legitimizes research and promotes duty of care for all living organisms; and public engagement and education helps build public trust and accurate understanding necessary for the field’s long-term survival. The other governance options, on the other hand, while important, are not critical. They should be encouraged as best practices as they address less immediate risks.
Reflecting on what you learned and did in class this week, outline any ethical concerns that arose, especially any that were new to you. Then propose any governance actions you think might be appropriate to address those issues. This should be included on your class page for this week.
This week’s discussion on a collaborative bio-future and the role of trust was interesting. I agree trust is essential for ethical progress, but it raised a practical concern for me: I realized I don’t fully understand the current, specific mechanisms and laws for it. I wonder what specific laws, committees, and step-by-step procedures actually check research ethics today? To address this knowledge gap I think there should be more scientific communication around this. It would be a road to strengthen trust and general understanding of ethics as a key priority for scientific research.
Sarkar, K., Bonnerjee, D., & Bagh, S. (2021). Engineered Bacteria Computationally Solve Chemically Generated 2X2 Maze Problems. Homi Bhabha National Institute (HBNI). https://doi.org/10.1101/2021.06.16.448778
Week 2 Lecture Prep
Homework Questions from Professor Jacobson:
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
According to Albertson & Preston (2006), the estimation for errors that error-prone DNA polymerase is once every 104–105 nucleotides polymerized, it can be lower for polymerases that have proofreading activity and can correct mistakes. For example, “twelve of the 15 known human DNA polymerases have no proofreading activity and are error-prone” (Albertson & Preston, 2006). Compared to the human genome, which is 3.2 billion base pairs long, an error-prone polymerase would make approximately 32,000 errors per cell division. However, there are ways to correct mistakes and significantly lower this statistic: error correcting polymerases, mismatch repair, recombination repair, or double-strand break repair (Dav University, n.d.).
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
In double-stranded DNA, there are six possible reading frames: three reading from the top strand, and three reading from the bottom strand. However, just one of the six frames is used to code for a protein, the rest of them do not work because a start codon is necessary to define the frame, and the ribosome binds specifically to the correct initiation site, determining that reading frame for the gene.
Homework Questions from Dr. LeProust:
What’s the most commonly used method for oligo synthesis currently?
Phosphoramidite synthesis
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Chemical synthesis methods, including the phosphoramidite process, cannot reliably produce oligonucleotides longer than 200 nucleotides. This limitation is due to accumulating errors with each synthetic cycle (Hoose et al. 2023, cited in Yin et al., 2024).
Why can’t you make a 2000bp gene via direct oligo synthesis?
This is because the length is superior to the 200nt that can be reliably created during phosphoramidite synthesis. So to achieve the 2000bp gene, you would have to do multiple rounds of smaller oligos and then stitch them together.
Homework Question from George Church:
Choose ONE of the following three questions to answer; and please cite AI prompts or paper citations used, if any.
What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The main 10 aminoacids are: Arginine, Isoleucine, lysine, Methionine, Phenylalanine, Histidine, Leucine, Threonine, Tryptophan and Valine.
Now, according to the Jurassic Park Wiki, the Lysine Contingency “is intended to prevent the spread of the animals in case they ever got off the island. Dr. Wu inserted a gene that creates a single faulty enzyme in protein metabolism. The animals can’t manufacture the amino acid lysine. Unless they’re continually supplied with lysine by us, they’ll slip into a coma and die”
It seems logical, because it is a second barrier of security in case the animals escape off the island. However, it is mostly a flawed hypothesis. Lysine is already an essential amino acid for all animals, meaning it must be obtained through diet, not synthesized internally. The dinosaurs would have needed to consume lysine-rich foods (meat, legumes, etc.) regardless of their engineering. So in the case of the dinosaurs escaping, other animals or plants would provide them with the necessary lysine, allowing them to survive. Although, maybe another hypothesis could be that the genetic modification may have created an exaggerated dependency on lysine, requiring amounts far greater than any natural diet could provide. In this scenario, Dr. Wu could have supplied a specially concentrated lysine supplement on the island to meet this particular need. If they escaped, even consuming lysine-rich foods in the wild would fail to meet their requirement, which would be a more clever (yet still very science-fiction oriented) option.
References:
Albertson, T. M., & Preston, B. D. (2006). DNA Replication Fidelity: proofreading in Trans. Current Biology, 16(6), R209–R211. https://doi.org/10.1016/j.cub.2006.02.031
Hoose A. Vellacott R. Storch M. Freemont P. S. Ryadnov M. G. DNA synthesis technologies to close the gene writing gap. Nat. Rev. Chem. 2023;7:144–161. doi: 10.1038/s41570-022-00456-9. https://dx.doi.org/10.1038/s41570-022-00456-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Yin Y, Arneson R, Yuan Y, Fang S. Long oligos: direct chemical synthesis of genes with up to 1728 nucleotides. Chem Sci. 2024 Dec 18;16(4):1966-1973. doi: 10.1039/d4sc06958g. PMID: 39759933; PMCID: PMC11694485.
Week 2 HW: DNA Read, Write, & Edit
Part 1: Benchling & In-silico Gel Art
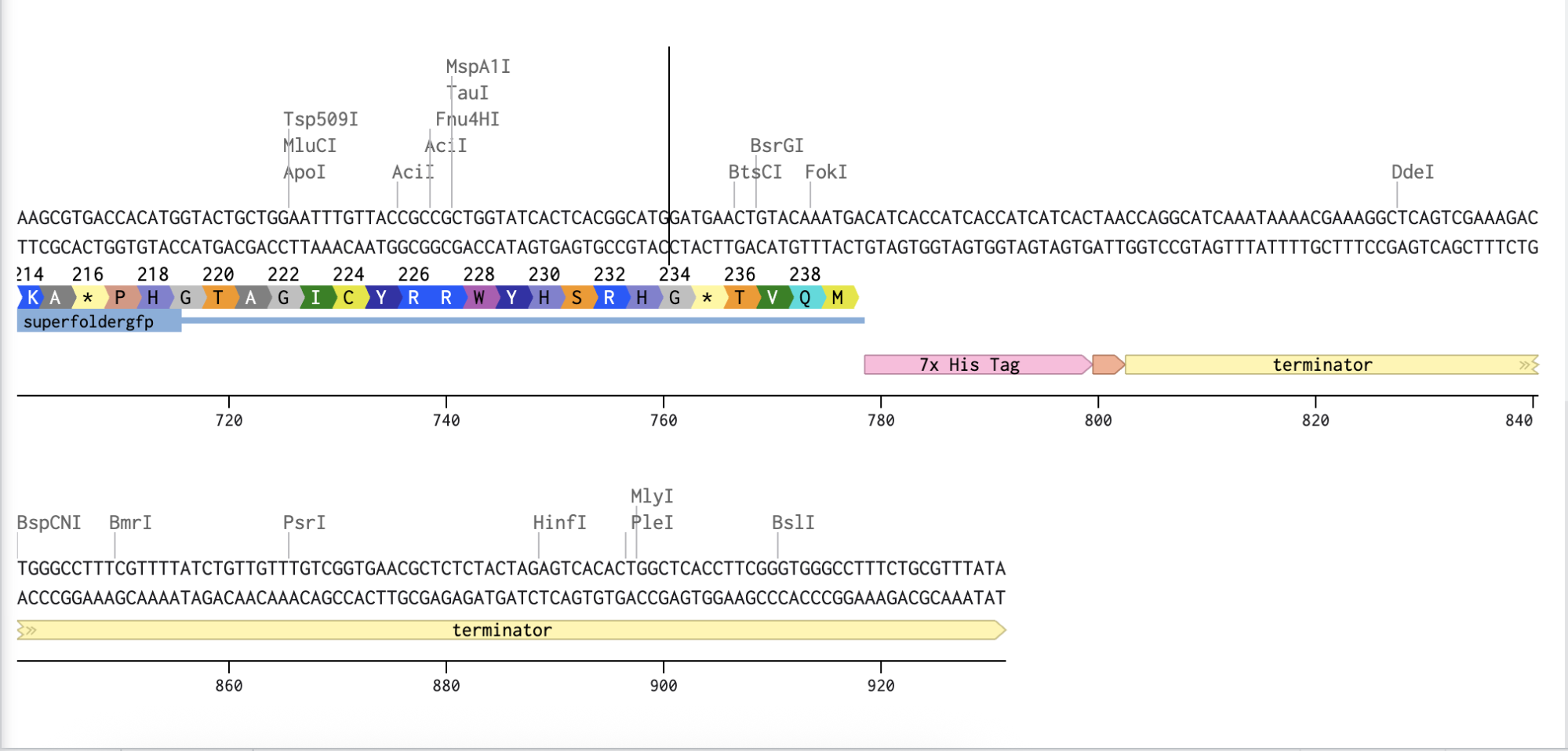
My original idea was to create two sister chromatids, since most of the patterns from the Enzymes were scattered vertical lines, and they kind of looked like alleles inside a chromosome. I had some trouble creating the centromere of the chromosome because none of the enzymes alone created just one line in the middle of the ladder (so around 800 bp), so I picked SacI and SalI and ignored the top line at 12.0 kb.
Original design using restriction enzymes
Sister chromatids highlighted from the original design
Part 3: DNA Design Challenge
3.1. Choose your protein.
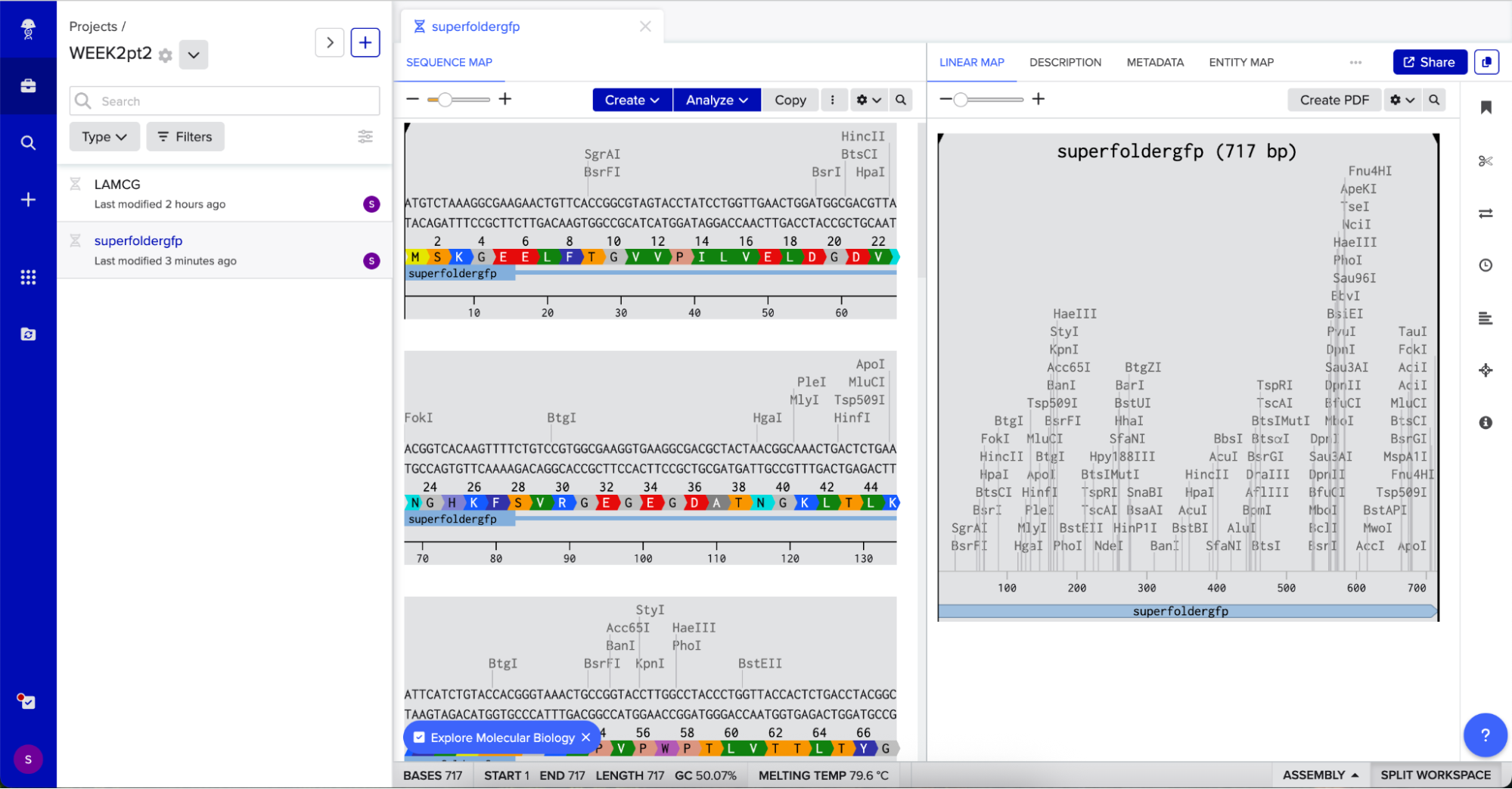
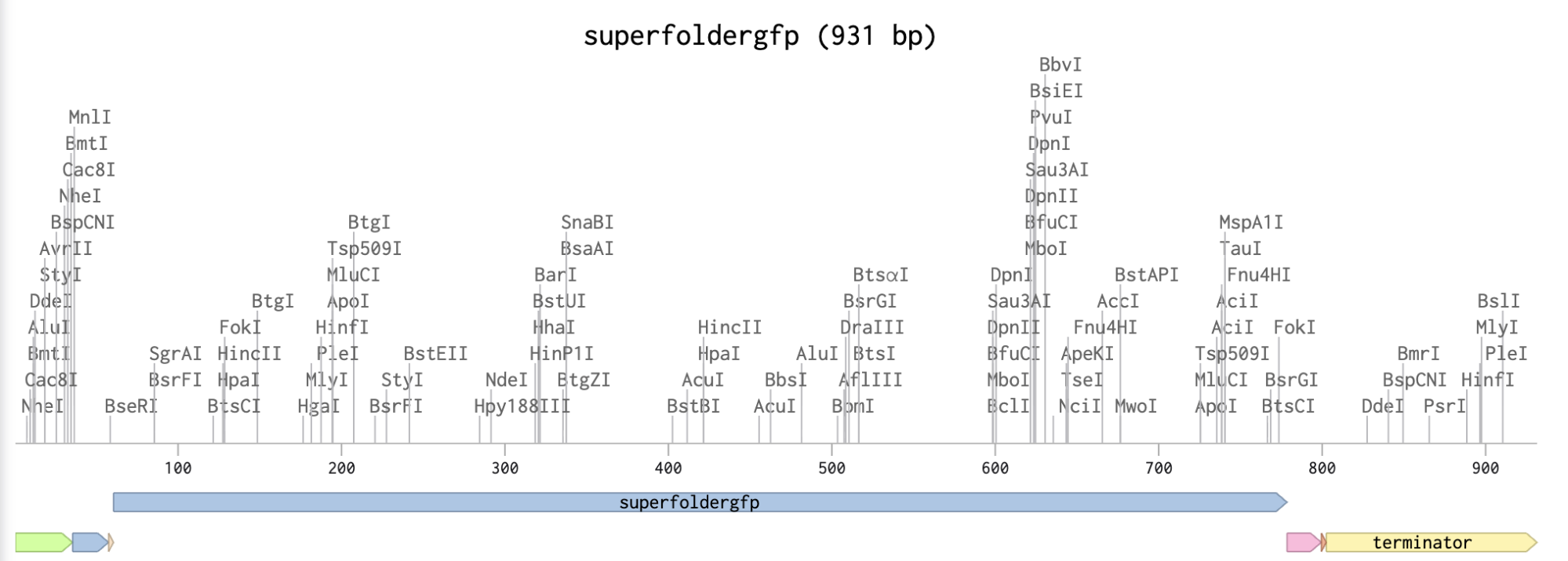
In relation to my interest in genetic logic gates, Sarkar et al. ’s (2021) research uses a 4-output genetic logic where each output is a fluorescent readout corresponding to a maze solution pattern. The protein I will be using is sfGFP (superfolder GFP) because it is widely used, monomeric, very well characterized, and has a strong fluorescence (Chiu & Jiang, 2017).
This is the protein’s sequence obtained from FPbase:
Codon optimization is important because organisms can have different preferences for codon usage, which means that when introducing a gene sequence on a host organism, its own codon usage preferences may affect gene expression or protein synthesis. When doing codon optimization, you are modifying the sequence to enhance protein expression in the host organism.
For this codon optimization I chose E.coli, because most genetic logic gates experiments involve this bacteria due to its simplicity to engineer and wide usage.
3.4. You have a sequence! Now what? What technologies could be used to produce this protein from your DNA?
Technologies like Twist’s Silicon-based DNA Synthesis allows for high precision protein synthesis. You just design a custom sequence on the Twist’s website and order the custom gene synthesis from them.
Part 4: Prepare a Twist DNA Synthesis Order
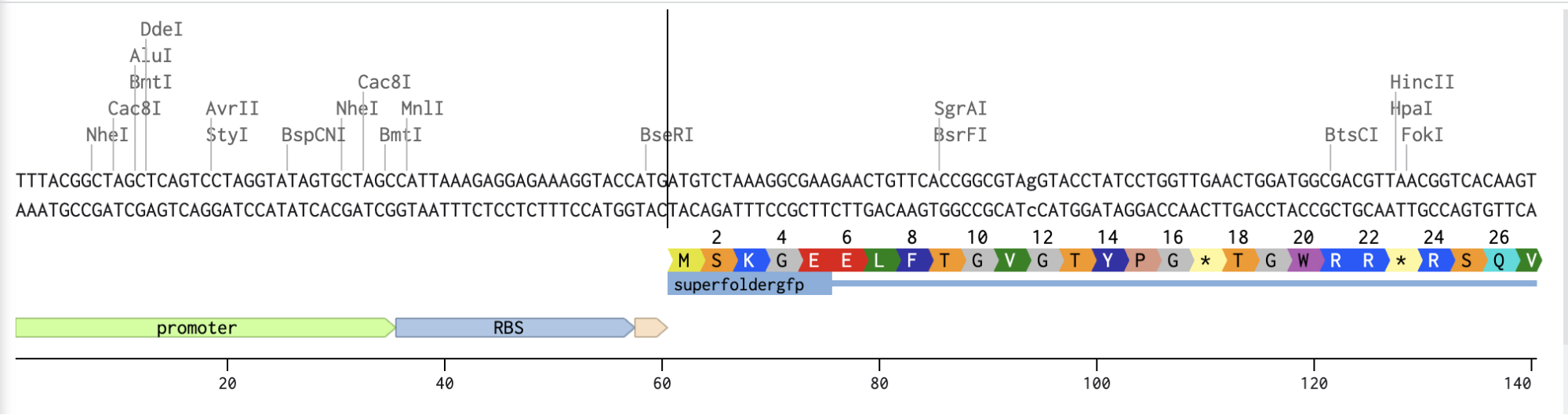
Checking the protein is going to express correctly
Adding the Promoter, RBS, Start Codon, Coding Sequence, His Tag, Stop Codon, and Terminator sequences in the beginning and end of my optimized sequence.
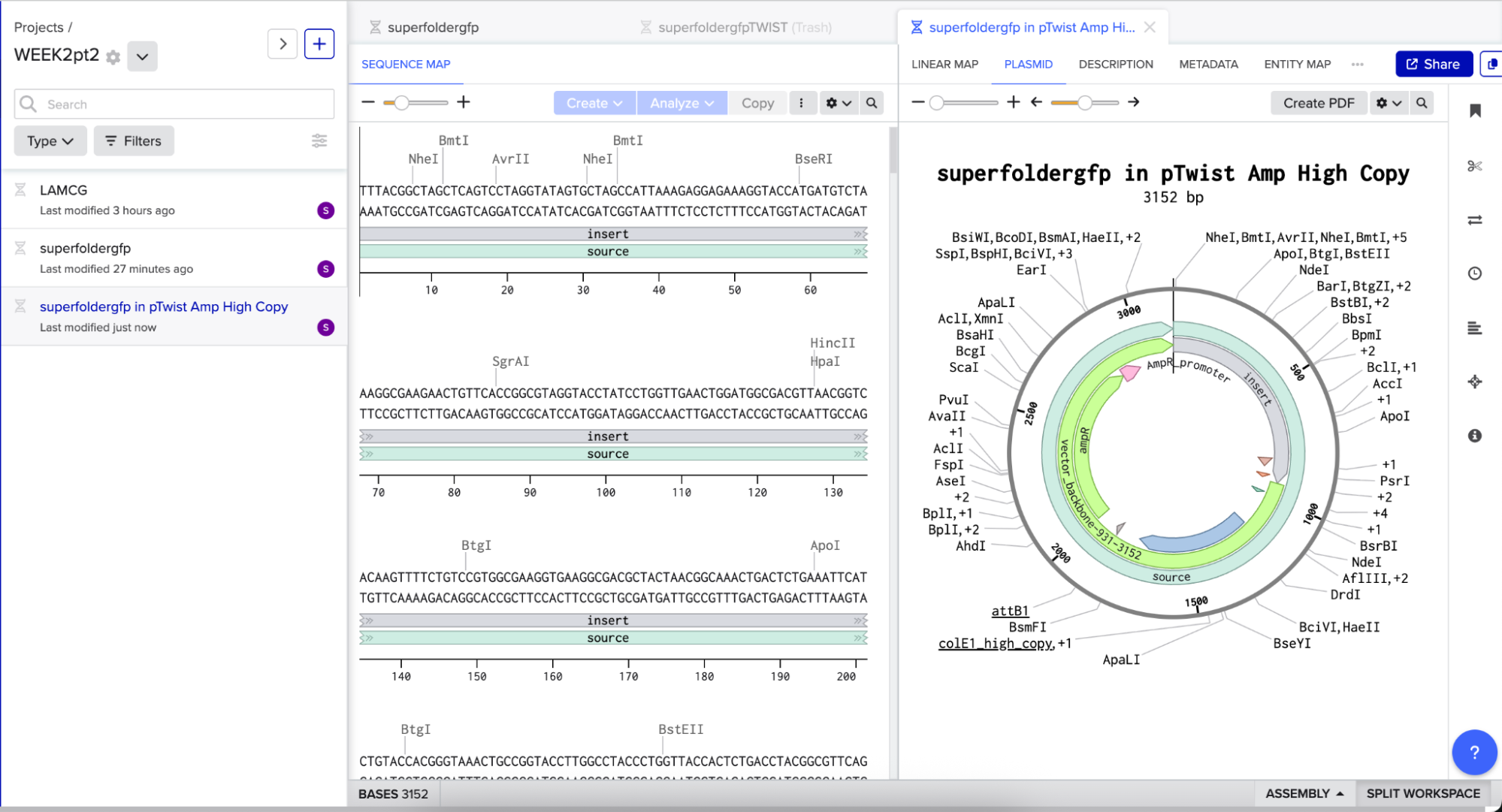
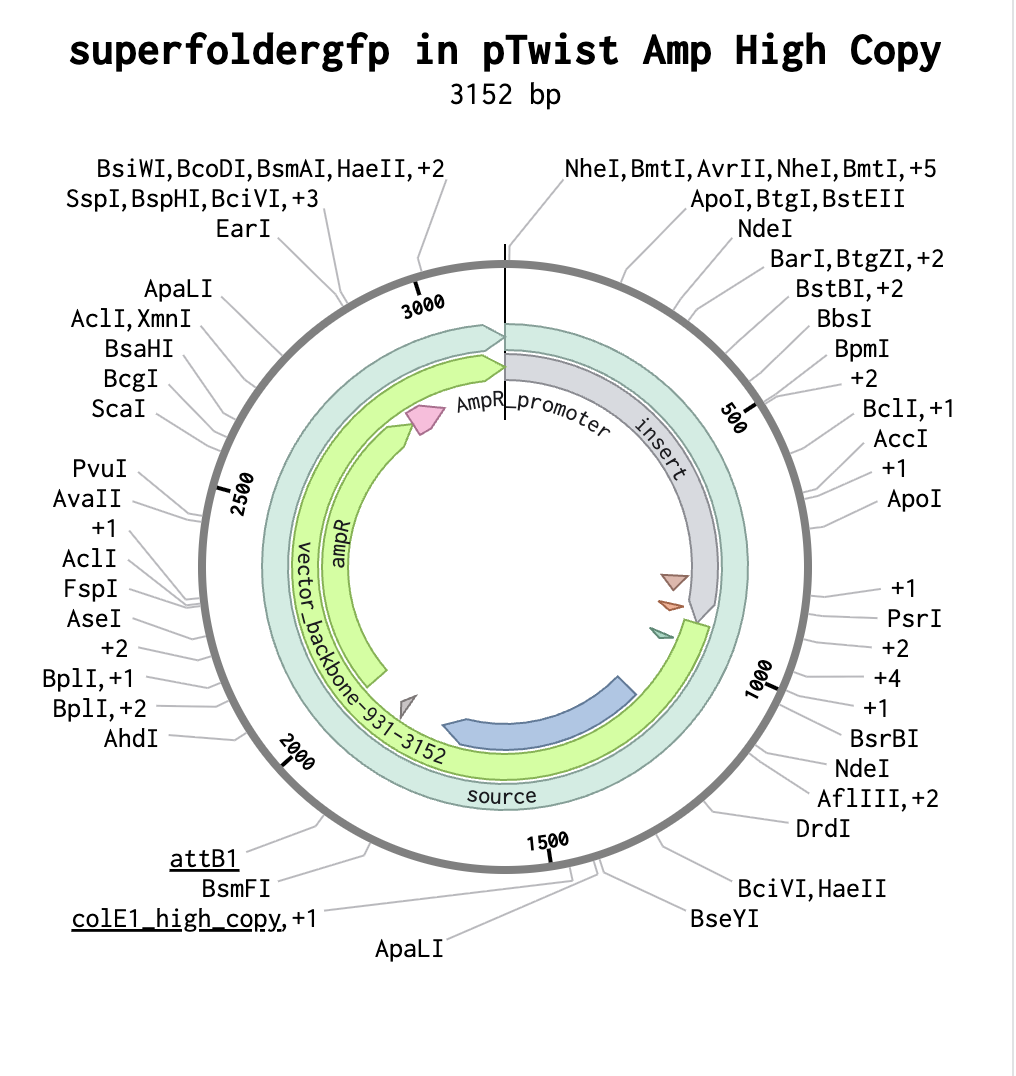
I then downloaded the fasta file for the sequence and uploaded it on Twist. Then, I picked the pTwist Amp High Copy - (2221bp) circular vector.
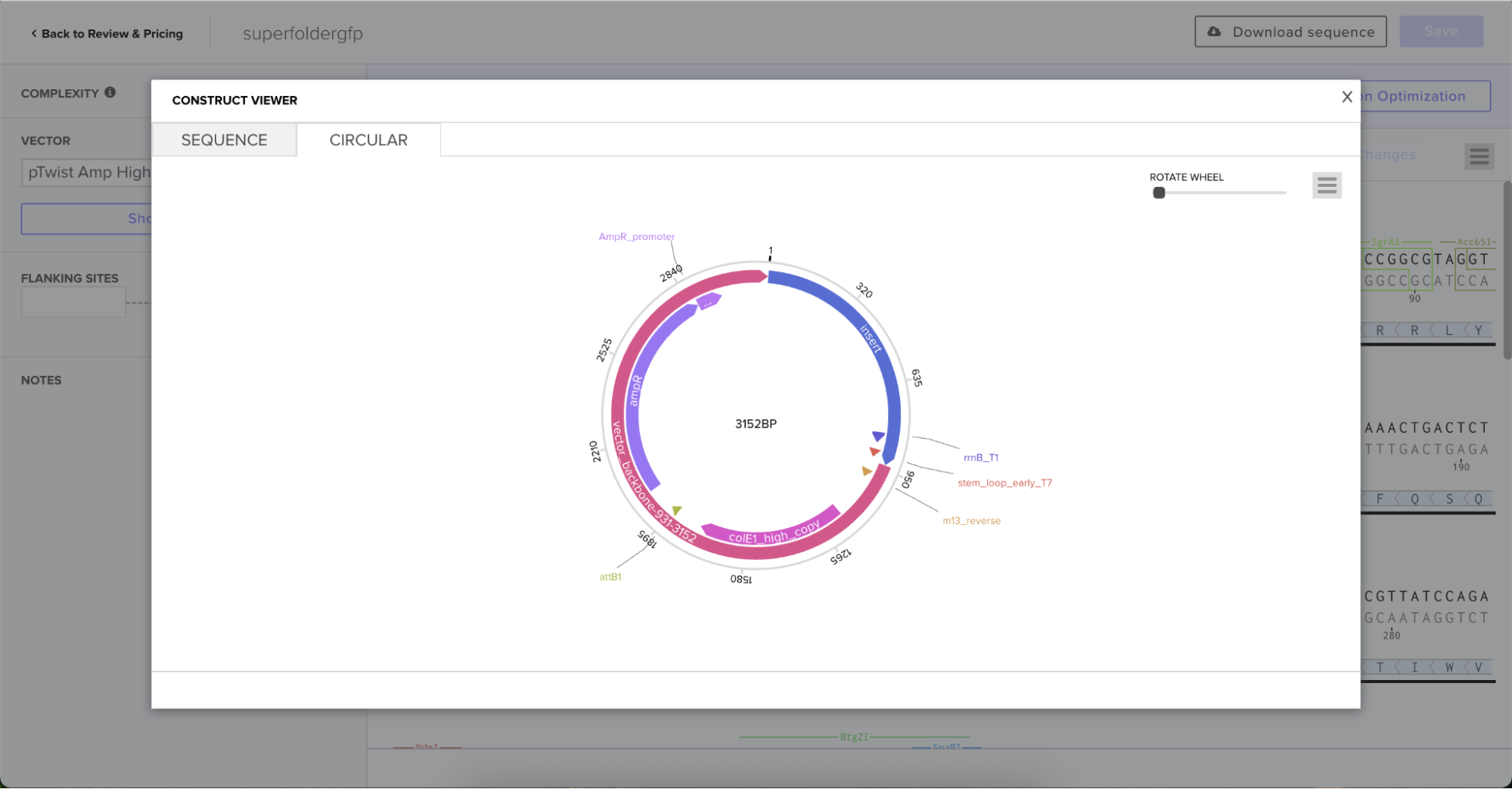
And here’s the circular construct viewer of the sequence + the vector
And here’s the plasmid on Benchling after uploading the downloaded construct from Twist:
Plasmid close-up:
5.1 DNA Read
1. What DNA would you want to sequence (e.g., read) and why?
I would like to sequence the plasmids from three engineered bacterial strains: green, red and yellow responder. This is the first step to verify that the genetic circuits are assembled correctly, and they don’t have mutations, premature stop codons, or show unwanted recombination errors. The sequences will come from E.coli’s DNA, specifically engineered E. coli DH5α strains.
2. In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Ilumina MiSeq, which offers high accuracy at a lower cost for verifying multiple plasmids. In theory, the short reads from this method are okay because I would know the expected sequence and just need to confirm it before continuing with the project methodology.
Also answer the following questions:
3. Is your method first-, second- or third-generation or other? How so?
Second generation because it can sequence multiple DNA fragments simultaneously, so it is more efficient instead of doing multiple runs to sequence the plasmids DNA.
4. What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
The DNA sequence of the E. coli DH5α strains (green, red and yellow responders) would be the input. In order to prepare it, I would need to:
Extract the plasmid DNA from each strain.
Quantify the DNA (can be done using Nanodrop)
Fragment the DNA (can be done using enzymes) to approximately 500 bp
Repair the sticky ends and create DNA with blunt ends
Prevent the fragments from ligating to each other during the adapter ligation reaction by A-tailing
Add sequencing adapters with barcodes by adapter ligation
Amplify the sequence using PCR
Pool multiple libraries into a flow cell
5. What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
After preparing the input, the steps for sequencing are:
Binding DNA fragments to the flow cell, the bridge amplification creates clusters of the identical copies of DNA.
Add fluorescently labeled reversible terminators
Capture which base was added to each cluster of identical copies by laser excitation
Use a software for base calling (could be Dorado by Oxford Nanopore Technologies)
Assign Phred quality scores to each base
6. What is the output of your chosen sequencing technology?
After the sequencing, I would have multiple FASTQ files with the raw reads with the Phred quality scores and BAM files showing variants, to see any mutations in the sequences.
5.2 DNA Write
1. What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
I would synthesize three genetic circuits for the bacterial pattern recognizer:
Green responder:
Combining the green + red responder plasmids in one cell
By synthesizing circuits instead of assembling, I could ensure more accuracy in the sequences.
2. What technology or technologies would you use to perform this DNA synthesis and why?
Twist could be very useful for this step, in order to achieve array-based oligo synthesis.
Also answer the following questions:
3. What are the essential steps of your chosen sequencing methods?
Key steps are:
Using FASTA format for the sequences
Using Twist to optimize codon usage, ensure higher accuracy, high parallelism and quality control
Do oligo synthesis on silicon chip
Cleave and release the oligos from the chip
Assemble the oligos into longer fragments using PCR or Gibson assembly
Clone longer fragments by introducing them into vectors
Do a full-length Sanger verification
4. What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
This sequencing methodology has many steps and could take weeks to do, especially because it depends on multiple steps with different shipping times. Also, the cost increases when creating longer fragments.
5.3 DNA Edit
1. What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
For my final project, I would need to edit the genome of the E. coli DH5α strain because it has LacI and AraC genes, which could intervene with the synthetic circuits that will be introduced later on. Also, removing these genes allow for real-world applications, as antibiotic-free systems are widely used for environmental or medical uses.
2. What technology or technologies would you use to perform these DNA edits and why?
CRISPR-Cas9 because it can cut the genome in precise places, facilitating the extraction of the unwanted genes, and then can stitch back the fragments together.
Also answer the following questions:
3. How does your technology of choice edit DNA?
First, sgRNA guides the Cas9 enzyme to target the DNA sequence. Then, Cas9 creates a double-strand cut in the desired place of the unwanted genes. Finally, the cell repairs the break through NHEJ or HDR.
4. What are the essential steps?
First, design the RNA map to highlight and target the prophages. Second, prepare the DNA template to be edited. Third, introduce the RNA map and the DNA template in E.coli cells.
5. What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
Steps for editing the DNA:
Prepare the input: E.coli DH5α strain, create the pCas plasmid (that encodes Cas9, Lambda Red and sgRNA for the CRISPR-Cas reaction), donor DNA fragments (from the synthesized PCR products), and the editing oligos for sgRNA cloning
Clone the sgRNAs
Transform pCas into target strain by electroporation and selection with kanamycin
Induce Lambda Red by growth with arabinose (which induces recombination proteins)
Add donor DNA and the transformed pCas, then electroporate
Amplify sequence using colony PCR
Grow pCas plasmid and test for loss of kanamycin resistance, to ensure it grew without antibiotic resistance
6. What are the limitations of your editing methods (if any) in terms of efficiency or precision?
CRISPR-Cas9 is not always 100% effective, as there is a small risk it will accidentally cut the DNA in the wrong places (these are called off-target effects). To avoid this, the guide RNA has to be very carefully designed. Also, another “limiting factor is the fact that dCas9 is a shared resource amongst the different gates which needs to be continuously expressed at very high concentrations, and this leads to high toxicity for the host cells” (Al-Radhawi et al., 2020).
References:
Chiu TY, Jiang JR. Logic Synthesis of Recombinase-Based Genetic Circuits. Sci Rep. 2017 Oct 9;7(1):12873. doi: 10.1038/s41598-017-07386-3. PMID: 28993615; PMCID: PMC5634492.
Sarkar, K., Bonnerjee, D., & Bagh, S. (2021). Engineered Bacteria Computationally Solve Chemically Generated 2X2 Maze Problems. Homi Bhabha National Institute (HBNI). https://doi.org/10.1101/2021.06.16.448778
Zhang, H., Lin, M., Shi, H. et al. Programming a Pavlovian-like conditioning circuit in Escherichia coli. Nat Commun 5, 3102 (2014). https://doi.org/10.1038/ncomms4102
Chen J, Li Y, Zhang K, Wang H2018.Whole-Genome Sequence of Phage-Resistant Strain Escherichia coli DH5α. Genome Announc6:10.1128/genomea.00097-18.https://doi.org/10.1128/genomea.00097-18
Rath, D., Amlinger, L., Rath, A., & Lundgren, M. (2015). The CRISPR-Cas immune system: biology, mechanisms and applications. Biochimie, 117, 119-128.
Al-Radhawi, M. A., Tran, A. P., Ernst, E. A., Chen, T., Voigt, C. A., & Sontag, E. D. (2020). Distributed implementation of boolean functions by transcriptional synthetic circuits. ACS Synthetic Biology, 9(8), 2172-2187.
Week 3 HW: Lab Automation
Opentrons Artwork
For this activity, I decided to do Majora’s Mask from The Legend of Zelda:
1. Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Fedorec et al. (2024) developed a biocomputer where bacterial colonies perform logic operations, eliminating the need for complex genetic engineering of individual cells: instead of building circuits inside the bacteria, they engineered “receiver” strains that respond to chemical concentration thresholds, then located the engineered strains at specific distances from some chemical input sources. The chemical gradients that overlap create concentrations at each colony location, and by changing the bacteria colony’s locations, they are programming them to perform AND and OR logic gates. The researchers used Opentrons OT2 handling robot to dispense the cultures onto agar plates and then add the chemical gradients. This approach is very interesting, because it treats physical space as the programmable medium.
Reference:
Fedorec, A. J., Treloar, N. J., Wen, K. Y., Dekker, L., Ong, Q. H., Jurkeviciute, G., … & Barnes, C. P. (2024). Emergent digital bio-computation through spatial diffusion and engineered bacteria. Nature Communications, 15(1), 4896.
2. Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
Since we had to think about two other final project ideas to add to the slide deck, here are my three final project ideas and how I could use automation tools as methodology:
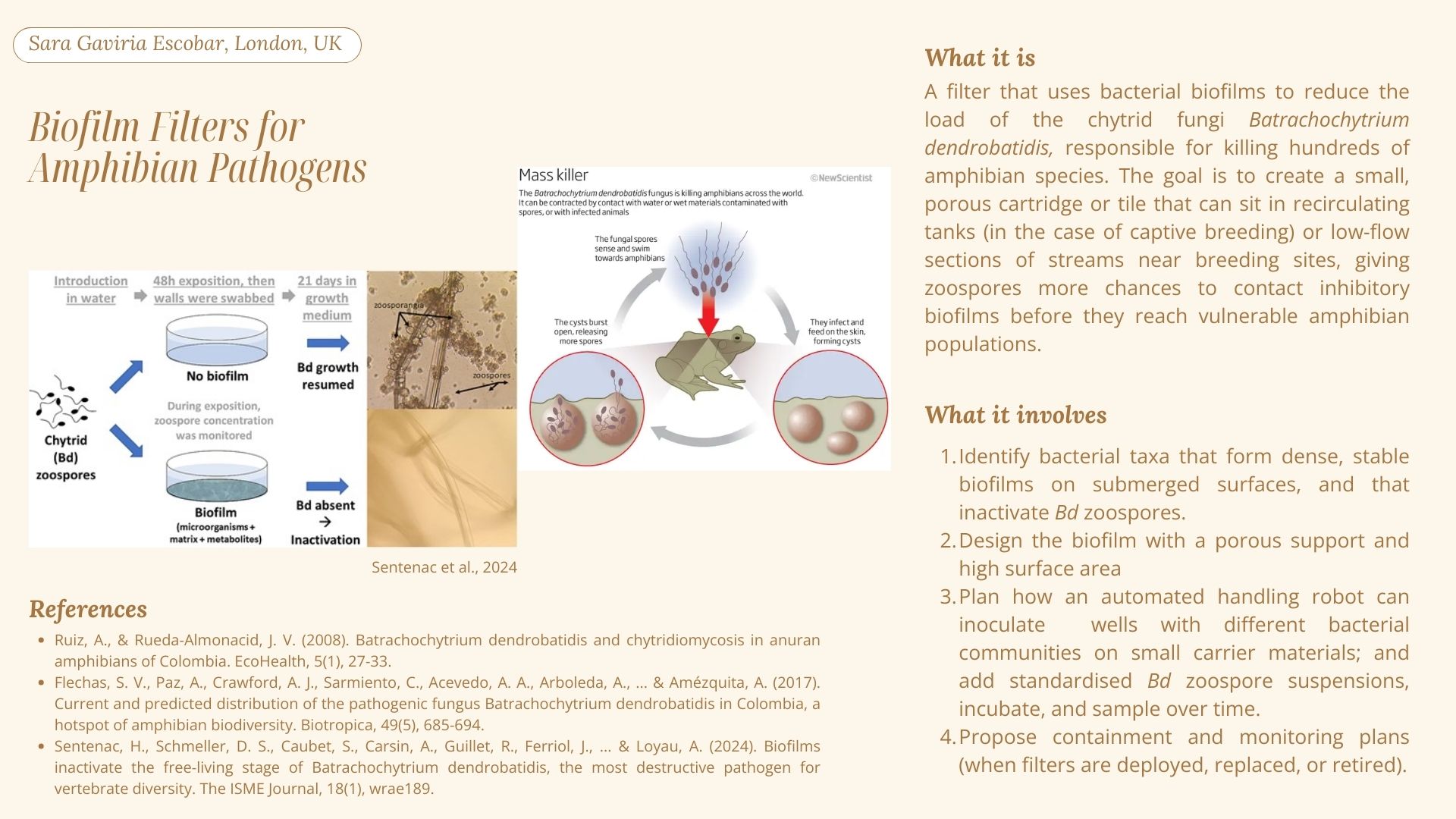
1. Biofilm Filters for Amphibian Pathogens
For the automation tools, a handling robot can inoculate wells with different bacterial communities on small carrier materials; and add standardised Bd zoospore suspensions, incubate, and sample over time.
2. Bacterial Pattern Recognition
For the automation tools, a handling robot (such as the OT2) could be used to dispense the E.coli cultures and to then add the chemical gradients.
3. Beneficial Biofilm Coats for Corals
In terms of automation, an automated handling robot can assemble different combinations of candidate strains in wells with CaCO₃ chips or shell fragments to act as the coral.
Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Because consuming something as a way to obtain energy does not mean we are assimilating it as part of ourselves. When humans eat meat (or anything, really), we are breaking it down into smaller pieces, and the molecule’s chemical bonds break down. The energy stored between those chemical bonds is released as energy for our bodies, and the nutrients from the food source are absorbed. In this process, our human cells are not acquiring, reading or translating any DNA from foreign organisms (a cow, a fish, a plant).
3. Why are there only 20 natural amino acids?
“The selection of the 20 standard residue types was made early on in evolution–their appearance predates RNA and DNA and it is highly likely that they already played a vital role throughout prebiotic chemical evolution (~4 Gyrs ago)” (Bywater, 2018). However, the question until this day is why 20 and not another number? According to Bywater (2018), our living systems’ DNA is able to cater for 64 possible amino acids types. He explains that there is a key factor that explain the number 20: those amino acids show “energetically well-separated conformers”.
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes, you can. They are referred to as noncanoninal amino acids (ncAAs) introduced into proteins. They can modify the protein backbone or the amino acid side chains (Budisa, 2025).
5. Where did amino acids come from before enzymes that make them, and before life started?
Miller and Urey proved that amino acids can form from a concoction of gases and electricity, without the existence of enzymes and ribozymes, which is what scientist say likely happened in primitive Earth, which was abundant in hydrogen, a key element for amino acid formation. RNA and enzymes are relatively recent in Earth’s history (Bywater, 2018).
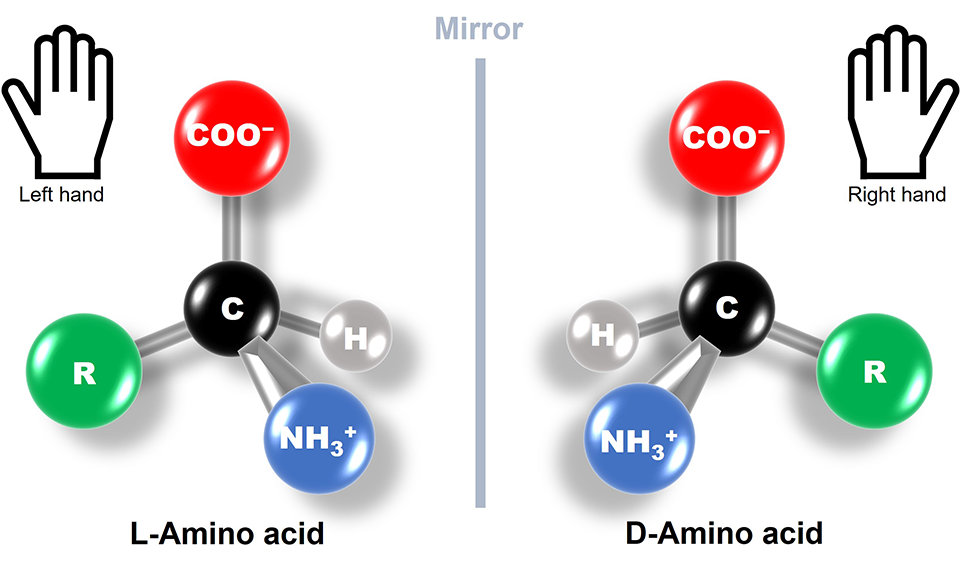
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
I understand that L-amino acids form right-handed α-helix conformations. So a α-helix made from D-amino acids would have the opposite: left-handed
7. Can you discover additional helices in proteins?
Yes, mainly identifiable by different patterns in the hydrogen bonds, creating tighter or wider geometries in the helices. For example, π-helices have “been described as α-aneurisms, α-bulges, or π-bulges” (Kumar & Bansal, 2015).
8. Why are most molecular helices right-handed?
Because of energy components. Left-handed helices tend to have unusual and weaker interactions than right-hand helices (Rzepa, n.d).
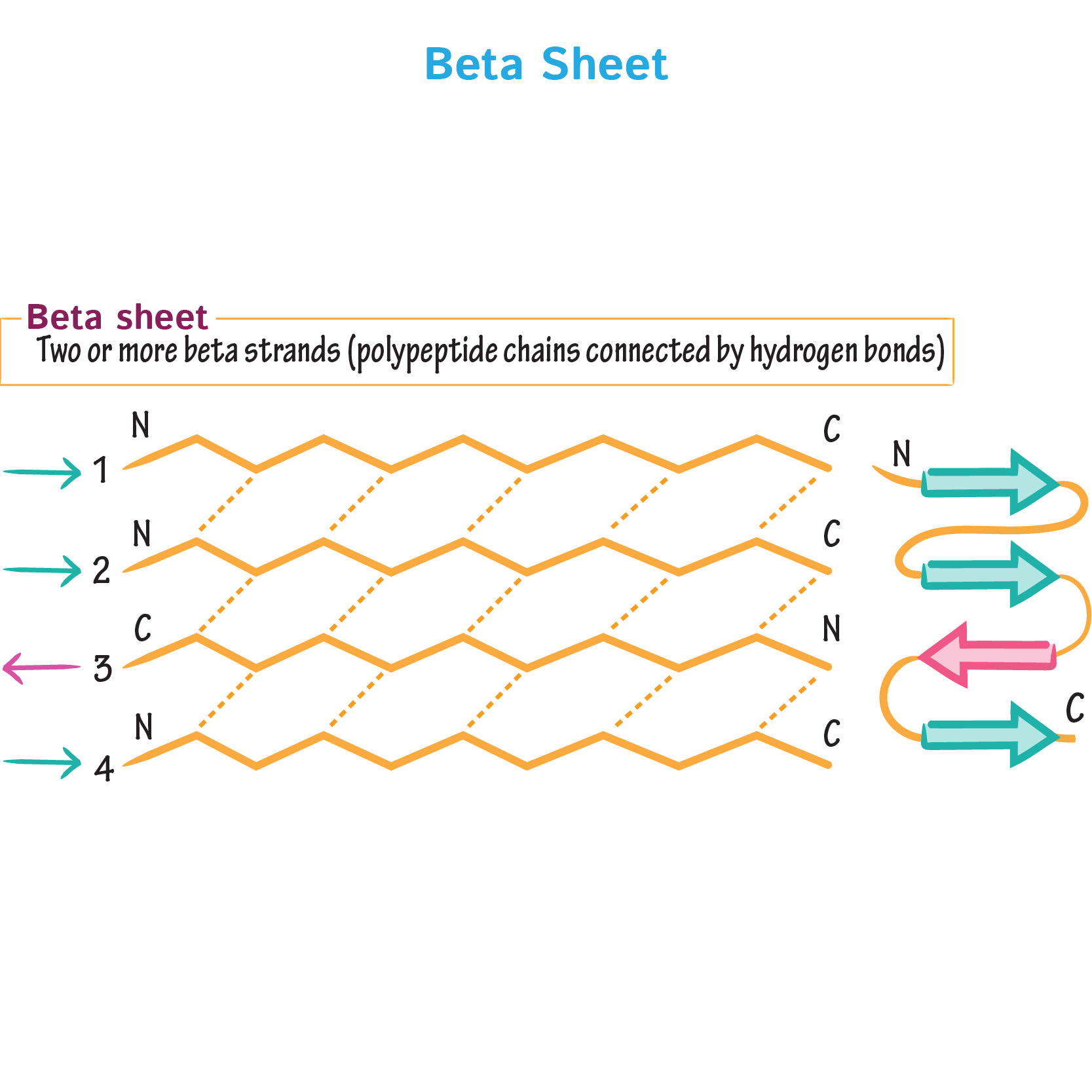
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets have hydrogen-bonding edges that are facing one way, which allows them to interact, bond and aggregate with other β-sheets.
Ditki (2017)
References:
Bywater, R. P. (2018). Why twenty amino acid residue types suffice (d) to support all living systems. Plos one, 13(10), e0204883.
Budisa, N. (2025). Introduction:“Noncanonical Amino Acids”. Chemical Reviews, 125(4), 1659-1662.
Kumar, P., & Bansal, M. (2015). Dissecting π‐helices: sequence, structure and function. The FEBS Journal, 282(22), 4415-4432.
1. Briefly describe the protein you selected and why you selected it.
My main final project idea is to develop a filter that uses bacterial biofilms to reduce the load of the chytrid fungi Batrachochytrium dendrobatidis (Bd), responsible for killing hundreds of amphibian species. According to Abramyan & Stajich (2012), Bd has a lot of chitin-binding modules in its genome, which could potentially be a reason for its high pathogenicity. There are potent antifungal chitinases that damage the cell walls of the pathogenic fungi that have been modified to be more thermostable, such as GH19 chitinase from Ficus microcarpa latex (Kozome et al., 2022). For my biofilm filter, this protein could be a great addition.
2. Identify the amino acid sequence of your protein.
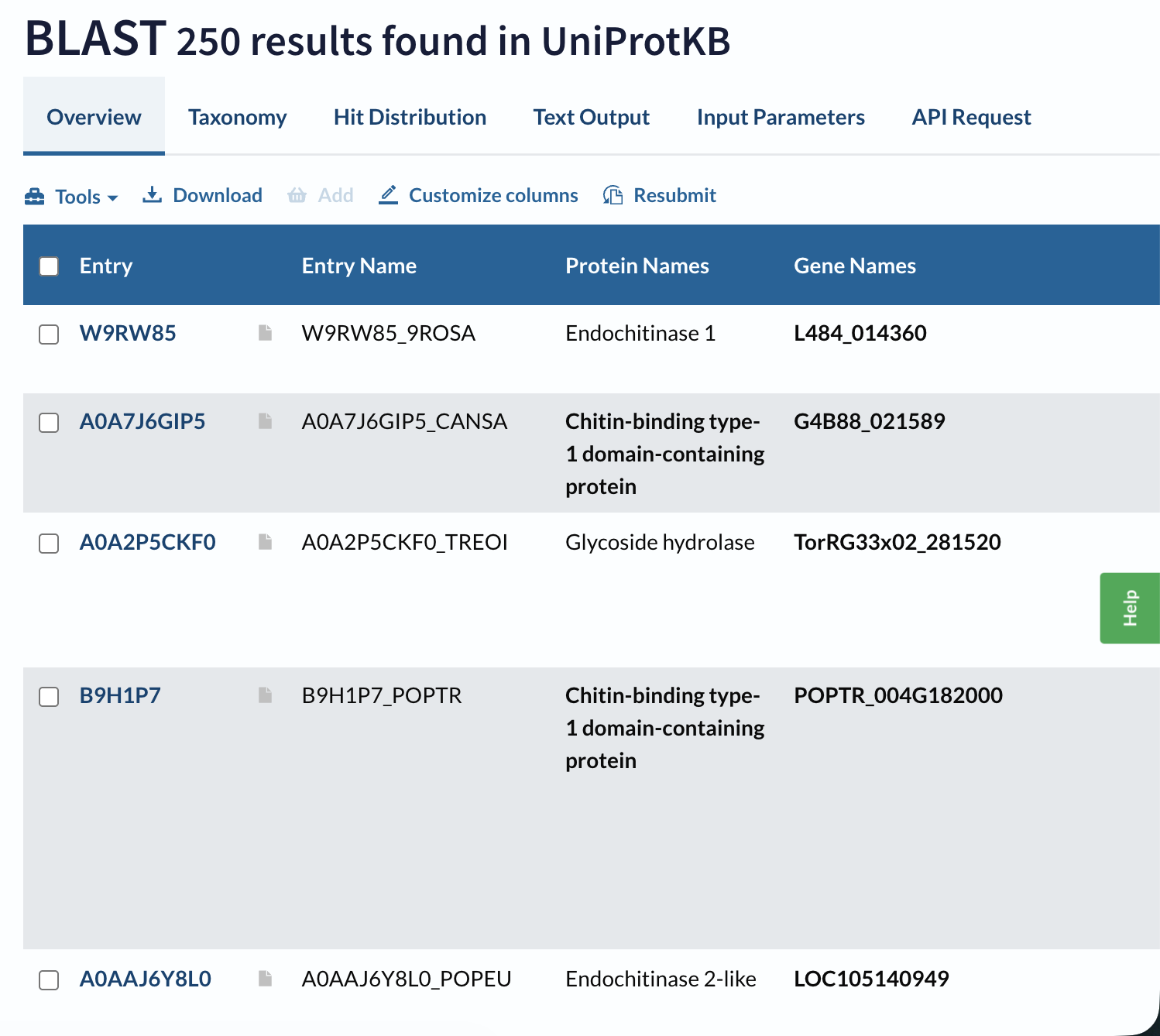
A BLAST search of A0A915Q9K7 against UniProt returns 250 homologous sequences above the default significance threshold, mainly GH19 chitinases from other flowering plants, especially within the Moraceae and related taxa.
Domain databases classify A0A915Q9K7 as a member of glycoside hydrolase family 19 (GH19), within a lysozyme‑like endochitinase superfamily. It carries the canonical GH19 catalytic motifs (CHITINASE_19_1 and CHITINASE_19_2) and is grouped in the “Endochitinase (Chitinase)” CATH superfamily.
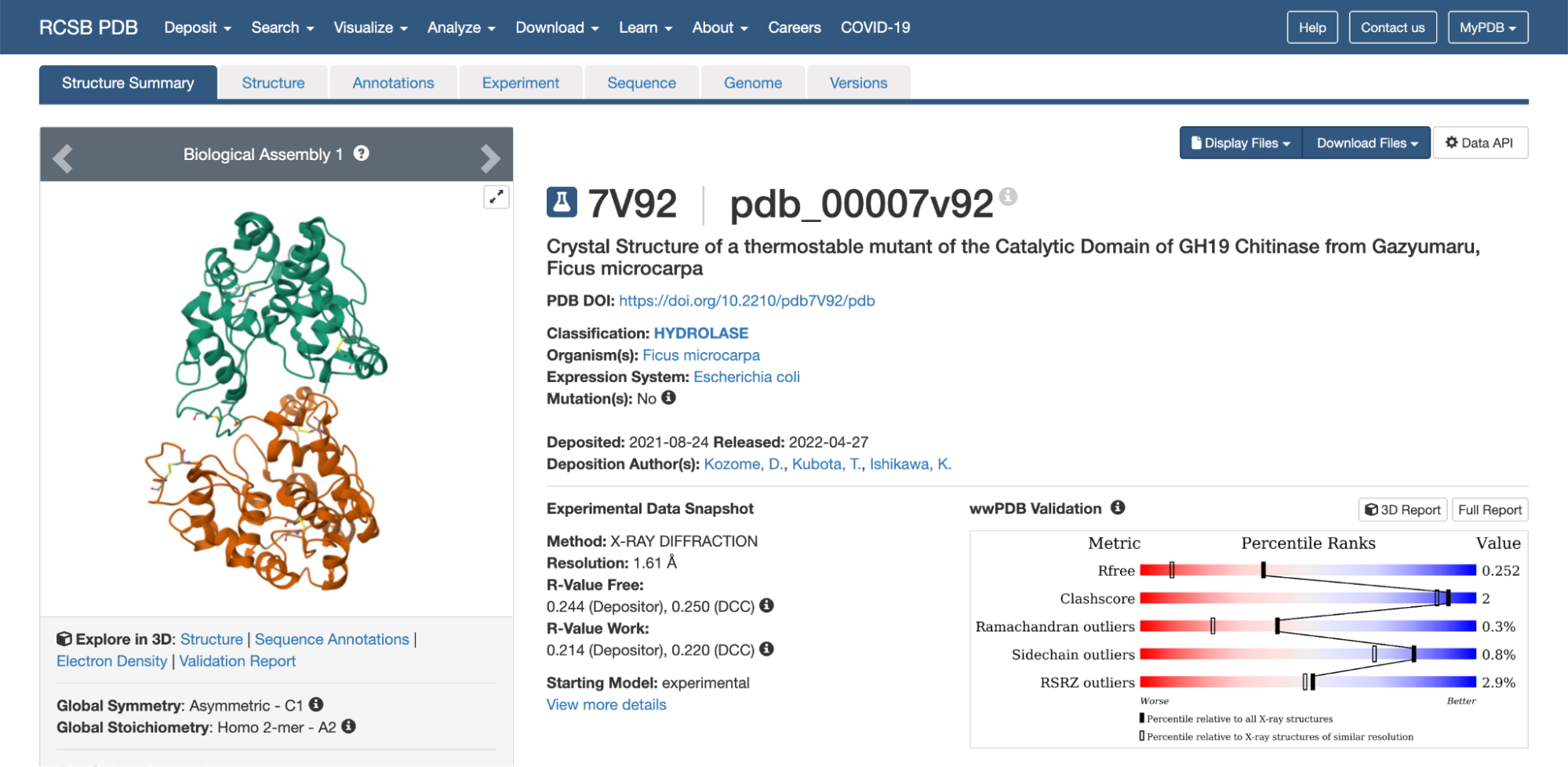
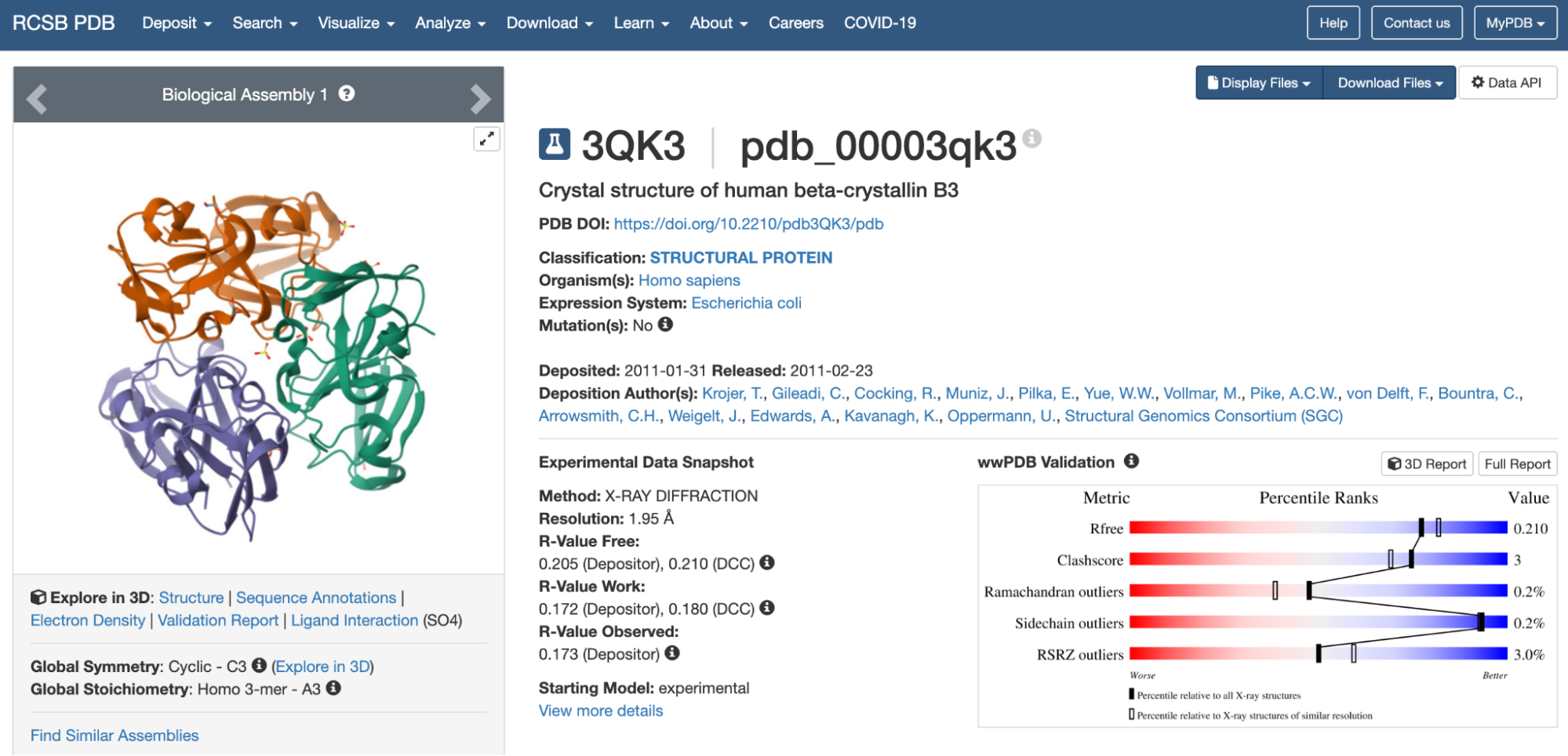
3. Identify the structure page of your protein in RCSB
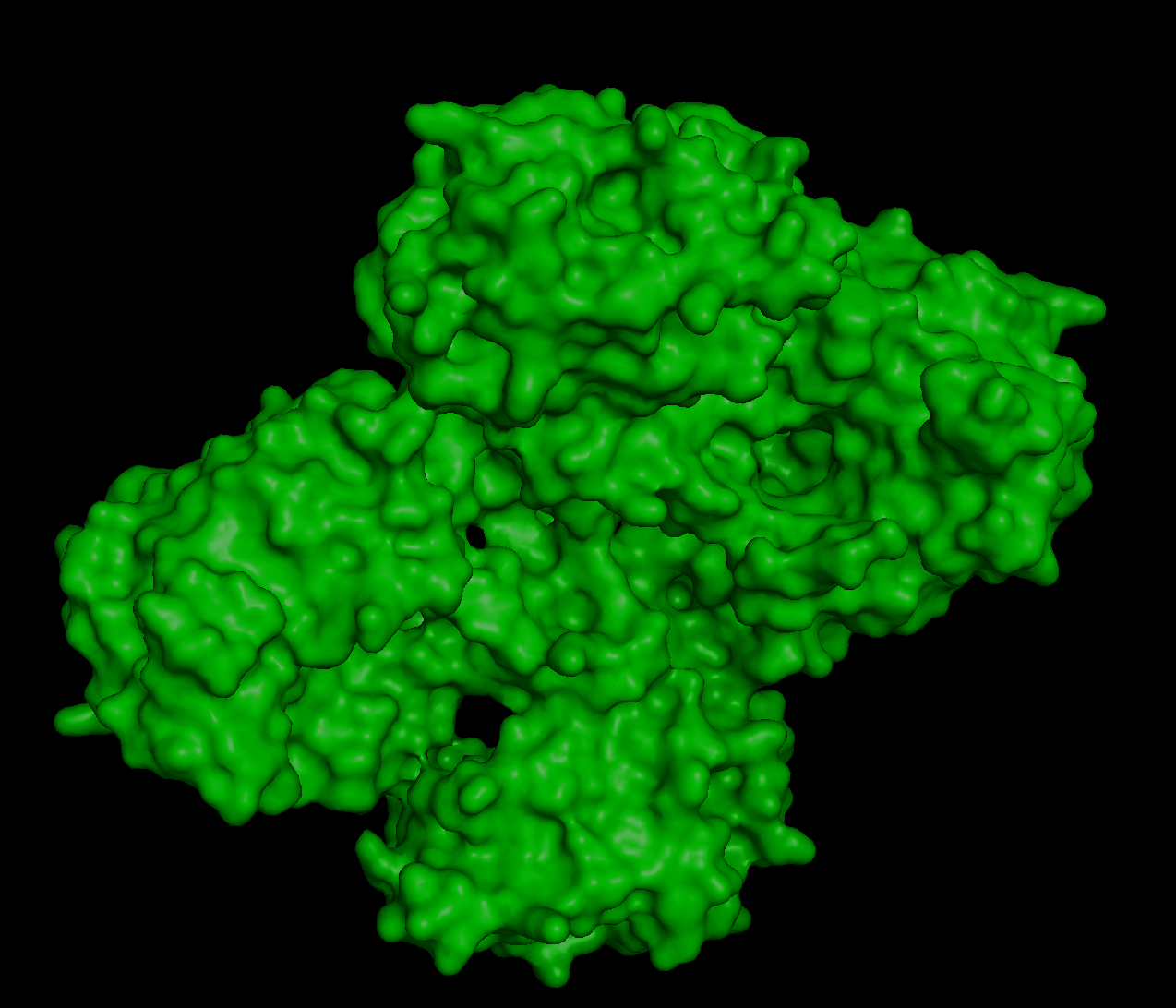
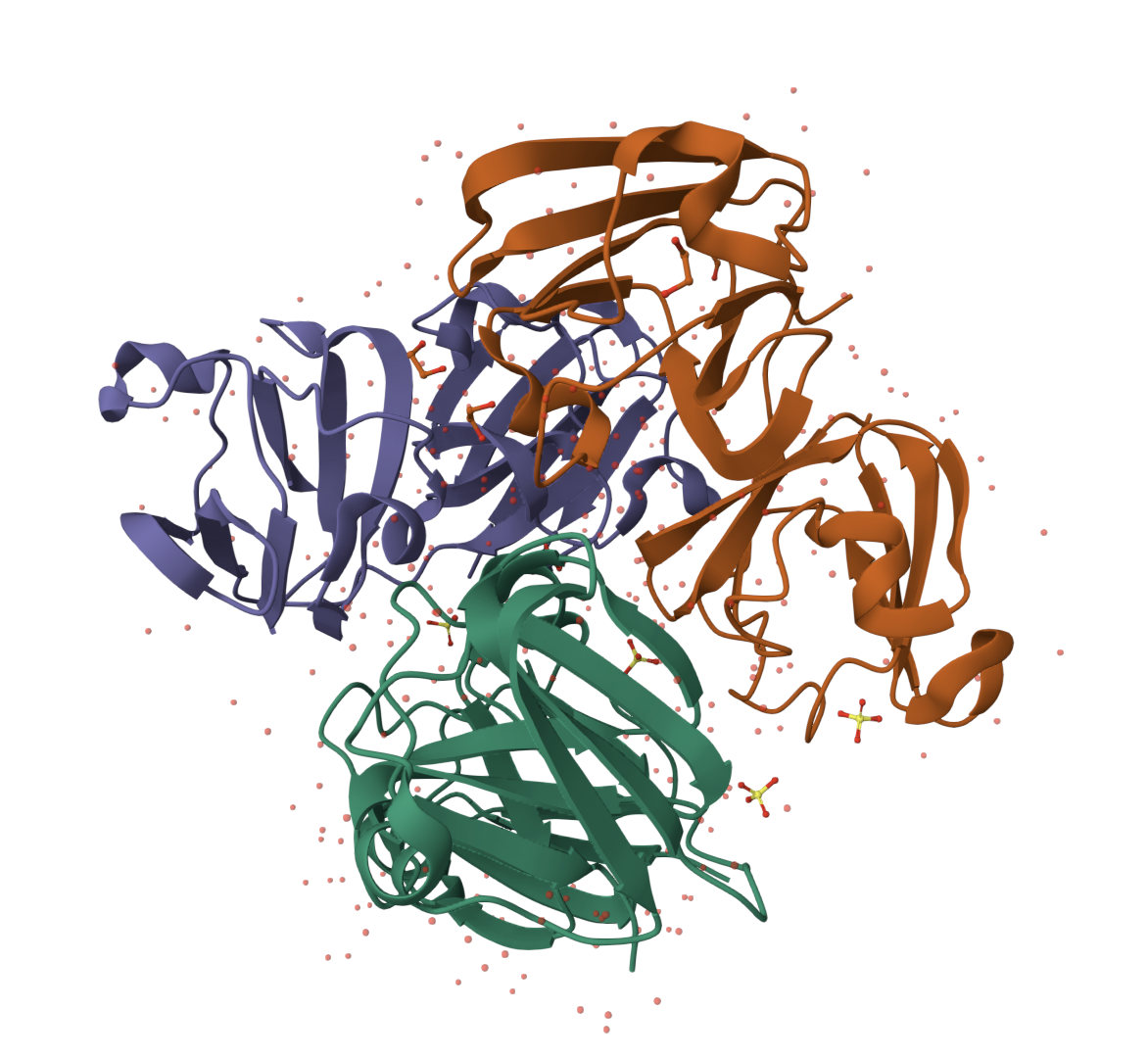
The 3D structure of the protein was solved by X‑ray crystallography at 1.61 Å resolution, which is very high quality.
When exploring the 3D view, I see four protein chains in the asymmetric unit (A–D). Around the structure, I also see smaller water molecules.
The 7V92 structure belongs to the Endochitinase / GH19 chitinase superfamily in the lysozyme‑like (SSF53955) fold class.
4. Open the structure of your protein in any 3D molecule visualization software:
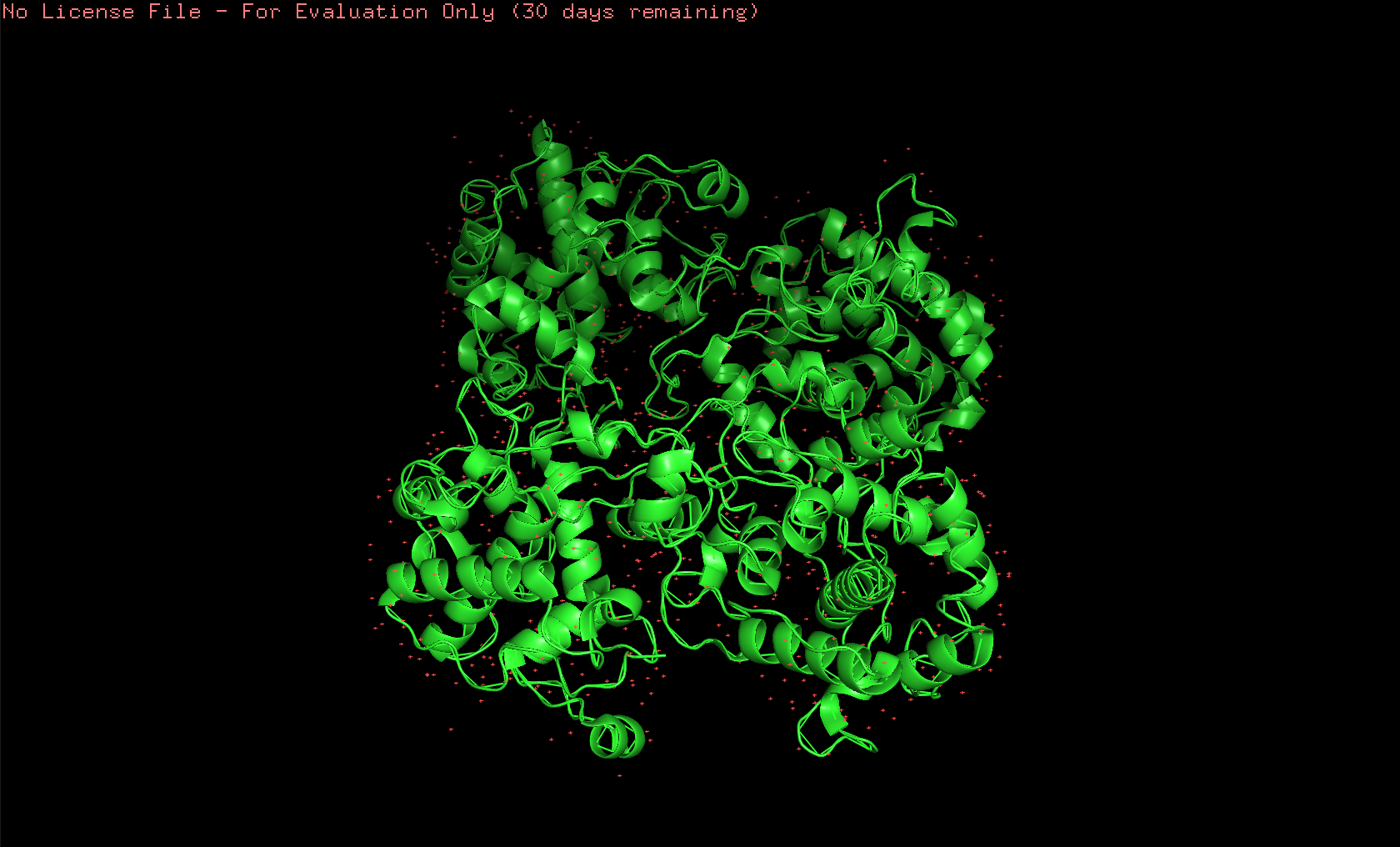
Here’s the first look of the protein in PyMOL:
Here’s the visualization of the protein as “cartoon”:
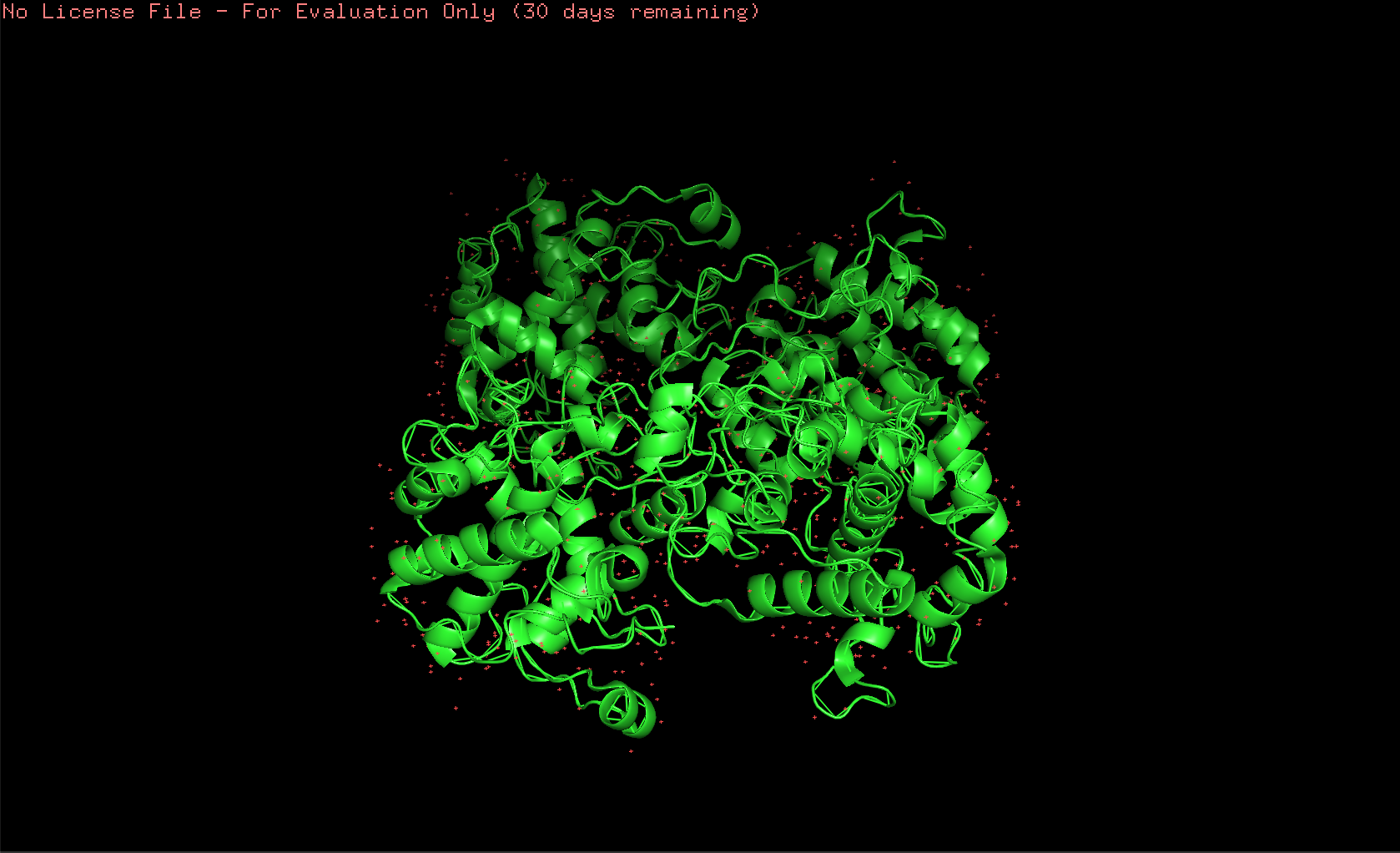
Here’s the visualization of the protein as “ribbon”:
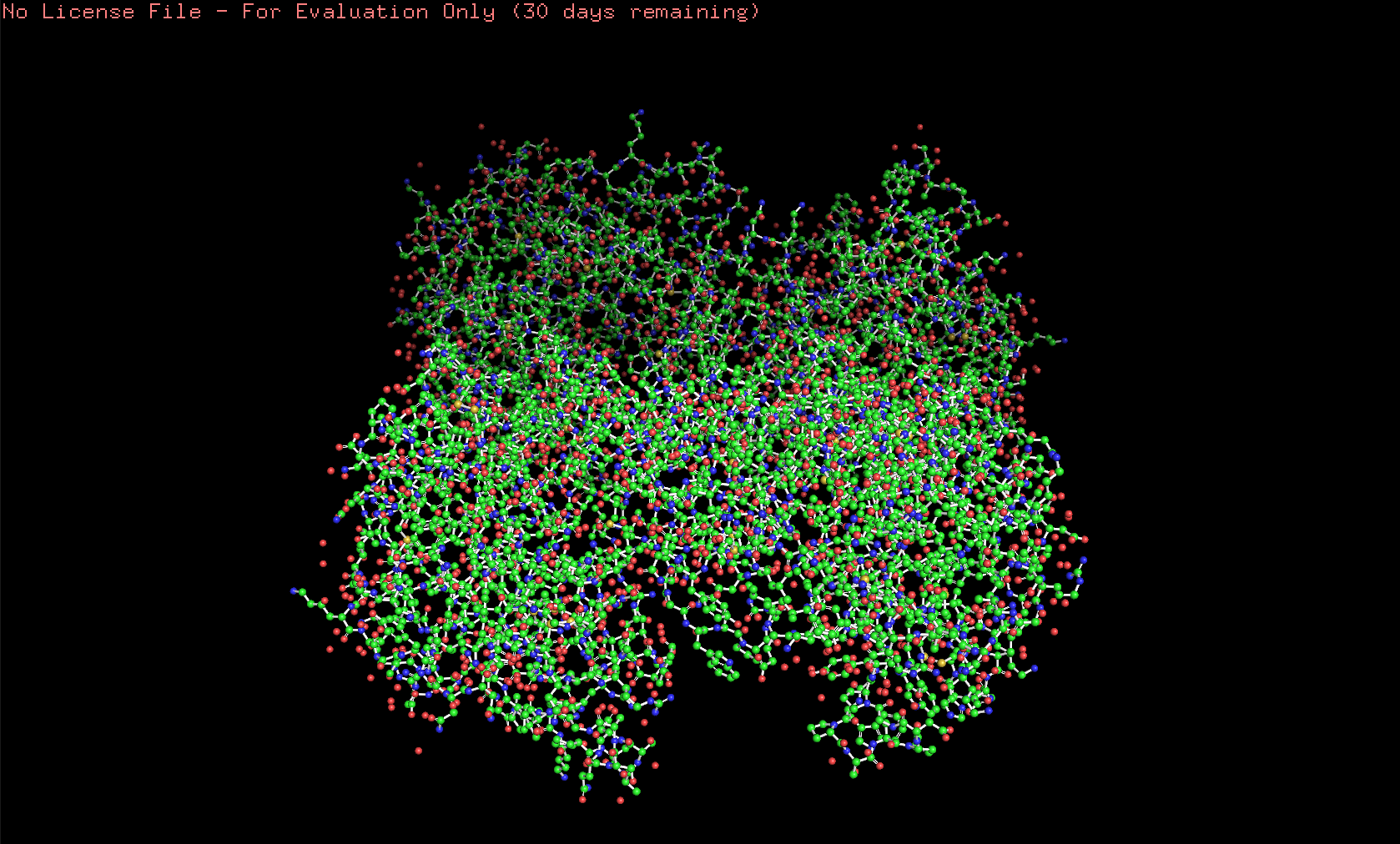
Here’s the visualization of the protein as “ball and stick”:
Here’s the visualization of the protein as “spheres”:
Here’s the visualization of the protein as “surface”:
And this is how it looks like when coloring it by secondary structure:
The protein has more helices than loops/coils.
To visualize the protein by residue class, I used this code created with ChatGPT:
{
# Acidic (Asp, Glu) – red
color red, resn ASP+GLU
# Basic (Lys, Arg, His) – blue
color blue, resn LYS+ARG+HIS
# Polar uncharged (Ser, Thr, Asn, Gln) – green
color green, resn SER+THR+ASN+GLN
# Hydrophobic (Ala, Val, Leu, Ile, Met, Phe, Trp, Pro) – yellow
color yellow, resn ALA+VAL+LEU+ILE+MET+PHE+TRP+PRO
# Cysteine – orange
color orange, resn CYS
# Glycine – cyan
color cyan, resn GLY
}
And this is the image produced by that code:
Hydrophobic residues are mainly in the interior of the protein structure,while hydrophilic and charged residues are mainly on the surface. This is a typical distribution of hydrophobic and hydrophilic residues on a soluble enzyme.
When inspecting for “holes”, I found these:
Although they are multiple holes, I would also say they are small.
References:
Kozome, D., Uechi, K., Taira, T., Fukada, H., Kubota, T., & Ishikawa, K. (2022). Structural analysis and construction of a thermostable antifungal chitinase. Applied and Environmental Microbiology, 88(12), e00652-22.
Abramyan, J., & Stajich, J. E. (2012). Species-specific chitin-binding module 18 expansion in the amphibian pathogen Batrachochytrium dendrobatidis. MBio, 3(3), 10-1128.
Part C. Using ML-Based Protein Design Tools
For this exercise, I chose the protein human beta-crystallin B3, a protein encoded by the gene CRYBB3. I find this protein super interesting because it is found in our eyes, helping maintain transparency and refractive index of the lens. It is crazy to me how a protein can be so transparent, flexible, and stable at the same time, adapting to the changes in the eye through aging. Mutations in the gene can cause cataracts and other eye diseases (NCBI, 2026).
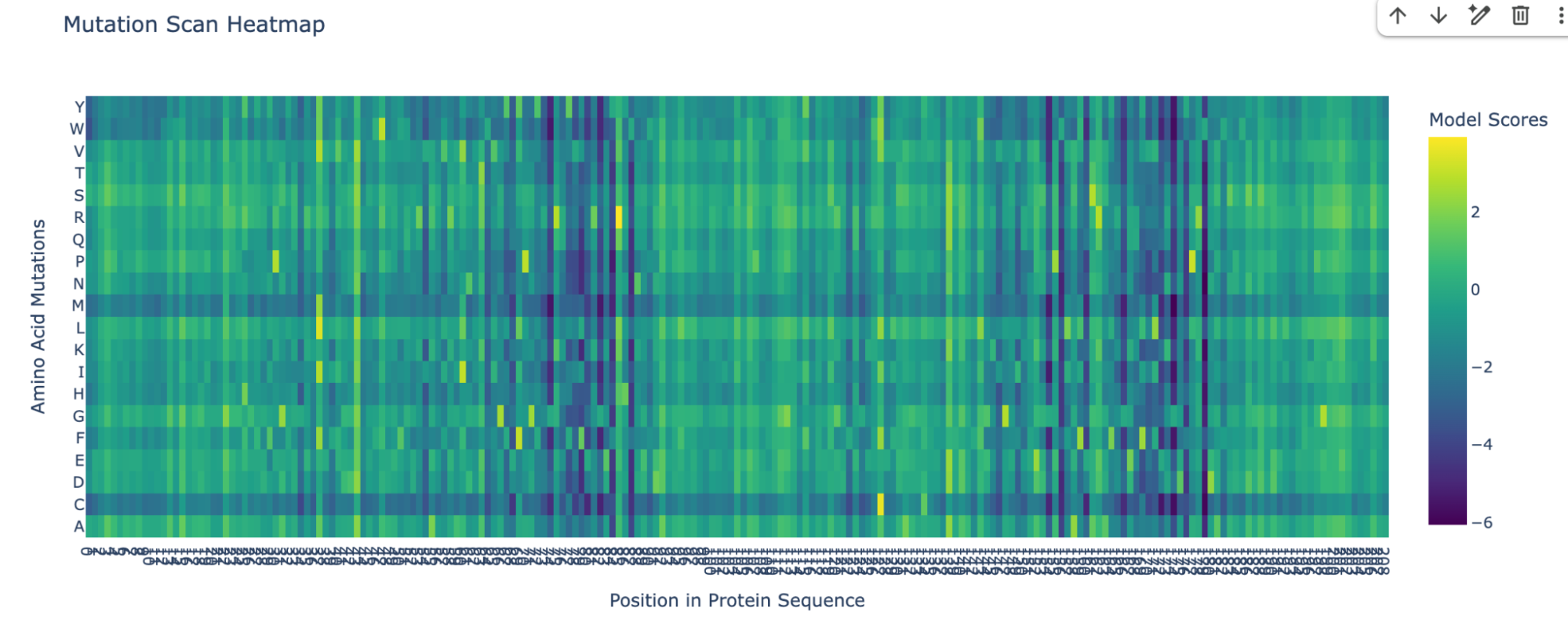
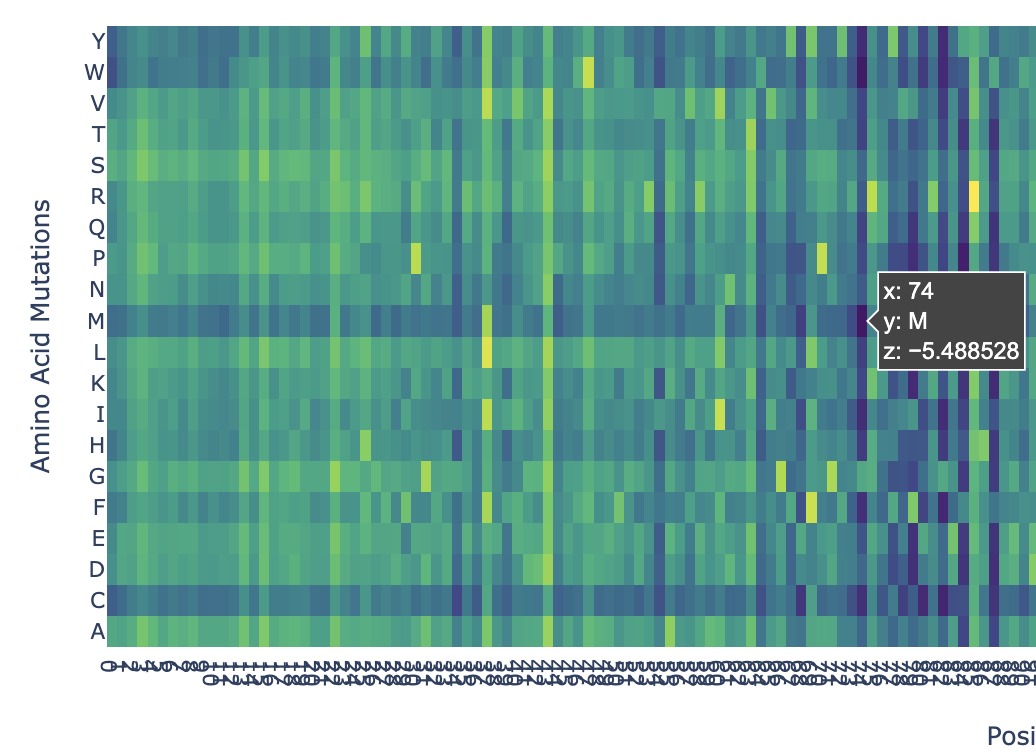
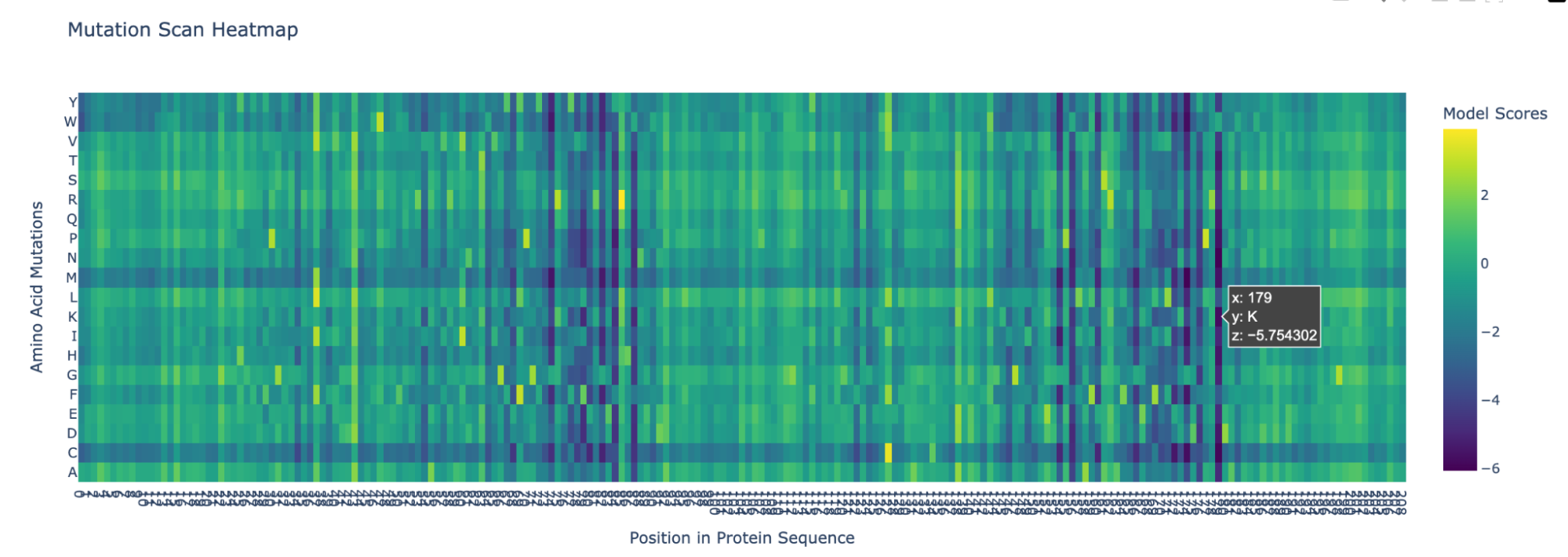
I pasted the sequence in the “Mutation Scans” section in the Google Colab, and run the code. This is the mutation scan heatmap obtained:
In the heatmap we can see that there are some vertical darker lines, meaning that position is important. For example, at position 74, the mutation of phenylalanine (F), tryptophan (W), methionine (M), isoleucine (I), and cysteine (C) shows dark purple, meaning this mutation would destabilize the protein because those amino acids are important for the protein’s core structure.
Here’s an example of how the methionine mutation is highlighted in position 74:
Position 179 also seems highly mutated:
2. Latent Space Analysis
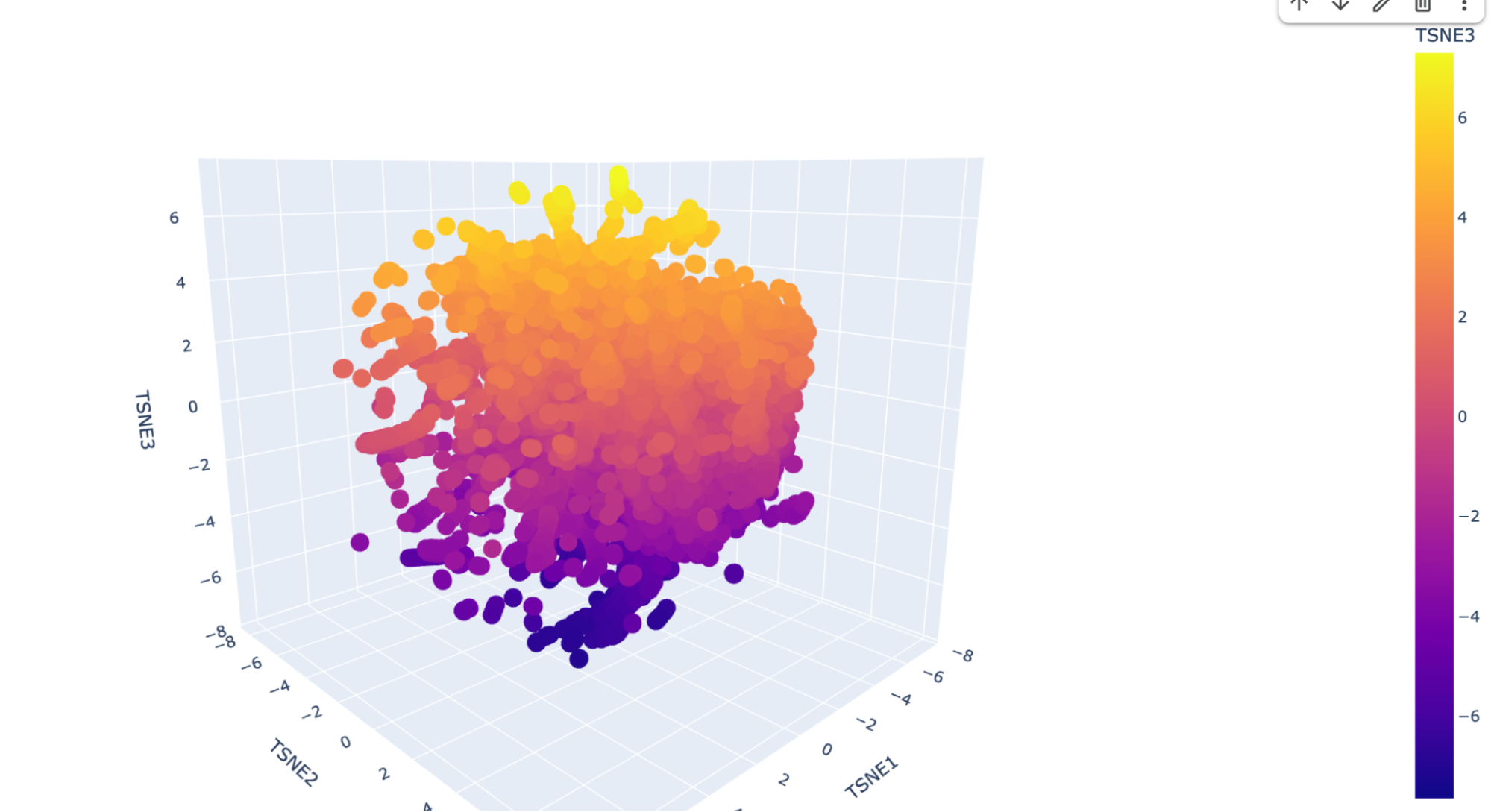
I ran the code as is and this is the visualization for the TSNE scatter plot:
It has a lot of data, and is very difficult to see where my protein (beta-crystallin B3) is. I checked multiple dots but could not find it, so I decided to ask Deepseek AI the following prompt: “I have the following code to do a Latent Space Analysis, but when I visualize the 3D scatter plot there are a lot of dots. Is there a way to find my protein, which is the human beta-crystallin B3 (sequence: MAEQHGAPEQAAAGKSHGDLGGSYKVILYELENFQGKRCELSAECPSLTDSLLEKVGSIQVESGPWLAFESRAFRGEQFVLEKGDYPRWDAWSNSRDSDSLLSLRPLNIDSPHHKLHLFENPAFSGRKMEIVDDDVPSLWAHGFQDRVASVRAINGTWVGYEFPGYRGRQYVFERGEYRHWNEWDASQPQLQSVRRIRDQKWHKRGRFPSS) {pasted code from the Google Colab }” and it suggested to add a new code that highlights my protein. This is the code:
{
# Find your protein in the embeddings
your_sequence = "MAEQHGAPEQAAAGKSHGDLGGSYKVILYELENFQGKRCELSAECPSLTDSLLEKVGSIQVESGPWLAFESRAFRGEQFVLEKGDYPRWDAWSNSRDSDSLLSLRPLNIDSPHHKLHLFENPAFSGRKMEIVDDDVPSLWAHGFQDRVASVRAINGTWVGYEFPGYRGRQYVFERGEYRHWNEWDASQPQLQSVRRIRDQKWHKRGRFPSS"
# Tokenize your sequence
your_tokens = tokenizer(your_sequence, return_tensors="pt")
with torch.no_grad():
your_outputs = esm2(input_ids=your_tokens['input_ids'], attention_mask=your_tokens['attention_mask'], output_hidden_states=True)
your_embedding = your_outputs.hidden_states[-1][0].mean(dim=0).cpu().numpy()
# Transform it with the same t-SNE
your_embedding_3d = tsne_3d.fit_transform(np.vstack([embeddings_array, your_embedding]))[-1]
# Print its coordinates
print(f"Your protein is at coordinates: TSNE1={your_embedding_3d[0]:.2f}, TSNE2={your_embedding_3d[1]:.2f}, TSNE3={your_embedding_3d[2]:.2f}")
# Find nearest neighbors
from scipy.spatial.distance import cdist
distances = cdist([your_embedding_3d], embeddings_3d)[0]
nearest_5 = np.argsort(distances)[:5]
print("\nYour 5 closest proteins in the plot:")
for i, idx in enumerate(nearest_5):
print(f"{i+1}. {protein_sequence_annotations[idx]} (distance: {distances[idx]:.3f})")
}
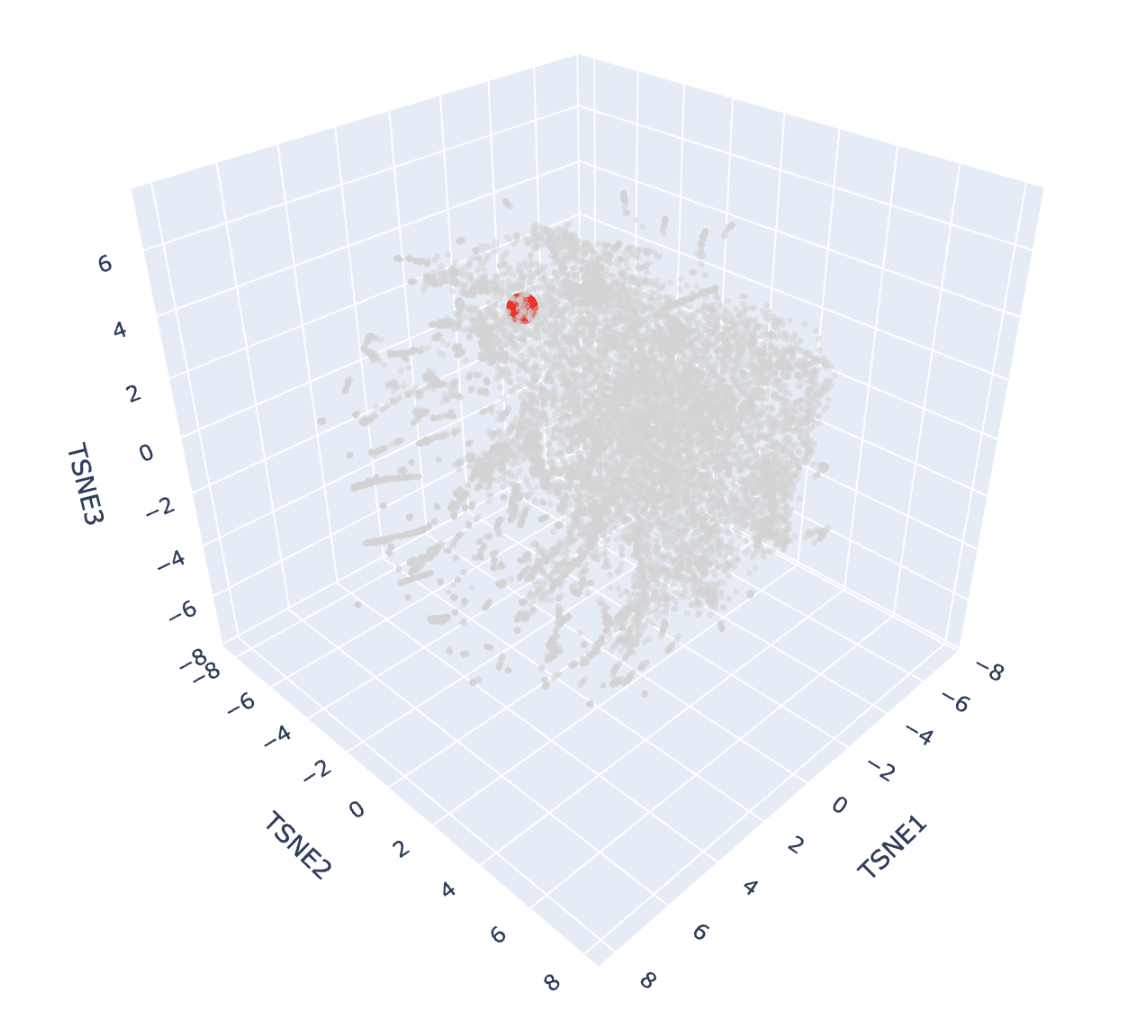
And here’s the scatter plot from that code:
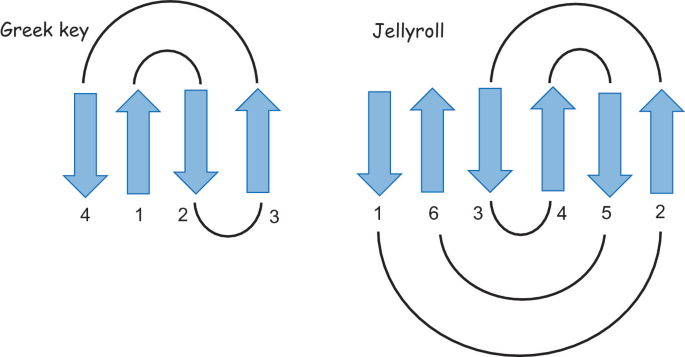
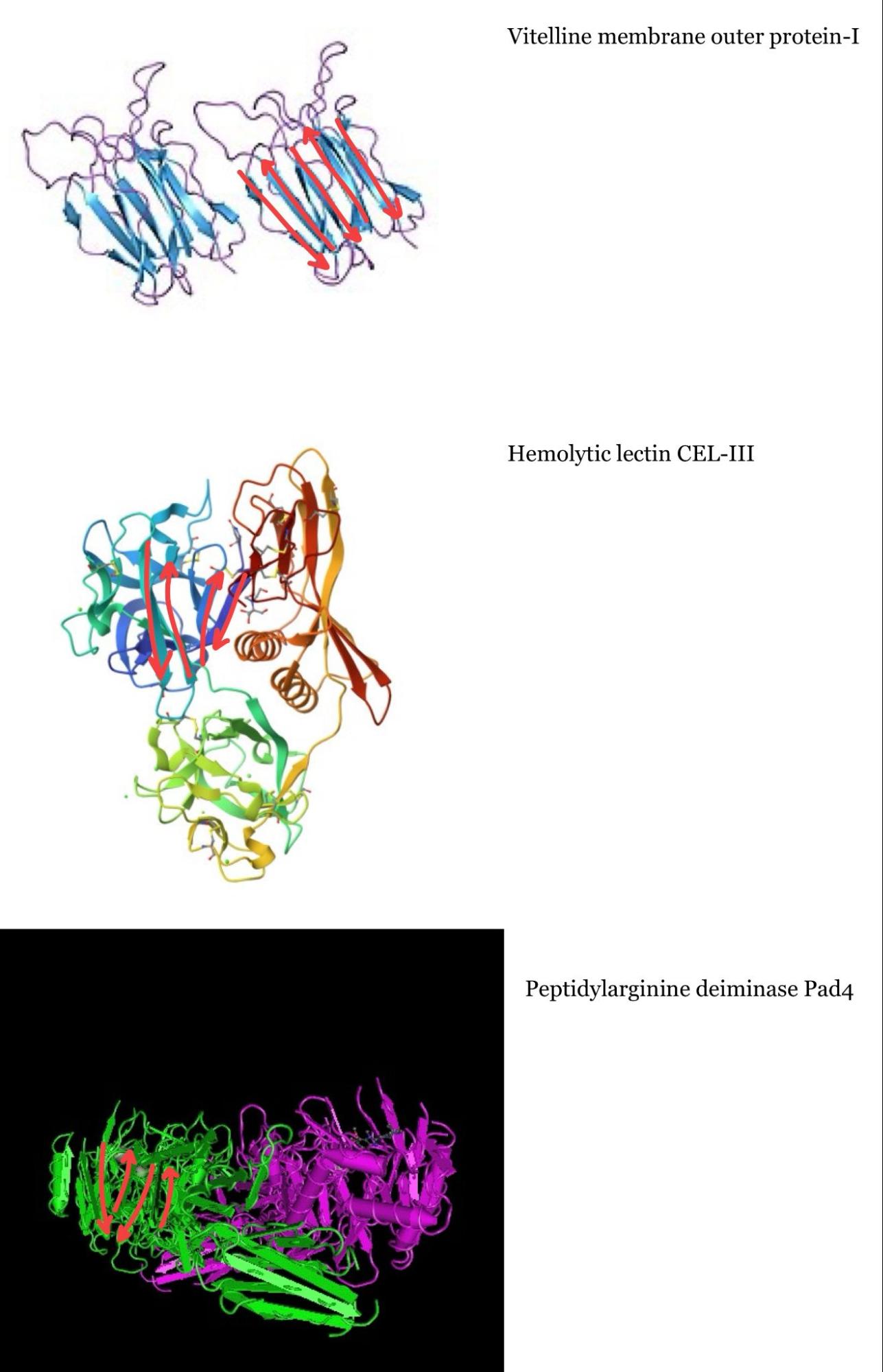
The protein is highlighted with the bright red dot. Thanks to this, I can now see better the proteins next to the one I picked. These are, for example: Vitelline membrane outer protein-I, a protein found on the vitelline membrane in a chicken egg; Hemolytic lectin CEL-III, a protein that binds to carbohydrates found in sea cucumbers; and Peptidylarginine deiminase Pad4, an enzyme that catalyzes post-translational modifications of proteins. It is interesting how different, at least in function, each of these proteins are with beta-crystallin B3. However, all of them have a structural similarity called the Greek key fold (see image below, taken from Piumetti, 2022).
I tried to identify the Greek key fold in each of the three proteins similar to mine, but it was more challenging than I thought, so maybe my drawings are incorrect:
C2. Protein Folding
Folding a protein
1. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
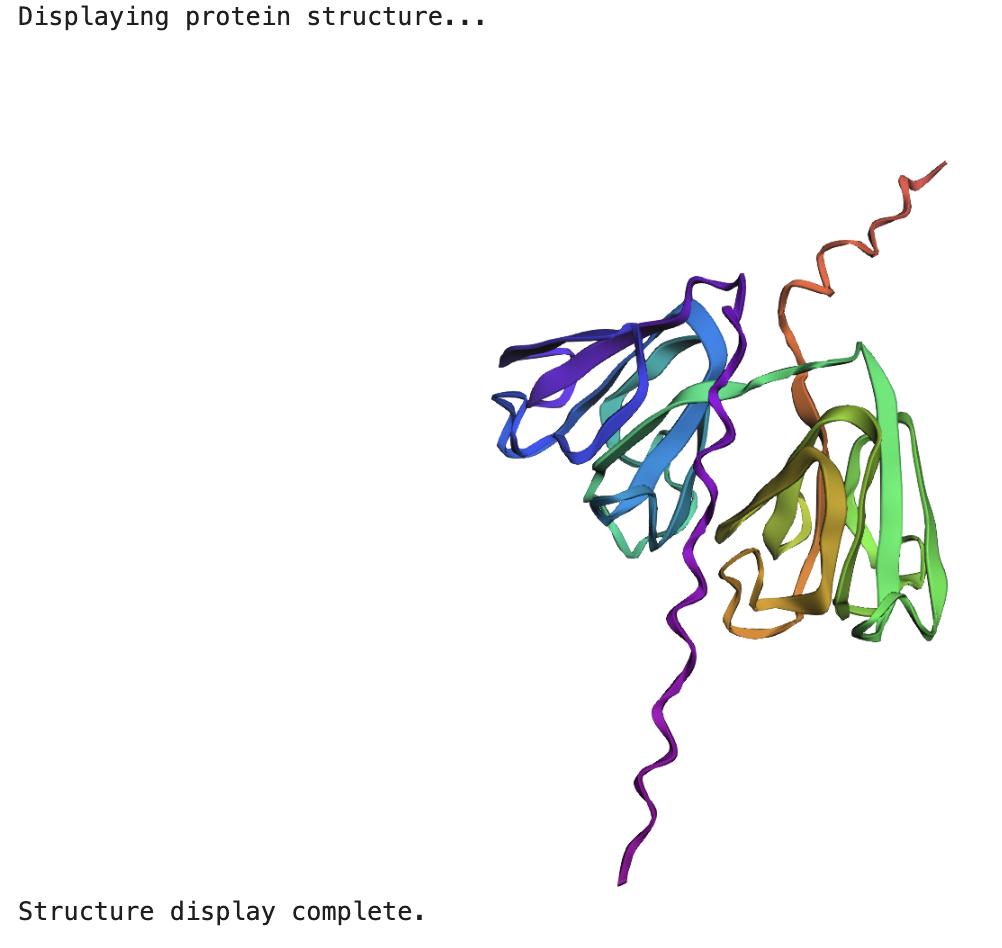
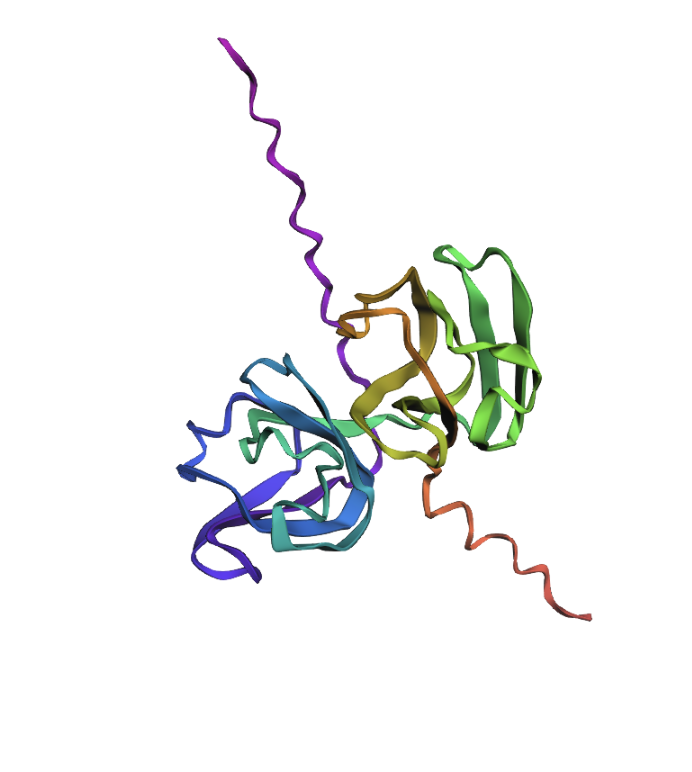
When running the ESMFold Protein Folding cells, this is the image for the protein:
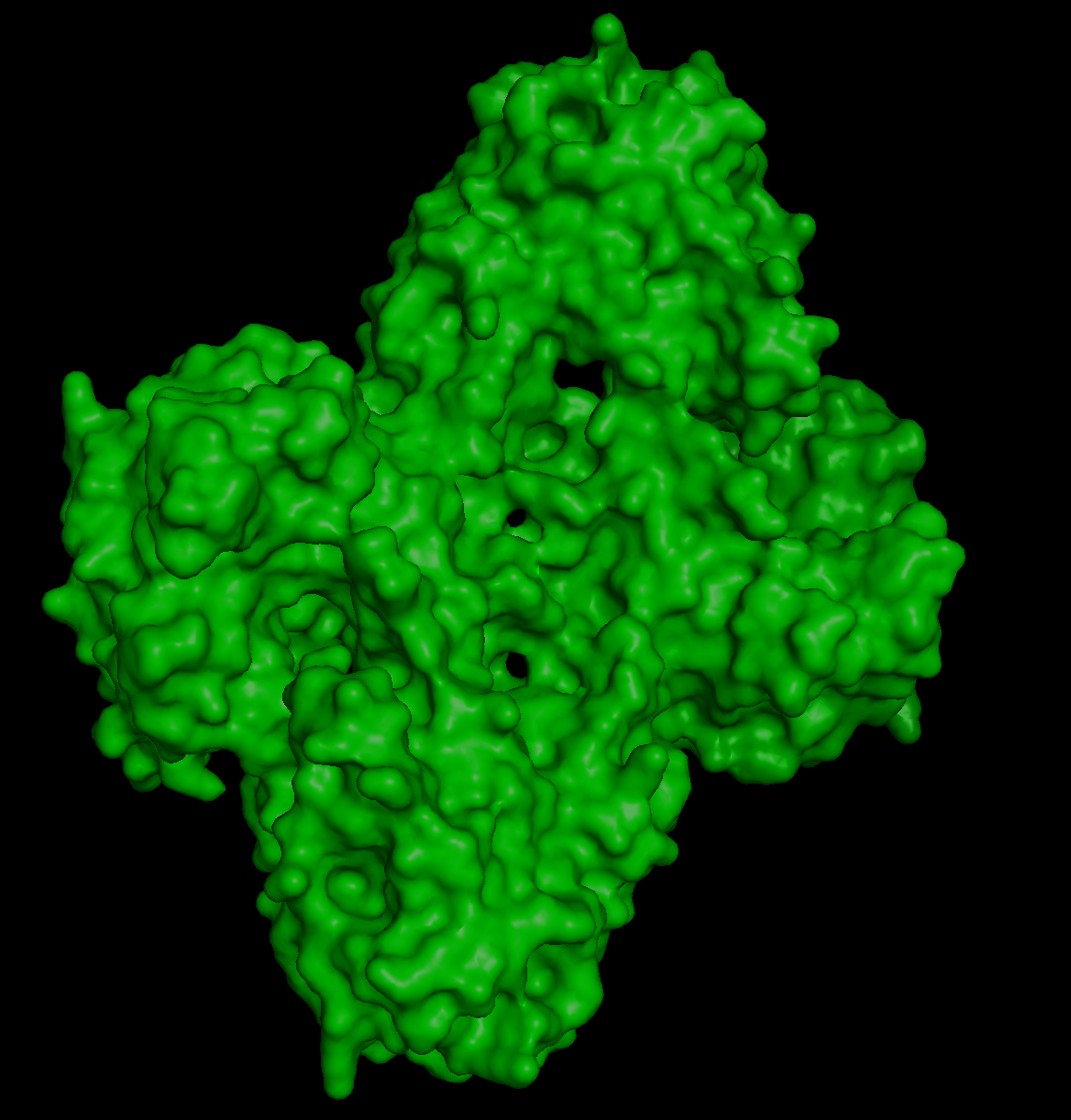
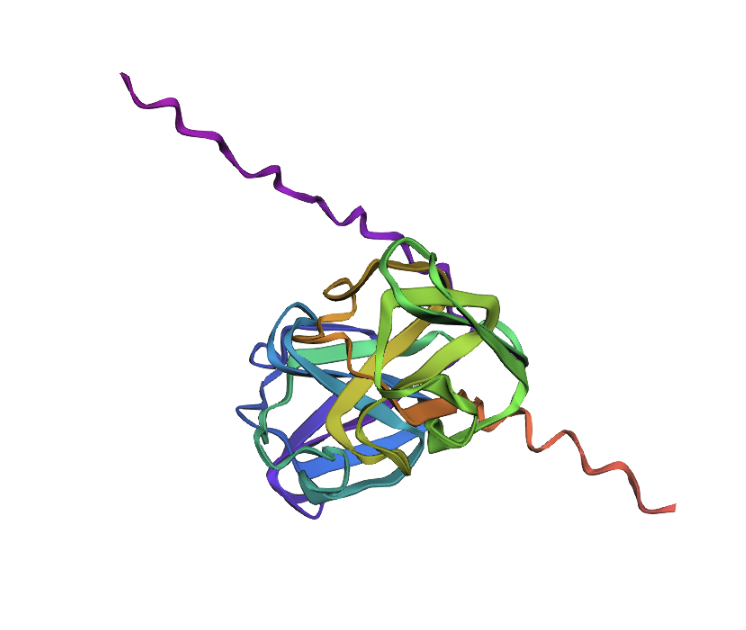
And this is the 3D structure in RCSB:
Both are different from each other. The experimental structure in RCSB (ID: 3QK3) contains 184 amino acids. However, the full human beta-crystallin B3 sequence from UniProt is 211 amino acids long.
I researched more about the actual structure of beta-crystallin B3, and it seems like it should have two beta-sheet domains. When I folded the complete sequence with ESMFold, the prediction shows the beta-crystallin B3 with those two beta-sheet domains. The first domain could match the 3QK3 structure, so the ESMFold model could be very accurate in this sense. The second domain is lacking, which could be due to the experimental approach in the RCSB structure, which is X-ray diffraction. Maybe the second domain was not achieved with the experimental approaches, only the first one crystallized successfully.
2. Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
Then, I tried to mutate the amino acid sequence by changing just one of them first. Here’s how the sequence starts: “MAEQHGAPEQAAAGKSH…”
I changed it to this: “MAEQHGAPEQRAAGKSH…” So the alanine (A) in position 11 was changed to a arginine (R). The structure looks the same as before:
I now want to add another two mutations to have three total. This is the new mutated sequence: ““MAEQHGAPEQRNEGKSH…” and the structure still looks the same:
So I would conclude that the model was quite resilient to mutations (for these positions at least).
C3. Protein Generation
Inverse-Folding a protein:
Here are the input options I introduced in the Helper section of the code, which are identifiers and characteristics of my protein, human beta-crystallin B3:
This is the output:
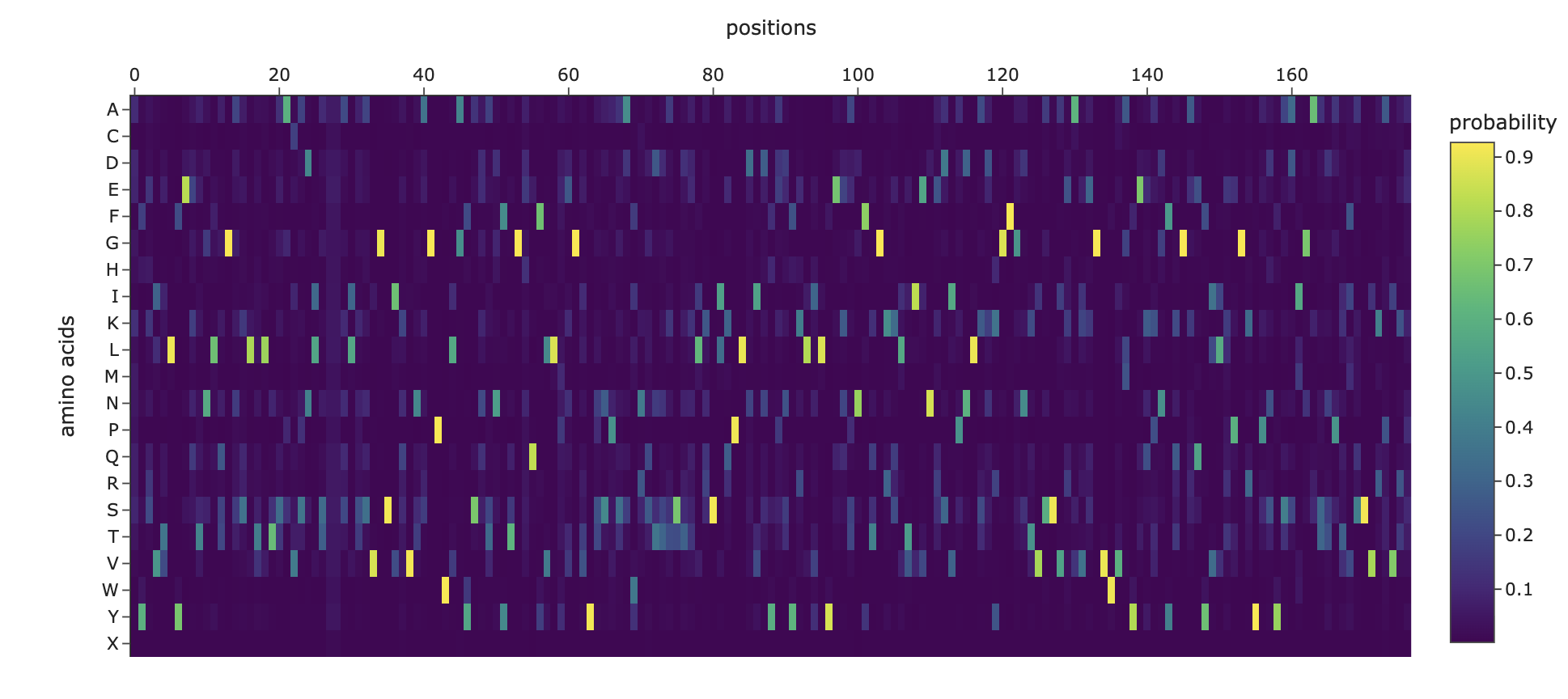
1. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
For the original protein sequence the score is 1.5725, and for the new generated sequence the score is 0.8219, which is lower. Lower means the model is more confident in this new generated sequence. Also, the seq_recovery is at 0.4000 which means 40% of the amino acids match the original sequence.
And this is the heat map for the amino acid probabilities:
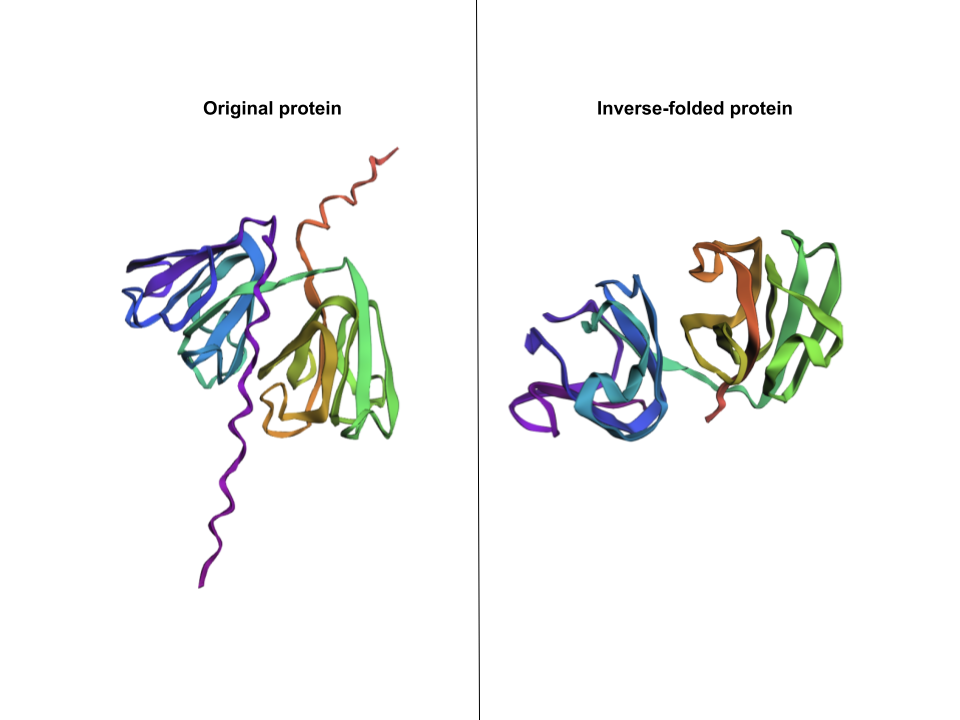
2. Input this sequence into ESMFold and compare the predicted structure to your original.
The new generated sequence is the following: “DYEIILYEKENLQGNSLTLTSAVSDLSXXKLSSVGSIKVVKGPWLAYSNKNYTGEQFVLPEGVYNSISDIRQDTSSTEIKSIKPLDIDYDTFELVLYEEENFQGKKLTIVNESVPNLADKGFGNTVSSAEAKKGVWVLYEKPNYQGRQFVLEPGKYPNYKDMGMSTPTVSSVKPVKK”
I pasted that one onto ESMFold and here’s the comparison:
This structure does capture beta-crystallin B3’s two beta-sheet domains, but is missing the two coils that go through the protein.
Piumetti, M. (2022). Structure of Proteins. In: Molecular Dynamics and Complexity in Catalysis and Biocatalysis. Springer, Cham. https://doi.org/10.1007/978-3-030-88500-7_1
Part D. Group Brainstorm on Bacteriophage Engineering
Proposal: Engineering the MS2 Lysis Protein L to Enhance Stability
Background
The MS2 bacteriophage lysis (L) is a 75 amino acid long-protein, and it is responsible for triggering host cell lysis, this is why it is also called a toxin from the group of bacteriophages (Mezhyrova, 2023). It is a powerful protein that has been used widely in studies where researchers seek to control cell death, but it is difficult to do so due to its instability (Mylon, 2010).
Objectives
To use computational protein design tools to engineer possible stable variants of the MS2 L-protein.
To identify variants where structure is preserved but higher stability is shown
Analyze if the variants can fold like the original protein and if they interact correctly with DnaJ, its chaperone
Methods
Obtain an initial protein backbone for MS2 L-protein using ESMFold
Obtain alternative sequences to the protein using ProteinMPNN
Mutate the alternative sequences to the protein using ESM-2
Model the variants using ESMFold and analyze if the folding is maintained in comparison to the initial protein (MS2 L-protein). Create a ranking based on confidence metrics
Assess the top 3 variant’s interactions with DnaJ using AlphaFold-Multimer to predict 3D structures of protein complexes (co-folding multiple chains)
Expected Outcomes
Hopefully, these methods are able to identify some stabilized MS2 L-protein variants with correct folding and interaction with its chaperone, DnaJ. If successful, these designs could serve as templates for further experimental testing in E. coli and provide a methodology adaptable to other phage‑derived membrane proteins that also show decreased stability.
Potential Challenges
The main concern would be generating variants with correct folding, but incorrect interaction with DnaJ, as computational models sometimes can not predict the dynamics of those interactions (Chamakura et al., 2017 & Mondal et al., 2024), thus disrupting the lysis mechanism.
References
Mezhyrova, J., Martin, J., Börnsen, C., Dötsch, V., Frangakis, A. S., Morgner, N., & Bernhard, F. (2023). In vitro characterization of the phage lysis protein MS2-L. Microbiome Research Reports, 2(4), 28.
Mylon, S. E., Rinciog, C. I., Schmidt, N., Gutierrez, L., Wong, G. C., & Nguyen, T. H. (2010). Influence of salts and natural organic matter on the stability of bacteriophage MS2. Langmuir, 26(2), 1035-1042.
Chamakura, K. R., Tran, J. S., & Young, R. (2017). MS2 lysis of Escherichia coli depends on host chaperone DnaJ. Journal of Bacteriology, 199(12), 10-1128.
Mondal, A., Singh, B., Felkner, R. H., De Falco, A., Swapna, G. V. T., Montelione, G. T., … & Perez, A. (2024). A Computational Pipeline for Accurate Prioritization of Protein‐Protein Binding Candidates in High‐Throughput Protein Libraries. Angewandte Chemie International Edition, 63(24), e202405767.
Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design
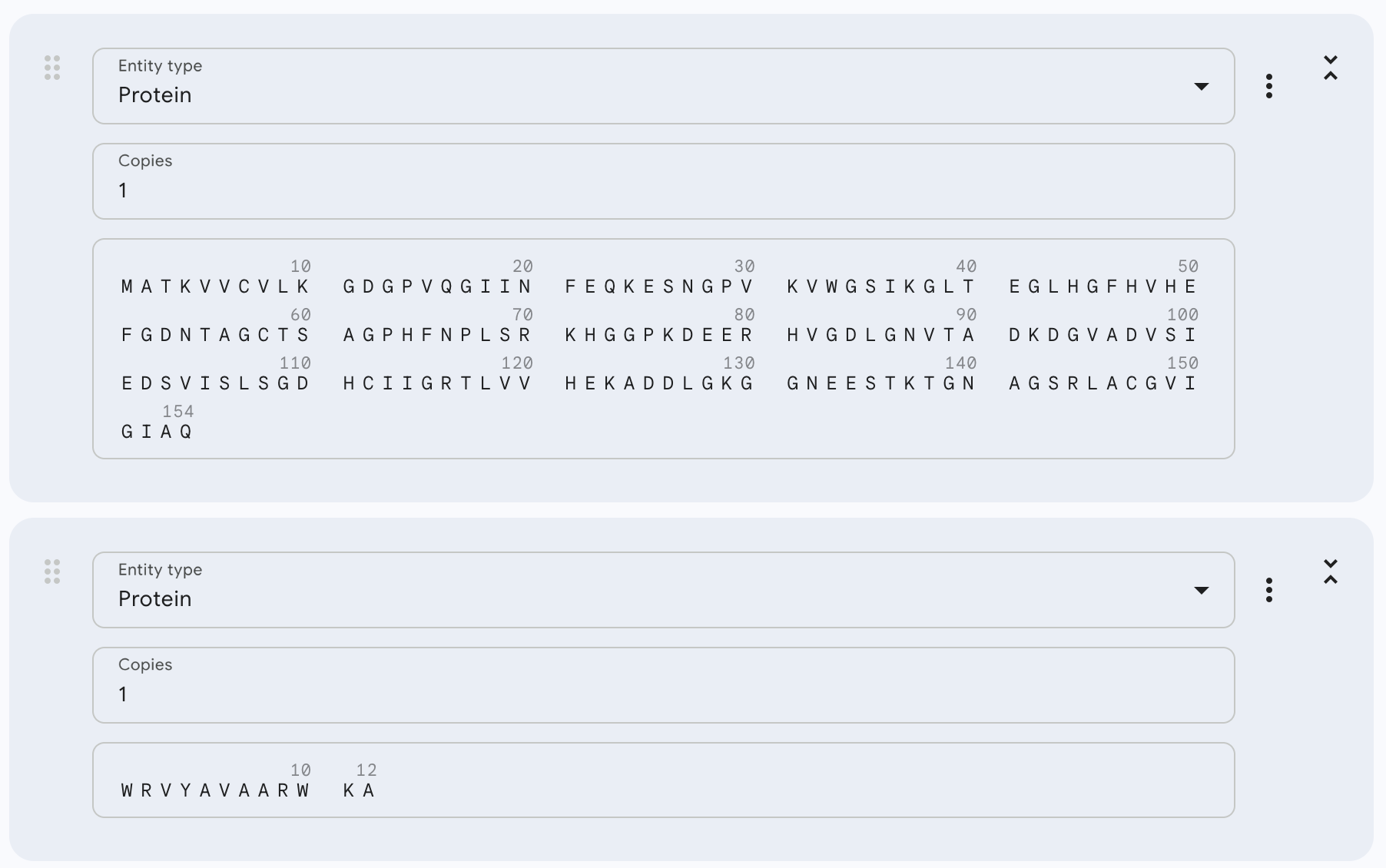
Part 1: Generate Binders with PepMLM
To get the human SOD1 sequence, I went to UniProt. The ID for this protein is P00441 and the sequence is the following:
That mutated sequence was introduced in the PepMLM-650M Colab, changing the peptide length to 12 and the number of binders to 4:
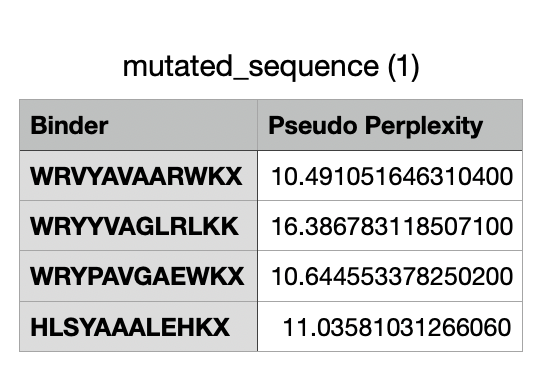
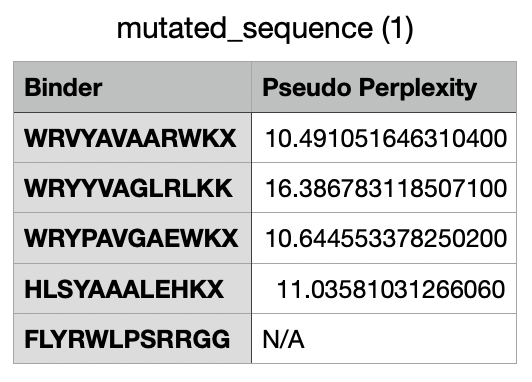
And after generating the peptides, here’s the results:
For the four generated binders, perplexity values vary. But, in general, a lower perplexity means a more confident model of the protein.
And this would be the list of all the binders, including the known binder last.
Part 2: Evaluate Binders with AlphaFold3
Before entering the mutated SOD1 sequence with each binder, I need to modify the sequence for each one because AlphaFold3 does not read B, J, O, U, X, Z characters, which are unknown in the sequence. I googled if there’s a way to solve this, and found in a Reddit post that AlphaFold suggests changing these to “A” (alanine), so these would be the new binder sequences:
WRVYAVAARWKA
WRYYVAGLRLKK
WRYPAVGAEWKA
HLSYAAALEHKA
FLYRWLPSRRGG (known binder)
Now, in AlphaFold, I introduced the mutant SOD1 sequence and then added the binder sequence like this:
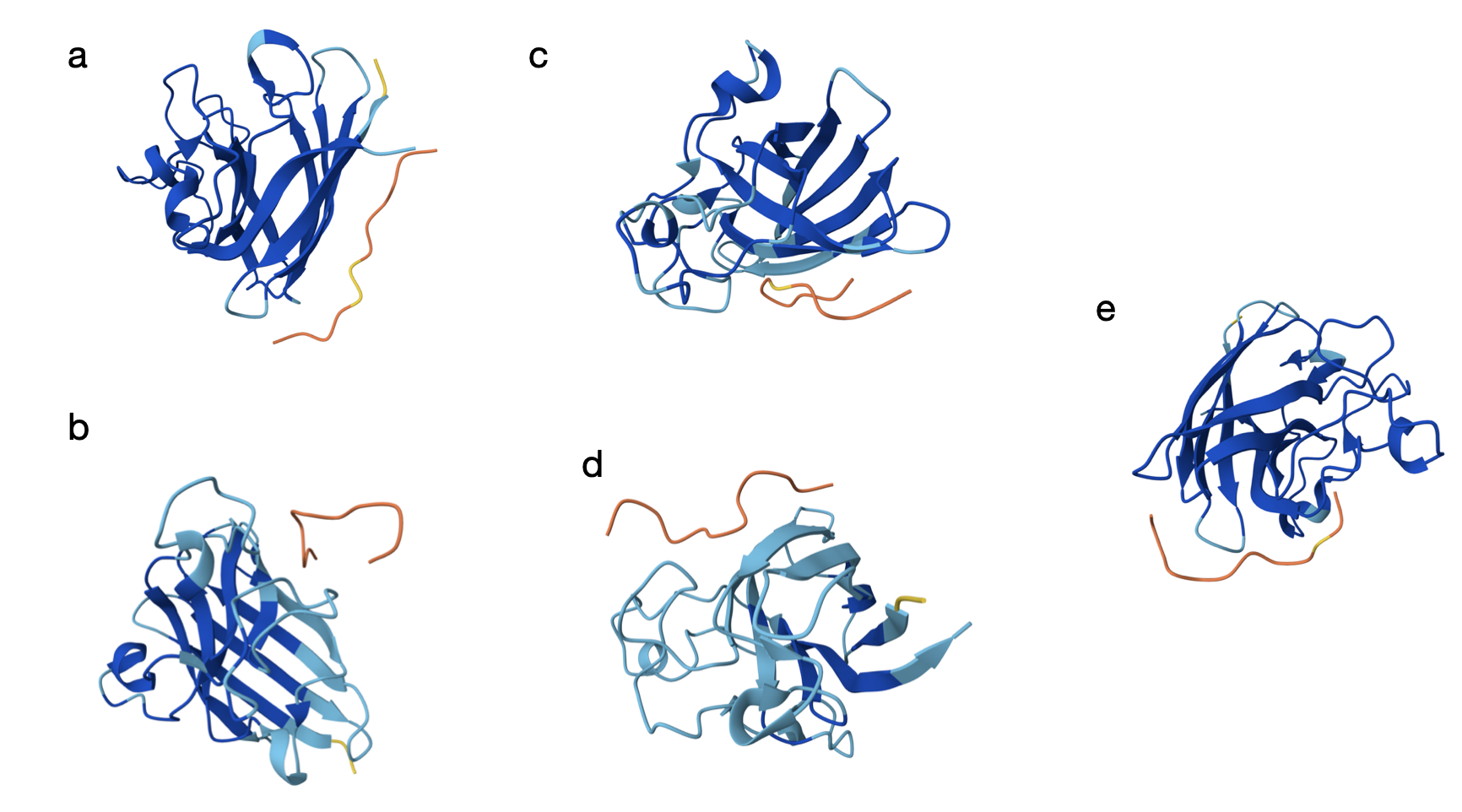
After running the sequence with each binder, the AlphaFold Server creates an entry with an ipTM score, a pTM score, the 3D model of the protein with its binder, and a graph.
Here are the ipTM scores for all peptides:
a (WRVYAVAARWKA) = 0.34
b (WRYYVAGLRLKK) = 0.3
c (WRYPAVGAEWKA) = 0.36
d (HLSYAAALEHKA) = 0.28
e(FLYRWLPSRRGG - known binder) = 0.35
Here are the models generated by AlphaFold for each:
From the models, the orange and yellow segments (the peptides) appear to bind on the outer surface of the SOD1 monomer rather than in the core. In most panels (a–c and e), it seems the peptide is located along the edge of the β-barrel, but not inserting into the barrel. The binding site does not look centered directly at the extreme N-terminus where the A4V mutation lies, but it is still in the same general surface region. As shown in the models, they seem mostly surface-bound with only partial shallow contacts (which is a bit discouraging).
The ipTM scores are all fairly modest and clustered in a narrow range (0.28–0.36), which suggests weak-to-moderate confidence in the predicted complexes. The best PepMLM-generated peptide is c (WRYPAVGAEWKA) with 0.36, which slightly exceeds the known binder (FLYRWLPSRRGG, 0.35) used as a control for this particular test. Peptides a (0.34) and b (0.30) are similar but slightly lower, while d (0.28) is the weakest. At least one generated peptide (c) performs a bit better than the known binder based on ipTM.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
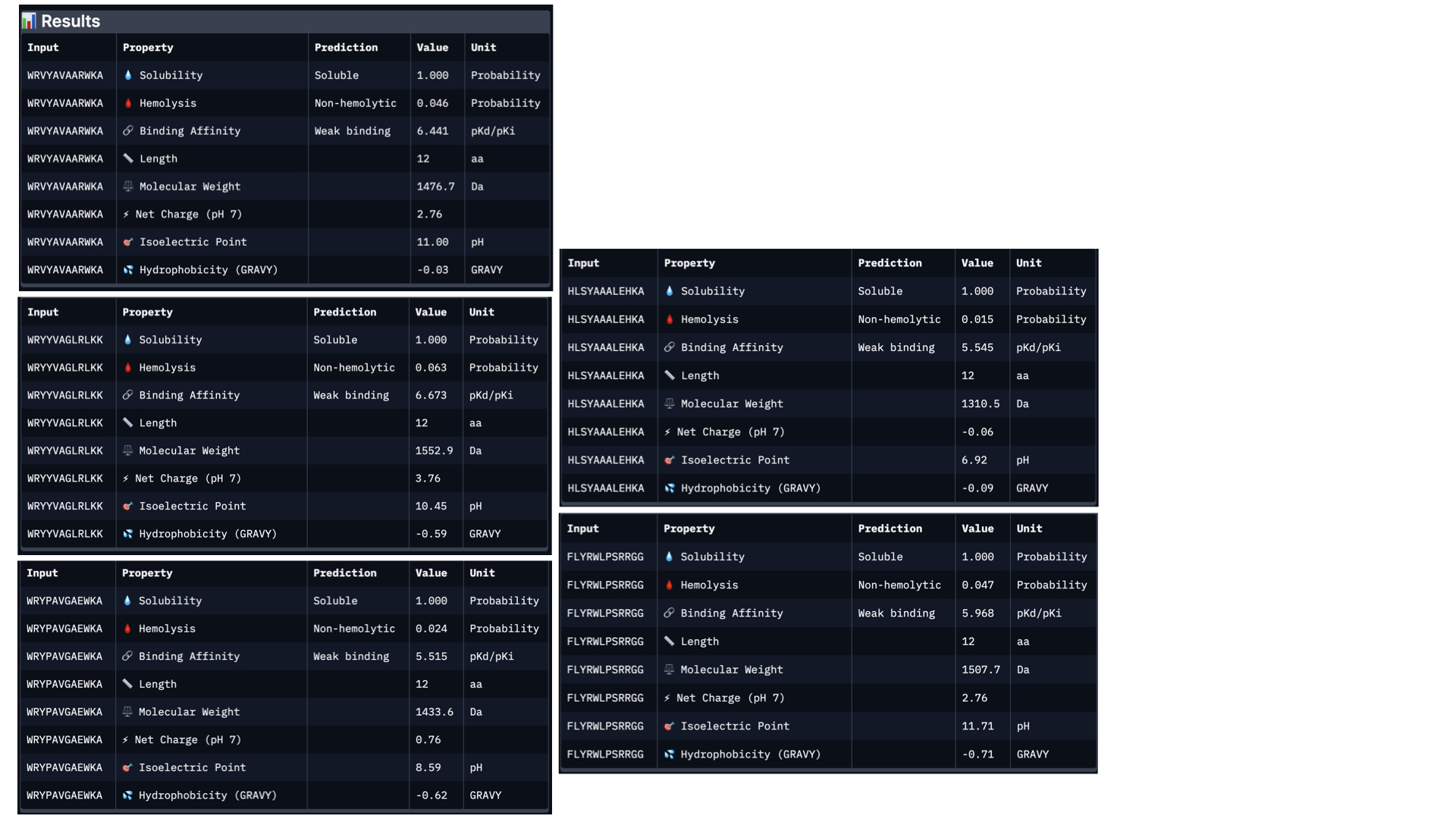
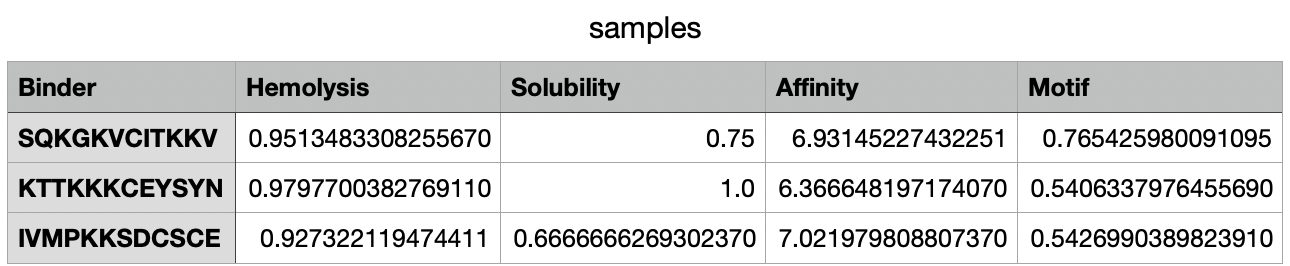
In PeptiVerse, I introduced the mutated human SOD1 sequence with each one of the five binders. Here are the results for each of them:
Analysing all peptides, it seems that all of them are predicted to be soluble, which is therapeutically good. Additionally, the hemolysis probability indicates how likely is the peptide able to destroy red blood cells; the lower the value, the better. All peptides show a relatively low hemolysis probability, being peptide #d the safest (sequence HLSYAAALEHKA). In terms of binding ability, a higher value indicates a stronger predicted binging; being peptide #b (sequence WRYYVAGLRLKK) the strongest. Finally, lower hydrophobicity levels are better because the peptides are more soluble. Just like with solubility, all have acceptable hydrophobicity levels.
In conclusion, the best peptide could be peptide #a (WRVYAVAARWKA), showing good solubility, low hydrophobicity, relatively strong predicted binding affinity, and low hemolysis risk. Overall, this peptide represents the most balanced option for further development, taking into account the results from PeptiVerse alone.
When comparing the two prediction methods (AlphaFold and PeptiVerse), there is no clear link between them. Peptide b had the strongest predicted binding affinity in PeptiVerse but a lower score in AlphaFold3, while peptide c scored highest in AlphaFold3 but did not have the strongest predicted affinity. This proves the two tools are measuring different things.
On the positive side, none of the peptides raised safety concerns, as all were predicted to be soluble and had low risk of harming red blood cells.
Taking everything into account, peptide a (WRVYAVAARWKA) offers the best overall balance. It performs reasonably well in both prediction methods and has good safety properties.
Part 4: Generate Optimized Peptides with moPPIt
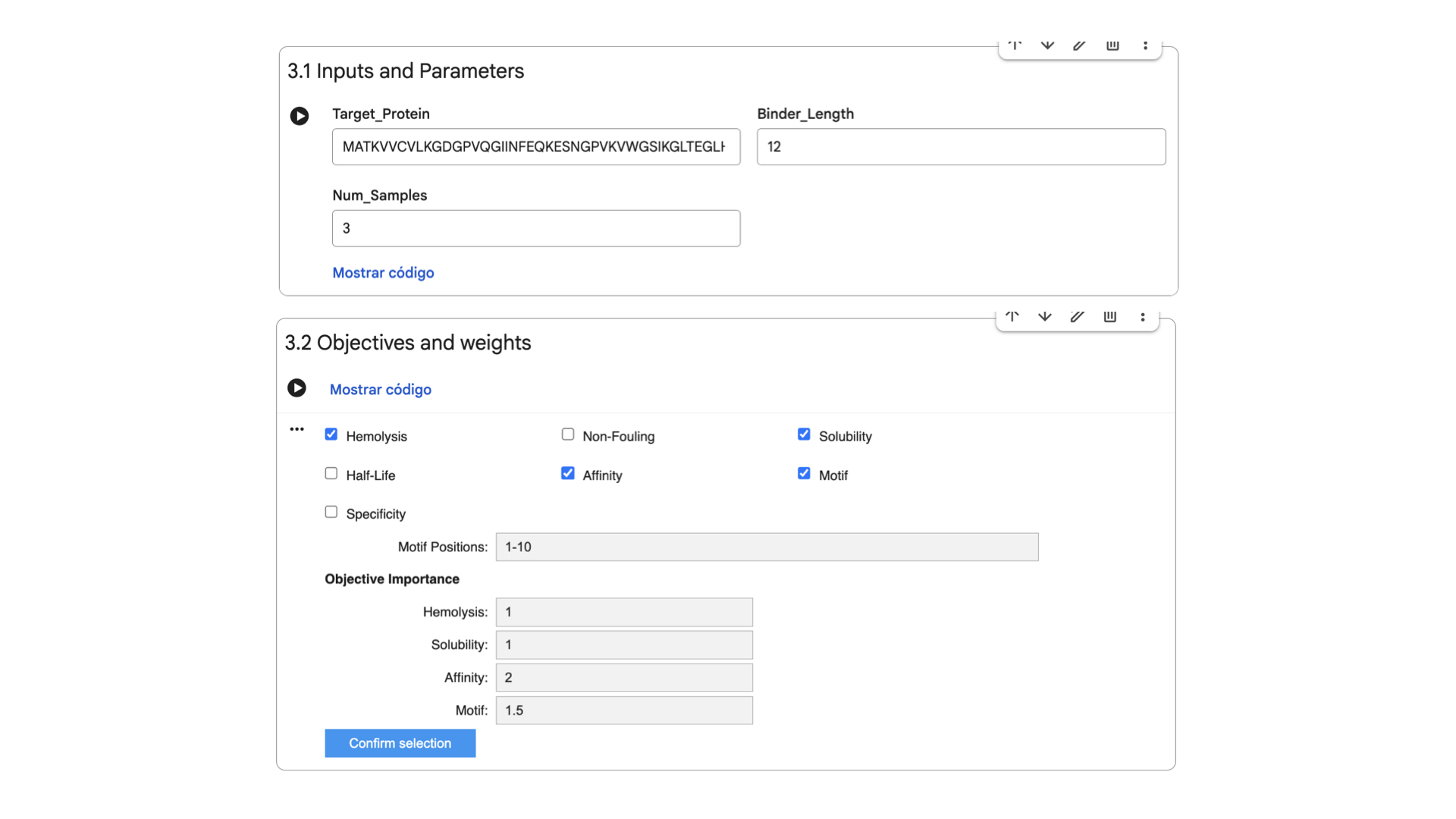
These are the settings for the moPPit Colab I adjusted:
Hemolysis and Solubility have the same weight to maintain therapeutic properties (as discussed in Part 3). Affinity has the highest weight to achieve strong binding, while Motif maintains a medium weight to ensure binding near the mutation.
After 38 minutes running, the .csv with the results was created:
Part C: Final Project: L-Protein Mutants
For this part, I decided to follow the step by step for L-Protein Engineering, which is Option 1: Mutagenesis.
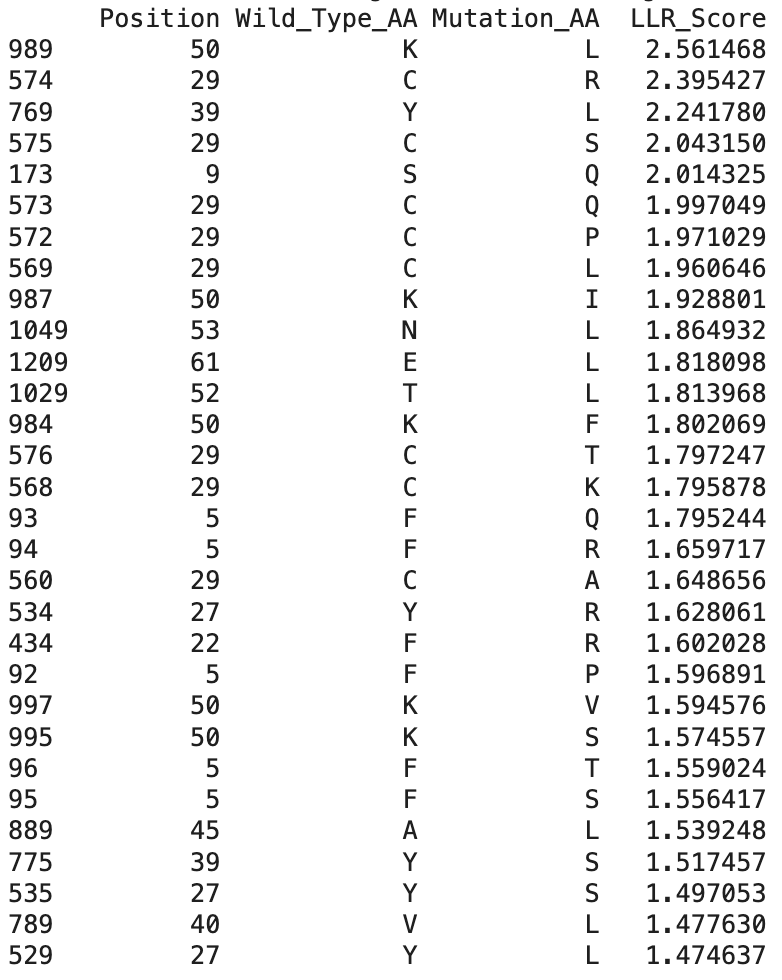
After running the ESM-2 Colab, I downloaded the L-protein mutants dataset as a csv and uploaded it to the Colab; and then obtained the file top_30_protein_mutations_scores.csv.
These are the top 30 protein mutations that come from the ESM-2 model. The model produces an LLR score for each mutation type at each residue position. If the LLR score is higher than 2, the mutation is strongly supported, between 1 and 2 is tolerated, between 0 and 1 is weakly tolerated, and below 0 is most probably damaging.
And here are the identified mutations from the experimental results:
When comparing ESM-2 model predictions with the data from the real experiments, I can see that several top LLR mutations occur at the same positions experimentally, which means that the mutations are tolerated:
Position 29 (C)
Position 50 (K)
Position 39 (Y)
The highest scoring mutation in the ESM-2 model predictions was K50L, with a LLR score of 2.56. This is a position in the transmembrane region, and is a change from lysine to leucine.
Also, some experimentally functional mutations from the L-Protein mutants dataset correspond to positive LLR scores from the ESM-2 model predictions. This means that the evolutionary model partially successfully identifies mutations that maintain protein stability and function.
Now, moving onto picking the mutations, I wanted to pick the mutations that are not the most obvious, so: that come from experimentally validated data, that are not the most repeated mutations in the table, and that can affect different biochemical properties.
The first thing to check is that the mutation shows lysis activity (so lysis = 1 on the table), and then check the region (soluble/transmembrane) and reflect on the type of amino acid. Here are the mutations I picked:
1. P13L
The experimental dataset shows Lysis = 1 and Protein = 1, the mutation is in the soluble region, and changing proline to leucine may increase folding flexibility (Yu et al., 2015) in the soluble domain of the protein.
2. R19H
The experimental dataset shows Lysis = 1, the mutation is in the soluble region, and changing arginine to histidine may modify electrostatic interactions (according to Muller et al., 2019), that could be interesting to see.
3. R20W
The experimental dataset shows Lysis = 1, the mutation is in the soluble region, and changing arginine to tryptophan may alter protein to protein interactions because of its hydrophobic characteristics, maybe stabilizing the protein further (Swift & Stewart, 1991).
4. A45P
The experimental dataset shows Lysis = 1, the mutation is in the transmembrane region, and changing alanine to proline could help in forming or changing the pore structure (Lee et al., 2003)
5. R18G
The experimental dataset shows Lysis = 1, the mutation is in the soluble region, and changing arginine to glycine could help in flexibility and folding of the protein (Gekko et al., 1994).
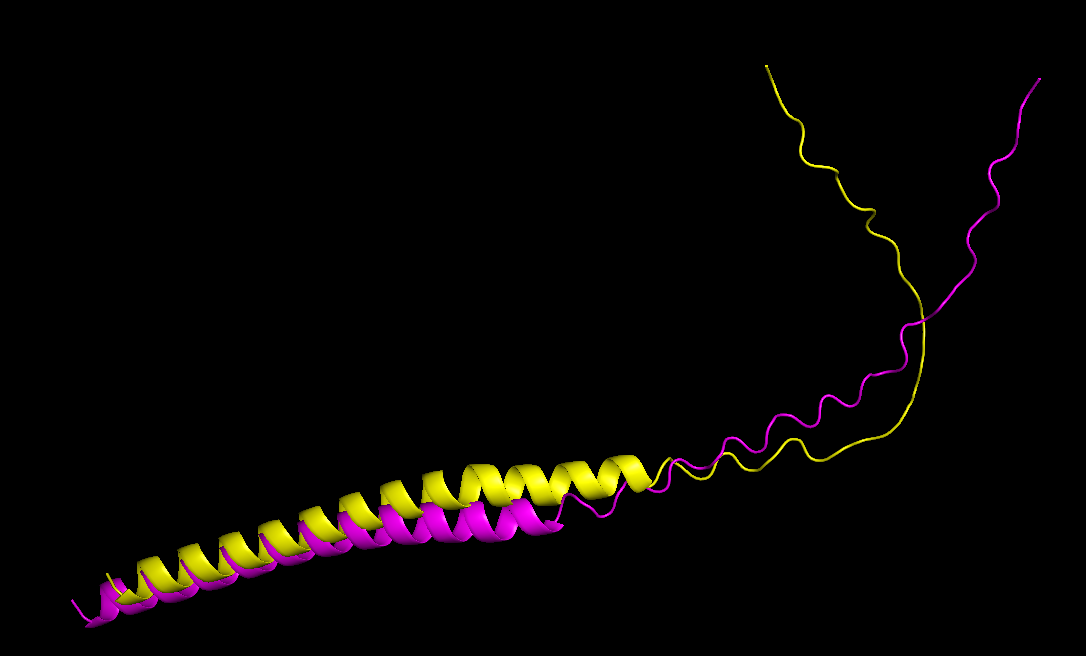
After choosing these 5 mutations, I will predict the 3D structure of the mutated L-protein and comparing it to the wild type.
I then went to ESM Fold and downloaded the PDB file to the predicted protein structure for the wild type sequence, and the predicted protein structure for my mutant sequence:
I uploaded those PDB files to PyMOL to visualize both structures better:
The mutant sequence is yellow, and the wild type is pink.
Comparing both structures, the mutant model maintained a similar overall fold, only with a more folded coil, which could mean that the mutations are unlikely to disrupt protein structure.
References:
Muller, L., Jackson, S. N., & Woods, A. S. (2019). Histidine, the less interactive cousin of arginine. European Journal of Mass Spectrometry, 25(2), 212-218.
Yu, H., Zhao, Y., Guo, C., Gan, Y., & Huang, H. (2015). The role of proline substitutions within flexible regions on thermostability of luciferase. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics, 1854(1), 65-72.
Swift, S., & Stewart, G. S. (1991). The molecular biology of tryptophan synthase: A model for protein-protein interaction. Biotechnology and Genetic Engineering Reviews, 9(1), 229-294.
Lee, D. J. S., Keramidas, A., Moorhouse, A. J., Schofield, P. R., & Barry, P. H. (2003). The contribution of proline 250 (P-2′) to pore diameter and ion selectivity in the human glycine receptor channel. Neuroscience letters, 351(3), 196-200.
Gekko, K., Kunori, Y., Takeuchi, H., Ichihara, S., & Kodama, M. (1994). Point mutations at glycine-121 of Escherichia coli dihydrofolate reductase: important roles of a flexible loop in the stability and function. The Journal of Biochemistry, 116(1), 34-41.
Week 6 HW: Genetic Circuits Part I: Assembly Technologies
Assignment: DNA Assembly
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion High-Fidelity PCR Master Mix contains all the core reagents necessary for accurate and efficient DNA amplification:
Phusion DNA Polymerase: A high-fidelity enzyme with 3′→5′ exonuclease proofreading activity that minimizes errors during DNA synthesis, especially important for mutation-based cloning (NEB, 2023).
dNTPs (deoxynucleotide triphosphates): Provide the nucleotide building blocks (A, T, G, C) for DNA strand elongation.
Reaction Buffer (with Mg²⁺): Maintains the ionic strength and conditions needed for optimal enzyme activity and DNA strand stability.
Stabilizers & enhancers: Help maintain enzyme performance across temperature ranges and buffer pH changes during thermocycling.
(New England Biolabs (NEB), 2023).
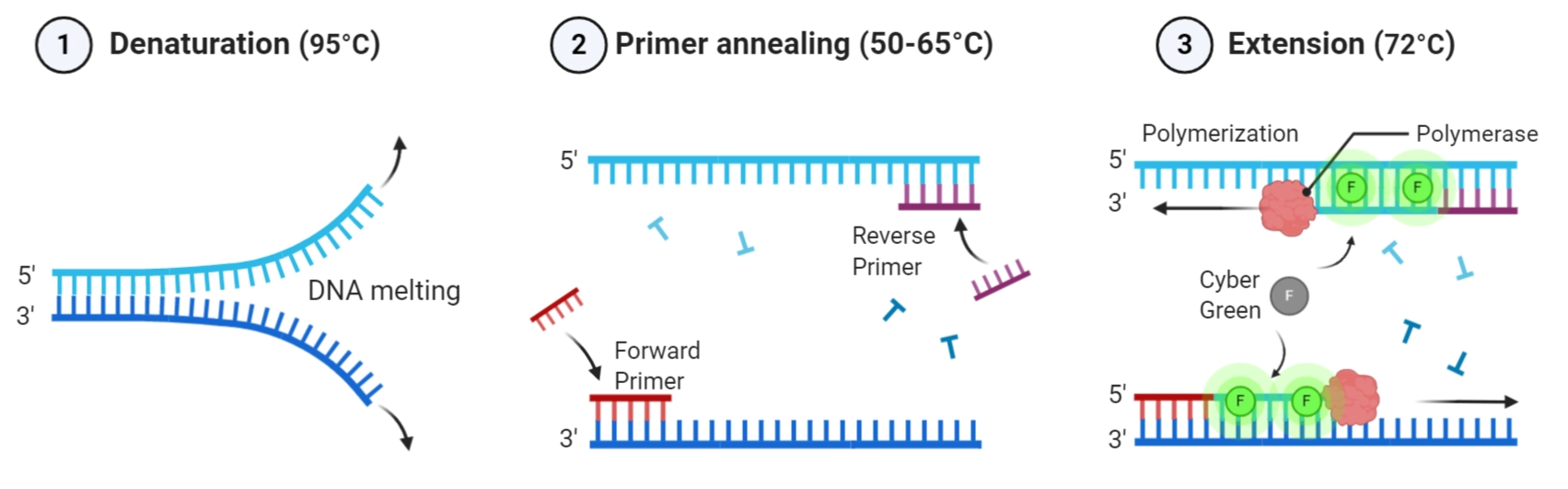
2. What are some factors that determine primer annealing temperature during PCR?
The annealing temperature (Ta) determines primer binding specificity. It depends mainly on:
Primer length: Longer primers (18–22 bp typical) increase Tm (melting temperature).
GC content: G–C pairs form three hydrogen bonds (vs. the two hydrogen bonds in A–T pairs), making high GC content primers bind more tightly (raising Tm).
Salt concentration (especially Mg2+): Stabilizes primer-template binding.
Secondary structures: Hairpins or dimers can lower effective primer availability, thus altering temperature behavior.
Complementarity between primers and template: Mismatches like the ones introduced for mutagenesis in this particular lab can reduce effective binding and could require lower Tas.
(Addgene Primer Design Guide; Primer 3 Manual, 2022.)
Primers in this lab are designed for melting temperatures between 52–58 °C, within 5 °C of each other for optimal pairing.
3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
In this lab, PCR is used to create fragments with intentional mismatches and overlapping ends for Gibson Assembly, which restriction digestion alone cannot provide.PCR uses a DNA copying machine to make many copies of a specific DNA piece, with the start and end points controlled and chosen by the user. This method is very flexible because it allows adding changes to the DNA sequence, like mutations or tags, and works even when there are no natural cutting points present. In contrast, restriction enzyme digestion uses proteins that cut DNA only at specific, short sequences. This method is very precise and predictable at those cut points, but it is limited to locations where these natural cutting sequences exist. PCR is the go-to choice when you need to customize the DNA or when natural cutting sites aren’t available, but digestion is preferred for routine cutting and pasting of DNA fragments when convenient cut sites exist (Green & Sambrook, 2023).
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To ensure PCR and digested DNA fragments are suitable for Gibson Assembly:
Verify that each fragment has 20–40 bp homologous overhangs complementary to adjacent fragments (as introduced by designed primers).
Confirm that fragments are designed in the correct 5′→3′ orientation for annealing and ligation.
Check purity and concentration: Check the concentration of the plasmid DNA and the purity of the fragments. DpnI digestion is useful to remove template plasmid DNA and Zymo cleanup to purify fragments. Nanodrop or Qubit can be useful to measure concentration.
Ensure the fragment size matches the predicted outcome from Benchling simulation before assembly.
Ensure there are no sequence errors by using a high-fidelity polymerase, and verify that overlapping regions match perfectly for efficient Gibson reaction.
(Gibson et al., 2009; NEBuilder HiFi DNA Assembly Guide, 2023).
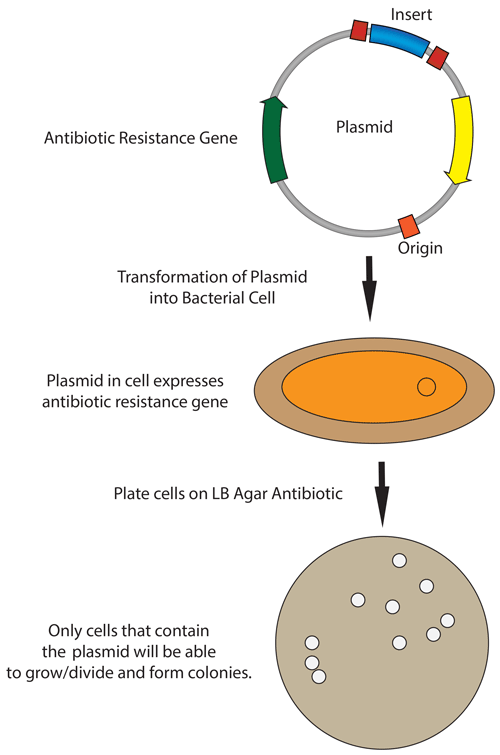
5. How does the plasmid DNA enter the E. coli cells during transformation?
In the protocol from the lab, during heat-shock transformation the plasmid DNA enters the E.coli cells in the following steps:
Competent E. coli cells are chilled to stabilize the membrane.
A 42 °C heat shock creates temporary pores in the membrane by rapidly increasing membrane fluidity.
The plasmid DNA enters the cell through diffusion across the pores.
Cells are then returned to ice or the nutrient-rich SOC medium for recovery. The membrane then reseals and the plasmid begins replication.
During plating on chloramphenicol LB agar, only transformed cells containing the plasmid survive due to antibiotic selection.
6. Describe another assembly method in detail (such as Golden Gate Assembly)
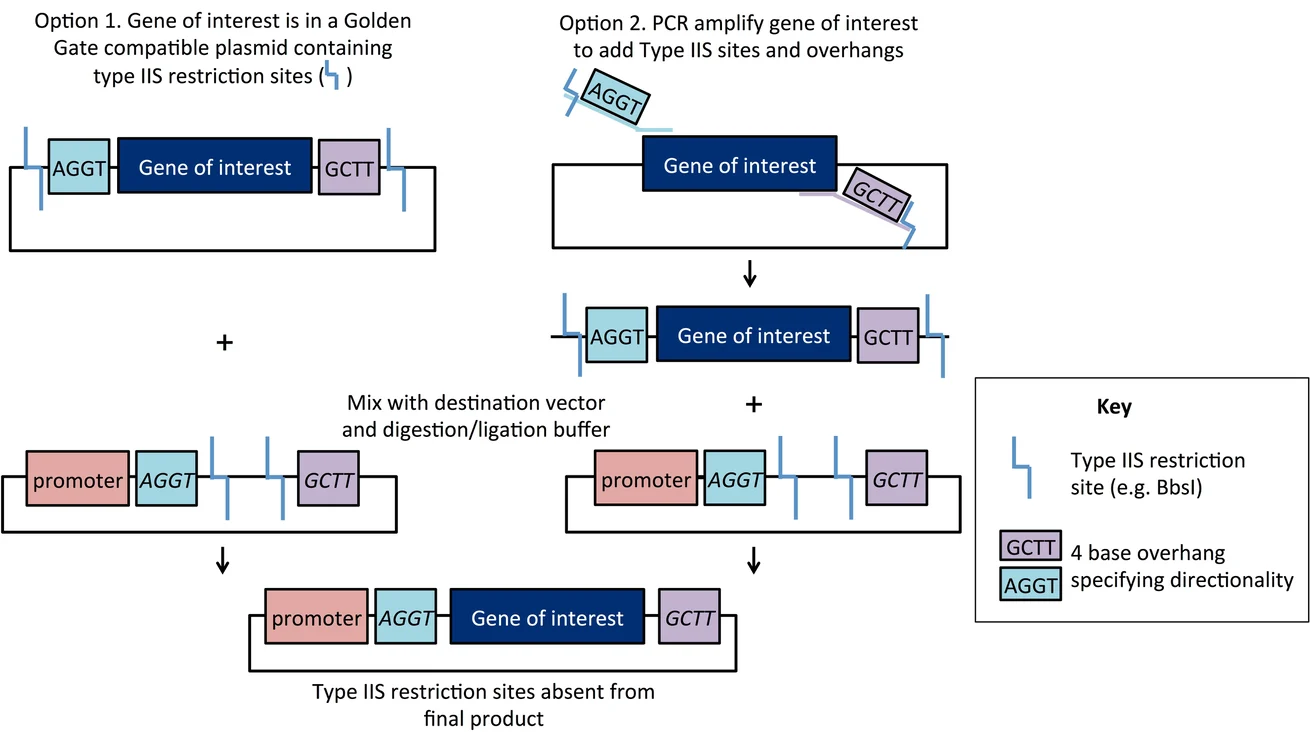
Golden Gate Assembly is a molecular cloning method that uses Type IIS restriction enzymes (for example BsaI) and DNA ligase in a single tube reaction to assemble multiple DNA fragments in one step and with high precision.
This is the step by step mechanism:
Type IIS enzymes cut DNA outside their recognition sites, creating 4-base overhangs that can be custom-designed for assembly.
The digestion and ligation occur in a thermocycling reaction that alternates between 37 °C (cutting) and 16 °C (ligation) steps.
Because recognition sites are removed in the process, the resulting construct lacks unwanted “scar” sequences.
This method has advantages. First, it can enable efficient one-pot assembly of multiple fragments, and it’s speedy and has high accuracy (so no need for overlapping sequences as in Gibson assembly).
(Engler et al., 2008; NEB Golden Gate Assembly Technical Resource).
A. Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Here’s a diagram made by Mary Gearing that I think illustrates Golden Gate Assembly pretty clearly:
B. Model this assembly method with Benchling or Asimov Kernel!
To model it in Benchling, I decided to use pUC19 because it is a common, well-studied cloning vector. It has a high copy number, an ampicillin resistance gene for selection, and a multiple cloning site (MCS) where I can insert new DNA.
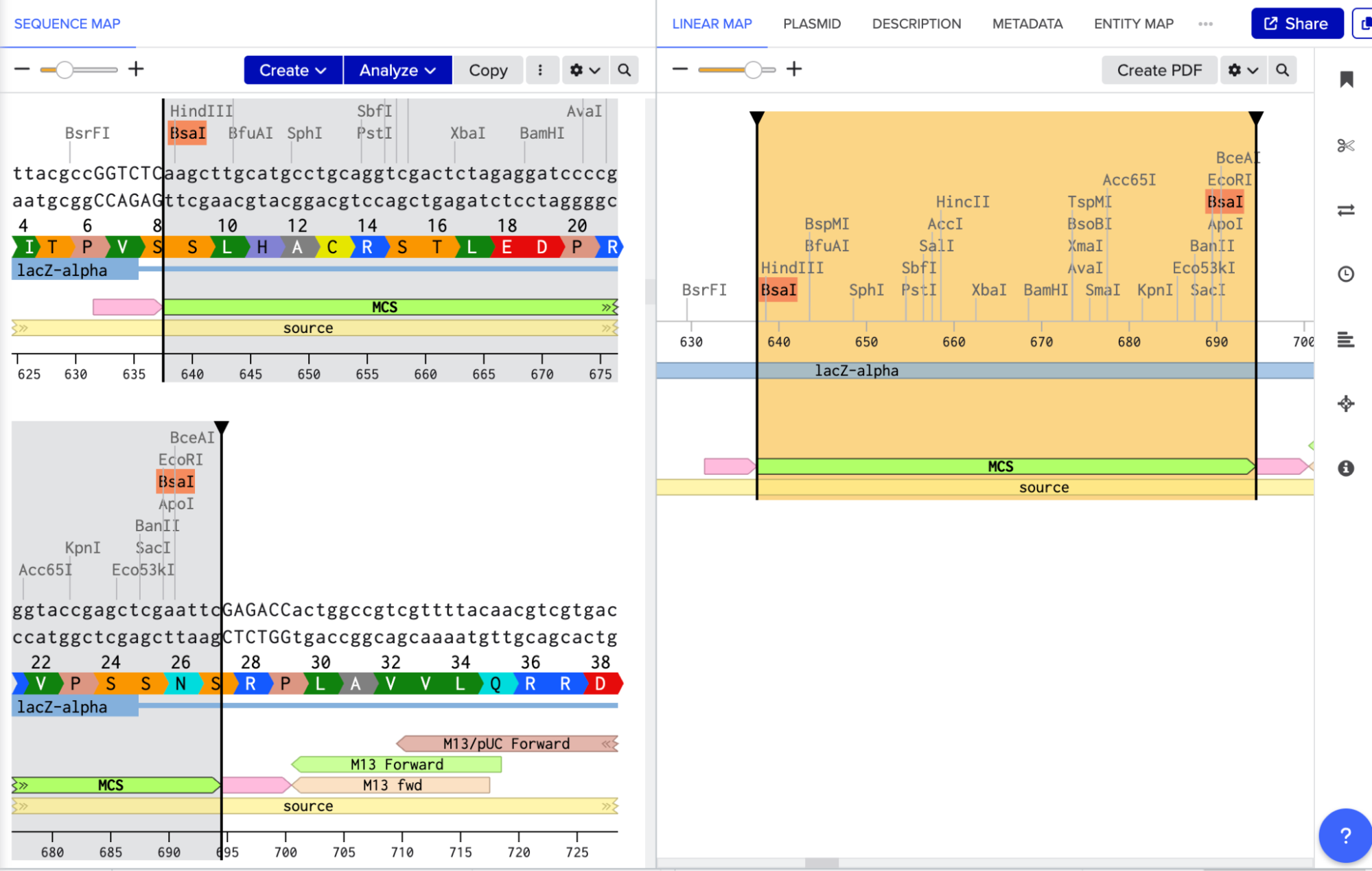
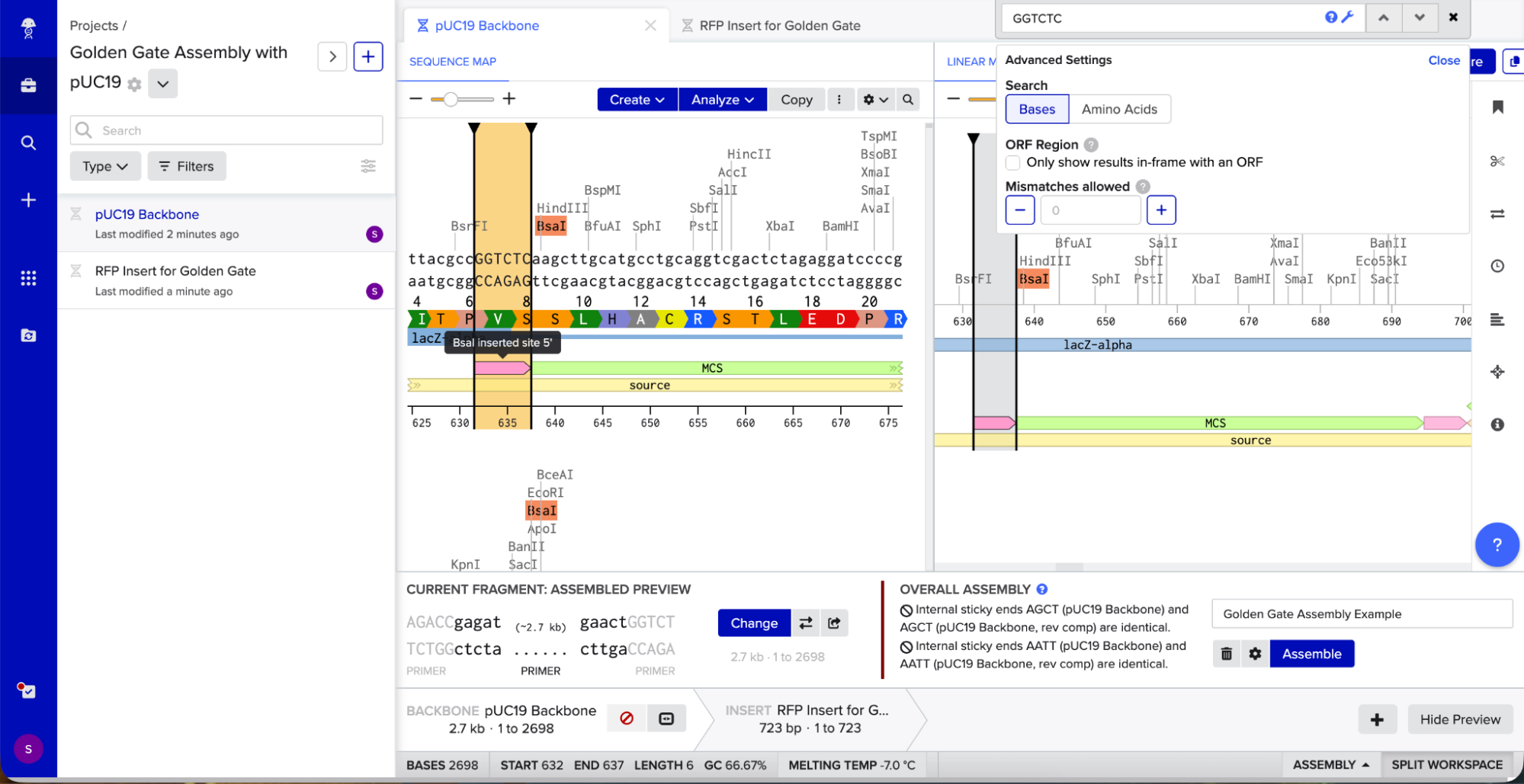
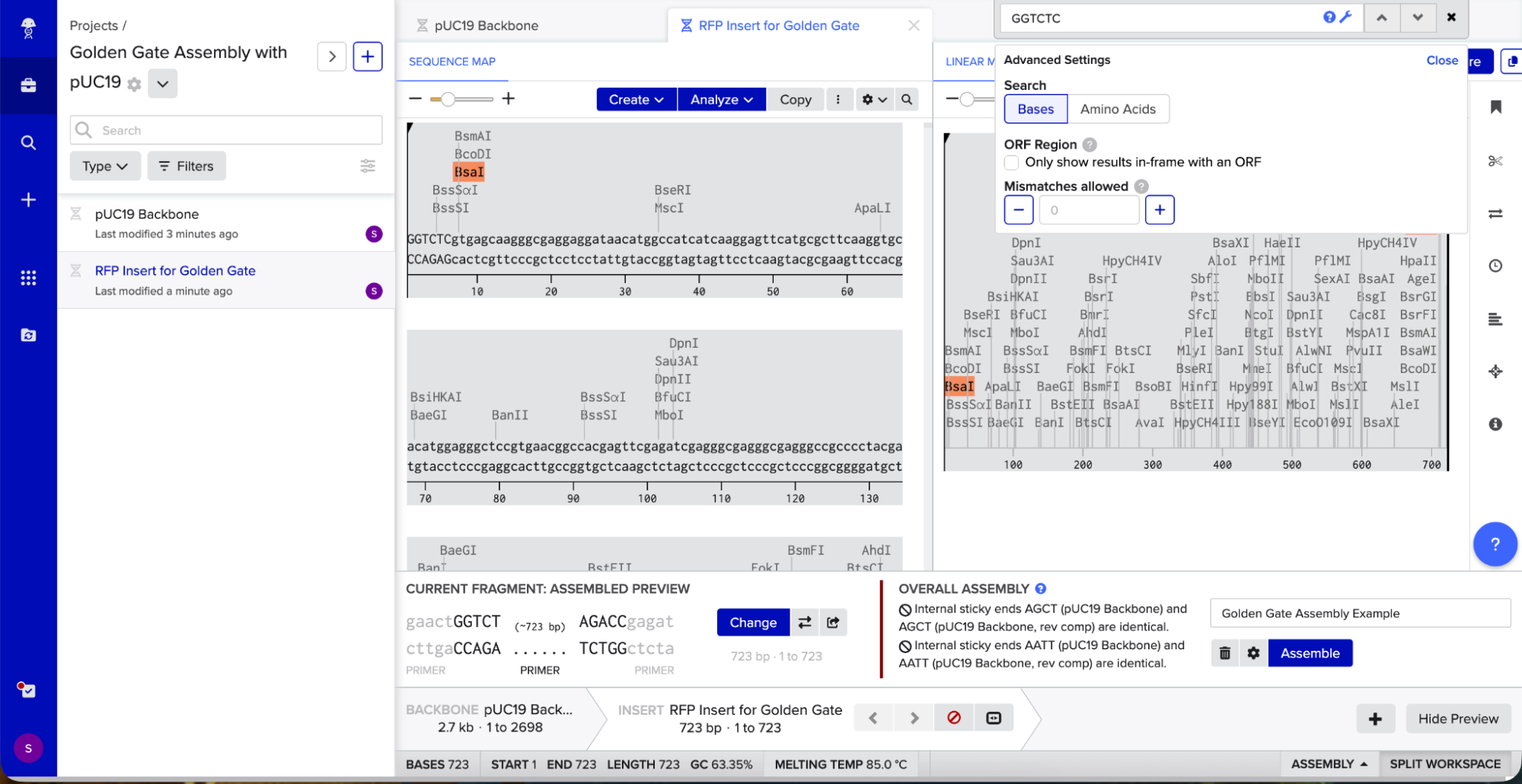
Golden Gate Assembly uses Type IIS enzymes like BsaI. So after importing the pUC19 sequence, I located the multiple cloning site (MCS) in pUC19 and added GGTCTC before it and GAGACC after it (see pink annotations on the image below - these are the inserted BsaI cut sites). These are the recognition sites where BsaI will cut.
I needed a gene to insert, so I chose RFP (red fluorescent protein) as my insert. I added GGTCTC at the start and GAGACC at the end so it would be compatible with my backbone.
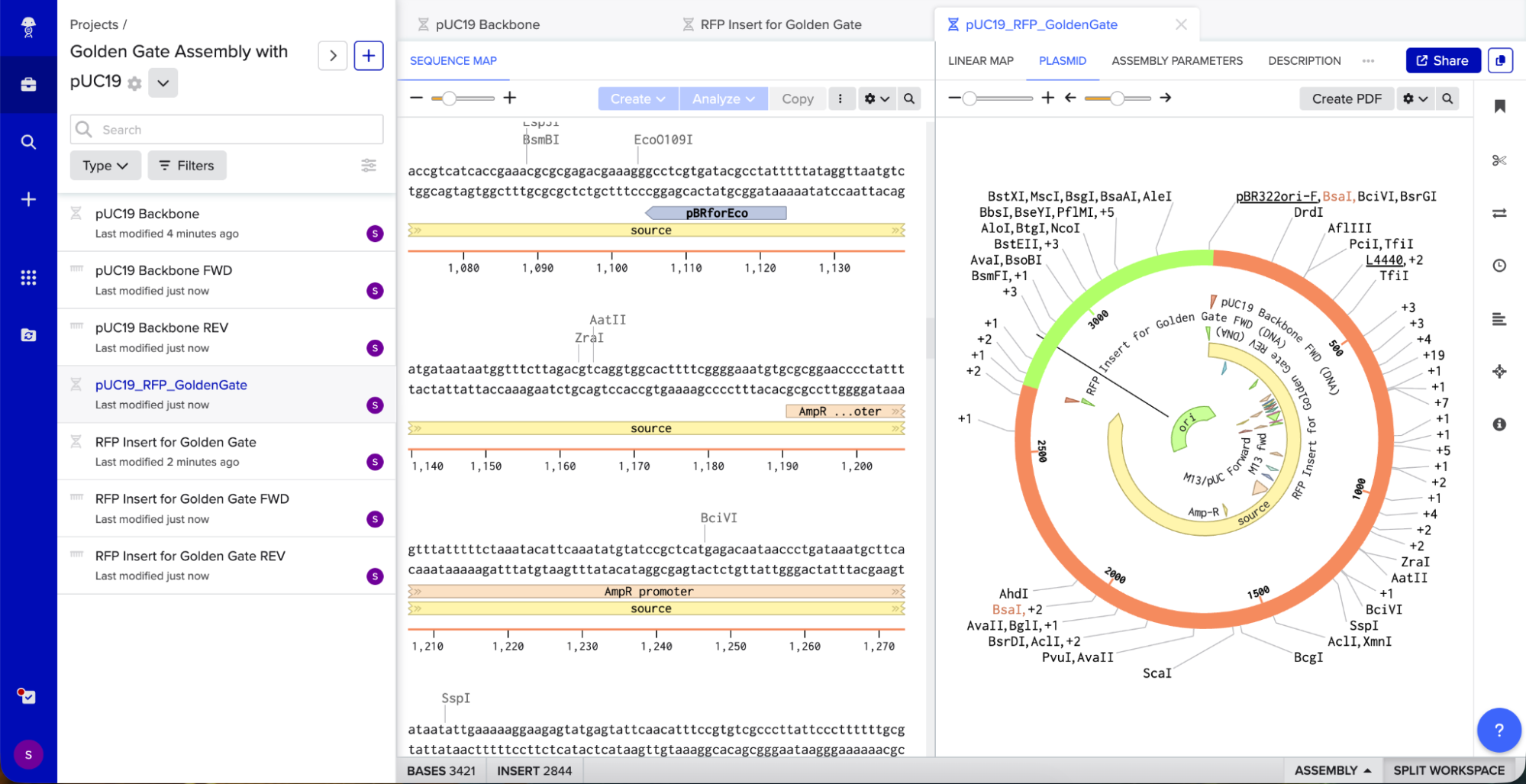
I opened Benchling’s Golden Gate Assembly tool. I selected my backbone fragment and my insert fragment, set the enzyme to BsaI, and clicked “Assemble.”
Benchling created a new circular plasmid with my RFP insert now inside the pUC19 backbone. I also checked where the backbone meets the insert, and the BsaI sites (GGTCTC and GAGACC) were gone. This is the proof that Golden Gate Assembly worked because the recognition sites are removed during assembly, leaving no “scar” sequence.
New England Biolabs (NEB) Phusion® High-Fidelity DNA Polymerase—Product Manual, 2023
Addgene Primer Design Guide; Primer 3 Manual, 2022.
Green & Sambrook, Molecular Cloning: A Laboratory Manual, 4th ed.; NEB Cloning Guide (2023).
Gibson et al., Nature Methods, 2009, 6(5):343–345.
NEBuilder HiFi DNA Assembly Guide, NEB 2023.
NEB Transformation Protocol, 2023.
Hanahan, D. (1983). “Studies on transformation of Escherichia coli with plasmids.” J. Mol. Biol., 166(4):557–580.
Engler, C., et al. (2008). “A one pot, one step, precision cloning method with high throughput capability.” PLoS ONE, 3(11): e3647.
NEB Golden Gate Assembly Technical Resource.
Week 7 HW: Genetic Circuits Part II: Neuromorphic Circuits
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Characteristic
Intracellular Artificial Neural Networks
Traditional Genetic Circuits (that use Boolean functions)
Input-output mapping
Continuous logic that can sum multiple inputs with determined importance or “weights”. This allows for classification of complex patterns.
Discrete simple logic (AND, OR, NAND) with ON/OFF behaviors.
Vulnerability to noise
Since they rely on graded responses, they can average across inputs. This makes them less vulnerable to change output when exposed to noise.
Sensitive to noise around thresholds. If there are small fluctuations the ON/OFF gate can be flipped.
Decision-making
They classify inputs into categories at once and produce signals to different “effector modules” (also called “winner-take-all decisions” in mammalian cells, as mentioned in Chen et al., 2024). This also allows for higher adaptive behavior.
They often produce a single binary output per circuit. This makes them less adaptable.
Table created using information taken from:
Chen, Z., Linton, J. M., Xia, S., Fan, X., Yu, D., Wang, J., … & Elowitz, M. B. (2024). A synthetic protein-level neural network in mammalian cells. Science, 386(6727), 1243-1250.
Gentili PL, Stano P. Chemical Neural Networks Inside Synthetic Cells? A Proposal for Their Realization and Modeling. Front Bioeng Biotechnol. 2022 Jun 6;10:927110. doi: 10.3389/fbioe.2022.927110. PMID: 35733531; PMCID: PMC9208290.
2. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Microbial biosensors to detect mercury in water already exist and have been successful at being an eco-friendly and cost-effective alternative to other methods (Zevallos-Aliaga et al., 2024 & Roointan et al., 2015). They tend to use MerR transcription factor and its cognate promoter (Pmer) to drive a fluorescent or luminescent reporter in response to Hg 2+ (Zevallos-Aliaga et al., 2024), which means that the sensor works by a single input → single output. A useful application could be to turn this into an intracellular neural‑network‑like classifier that can receive several inputs inside the same cell to determine if a particular food matrix is likely above a regulatory mercury given limit.
The inputs to the intracellular neural network would be analog signals whose expression levels vary with concentration:
Pmer drives the expression of a regulator, protein A (for example, MerR fused to an activation domain), in response to bioavailable Hg2+.
A separate metal‑induced stress promoter activated by oxidative stress generated during mercury exposure drives regulator B, capturing more general toxicity associated with high mercury levels rather than Hg 2+ alone.
The network using protein A and regulator B would involve creating two layers. The IANN uses A and B as the outputs of two input neurons, which feed into a final decision node. A synthetic promoter (let’s call it Poutput) is designed with binding sites for both protein A and regulator B. Poutput could approximate a weighted sum of variables and concentrations and a threshold, where it would only activate a fluorescent protein when detecting that the combined levels of A and B exceed a defined level. This corresponds to the network classifying the sample as “above the safe mercury limit,” whereas lower or unbalanced inputs keep the output near baseline.
In practice, you could incubate a small portion of a fish fillet (or any other food matrix) with the engineered bacteria. If the intracellular ANN determines that the pattern of Hg 2+ and stress response lies in the “unsafe” region of its input space, the cells switch ON the fluorescent color, indicating that the food sample likely exceeds regulatory mercury limits. If not, the reporter remains OFF, indicating the sample is probably within safe limits to consume.
This design could face an important limitation within metal‑responsive regulators. Sometimes, they show cross‑reactivity with other metals, which may lead to false positives. Also, since response times are affected by transcription, translation, and protein synthesis, an answer to the test could take a long time (more in the hours than in the minutes range), which could be complicated when testing large amounts of samples for human consumption.
References:
Zevallos-Aliaga D, De Graeve S, Obando-Chávez P, Vaccari NA, Gao Y, Peeters T, Guerra DG. Highly Sensitive Whole-Cell Mercury Biosensors for Environmental Monitoring. Biosensors (Basel). 2024 May 13;14(5):246. doi: 10.3390/bios14050246. PMID: 38785720; PMCID: PMC11117708.
ROOINTAN, A, SHABAB, N, KARIMI, J, RAHMANI, A, ALIKHANI, M. Y, & SAIDIJAM, M (2015). Designing a bacterial biosensor for detection of mercury in water solutions. Turkish Journal of Biology 39 (4): 550-555. https://doi.org/10.3906/biy-1411-49
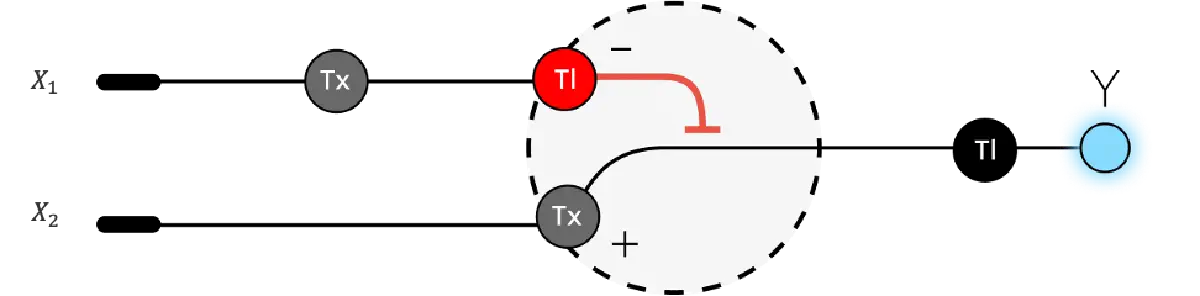
3. Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
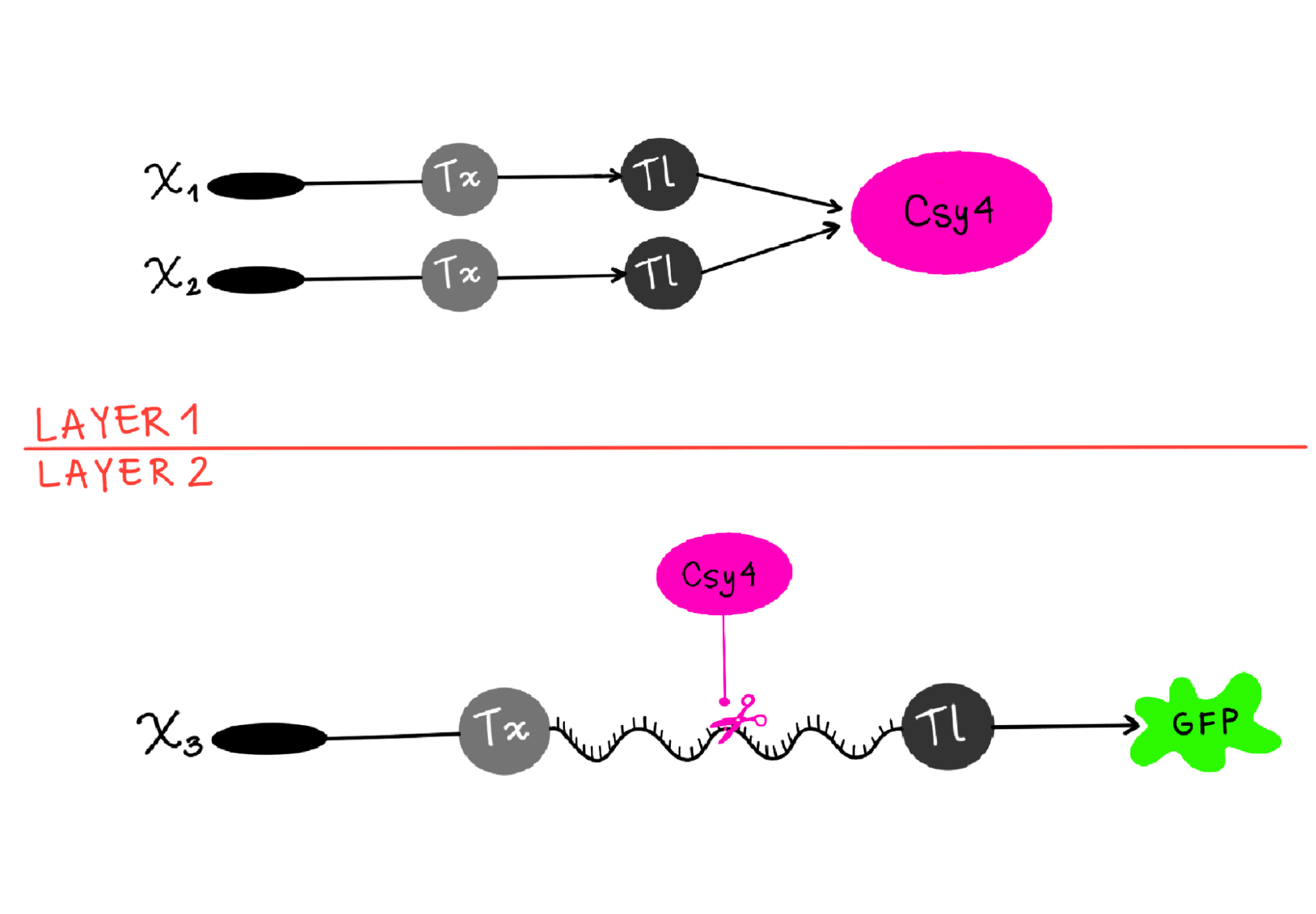
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
I’m not sure this diagram is correct, but I tried to show how X1 and X2 are genes, which go through transcription (Tx) and translation (Tl) to then produce the endoribonuclease (Csy4 like in the example) as output in layer 1. And then in Layer 2, Csy4 recognizes the endoribonuclease site in the mRNA of the fluorescent protein (GFP) and cuts it, so when it goes through Translation, the output is a regulated GFP.
Assignment Part 2: Fungal Materials
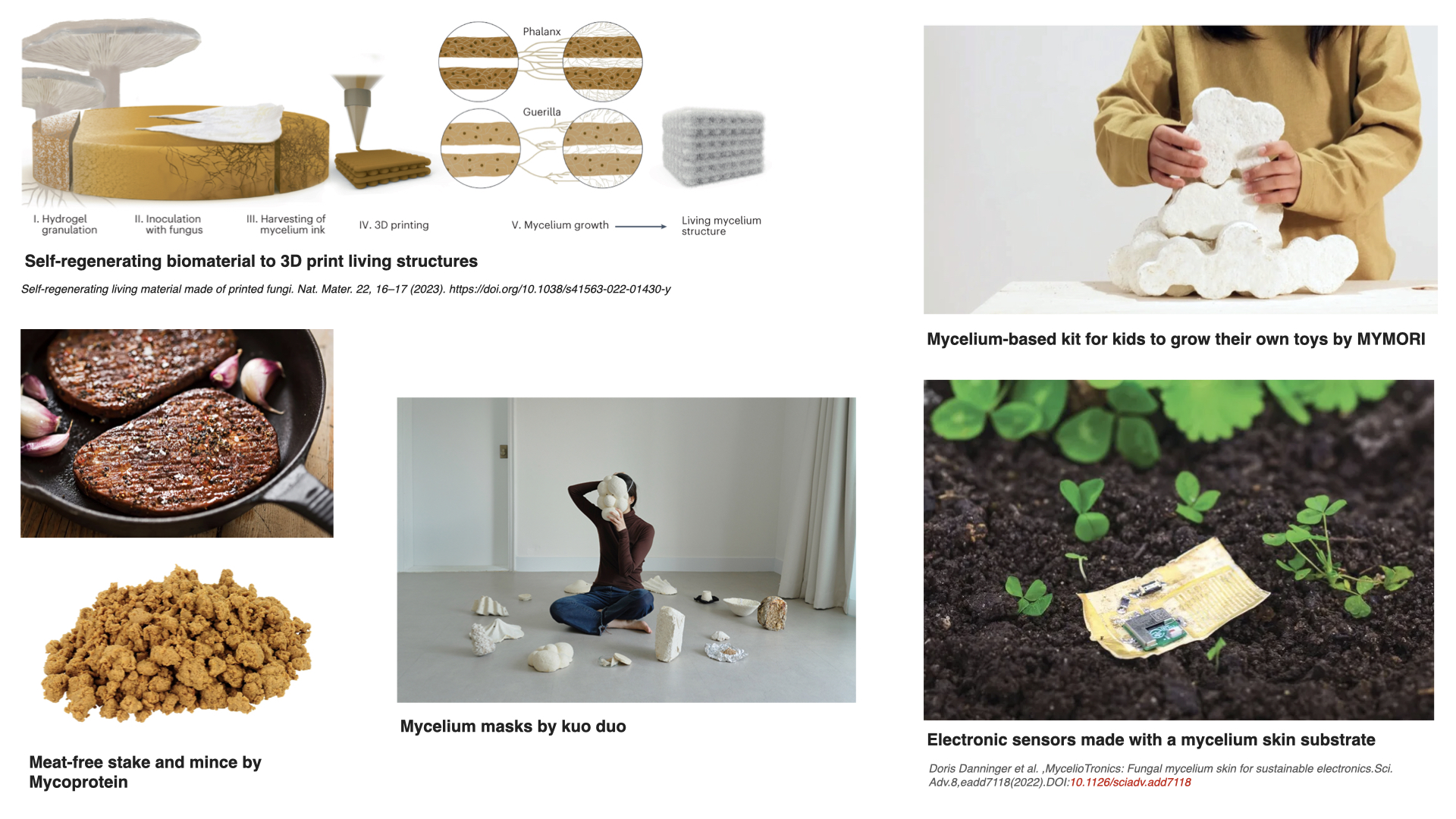
1. What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Fungal materials are becoming very popular very fast. I have seen many different uses: construction materials, clothing and jewelry, household objects… But I wanted to find other possibilities, maybe less common ones. These are some I found particularly interesting:
In most examples, ranging from construction materials to meat-free food alternatives, fungi are used because they are easily grown by using cheap plant waste (straw, rye, cardboard, paper, or others), relatively little energy, and more sustainable than the more common industrial alternatives (plastics, animal leather, polystyrene foams, etc.). Also, because their mycelium can easily grow into complex 2D or 3D shapes, bind loose materials into solid composites, and form flexible sheets. In short, its advantages are: renewability, biodegradability, and the ability to be grown with specific textures and forms. Nevertheless, many of these mycelium products need improvements. Some show lower mechanical performance, more vulnerability to moisture, and limited long‑term durability compared to their industrial counterparts. This makes them a bit niche in the beginning and difficulties their direct implementation.
2. What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
I find the electronic sensors made with mycelium skin substrate by MycelioTronics very interesting, and I immediately thought of the infamous Arduino breadboards. I would engineer fungi to grow mycelium skins that act as biodegradable Arduino‑style breadboards, where the mycelium itself helps carry electrical signals between components. The engineered mycelium could make more conductive substances (for example, metal‑binding molecules) along certain growth paths in the breadboard, so parts of the sheet become “tracks” that electricity can flow through, while the rest stays more insulating and structural. This way, the mycelium acts as the material that supports the components connected on the breadboard but also acts as the network that helps connect the different holes and contact points.
For this idea, fungi are more advantageous than bacteria because they naturally grow continuous 2D and 3D structures, they can be grown in cheaper substrates and sometimes under more simple conditions, and as eukaryotes they are better at more complex engineering. Engineering their DNA could potentially change how the grown sheet behaves as a structural material and as a bioelectric material.
Week 9 HW: Cell-Free Systems
Homework Part A: General and Lecturer-Specific Questions
General homework questions
1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Synthesizing proteins outside living cells allows for more control over the environmental variables (pH, temperature, particular concentrations of ions and molecules, etc.), as the cell is not interacting with complex surroundings related to metabolic processes, resources or cofactors. This control is beneficial for experiments such as those that involve artificial aminoacids, since they can’t be assembled inside natural cells; or those that could involve toxic proteins that can’t be produced inside living natural cells.
2. Describe the main components of a cell-free expression system and explain the role of each component.
DNA/RNA template: encodes the target protein; tells the system what to make
RNA polymerase: transcribes DNA into mRNA
Ribosomes: read the mRNA and build the protein
tRNAs: carry amino acids to the ribosome, matching each codon
Amino acids: the building blocks of the protein
Nucleotides (ATP, GTP, etc.): building blocks for RNA and energy for the reactions
Energy regeneration system: continuously regenerates ATP so the reaction doesn’t stop early
Cofactors/coenzymes (Mg²⁺, K⁺, etc.): stabilize ribosomes and support enzymatic activity
Buffer: maintains stable pH
Cell extract (lysate): the practical source of most of the already mentioned components; provides ribosomes, polymerases, and translation factors all at once
3. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Transcription and translation are processes that consume large amounts of ATP and GTP. Since there’s no living cell to continuously produce new energy, the reaction will stop as soon as the initial ATP runs out. Also, the phosphate byproducts that accumulate can actually inhibit the reaction.
A solution for this is to include 3-PGA in the reaction. The enzymes already present in the cell extract will convert 3-PGA into ATP over time, maintaining energy levels throughout the experiment so the reaction doesn’t stop.
4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic cell-free expression systems are cheaper, faster, and give higher yields; they are especially useful for simple proteins that don’t need post-translational modifications. For example, GFP (green fluorescent protein) is a protein that does not require post-translational modifications and is perfect for E.coli based cell-free expression systems.
Eukaryotic systems, on the other hand, are more expensive and slow, but support proper folding and post-translational modifications needed by more complex proteins. For example, a human hormone requires specific disulfide bonds and glycosylation to be functional, so an eukaryotic cell-free system is necessary.
5. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Membrane proteins are hydrophobic, so without a membrane-like environment they tend to fold incorrectly or clump together as they’re synthesized.
To avoid this, membrane mimics (liposomes, nanodiscs, detergents, etc.) could be directly added to the reaction so the protein has somewhere to insert as it’s being made. Then, I could run optimization experiments of varying type and concentration of the chosen membrane mimic and temperature, checking protein yield (via gel or western blot) and functionality (via an activity assay) to confirm the protein is folding correctly.
6. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
The mRNA or the protein is being broken down by contaminating enzymes in the extract. To fix this, you could add RNase inhibitors to protect the mRNA, or use a protease-deficient extract.
The reaction is running out of energy too quickly. To fix this, you could optimize or add more energy regeneration system (for example, increase 3-PGA concentration or use a creatine phosphate system).
The DNA concentration is too low, the quality is poor, or the promoter isn’t efficient enough. To fix this, you could increase the DNA input, check the purity, and/or use a stronger or more appropriate promoter.
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
1. Pick a function and describe it.
An example could be a synthetic minimal cell that detects an antibiotic and reports its presence, which could be useful to evaluate clinical or environmental samples. More specifically, the cell detects tetracycline, a broad-spectrum antibiotic, and produces a visible signal with GFP (green fluorescent protein) fluorescence.
1.1 What would your synthetic cell do? What is the input and what is the output?
Input: tetracycline
Output: GFP fluorescence
1.2 Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Technically yes, because the sensing of tetracycline and GFP expression could happen in a test tube. However, without encapsulation there is no control over what is entering the system, and the reaction can degrade quickly.
1.3 Could this function be realized by genetically modified natural cell?
Yes, because engineered bacteria with a tetracycline-responsive promoter that drives GFP could do this.
1.4 Describe the desired outcome of your synthetic cell operation.
At normal or zero tetracycline levels, no GFP signal. When tetracycline is present above a determined threshold concentration, the synthetic cell expresses GFP and produces a fluorescent output proportional to antibiotic concentration.
2. Design all components that would need to be part of your synthetic cell.
2.1 What would be the membrane made of?
Mainly phospholipids and cholesterol, as these form stable lipid vesicles that mimic natural cell membranes.
2.2 What would you encapsulate inside? Enzymes, small molecules.
Inside, there would be a DNA circuit, which would contain the GFP gene under the control of a tetracycline-responsive promoter, bacterial cell-free Tx/Tl machinery (so ribosomes, RNA polymerase, tRNAs, amino acids, and energy regeneration machinery), and a membrane pore gene (such as α-hemolysin) to allow tetracycline to enter the vesicle of the cell.
2.3 Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
Bacterial system is ok, since tetracycline-responsive promoters work well in prokaryotic systems (with the additional benefit that this also keeps the system simpler and cheaper). No mammalian system is needed here since there is no need of a mammalian transcription factor system.
2.4 How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
Tetracycline is kinda membrane-permeable, but to ensure its entry, you can induce expression of α-hemolysin (αHL) pores in the membrane. These pores allow small molecules (such as tetracycline) to enter the vesicle, triggering the genetic circuit inside.
3. Experimental details
3.1 List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
Lipids: phospholipids and cholesterol (for vesicle membrane)
Tx/Tl system: E. coli cell-free extract
Gene 1: aHL (alpha-hemolysin), which is the membrane pore to allow tetracycline entry
Gene 2: gfp (GFP reporter) under a tetracycline-responsive promoter
3.2 How will you measure the function of your system?
By using a plate reader or a fluorescence microscope, you can measure the GFP fluorescence as the tetracycline concentration is varied. After collecting this data, you can create a curve of fluorescence vs. tetracycline concentration to validate that the system activates at the right threshold. As a control, you can run the vesicles without αHL pores to confirm that tetracycline entry through the pore is required.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
1. Write a one-sentence summary pitch sentence describing your concept.
A reusable biosensor strip embedded with freeze-dried cell-free reactions that coastal fishermen can dip into seawater to detect mercury contamination in their fishing grounds, producing a visible color change as a signal.
2. How will the idea work, in more detail? Write 3-4 sentences or more.
The strip is a fabric-like material covered with freeze-dried cell-free reactions. When a fisherman dips the strip into seawater, the water rehydrates the reaction and activates a DNA circuit responsive to free mercury ions (Hg²⁺) in the ocean. At safe mercury levels, no color change occurs. Above a threshold concentration linked to seafood safety guidelines, the circuit drives expression of a chromoprotein, producing a visible color. The strips can be manufactured cheaply, packaged dry, and distributed to fishing communities along the Colombian Pacific and Caribbean coasts, where mercury contamination from illegal gold mining that happens upstream is a documented and growing problem (Palacios-Torres et al., 2018; Marrugo-Negrete et al., 2008). Farmers near rivers could use a similar strip design to test irrigation water for heavy metal contamination before it enters their crops (Marrugo-Negrete et al., 2017). This idea doesn’t require lab equipment, electricity, training or special skills.
3. What societal challenge or market need will this address?
Illegal gold mining in Colombia is becoming a pressing problem. It releases large amounts of mercury and other heavy metals into rivers and coastal waters, contaminating important water ecosystems and soils that are not only home for hundreds of key species, but are also a source of food for the Colombian population. Fishing communities in particular in regions like the Chocó, the Gulf of Morrosquillo, and the Magdalena River basin rely on these waters for both food and income, but they have no practical way to know whether the fish they catch or the water they use is contaminated (Palacios-Torres et al., 2018; Marrugo-Negrete et al., 2008). According to the World Health Organization, mercury is one of the ten most hazardous substances in the world, with the Agency for Toxic Substances and Disease Registry ranking it third. It causes serious neurological damage, especially in children; different types of cancer; endothelial dysfunction; gastric and vascular disorders; liver, kidney, and brain damage; hormonal imbalances, miscarriages, and reproductive disorders; skin lesions; vision damage; and even death (Charkiewicz et al., 2025). Commercial testing requires sending samples to labs outside of the country, which proves to be expensive, slow, and highly inaccessible to these rural communities. Designing this reusable biosensor strip would help put environmental monitoring directly in the hands of the people who need it most, allowing them to make informed decisions about where to fish, when to avoid certain areas, and when to alert authorities and the public. Community organizations or environmental NGOs could help distribute them with a small guide explaining a tiered color readout (light color = caution, strong color = danger) that can give semi-quantitative information without requiring any complex measuring instruments.
4. How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
The cells will be freeze-dried, and the seawater will activate them. Seawater is saline and contains ions, posing a challenge for the design of the strip, but the cell-free reaction can be optimized and buffered within the strip to function correctly when rehydrated with environmental water samples. During the engineering process multiple calibration tests would be needed to establish reliable thresholds.
Another challenge would be storage. The strips would be freeze-dried with trehalose, a sugar derived substance that has been widely used to stabilize proteins, mammalian cells and other cell-free systems (Olsso et al., 2016). Using trehalose and sealing them individually will allow the strips to be stored at room temperature for longer (hopefully months) to allow a wider window for shipping and the use itself when needed by the communities. The best case scenario is creating a reusable strip, and testing how many times one strip can be used without losing accuracy in the results; but another challenge this proposal poses is to make sure that all the materials used for the strip are not one-use and/or disposable, creating more trash.
References:
Palacios-Torres, Y., Caballero-Gallardo, K., & Olivero-Verbel, J. (2018). Mercury pollution by gold mining in a global biodiversity hotspot, the Chocó biogeographic region, Colombia. Chemosphere, 193, 421-430.
Marrugo-Negrete, J., Benitez, L. N., & Olivero-Verbel, J. (2008). Distribution of mercury in several environmental compartments in an aquatic ecosystem impacted by gold mining in northern Colombia. Archives of Environmental Contamination and Toxicology, 55(2), 305-316.
Marrugo-Negrete, J., Pinedo-Hernández, J., & Díez, S. (2017). Assessment of heavy metal pollution, spatial distribution and origin in agricultural soils along the Sinú River Basin, Colombia. Environmental research, 154, 380-388.
Charkiewicz, A. E., Omeljaniuk, W. J., Garley, M., & Nikliński, J. (2025). Mercury exposure and health effects: what do we really know?. International journal of molecular sciences, 26(5), 2326.
Olsson, C., Jansson, H., & Swenson, J. (2016). The role of trehalose for the stabilization of proteins. The Journal of Physical Chemistry B, 120(20), 4723-4731.
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
1. Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Astronauts (especially the ones that go on long-duration missions) face difficult psychological challenges: communication delays with Earth, profound feelings of isolation, disrupted circadian rhythms from irregular light exposure in orbit, poor sleep and the worsening of cognitive performance (Collins, 2003). Helping them regulate their circadian rhythms could be an important step for sleep mediation, stress resilience and mental peace during their missions. Melatonin is the hormone whose synthesis depends on enzymatic pathways sensitive to light cues. Understanding how melatonin works inside astronauts’ bodies could be key to protecting astronaut mental health.
2. Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
The enzyme AANAT (arylalkylamine N-acetyltransferase) is the rate-limiting enzyme in melatonin biosynthesis. Its upstream regulator is the CLOCK gene circadian transcription factor.
3. Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
AANAT enzymatic activity controls the nightly increase in melatonin that drives circadian rhythm synchronization. In space, the absence of normal 24-hour light/dark cycles suppresses AANAT expression, reducing melatonin production and fragmenting sleep (Zong et al., 2025). Using BioBits to express AANAT in space under simulated circadian promoter control can be an opportunity to test whether the cell-free Tx/Tl machinery can reliably produce the AANAT enzyme on demand. If results are positive, astronauts could use a biosynthesis system that supplements melatonin during their mission, avoiding pre-packed pharmaceuticals that can degrade over long space missions.
4. Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
Hypothesis: BioBits cell-free reactions aboard spacecraft will produce equal or slightly lower yields of AANAT protein in comparison to Earth, creating a successful biosynthesis system that supplies astronauts with melatonin during space travel.
Melatonin deficiency is already documented in astronauts, but it is unclear how much of this is due to light exposure versus a genuine suppression of the biosynthetic machinery itself (Zong et al., 2025). If cell-free reactions can produce AANAT in space at comparable yields to Earth, this validates the concept of on-demand biosynthesis of melatonin (and perhaps other psychoactive molecules) aboard spacecraft. If yields are lower, it points to key specific technical challenges (for example: DNA/mRNA damage due to space radiation) that could be solved before in-space biomanufacturing of therapeutics becomes a reality.
5. Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
First, create freeze-dried BioBits pellets that contain the AANAT gene attached to an sfGFP reporter, to allow fluorescence to signal if protein expression is successful. One set of pellets will be activated aboard the spacecraft, while an identical set is activated on Earth as the first control. The two additional controls are: (1) pellets with an always-on gene (positive control), and (2) pellets with no DNA (negative control). Astronauts will rehydrate one pellet every four hours over 24 hours to mimic a circadian cycle. Protein expression will be analyzed and measured using the fluorescence viewer for real-time results. Higher brightness means more AANAT protein was made.
References:
Collins, D. L. (2003). Psychological issues relevant to astronaut selection for long-duration space flight: a review of the literature. Journal of Human Performance in Extreme Environments, 7(1), 1.
Zong, H., Fei, Y., & Liu, N. (2025). Circadian disruption and sleep disorders in astronauts: a review of multi-disciplinary interventions for long-duration space missions. International Journal of Molecular Sciences, 26(11), 5179.
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
My project MycoBoard consists of:
Aim 1 - Experimental: Design and computationally validate the cmt overexpression construct for N. crassa, simulating expected silver biosorption and resistance outcomes
Aim 2 - Development: Translate the validated computational design into a physical prototype: grow engineered N. crassa mats inside molds and validate by working LED circuit powered through a biologically grown conductive track.
Aim 3 - Visionary: Replace physical molds with optogenetic control of cmt expression, using an interface to draw circuit layouts that the fungus then follows while growing.
Because my project currently focuses on the computational design (Aim 1), most of my measurements are predictions:
Sequence correctness and reading frame validation: See whether the designed cmt overexpression construct has the correct DNA sequence and maintains the proper reading frame. This would be on Benchling and SnapGene by virtually translating the DNA into protein and checking that the HA tag and cmt gene are fused without any unexpected stop codons in between.
Simulated silver binding capacity per square centimeter: Looking for published data on how much silver the metallothionein protein can bind per molecule, I can calculate the expected (or approximated) amount of silver that one square centimeter of my fungal mat could capture in experimental procedures. This is a mathematical prediction, not really a measurement, but is useful data for aim 2.
Predicted resistance heatmap: Based on published bioabsorption values for N. crassa I plan on creating a simulated heatmap showing expected electrical resistance across the mat. This is useful to know where the conductive tracks should work before growing a real fungal mat.
However, I have also planned measurements for a future wet lab phase (Aim 2) where I could physically grow the mats:
If the cmt gene was successfully inserted: After building the DNA construct, I can take a sample of the fungal colonies and run a colony PCR. Then, I can run those copies on gel electrophoresis, separating the extracted DNA by size. Knowing the length (around 600 and 700 bp) of the cmt gene, I can know if it’s in the sample I took. A plan B for this could also be to send the sample for Sanger DNA sequencing, and then see if it is actually the cmt gene sequence.
If the cmt protein is actually being produced: Having the gene does not mean the fungus makes the protein. Because my design adds a small HA tag to the cmt protein, I can use a western blot, where an antibody sticks to the HA tag. If there is presence of a dark blot at the right size, I can confirm the protein is present in engineered cells but not in wild-type controls.
If the engineered mat is more conductive than wild-type: This is the most important measurement. To do the comparison, I can grow engineered and wild-type mats in silver nitrate solution, dry them, and then use a standard multimeter to measure electrical resistance following a small 5×5 grid. Engineered mats should show lower ohm values than wild-type ones.
If the silver nanoparticles are depositing along the hyphae: To see the silver directly, I can put a small piece of the mat under a microscope. Using regular brightfield microscopy, silver nanoparticles look like dark spots or clusters along the “hyphae”. Or, on the other hand, using scanning electron microscopy, I could see the actual shape and size of the nanoparticles at much higher magnification.
If the mat decomposes in soil: To test the compostability of the mat, I can bury one used mat and one small piece of standard FR4 fiberglass circuit board in a container of soil, and take measurements (thickness, weight) and pictures each week for a period of a month or a couple of months.
Waters Part I — Molecular Weight
We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
1. Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
eGFP Sequence:
Note: This contains a His-purification tag (HHHHHH) and a linker (the LE before it).
The results from the Expasy calculator are the following:
2. Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
2.1 Determine z for each adjacent pair of peaks (n, n+1):
2.2 Determine the MW of the protein:
2.3 Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1
3. Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
Since the protein unfolded and is showing at its denatured state, the protein is in a larger charged state than the mass spectrometer can catch, so you can’t see it.
Waters Part II — Secondary/Tertiary structure
We will analyze eGFP in its native, folded state and compare it to its denatured, unfolded state on a quadrupole time-of-flight MS. We will be doing MS-only analysis (no liquid chromatography, also known as “direct infusion” experiments) on the Waters Xevo G3-QToF MS.
1. Based on learnings in the lab, please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses (Figure 2)?
Unfolded proteins tend to have many high charge states, which means that the spectrometer will produce peaks across low m/z, because there are more basic sites exposed where more protons can attach to the protein. Folded proteins, on the other hand, tend to have less lower charge states, which means that the spectrometer will produce peaks at high m/z, because the folded structure hides the protonatable sites. In Figure 2 you can see the peaks around 800-1800 m/z (top) for the unfolded protein and the fewer peaks around 2000-3000 m/z (bottom) for the folded protein.
2. Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 QTof MS (see Figure 3), can you discern the charge state of the peak at ~2800 What is the charge state? How can you tell?
Waters Part III — Peptide Mapping - primary structure
We will digest the eGFP protein standard into peptides using trypsin (an enzyme that selectively cleaves the peptide bond after Lysine (K) and Arginine (R) residues. The resulting peptides will be analyzed on the Waters BioAccord LC-MS to measure their molecular weights and fragmented to confirm the amino acid sequence within each peptide – generating a “peptide map”. This process is used to confirm the primary structure of the protein. There are a variety of tools available online to calculate protein molecular weight and predict a list of peptides generated from a tryptic digest. We will be using tools within the online resource Expasy (the bioinformatics resource portal of the Swiss Institute of Bioinformatics (SIB)) to predict a list of tryptic peptides from eGFP.
1. How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking the biochemical properties tab will show you a count for each amino acid).
2. How many peptides will be generated from tryptic digestion of eGFP?2.1 Navigate to https://web.expasy.org/peptide_mass/2.2 Copy/paste the sequence above into the input box in the PeptideMass tool to expected list of peptides.2.3 Use Figure 4 below as a guide for the relevant parameters to predict peptides from eGFP.
Here are the results from PeptideMass:
It reports 19 peptides
3. Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
According to Figure 5a, I can see are approximately 16 peaks above the 10% relative abundance between 0.5 and 6 minutes:
0.43, 0.61, 0.79, 1.20, 1.43, 1.80, 1.85, 1.93, 2.17, 2.26, 2.54, 2.78, 3.27, 3.53, 3.59, 3.70
4. Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
I see less peaks in the peptide map than the ones predicted. Maybe the prediction had more peptides because they are smaller, and they are retained.
5. Identify the mass-to-charge of the peptide shown in Figure 5b. What is the charge () of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide
6. Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
7. What is the percentage of the sequence that is confirmed by peptide mapping?
According to Figure 6, 88% of the eGFP sequence is confirmed by peptide mapping.
Waters Part IV — Oligomers
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
Now looking at the peaks in Figure 7, I see peaks at approximately 3.4 MDa, 8.33 MDa, 12.67 MDa and a smaller one around 16-17 MDa.
Waters Part V — Did I make GFP?
Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
Theoretical
Observed/measured on the Intact LC-MS
PPM Mass Error
Molecular weight (kDa)
27.960 kDa
27.990 kDa
787 ppm
The measured mass is close to the theoretical mass, confirming that the protein could be eGFP :)
Week 11 HW: Bioproduction & Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
1) Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST. 2) Make a note on your HTGAA webpages including: what you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”); what you liked about the project; and what about this collaborative art experiment could be made better for next year.
For the first Global Pixel Art, I contributed one green pixel (with GFP) in a corner of the artwork:
And for the second Global Pixel Art, I contributed four blue wells on the plate located at
Since this contribution required a hypothesis with the different provided chemical concentrations and DNA template concentrations for every well, my hypothesis would be that increasing HEPES‑KOH from 45 mM to 67.5 mM will increase fluorescence by better maintaining neutral pH throughout the reaction, which is important for folding fluorescent proteins slower like the ones used in the artwork.
Here are the final values I adjusted for every well:
Well
Condition
HEPES concentration
DNA Template
Well Q4 - E16
Baseline
45 mM (standard)
50 nM
Well Q4 - E17
Baseline replicate
45 mM
50 nM
Well Q4 - E18
High buffer
~67.5 mM (1.5x)
50 nM
Well Q4 - E19
High buffer replicate
~67.5 mM (1.5x)
50 nM
What I liked about this activity:
I really enjoyed the collaborative aspect and the idea that everyone in the class could contribute to a single shared artwork, even if just by changing one pixel (like I did). I also appreciated that you could only edit one pixel at a time between set time periods. I liked that the process was gradual.
What could be better for next year:
What could be improved:
The Part 2 instructions about concentrations and wells were a bit confusing, I wasn’t sure of what I had to do exactly even after watching the recitation.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
1. Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
a) E. coli Lysate
BL21 (DE3) Star Lysate (T7 RNA Polymerase included) Provides ribosomes , tRNAs and enzymes for transcription and translation . The mutation “Star” reduces the degradation of mRNA and the T7 RNA polymerase allows high level transcription from the DNA template .
b) Salts/Buffer
Potassium Glutamate: Contributes to the ionic strength and the potassium ions needed for activity of the ribosome and protein synthesis.
HEPES-KOH pH 7.5: buffer to keep the pH of the reaction at ~pH 7.5 to avoid acidification from metabolic by-products during long incubations.
Magnesium Glutamate: Magnesium ions are important cofactors for the stability of ribosomes, activity of RNA polymerase and NTP-requiring enzymes.
Potassium phosphate monobasic & dibasic: Provides inorganic phosphate for ATP formation through glycolysis. Also provides a secondary buffer system.
c) Energy / Nucleotide System
Ribose & Glucose: Are energy sources converted into ATP and GTP by the lysate’s metabolic pathways over 20+ hours.
AMP, CMP, GMP & UMP: Act as recyclable nucleotide precursors, which the lysate converts into active NTPs (ATP, CTP, GTP, UTP) for transcription.
Guanine: A nucleobase that the salvage pathway converts into GMP and then GTP when GMP is not supplied directly.
d) Translation Mix (Amino Acids)
17 Amino Acid Mix: Supplies the required building blocks for polypeptide chain assembly during protein synthesis.
Tyrosine: Added separately as it has low solubility at neutral pH and would precipitate if mixed with the other amino acids.
Cysteine: Added separately as it is prone to oxidation which can form unwanted disulphide bonds
e) Additives
Nicotinamide: Is a precursor for NAD⁺/NADH cofactors, necessary for metabolic energy flux over long incubations.
f) Backfill
Nuclease Free Water: Brings the reaction to final volume without adding RNases or DNases that could degrade DNA or RNA templates.
2. Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The PEP-NTP mix is ready to churn out proteins in its short time, providing pre-made NTPs and high-energy phosphoenolpyruvate for quick transcription and translation. The NMP-Ribose-Glucose mix is optimized for longer periods of time, using cheaper nucleotide precursors and sugars that are slowly converted to energy and NTPs by the lysate’s own metabolic pathways over many hours.
3. Bonus question: How can transcription occur if GMP is not included but Guanine is?
Transcription still occurs because the E. coli lysate has salvage pathway enzymes that add a phosphoribosyl group to convert Guanine to GMP. This GMP is then phosphorylated to GTP, which is the actual substrate for RNA polymerase.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
1. Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sf-GFP: It matures fast and folds well in cell-free systems, but needs molecular oxygen to form the chromophore.
mRFP1: Slow maturation time (less than 1h) means fluorescence lags translation and it is moderately acid sensitive so pH drops over a few hours can quench the signal.
mKO2: Bright orange protein, relatively fast maturation, but moderate acid sensitivity (to help manage it some use glycolysis driven acidification but this can gradually reduce its fluorescence).
mTurquoise2: Good pH stability and high quantum yield with fast maturation, it is one of the most forgiving reporters in cell-free systems even though it is oxygen dependent.
mScarlet-I: Bright red protein, faster maturation than previous red FPs, but very oxygen-dependent and needs maintained energy input during long incubations.
Electra2: Achieves efficient folding in E. coli cytoplasm and high stability, but needs two-step oxygen-dependent maturation.
2. Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
I picked mRFP1 because of its sensitivity to pH changes, so the target improvement would be to slow its maturation time and moderate its sensitivity to acidic conditions. This could be achieved by experimenting with HEPES-KOH and Magnesium Glutamate. So over approximately 36 hours, HEPES-KOH will be increased to pH 7.5, and Magnesium Glutamate will also increase from 6.975 mM to 8.5 mM. What I expect to see is a maintained pH near 7.5 for longer times, preserving fluorescence of matured mRFP1 protein, so it also folds properly. The Magnesium Glutamate increase can allow for chelation to keep the ribosomes active and maturation can continue.
3. The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
Based on my hypothesis for mRFP1, the master mix compositions would be increasing HEPES-KOH from 45 mM to 80 mM and Magnesium Glutamate from 6.975 mM to 8.5 mM.
4. The final phase of this lab will be analyzing the fluorescence data we collect to determine whether we can draw any conclusions about favorable reagent compositions for our fluorescent proteins. This will be due a week after the data is returned (date TBD!). The reaction composition for each well will be as follows:
6 μL of Lysate
10 μL of 2X Optimized Master Mix from above
2 μL of assigned fluorescent protein DNA template
2 μL of your custom reagent supplements
Total: 20 μL reaction
!! The final fluorescence data has not yet been returned, so analysis of whether my hypothesis was correct is not possible yet.