Week 2 HW: DNA Read, Write, & Edit
Part 1: Benchling & In-silico Gel Art
My original idea was to create two sister chromatids, since most of the patterns from the Enzymes were scattered vertical lines, and they kind of looked like alleles inside a chromosome. I had some trouble creating the centromere of the chromosome because none of the enzymes alone created just one line in the middle of the ladder (so around 800 bp), so I picked SacI and SalI and ignored the top line at 12.0 kb.

Original design using restriction enzymes
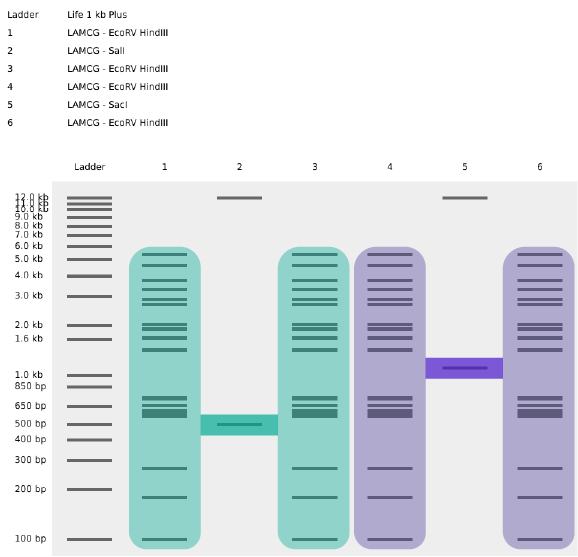
Sister chromatids highlighted from the original design
Part 3: DNA Design Challenge
3.1. Choose your protein.
In relation to my interest in genetic logic gates, Sarkar et al. ’s (2021) research uses a 4-output genetic logic where each output is a fluorescent readout corresponding to a maze solution pattern. The protein I will be using is sfGFP (superfolder GFP) because it is widely used, monomeric, very well characterized, and has a strong fluorescence (Chiu & Jiang, 2017).
This is the protein’s sequence obtained from FPbase:
MSKGEELFTG VVPILVELDG DVNGHKFSVR GEGEGDATNG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKR HDFFKSAMPE GYVQERTISF KDDGTYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNFNSHNV YITADKQKNG IKANFKIRHN VEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSVLSKD PNEKRDHMVL LEFVTAAGIT HGMDELYK
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
I introduced the Amino acid sequence in BioInformatics Sequence Manipulation Suite and obtained this:
reverse translation to a 714 base sequence of most likely codons.
atgagcaaaggcgaagaactgtttaccggcgtggtgccgattctggtggaactggatggc gatgtgaacggccataaatttagcgtgcgcggcgaaggcgaaggcgatgcgaccaacggc aaactgaccctgaaatttatttgcaccaccggcaaactgccggtgccgtggccgaccctg gtgaccaccctgacctatggcgtgcagtgctttagccgctatccggatcatatgaaacgc catgatttttttaaaagcgcgatgccggaaggctatgtgcaggaacgcaccattagcttt aaagatgatggcacctataaaacccgcgcggaagtgaaatttgaaggcgataccctggtg aaccgcattgaactgaaaggcattgattttaaagaagatggcaacattctgggccataaa ctggaatataactttaacagccataacgtgtatattaccgcggataaacagaaaaacggc attaaagcgaactttaaaattcgccataacgtggaagatggcagcgtgcagctggcggat cattatcagcagaacaccccgattggcgatggcccggtgctgctgccggataaccattat ctgagcacccagagcgtgctgagcaaagatccgaacgaaaaacgcgatcatatggtgctg ctggaatttgtgaccgcggcgggcattacccatggcatggatgaactgtataaa
3.3. Codon optimization. Using NovoProLabs Codon Optimization tool, here’s the optimized sequence:
Optimized for E.coli
ATGTCTAAAGGCGAAGAACTGTTCACCGGCGTAGTACCTATCCTGGTTGAACTGGATGGCGACGTTAACGGTCACAAGTTTTCTGTCCGTGGCGAAGGTGAAGGCGACGCTACTAACGGCAAACTGACTCTGAAATTCATCTGTACCACGGGTAAACTGCCGGTACCTTGGCCTACCCTGGTTACCACTCTGACCTACGGCGTTCAGTGTTTCTCCCGTTATCCAGACCATATGAAACGCCATGACTTCTTCAAAAGCGCGATGCCGGAAGGTTACGTACAGGAGCGTACCATCTCTTTCAAGGACGATGGCACCTACAAAACCCGTGCTGAAGTTAAGTTCGAAGGCGACACCCTGGTTAACCGTATCGAACTGAAGGGCATCGACTTCAAAGAAGACGGCAACATTCTGGGTCACAAGCTGGAGTACAACTTCAACAGCCACAACGTGTACATCACTGCCGACAAACAGAAAAACGGTATTAAAGCGAACTTCAAAATCCGTCATAACGTCGAAGATGGTTCCGTTCAACTGGCTGATCATTACCAGCAGAACACTCCGATCGGTGATGGCCCGGTTCTGCTGCCGGACAACCACTACCTGTCTACGCAATCCGTGCTGTCTAAAGACCCGAACGAAAAGCGTGACCACATGGTACTGCTGGAATTTGTTACCGCCGCTGGTATCACTCACGGCATGGATGAACTGTACAAATGA
Codon optimization is important because organisms can have different preferences for codon usage, which means that when introducing a gene sequence on a host organism, its own codon usage preferences may affect gene expression or protein synthesis. When doing codon optimization, you are modifying the sequence to enhance protein expression in the host organism.
For this codon optimization I chose E.coli, because most genetic logic gates experiments involve this bacteria due to its simplicity to engineer and wide usage.
3.4. You have a sequence! Now what? What technologies could be used to produce this protein from your DNA?
Technologies like Twist’s Silicon-based DNA Synthesis allows for high precision protein synthesis. You just design a custom sequence on the Twist’s website and order the custom gene synthesis from them.
Part 4: Prepare a Twist DNA Synthesis Order
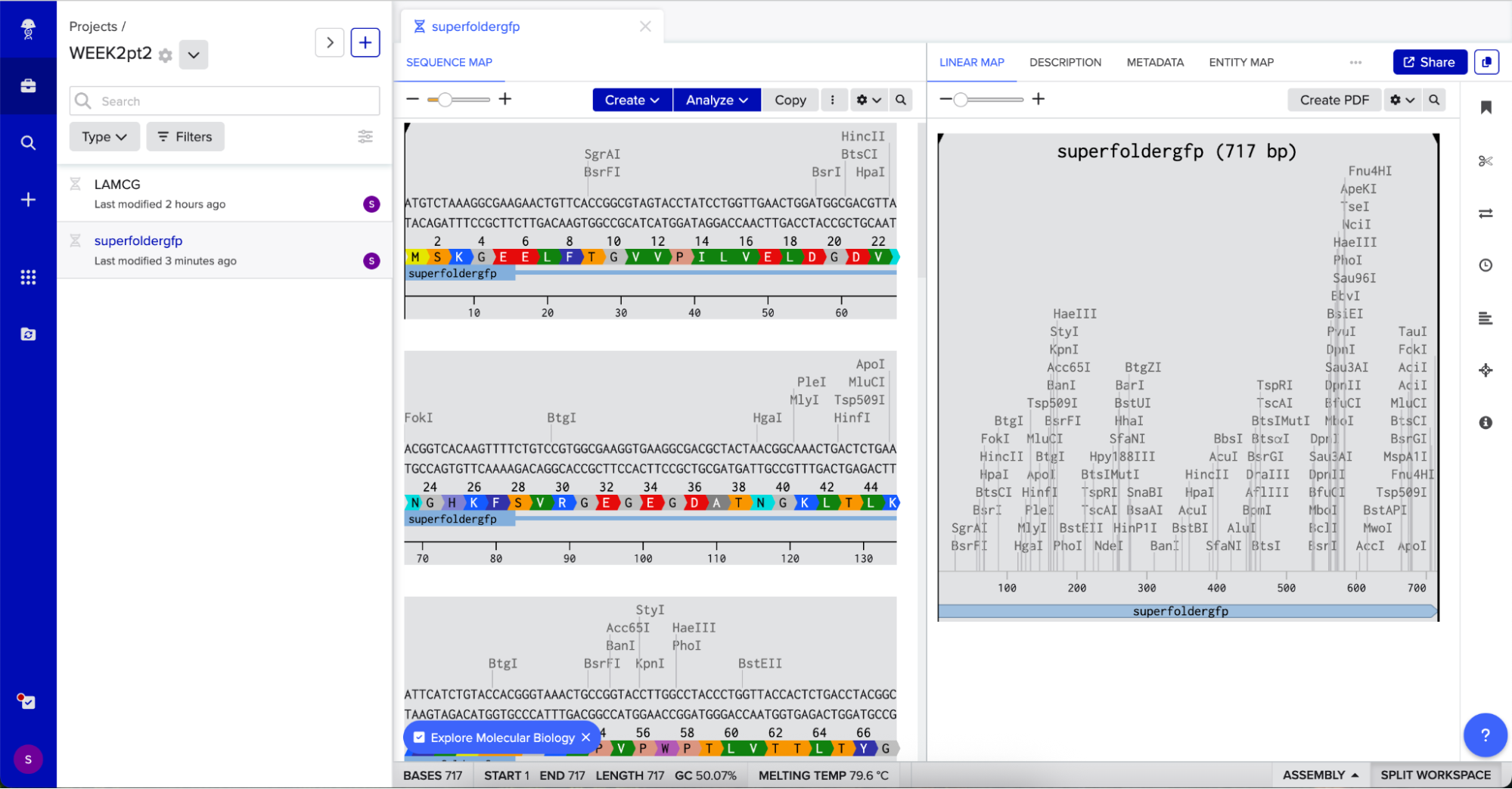
Checking the protein is going to express correctly
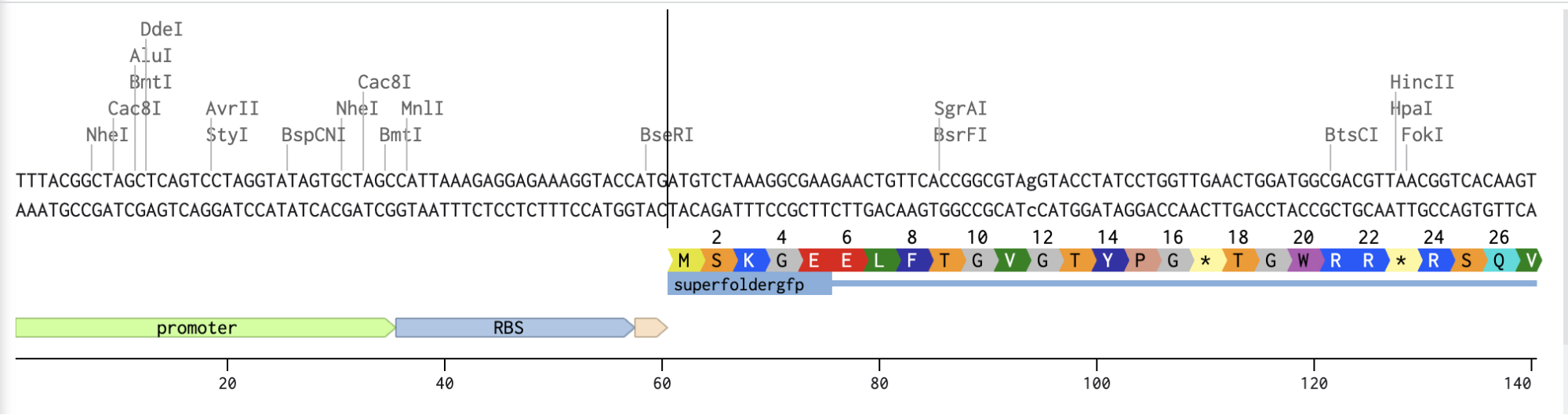
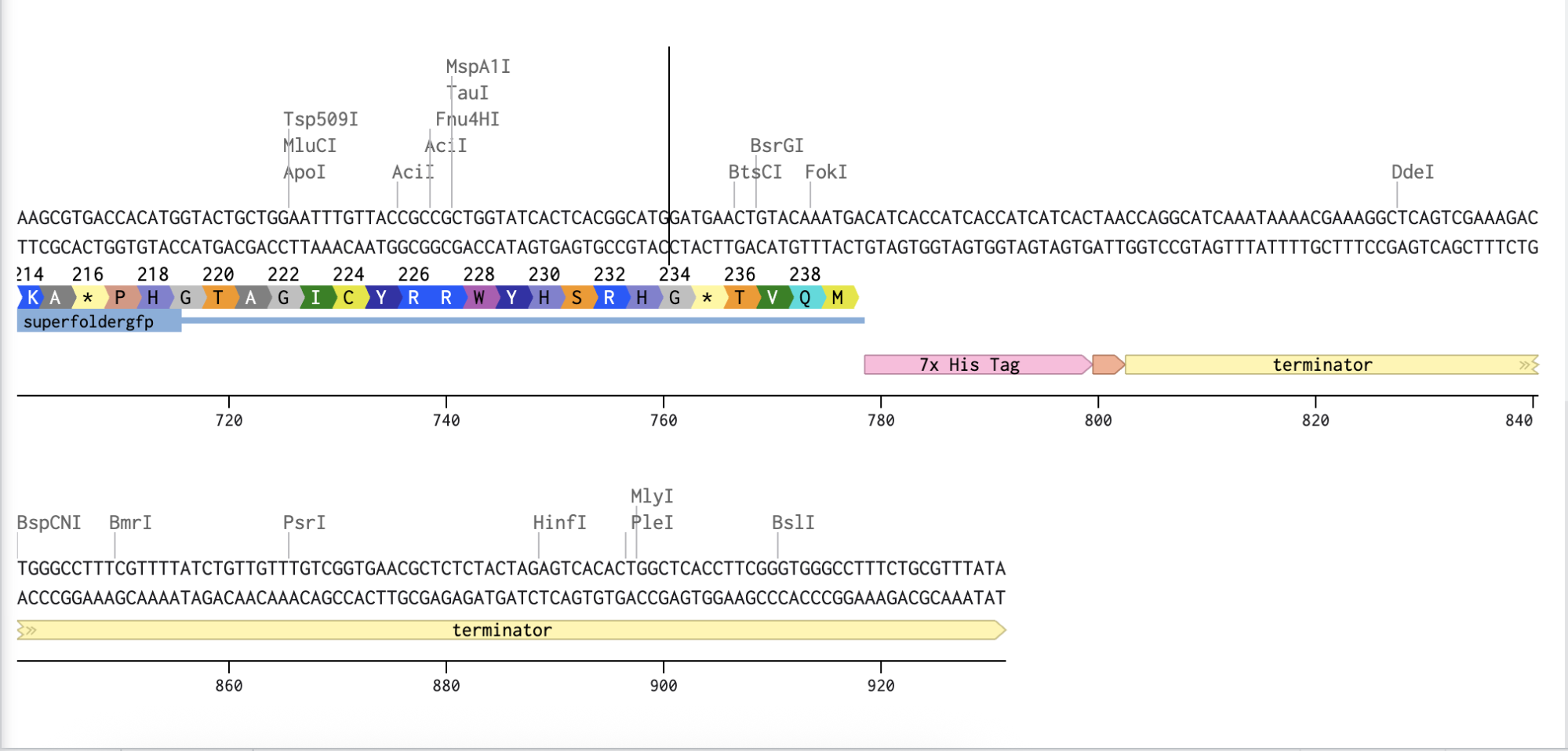
Adding the Promoter, RBS, Start Codon, Coding Sequence, His Tag, Stop Codon, and Terminator sequences in the beginning and end of my optimized sequence.
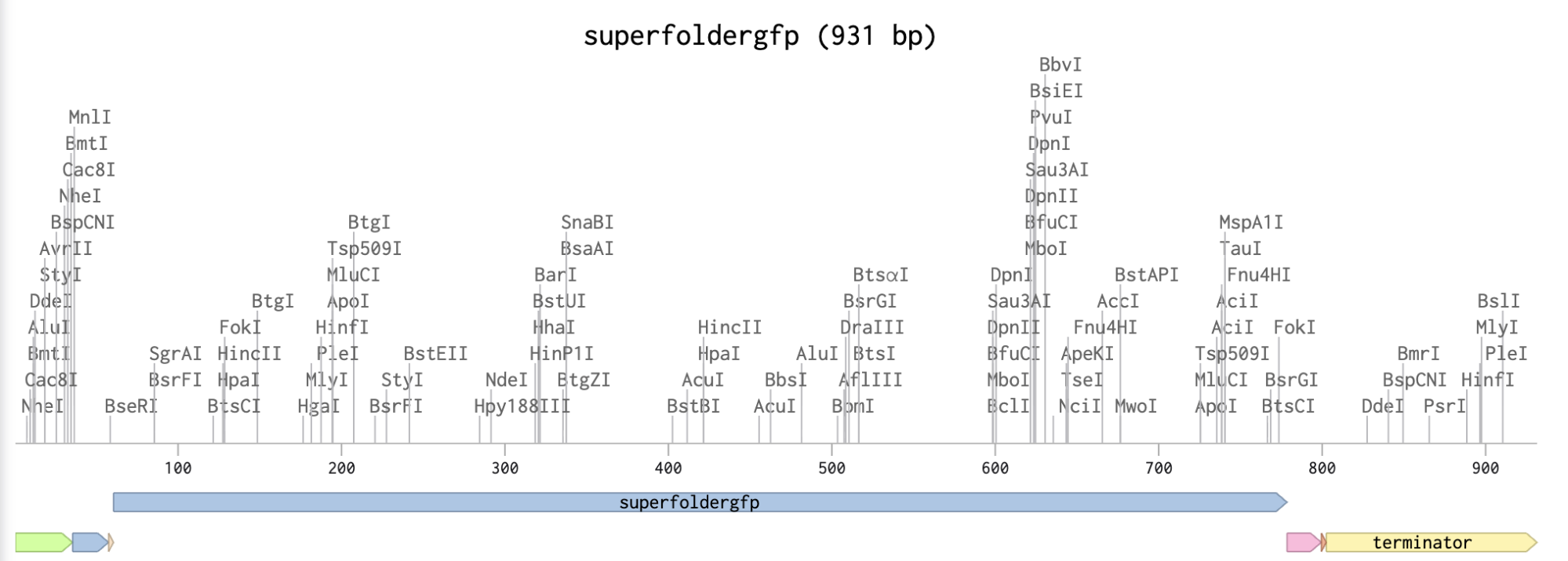
Linear Map of the annotated sequence. Here’s the shared link to it!
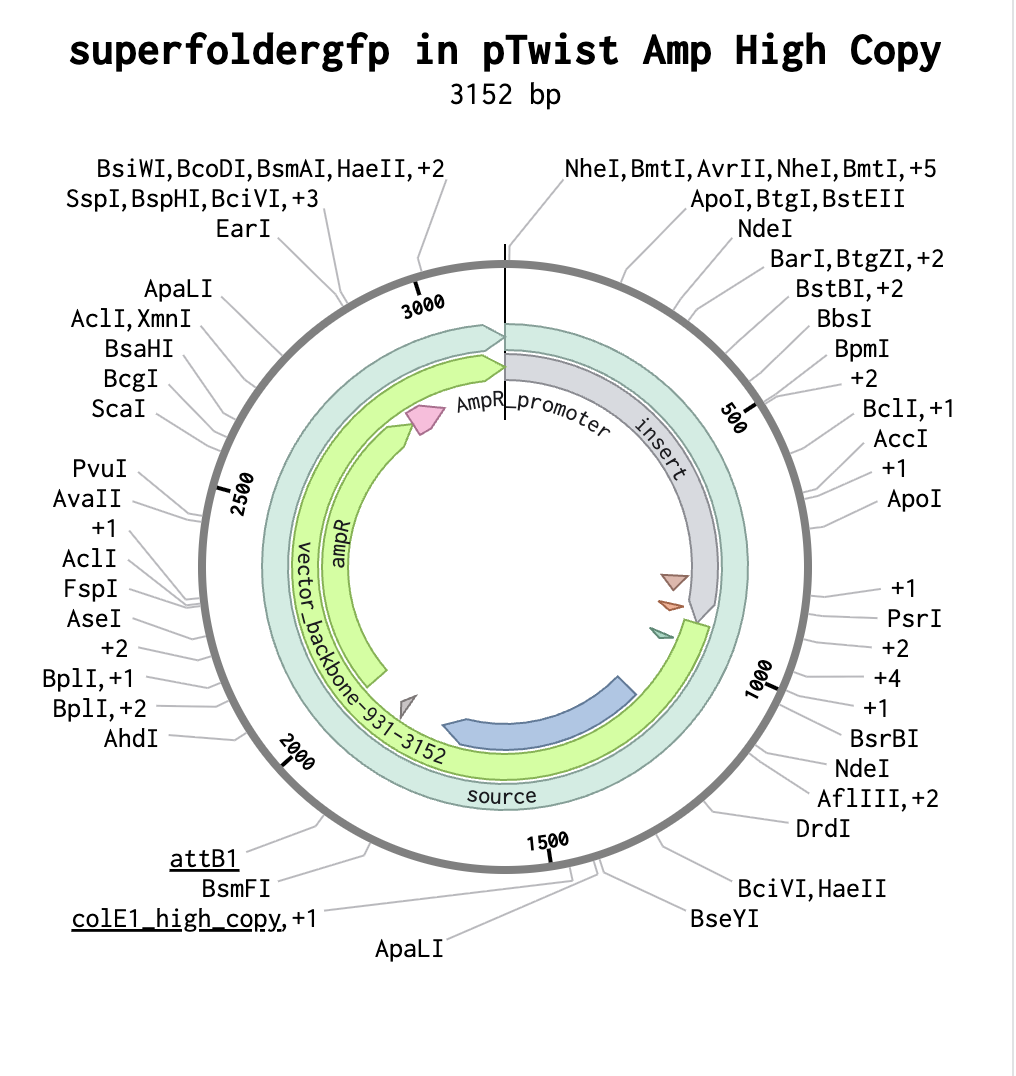
I then downloaded the fasta file for the sequence and uploaded it on Twist. Then, I picked the pTwist Amp High Copy - (2221bp) circular vector.
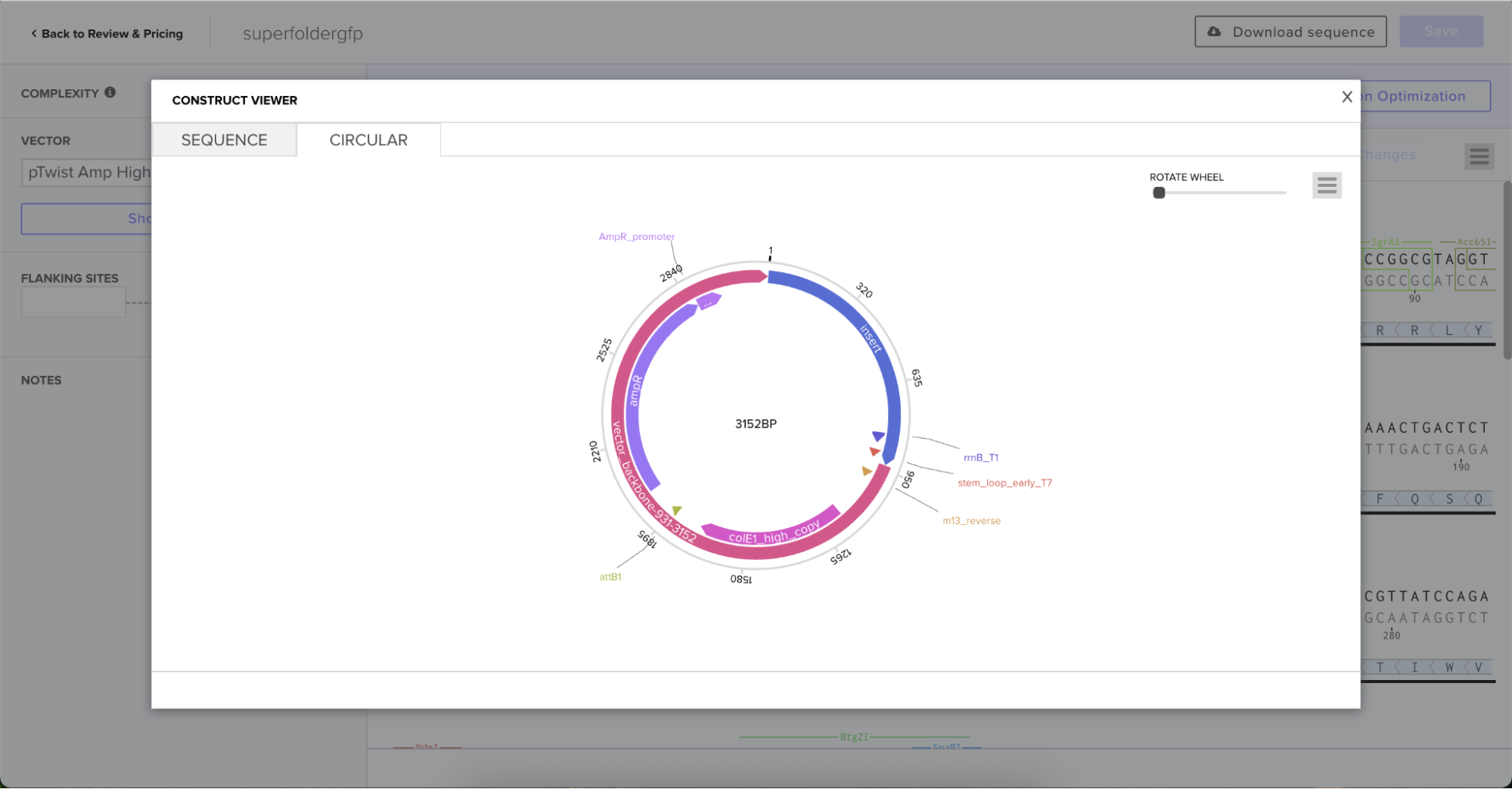
And here’s the circular construct viewer of the sequence + the vector
And here’s the plasmid on Benchling after uploading the downloaded construct from Twist:
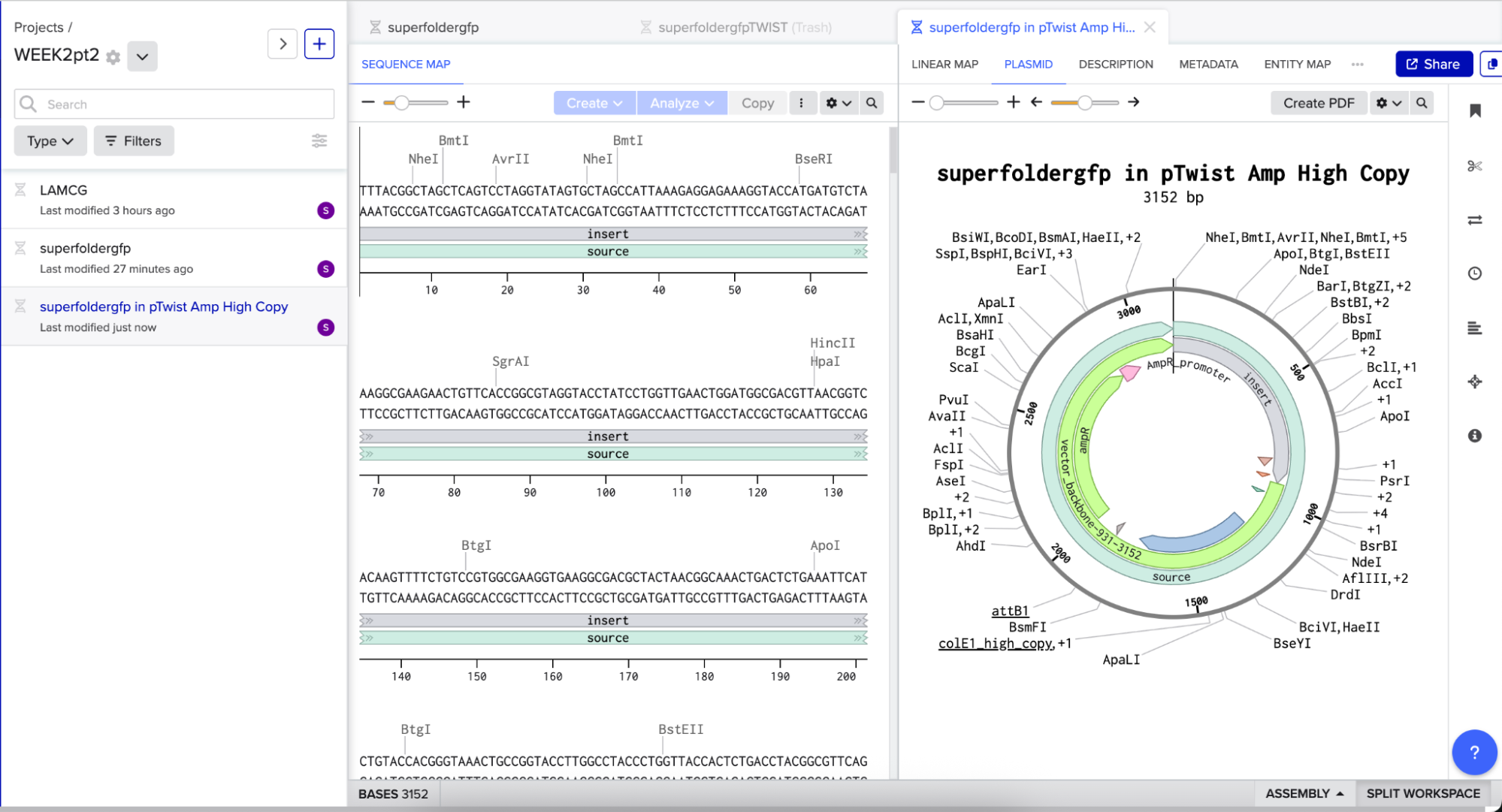
Plasmid close-up:
5.1 DNA Read
1. What DNA would you want to sequence (e.g., read) and why?
I would like to sequence the plasmids from three engineered bacterial strains: green, red and yellow responder. This is the first step to verify that the genetic circuits are assembled correctly, and they don’t have mutations, premature stop codons, or show unwanted recombination errors. The sequences will come from E.coli’s DNA, specifically engineered E. coli DH5α strains.
2. In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Ilumina MiSeq, which offers high accuracy at a lower cost for verifying multiple plasmids. In theory, the short reads from this method are okay because I would know the expected sequence and just need to confirm it before continuing with the project methodology.
Also answer the following questions:
3. Is your method first-, second- or third-generation or other? How so?
Second generation because it can sequence multiple DNA fragments simultaneously, so it is more efficient instead of doing multiple runs to sequence the plasmids DNA.
4. What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
The DNA sequence of the E. coli DH5α strains (green, red and yellow responders) would be the input. In order to prepare it, I would need to: Extract the plasmid DNA from each strain. Quantify the DNA (can be done using Nanodrop) Fragment the DNA (can be done using enzymes) to approximately 500 bp Repair the sticky ends and create DNA with blunt ends Prevent the fragments from ligating to each other during the adapter ligation reaction by A-tailing Add sequencing adapters with barcodes by adapter ligation Amplify the sequence using PCR Pool multiple libraries into a flow cell
5. What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
After preparing the input, the steps for sequencing are: Binding DNA fragments to the flow cell, the bridge amplification creates clusters of the identical copies of DNA. Add fluorescently labeled reversible terminators Capture which base was added to each cluster of identical copies by laser excitation Use a software for base calling (could be Dorado by Oxford Nanopore Technologies) Assign Phred quality scores to each base
6. What is the output of your chosen sequencing technology?
After the sequencing, I would have multiple FASTQ files with the raw reads with the Phred quality scores and BAM files showing variants, to see any mutations in the sequences.
5.2 DNA Write
1. What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
I would synthesize three genetic circuits for the bacterial pattern recognizer: Green responder:
5’- [EcoRI] - [pBAD promoter] - [RBS] - [GFP gene] - [Terminator] - [pLac promoter (reverse)] - [RBS] - [LacI gene] - [Terminator] - [Ampicillin resistance] - [pUC origin] - [XhoI] -3'
Red responder:
5’- [EcoRI] - [pLac promoter] - [RBS] - [RFP gene] - [Terminator] - [pBAD promoter (reverse)] - [RBS] - [AraC gene] - [Terminator] - [Chloramphenicol resistance] - [p15A origin] - [XhoI] -3'
Yellow responder:
Combining the green + red responder plasmids in one cell
By synthesizing circuits instead of assembling, I could ensure more accuracy in the sequences.
2. What technology or technologies would you use to perform this DNA synthesis and why?
Twist could be very useful for this step, in order to achieve array-based oligo synthesis.
Also answer the following questions:
3. What are the essential steps of your chosen sequencing methods?
Key steps are:
- Using FASTA format for the sequences
- Using Twist to optimize codon usage, ensure higher accuracy, high parallelism and quality control
- Do oligo synthesis on silicon chip
- Cleave and release the oligos from the chip
- Assemble the oligos into longer fragments using PCR or Gibson assembly
- Clone longer fragments by introducing them into vectors
- Do a full-length Sanger verification
4. What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
This sequencing methodology has many steps and could take weeks to do, especially because it depends on multiple steps with different shipping times. Also, the cost increases when creating longer fragments.
5.3 DNA Edit
1. What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
For my final project, I would need to edit the genome of the E. coli DH5α strain because it has LacI and AraC genes, which could intervene with the synthetic circuits that will be introduced later on. Also, removing these genes allow for real-world applications, as antibiotic-free systems are widely used for environmental or medical uses.
2. What technology or technologies would you use to perform these DNA edits and why? CRISPR-Cas9 because it can cut the genome in precise places, facilitating the extraction of the unwanted genes, and then can stitch back the fragments together.
Also answer the following questions:
3. How does your technology of choice edit DNA?
First, sgRNA guides the Cas9 enzyme to target the DNA sequence. Then, Cas9 creates a double-strand cut in the desired place of the unwanted genes. Finally, the cell repairs the break through NHEJ or HDR.
4. What are the essential steps?
First, design the RNA map to highlight and target the prophages. Second, prepare the DNA template to be edited. Third, introduce the RNA map and the DNA template in E.coli cells.
5. What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
Steps for editing the DNA:
- Prepare the input: E.coli DH5α strain, create the pCas plasmid (that encodes Cas9, Lambda Red and sgRNA for the CRISPR-Cas reaction), donor DNA fragments (from the synthesized PCR products), and the editing oligos for sgRNA cloning
- Clone the sgRNAs
- Transform pCas into target strain by electroporation and selection with kanamycin
- Induce Lambda Red by growth with arabinose (which induces recombination proteins)
- Add donor DNA and the transformed pCas, then electroporate
- Amplify sequence using colony PCR
- Grow pCas plasmid and test for loss of kanamycin resistance, to ensure it grew without antibiotic resistance
6. What are the limitations of your editing methods (if any) in terms of efficiency or precision?
CRISPR-Cas9 is not always 100% effective, as there is a small risk it will accidentally cut the DNA in the wrong places (these are called off-target effects). To avoid this, the guide RNA has to be very carefully designed. Also, another “limiting factor is the fact that dCas9 is a shared resource amongst the different gates which needs to be continuously expressed at very high concentrations, and this leads to high toxicity for the host cells” (Al-Radhawi et al., 2020).
References:
Chiu TY, Jiang JR. Logic Synthesis of Recombinase-Based Genetic Circuits. Sci Rep. 2017 Oct 9;7(1):12873. doi: 10.1038/s41598-017-07386-3. PMID: 28993615; PMCID: PMC5634492.
Sarkar, K., Bonnerjee, D., & Bagh, S. (2021). Engineered Bacteria Computationally Solve Chemically Generated 2X2 Maze Problems. Homi Bhabha National Institute (HBNI). https://doi.org/10.1101/2021.06.16.448778
Zhang, H., Lin, M., Shi, H. et al. Programming a Pavlovian-like conditioning circuit in Escherichia coli. Nat Commun 5, 3102 (2014). https://doi.org/10.1038/ncomms4102
Chen J, Li Y, Zhang K, Wang H2018.Whole-Genome Sequence of Phage-Resistant Strain Escherichia coli DH5α. Genome Announc6:10.1128/genomea.00097-18.https://doi.org/10.1128/genomea.00097-18
Rath, D., Amlinger, L., Rath, A., & Lundgren, M. (2015). The CRISPR-Cas immune system: biology, mechanisms and applications. Biochimie, 117, 119-128.
Al-Radhawi, M. A., Tran, A. P., Ernst, E. A., Chen, T., Voigt, C. A., & Sontag, E. D. (2020). Distributed implementation of boolean functions by transcriptional synthetic circuits. ACS Synthetic Biology, 9(8), 2172-2187.