Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
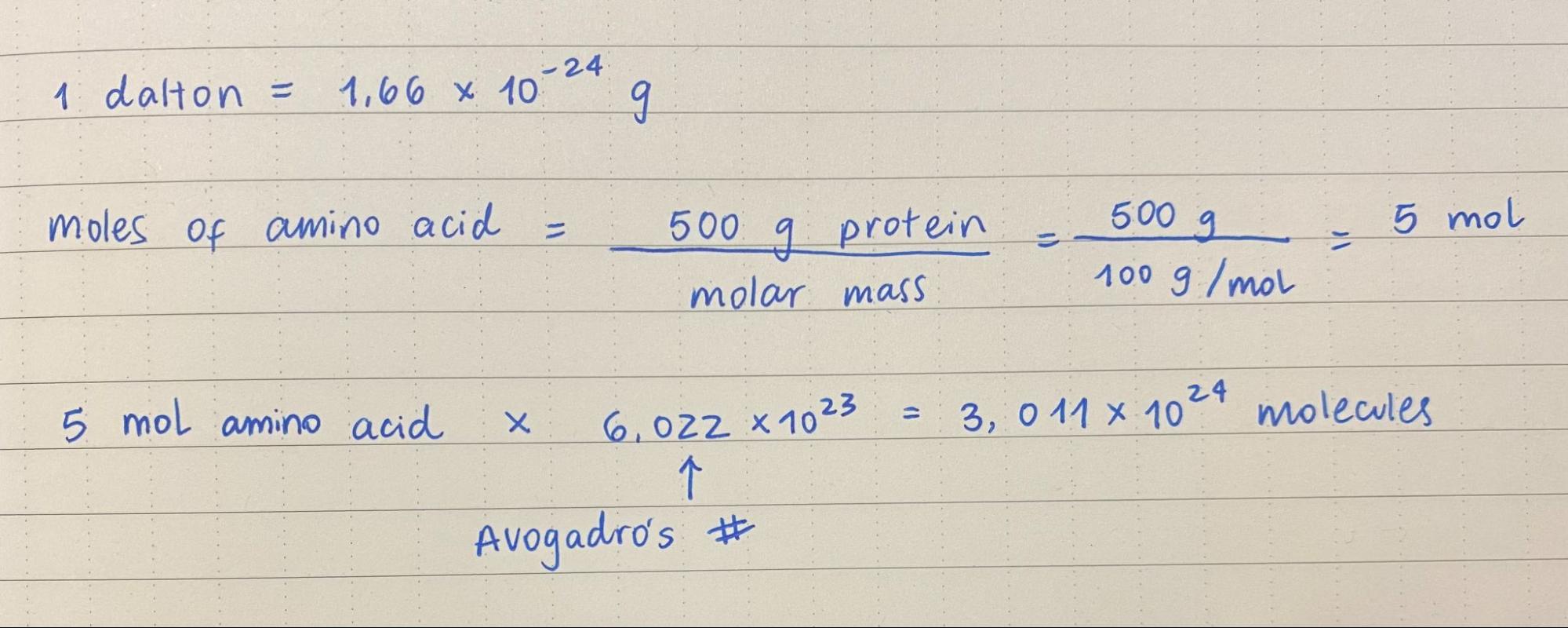
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Because consuming something as a way to obtain energy does not mean we are assimilating it as part of ourselves. When humans eat meat (or anything, really), we are breaking it down into smaller pieces, and the molecule’s chemical bonds break down. The energy stored between those chemical bonds is released as energy for our bodies, and the nutrients from the food source are absorbed. In this process, our human cells are not acquiring, reading or translating any DNA from foreign organisms (a cow, a fish, a plant).
3. Why are there only 20 natural amino acids?
“The selection of the 20 standard residue types was made early on in evolution–their appearance predates RNA and DNA and it is highly likely that they already played a vital role throughout prebiotic chemical evolution (~4 Gyrs ago)” (Bywater, 2018). However, the question until this day is why 20 and not another number? According to Bywater (2018), our living systems’ DNA is able to cater for 64 possible amino acids types. He explains that there is a key factor that explain the number 20: those amino acids show “energetically well-separated conformers”.
4. Can you make other non-natural amino acids? Design some new amino acids.
Yes, you can. They are referred to as noncanoninal amino acids (ncAAs) introduced into proteins. They can modify the protein backbone or the amino acid side chains (Budisa, 2025).
5. Where did amino acids come from before enzymes that make them, and before life started? Miller and Urey proved that amino acids can form from a concoction of gases and electricity, without the existence of enzymes and ribozymes, which is what scientist say likely happened in primitive Earth, which was abundant in hydrogen, a key element for amino acid formation. RNA and enzymes are relatively recent in Earth’s history (Bywater, 2018).
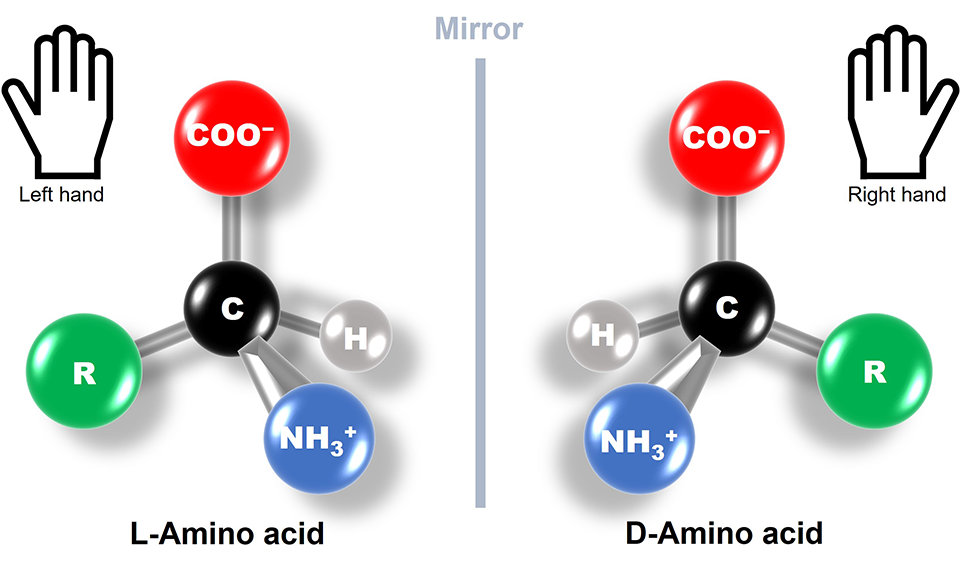
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
I understand that L-amino acids form right-handed α-helix conformations. So a α-helix made from D-amino acids would have the opposite: left-handed
7. Can you discover additional helices in proteins?
Yes, mainly identifiable by different patterns in the hydrogen bonds, creating tighter or wider geometries in the helices. For example, π-helices have “been described as α-aneurisms, α-bulges, or π-bulges” (Kumar & Bansal, 2015).
8. Why are most molecular helices right-handed?
Because of energy components. Left-handed helices tend to have unusual and weaker interactions than right-hand helices (Rzepa, n.d).
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets have hydrogen-bonding edges that are facing one way, which allows them to interact, bond and aggregate with other β-sheets.
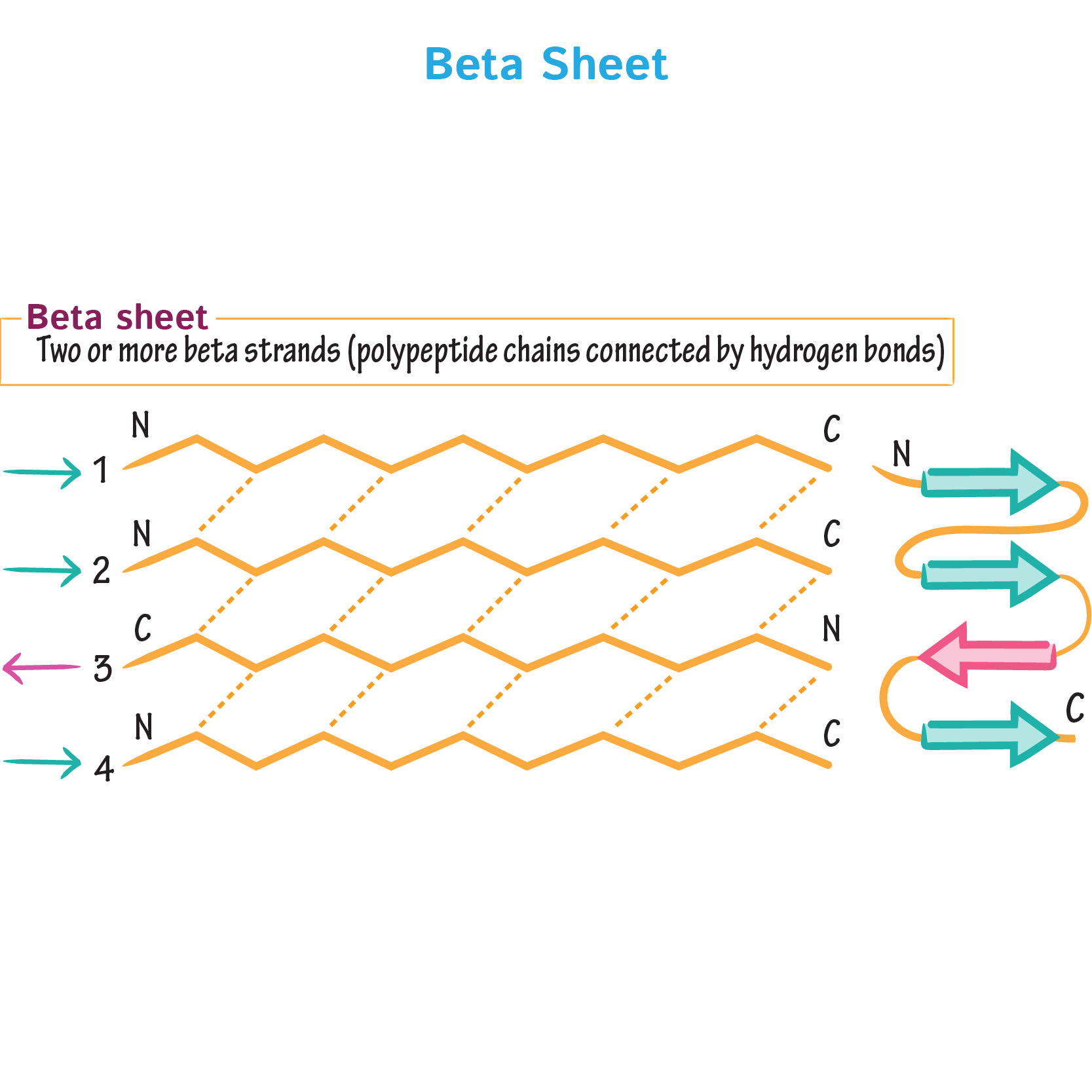 Ditki (2017)
Ditki (2017)
References:
- Bywater, R. P. (2018). Why twenty amino acid residue types suffice (d) to support all living systems. Plos one, 13(10), e0204883.
- Budisa, N. (2025). Introduction:“Noncanonical Amino Acids”. Chemical Reviews, 125(4), 1659-1662.
- Kumar, P., & Bansal, M. (2015). Dissecting π‐helices: sequence, structure and function. The FEBS Journal, 282(22), 4415-4432.
- Rzepa, H. (n.d). “Why are α-helices in proteins mostly right handed?”. Imperial College London. https://www.ch.ic.ac.uk/rzepa/blog/?p=3802
- Ditki (2017). Biochemistry Glossary: Protein Structure Class: 2(b). Secondary - Beta Sheets. Ditki Medical & Biological Sciences. https://ditki.com/course/biochemistry/glossary/biochemical-pathway/beta-sheets-aka-beta-pleated-sheets
Part B: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
My main final project idea is to develop a filter that uses bacterial biofilms to reduce the load of the chytrid fungi Batrachochytrium dendrobatidis (Bd), responsible for killing hundreds of amphibian species. According to Abramyan & Stajich (2012), Bd has a lot of chitin-binding modules in its genome, which could potentially be a reason for its high pathogenicity. There are potent antifungal chitinases that damage the cell walls of the pathogenic fungi that have been modified to be more thermostable, such as GH19 chitinase from Ficus microcarpa latex (Kozome et al., 2022). For my biofilm filter, this protein could be a great addition.
2. Identify the amino acid sequence of your protein.
The ID of the protein in UniProt is A0A915Q9K7, PDB 7V92: https://www.uniprot.org/uniprotkb/A0A915Q9K7/entry
 The sequence is 245 amino acids long, and the most common aminoacid is G, which appears 30 times.
The sequence is 245 amino acids long, and the most common aminoacid is G, which appears 30 times.
Complete sequence: “DISKLISRGTFDQMLKHRNDGACPAKGFYTYDAFIAAAKAFPGFGTTGDDATRKREIAAFLGQTSHETTGGWPSAPDGPYSWGYCFLREKNPSSSYCSPSPTYPCAPGKQYYGRGPIQLSWNYNYGPCGKAIGVDLLNNPDLVATDPVISFKTALWFWMTPQSPKPSCHNVITGIWKPSAADQSAGRVPGYGATTNIINGGLECGQGWKPQVEDRIGFYKRYCDIFKVGYGNNLDCYNQRPFGSG”
A BLAST search of A0A915Q9K7 against UniProt returns 250 homologous sequences above the default significance threshold, mainly GH19 chitinases from other flowering plants, especially within the Moraceae and related taxa.
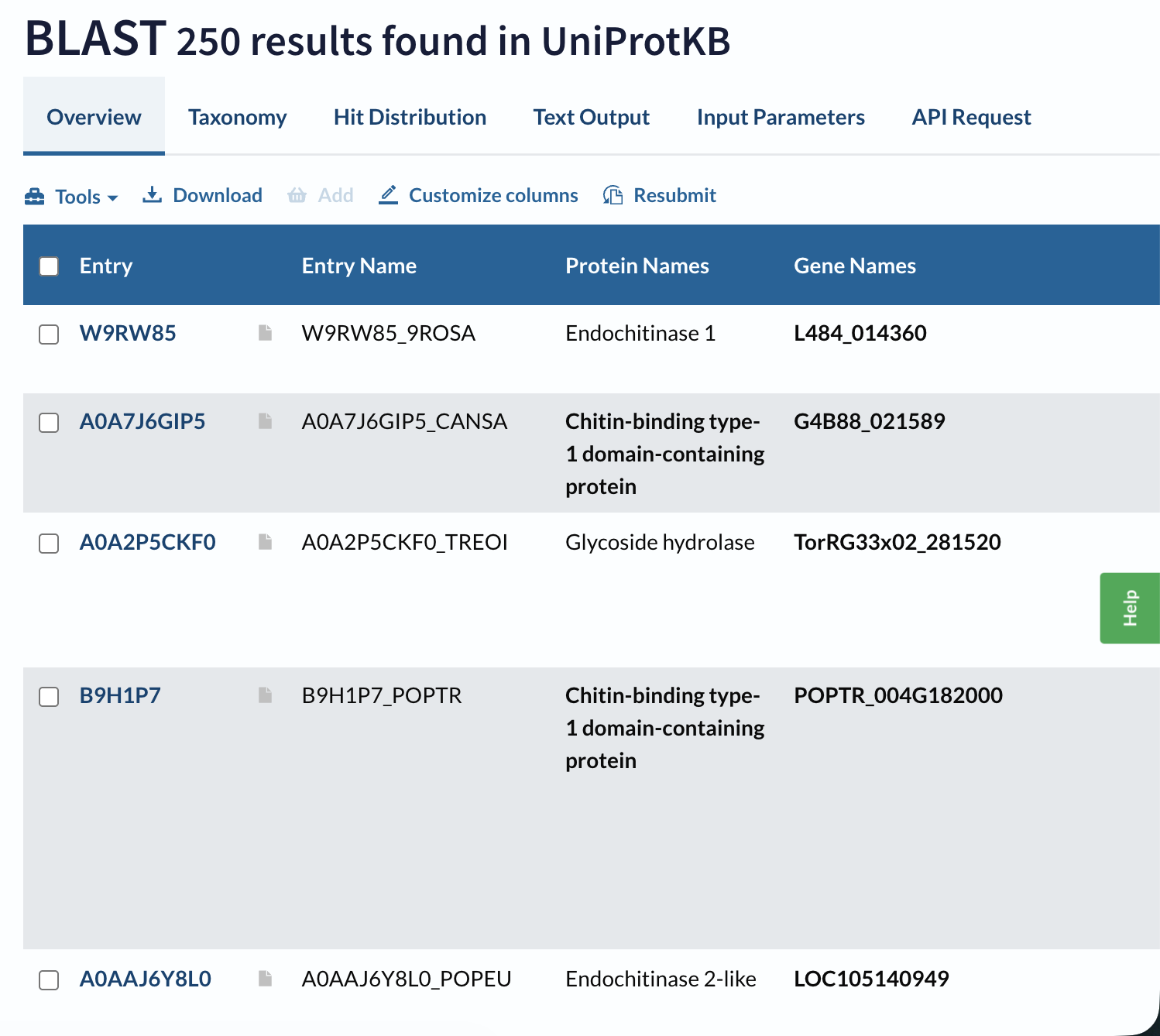 Domain databases classify A0A915Q9K7 as a member of glycoside hydrolase family 19 (GH19), within a lysozyme‑like endochitinase superfamily. It carries the canonical GH19 catalytic motifs (CHITINASE_19_1 and CHITINASE_19_2) and is grouped in the “Endochitinase (Chitinase)” CATH superfamily.
Domain databases classify A0A915Q9K7 as a member of glycoside hydrolase family 19 (GH19), within a lysozyme‑like endochitinase superfamily. It carries the canonical GH19 catalytic motifs (CHITINASE_19_1 and CHITINASE_19_2) and is grouped in the “Endochitinase (Chitinase)” CATH superfamily.
3. Identify the structure page of your protein in RCSB
In RCSB the protein 7V92 Chitinase can be found in this link: https://www.rcsb.org/structure/7V92
The 3D structure of the protein was solved by X‑ray crystallography at 1.61 Å resolution, which is very high quality.
When exploring the 3D view, I see four protein chains in the asymmetric unit (A–D). Around the structure, I also see smaller water molecules.
 The 7V92 structure belongs to the Endochitinase / GH19 chitinase superfamily in the lysozyme‑like (SSF53955) fold class.
The 7V92 structure belongs to the Endochitinase / GH19 chitinase superfamily in the lysozyme‑like (SSF53955) fold class.
4. Open the structure of your protein in any 3D molecule visualization software:
Here’s the first look of the protein in PyMOL:
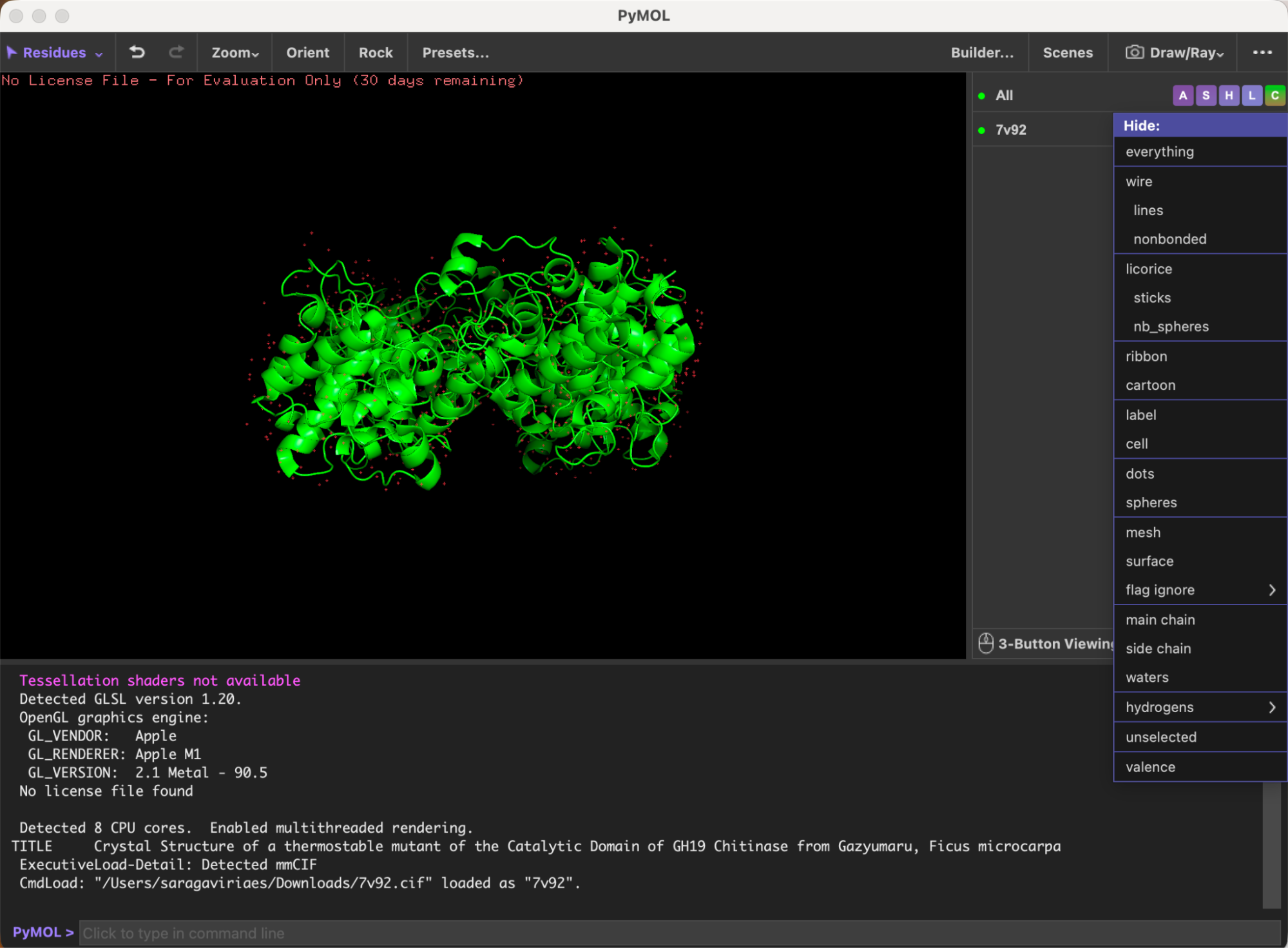
Here’s the visualization of the protein as “cartoon”:
 Here’s the visualization of the protein as “ribbon”:
Here’s the visualization of the protein as “ribbon”:
 Here’s the visualization of the protein as “ball and stick”:
Here’s the visualization of the protein as “ball and stick”:
 Here’s the visualization of the protein as “spheres”:
Here’s the visualization of the protein as “spheres”:

Here’s the visualization of the protein as “surface”:
And this is how it looks like when coloring it by secondary structure:
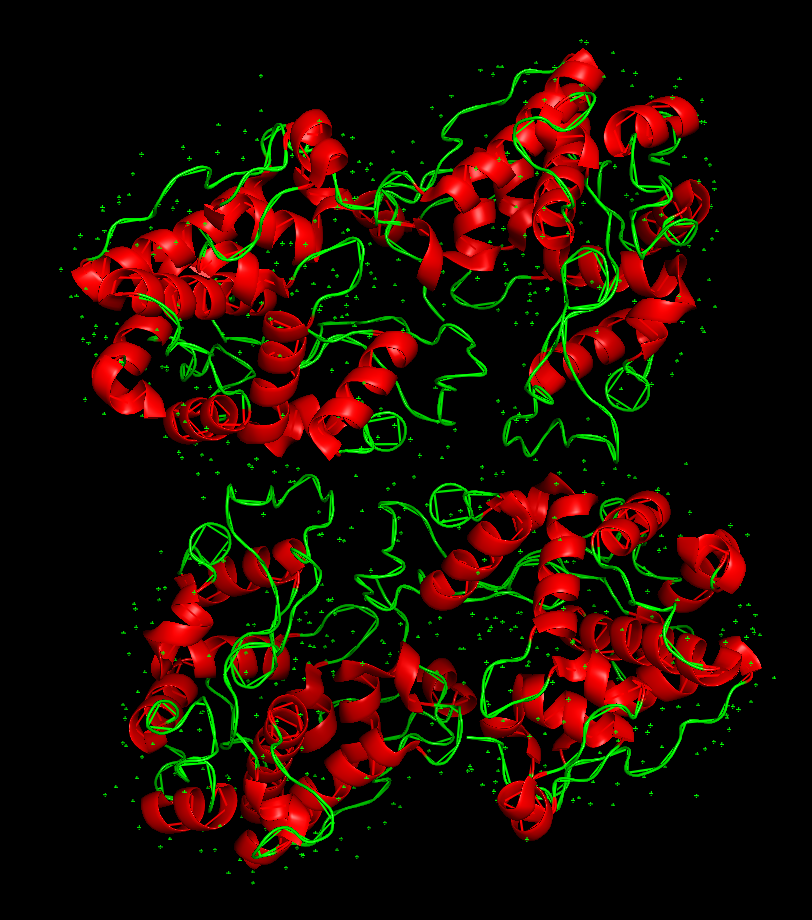 The protein has more helices than loops/coils.
The protein has more helices than loops/coils.
To visualize the protein by residue class, I used this code created with ChatGPT:
And this is the image produced by that code:
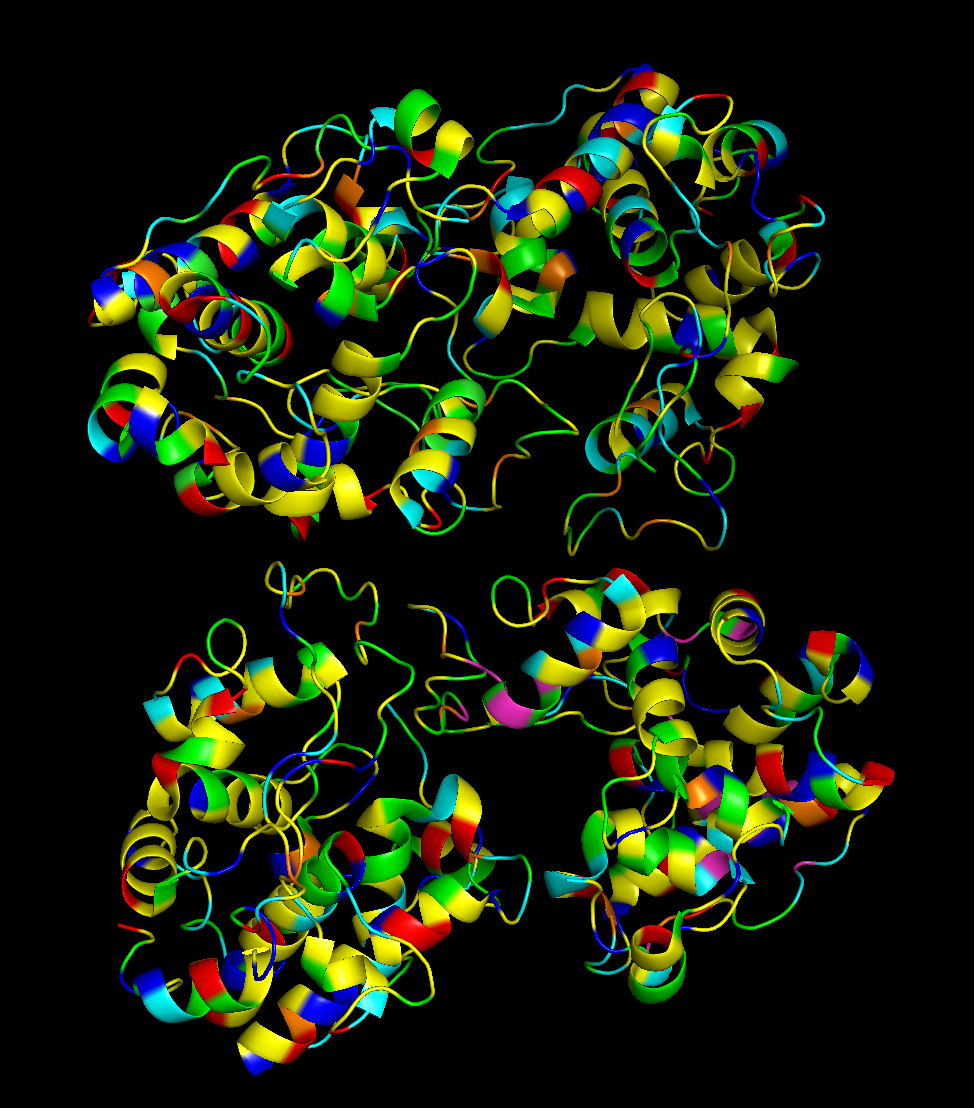 Hydrophobic residues are mainly in the interior of the protein structure,while hydrophilic and charged residues are mainly on the surface. This is a typical distribution of hydrophobic and hydrophilic residues on a soluble enzyme.
Hydrophobic residues are mainly in the interior of the protein structure,while hydrophilic and charged residues are mainly on the surface. This is a typical distribution of hydrophobic and hydrophilic residues on a soluble enzyme.
When inspecting for “holes”, I found these:
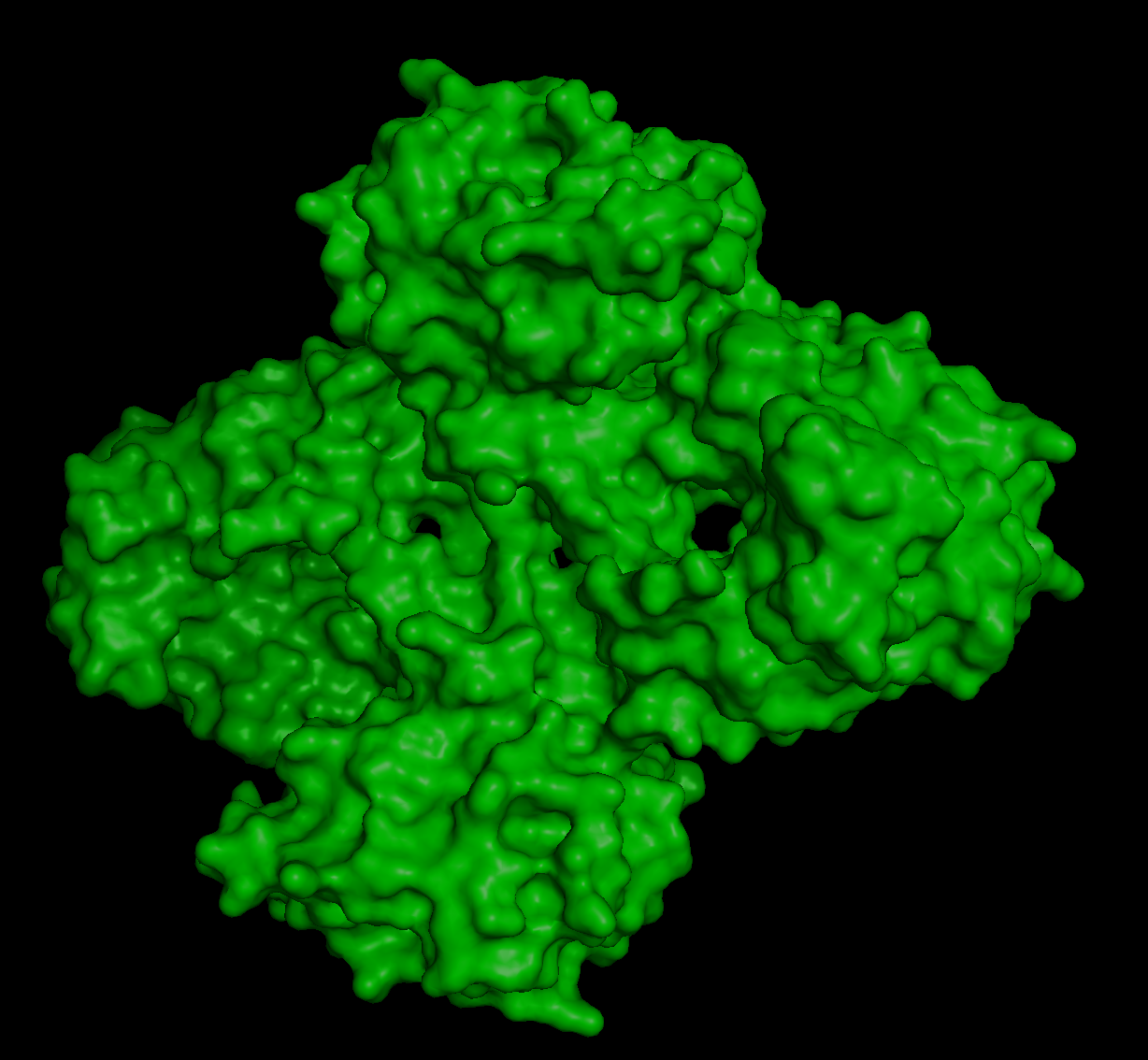
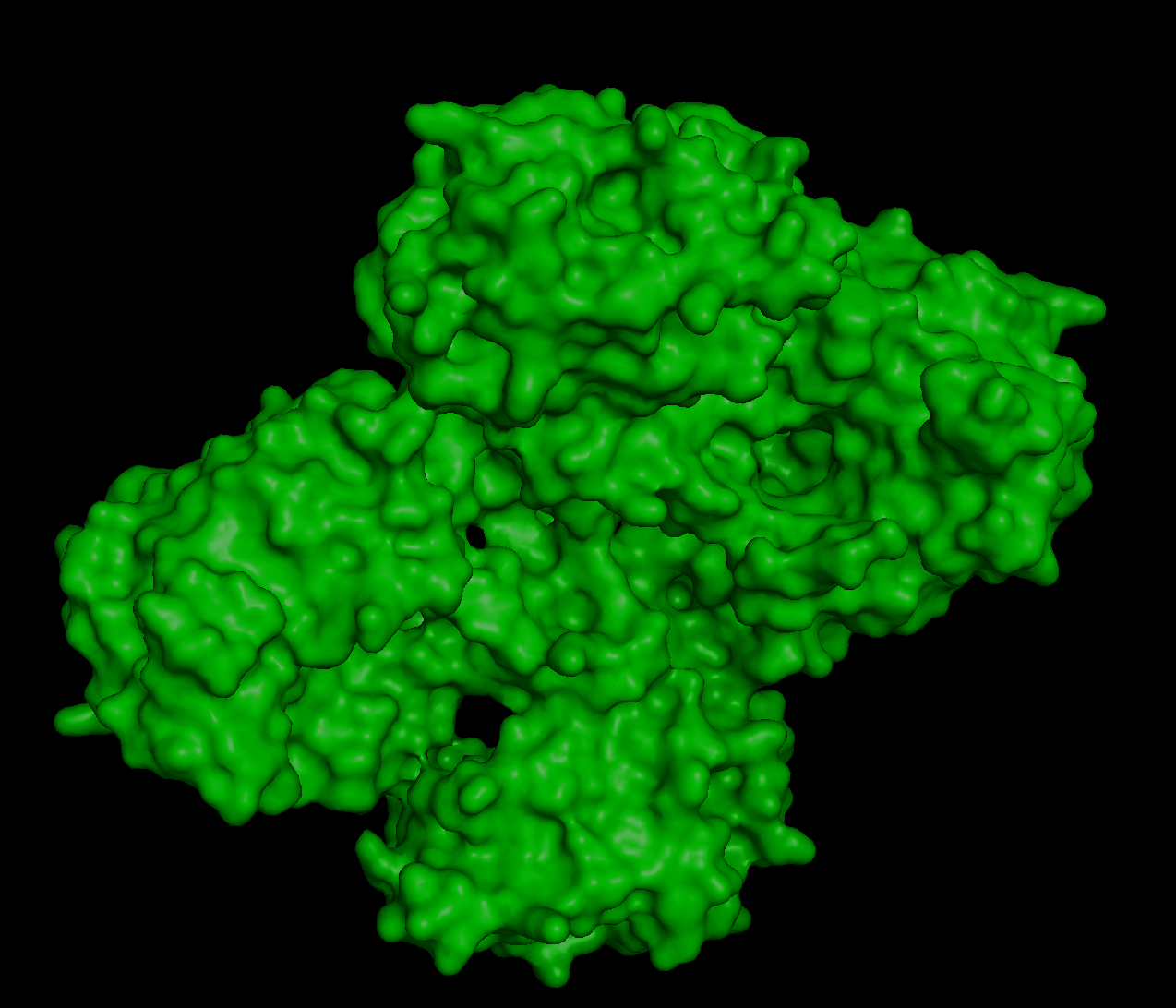
 Although they are multiple holes, I would also say they are small.
Although they are multiple holes, I would also say they are small.
References:
- Kozome, D., Uechi, K., Taira, T., Fukada, H., Kubota, T., & Ishikawa, K. (2022). Structural analysis and construction of a thermostable antifungal chitinase. Applied and Environmental Microbiology, 88(12), e00652-22.
- Abramyan, J., & Stajich, J. E. (2012). Species-specific chitin-binding module 18 expansion in the amphibian pathogen Batrachochytrium dendrobatidis. MBio, 3(3), 10-1128.
Part C. Using ML-Based Protein Design Tools
For this exercise, I chose the protein human beta-crystallin B3, a protein encoded by the gene CRYBB3. I find this protein super interesting because it is found in our eyes, helping maintain transparency and refractive index of the lens. It is crazy to me how a protein can be so transparent, flexible, and stable at the same time, adapting to the changes in the eye through aging. Mutations in the gene can cause cataracts and other eye diseases (NCBI, 2026).
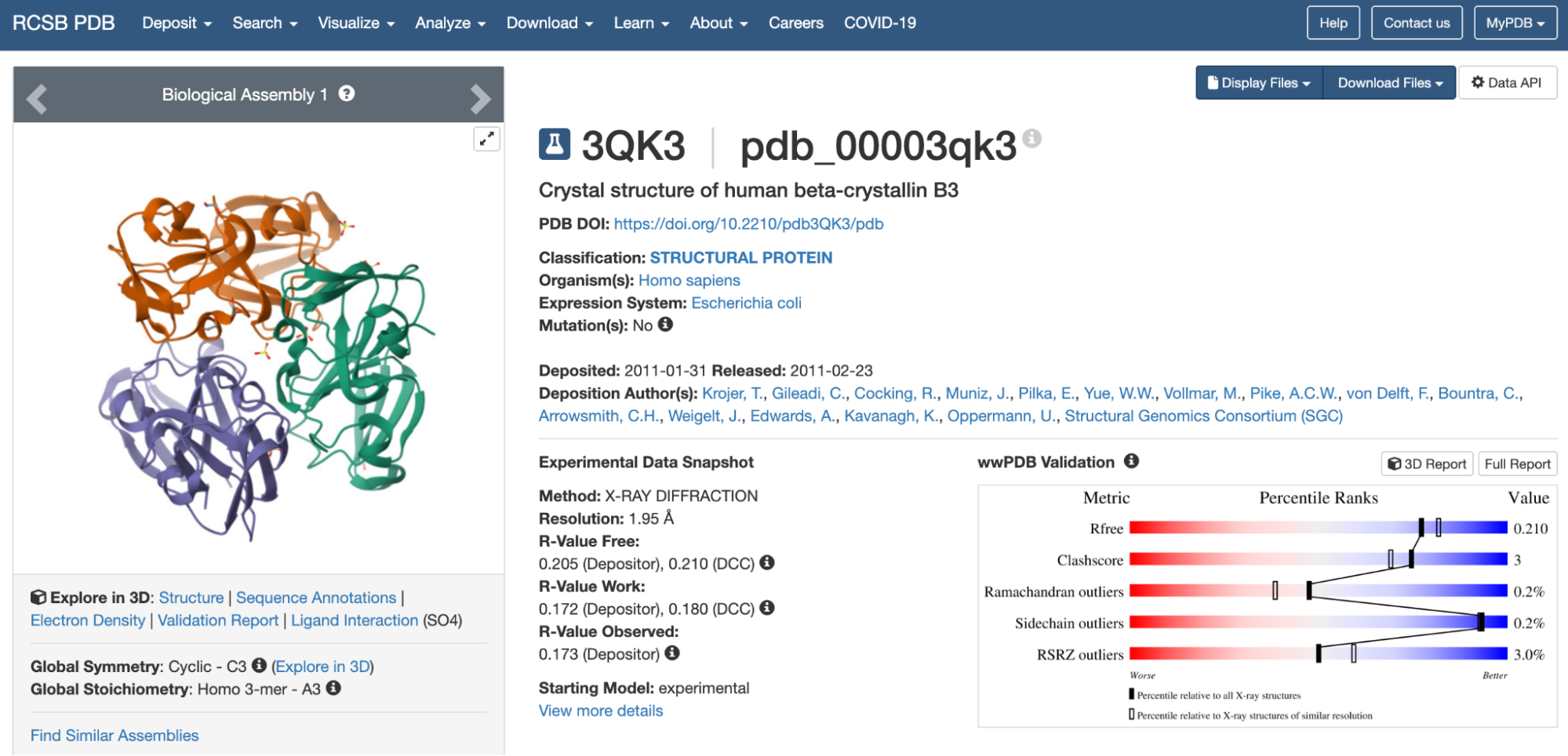 https://www.rcsb.org/structure/3QK3
https://www.rcsb.org/structure/3QK3
C1. Protein Language Modeling
1. Deep Mutational Scans
I looked for the protein in UniProt to see the sequence, and it appears with the ID 3QK3 .
The sequence for the protein human beta-crystallin B3 is the following:
MAEQHGAPEQAAAGKSHGDLGGSYKVILYELENFQGKRCELSAECPSLTDSLLEKVGSIQVESGPWLAFESRAFRGEQFVLEKGDYPRWDAWSNSRDSDSLLSLRPLNIDSPHHKLHLFENPAFSGRKMEIVDDDVPSLWAHGFQDRVASVRAINGTWVGYEFPGYRGRQYVFERGEYRHWNEWDASQPQLQSVRRIRDQKWHKRGRFPSS
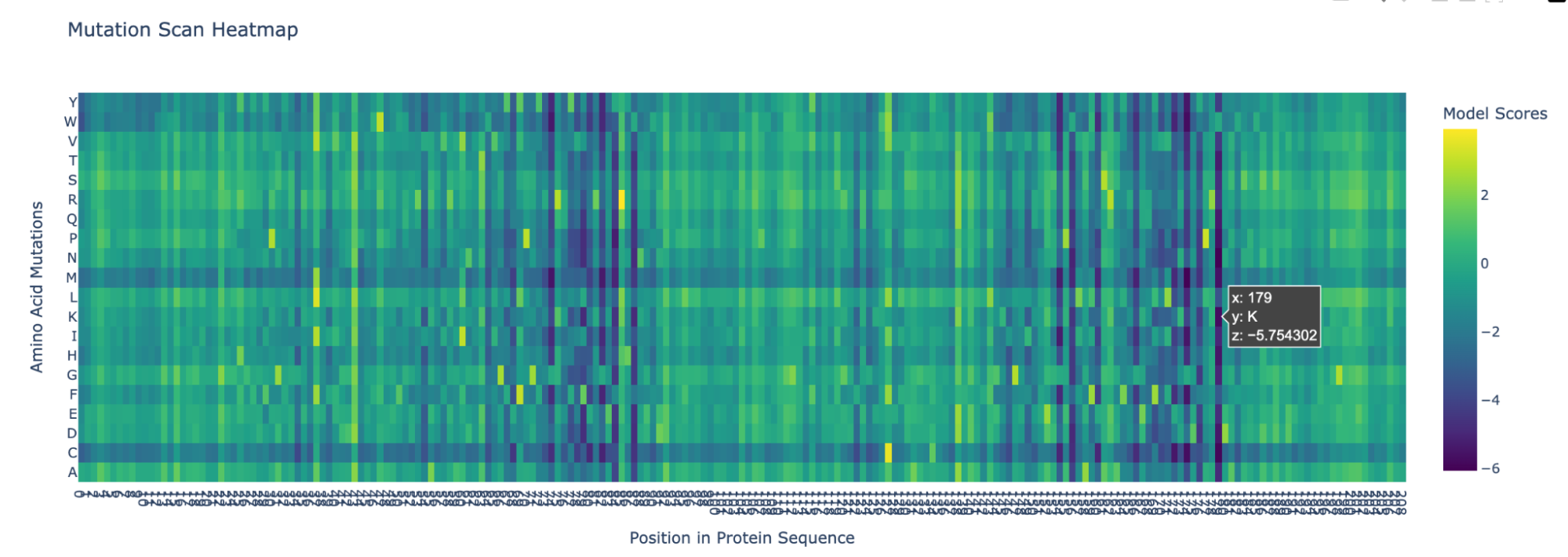
I pasted the sequence in the “Mutation Scans” section in the Google Colab, and run the code. This is the mutation scan heatmap obtained:
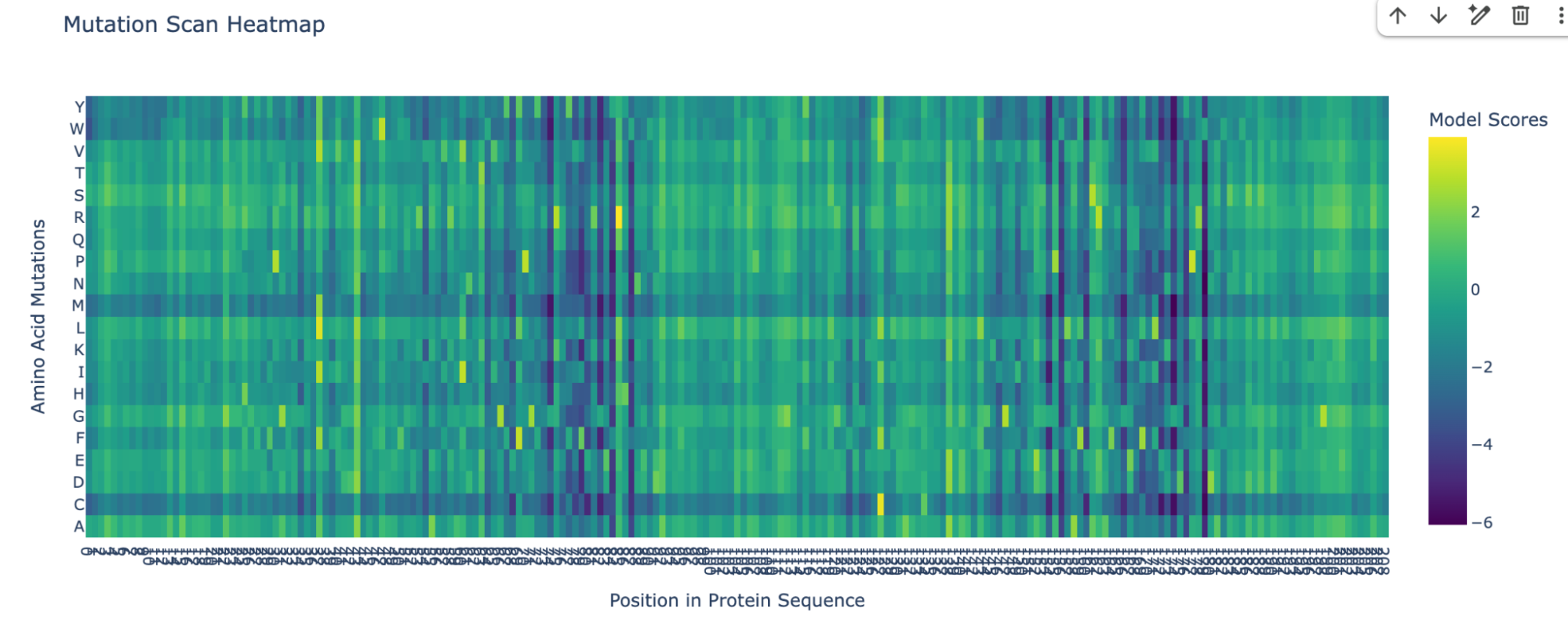
In the heatmap we can see that there are some vertical darker lines, meaning that position is important. For example, at position 74, the mutation of phenylalanine (F), tryptophan (W), methionine (M), isoleucine (I), and cysteine (C) shows dark purple, meaning this mutation would destabilize the protein because those amino acids are important for the protein’s core structure.
Here’s an example of how the methionine mutation is highlighted in position 74:
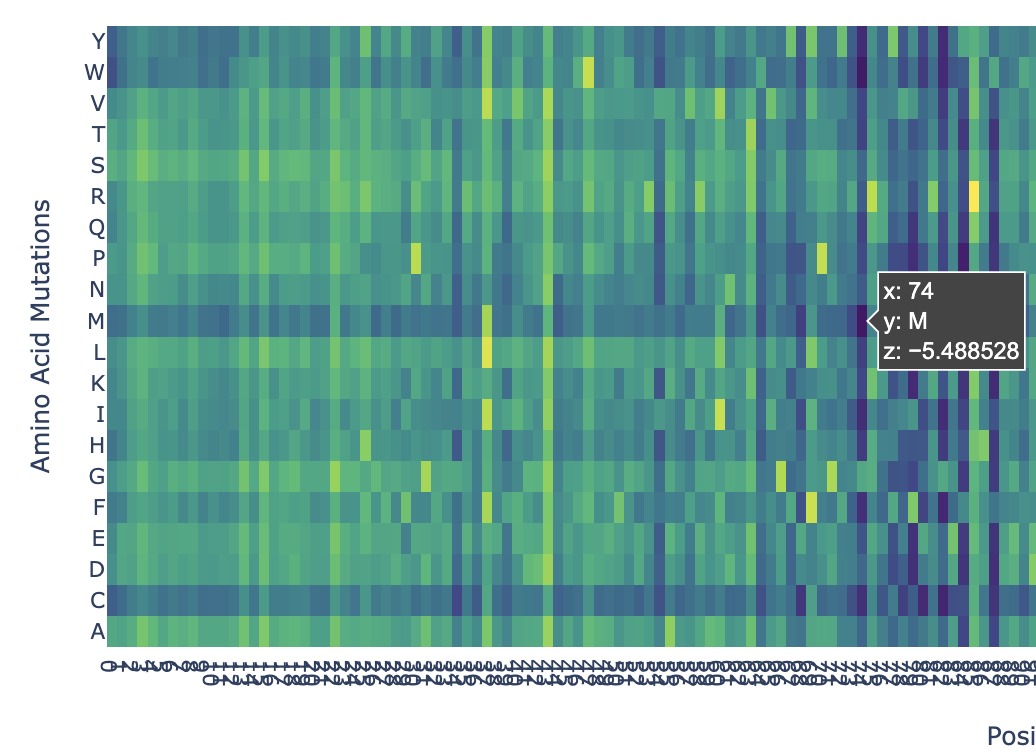
Position 179 also seems highly mutated:
2. Latent Space Analysis
I ran the code as is and this is the visualization for the TSNE scatter plot:
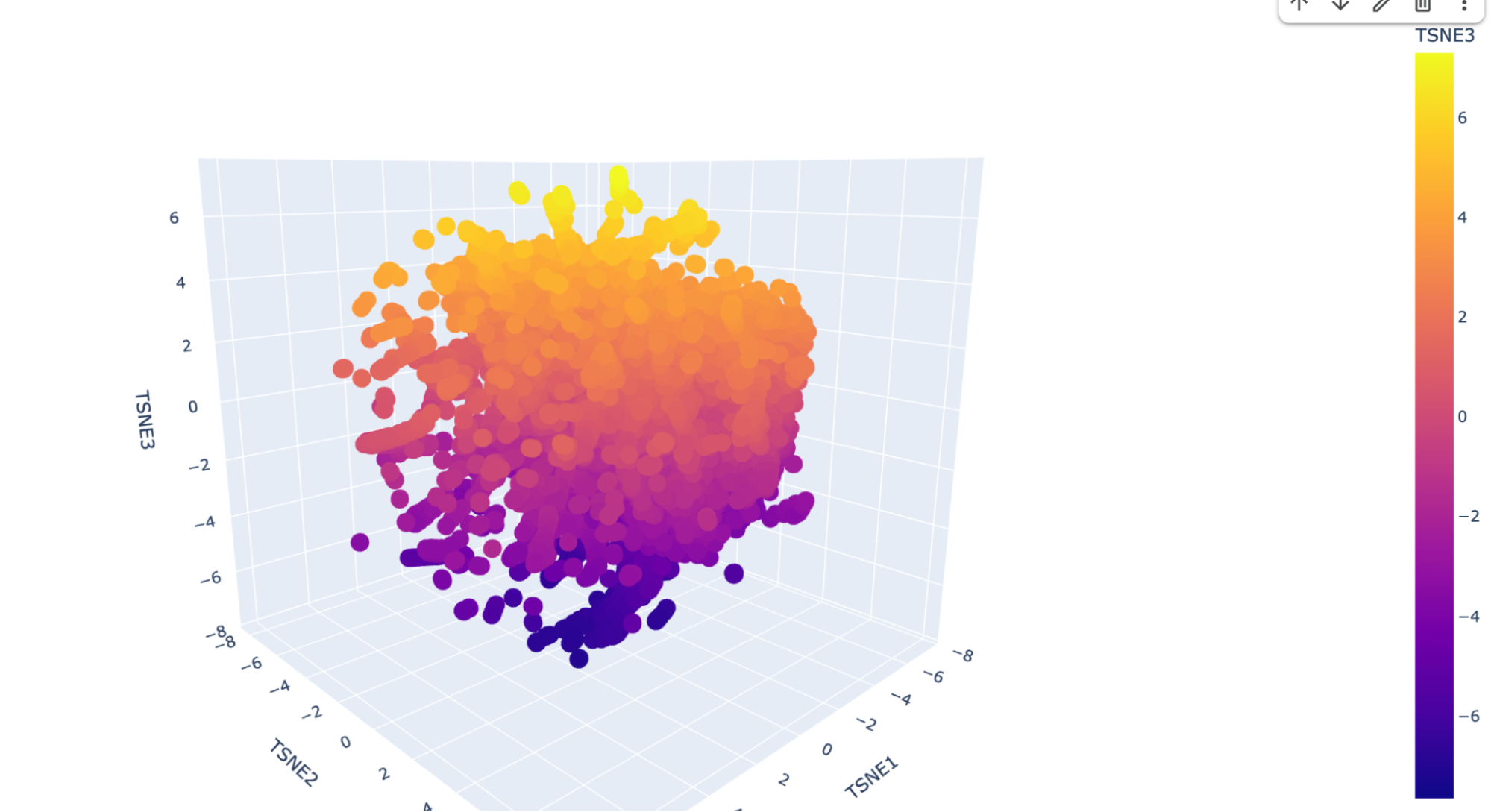
It has a lot of data, and is very difficult to see where my protein (beta-crystallin B3) is. I checked multiple dots but could not find it, so I decided to ask Deepseek AI the following prompt: “I have the following code to do a Latent Space Analysis, but when I visualize the 3D scatter plot there are a lot of dots. Is there a way to find my protein, which is the human beta-crystallin B3 (sequence: MAEQHGAPEQAAAGKSHGDLGGSYKVILYELENFQGKRCELSAECPSLTDSLLEKVGSIQVESGPWLAFESRAFRGEQFVLEKGDYPRWDAWSNSRDSDSLLSLRPLNIDSPHHKLHLFENPAFSGRKMEIVDDDVPSLWAHGFQDRVASVRAINGTWVGYEFPGYRGRQYVFERGEYRHWNEWDASQPQLQSVRRIRDQKWHKRGRFPSS) {pasted code from the Google Colab }” and it suggested to add a new code that highlights my protein. This is the code:
And here’s the scatter plot from that code:
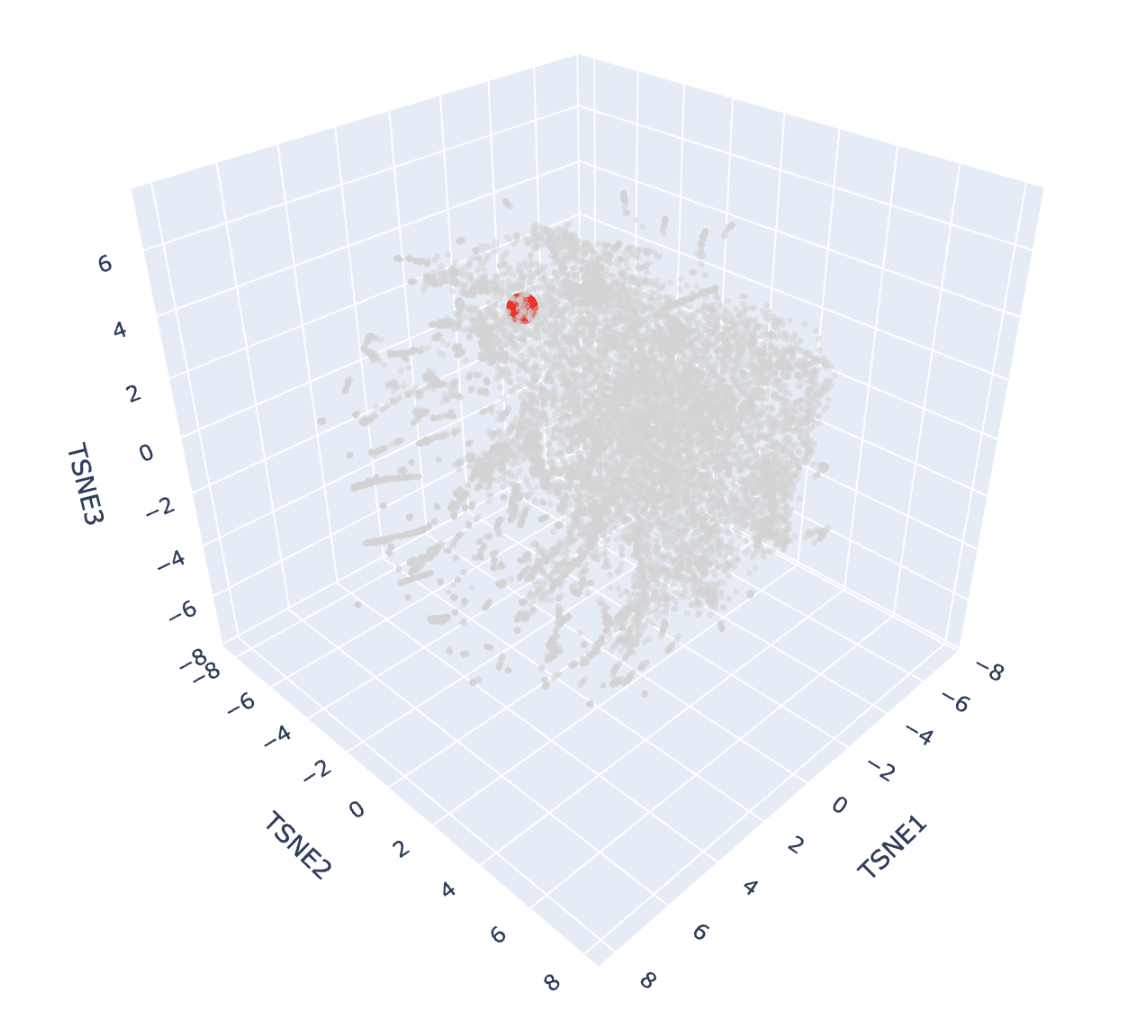
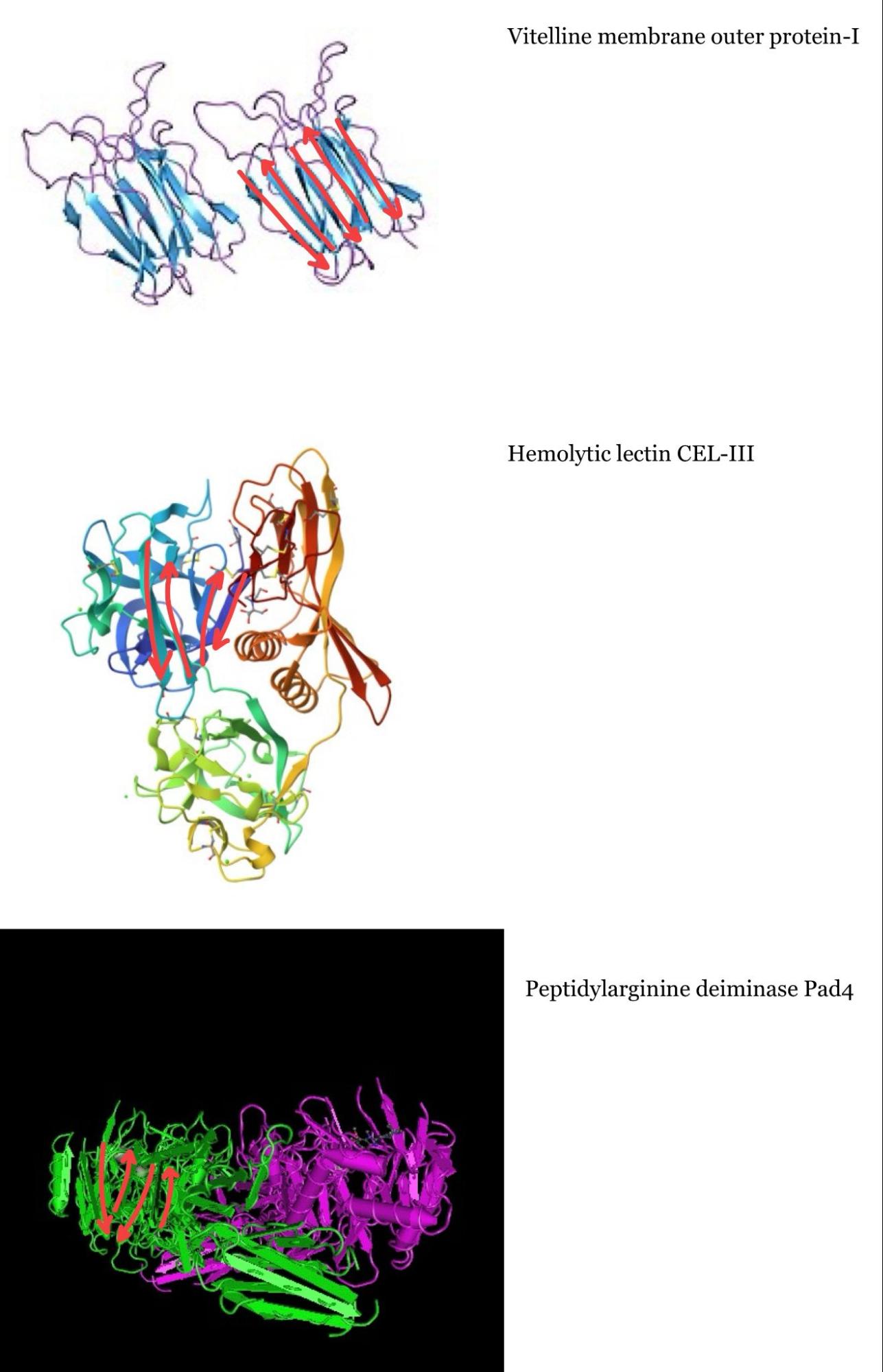
The protein is highlighted with the bright red dot. Thanks to this, I can now see better the proteins next to the one I picked. These are, for example: Vitelline membrane outer protein-I, a protein found on the vitelline membrane in a chicken egg; Hemolytic lectin CEL-III, a protein that binds to carbohydrates found in sea cucumbers; and Peptidylarginine deiminase Pad4, an enzyme that catalyzes post-translational modifications of proteins. It is interesting how different, at least in function, each of these proteins are with beta-crystallin B3. However, all of them have a structural similarity called the Greek key fold (see image below, taken from Piumetti, 2022).
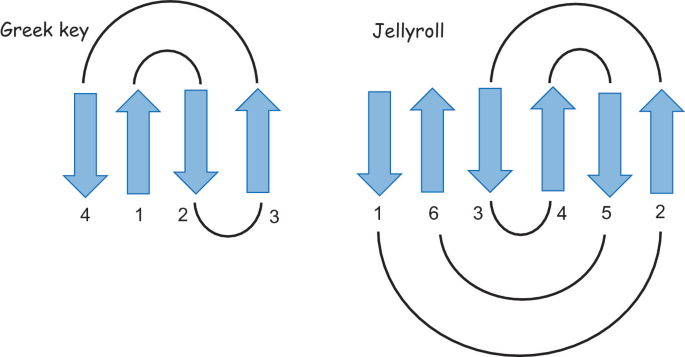
I tried to identify the Greek key fold in each of the three proteins similar to mine, but it was more challenging than I thought, so maybe my drawings are incorrect:
C2. Protein Folding
Folding a protein
1. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
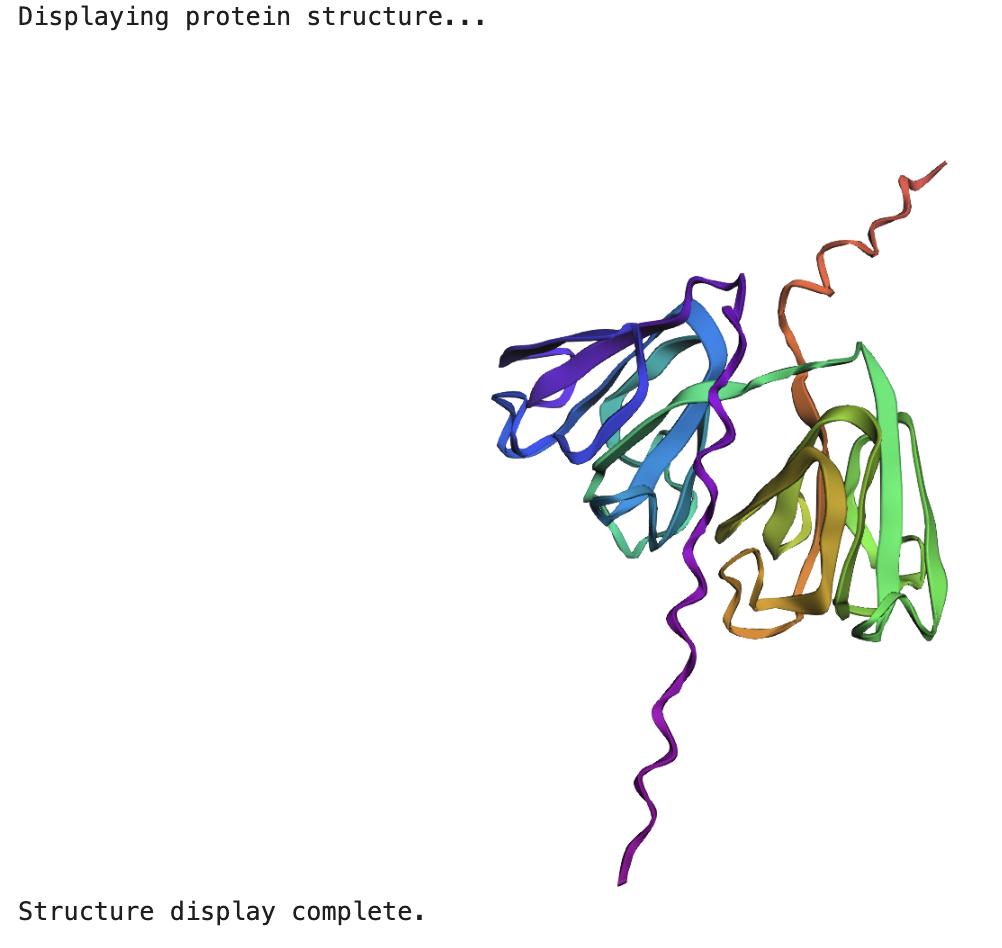
When running the ESMFold Protein Folding cells, this is the image for the protein:
And this is the 3D structure in RCSB:
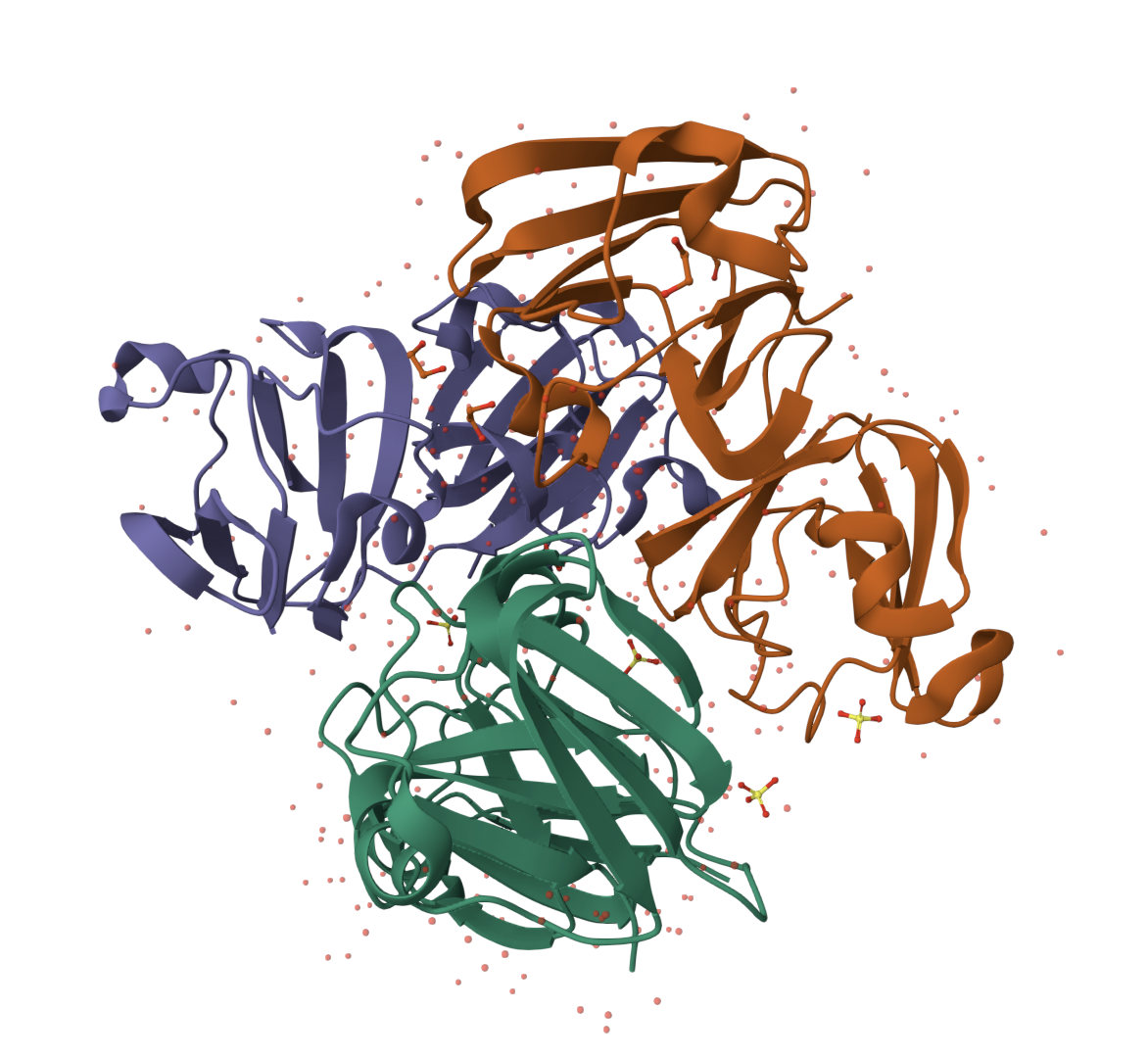
Both are different from each other. The experimental structure in RCSB (ID: 3QK3) contains 184 amino acids. However, the full human beta-crystallin B3 sequence from UniProt is 211 amino acids long.
I researched more about the actual structure of beta-crystallin B3, and it seems like it should have two beta-sheet domains. When I folded the complete sequence with ESMFold, the prediction shows the beta-crystallin B3 with those two beta-sheet domains. The first domain could match the 3QK3 structure, so the ESMFold model could be very accurate in this sense. The second domain is lacking, which could be due to the experimental approach in the RCSB structure, which is X-ray diffraction. Maybe the second domain was not achieved with the experimental approaches, only the first one crystallized successfully.
2. Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
Then, I tried to mutate the amino acid sequence by changing just one of them first. Here’s how the sequence starts: “MAEQHGAPEQAAAGKSH…” I changed it to this: “MAEQHGAPEQRAAGKSH…” So the alanine (A) in position 11 was changed to a arginine (R). The structure looks the same as before:
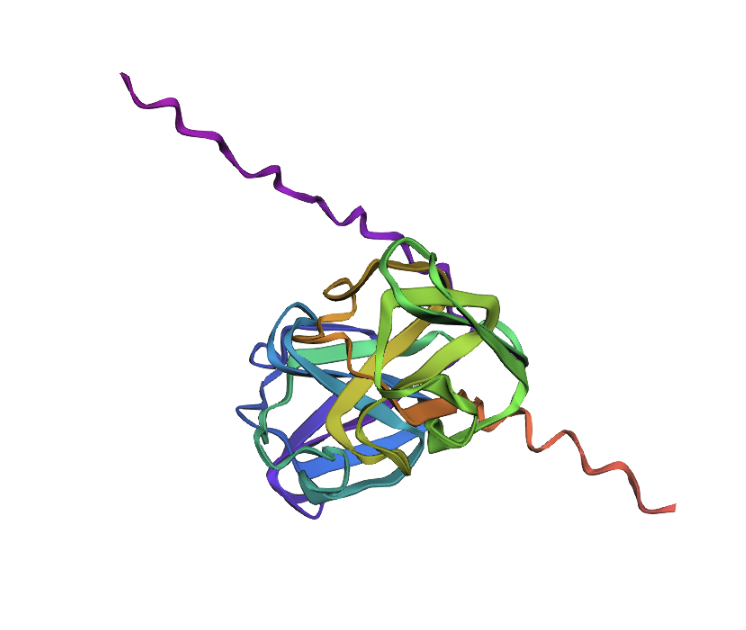
I now want to add another two mutations to have three total. This is the new mutated sequence: ““MAEQHGAPEQRNEGKSH…” and the structure still looks the same:
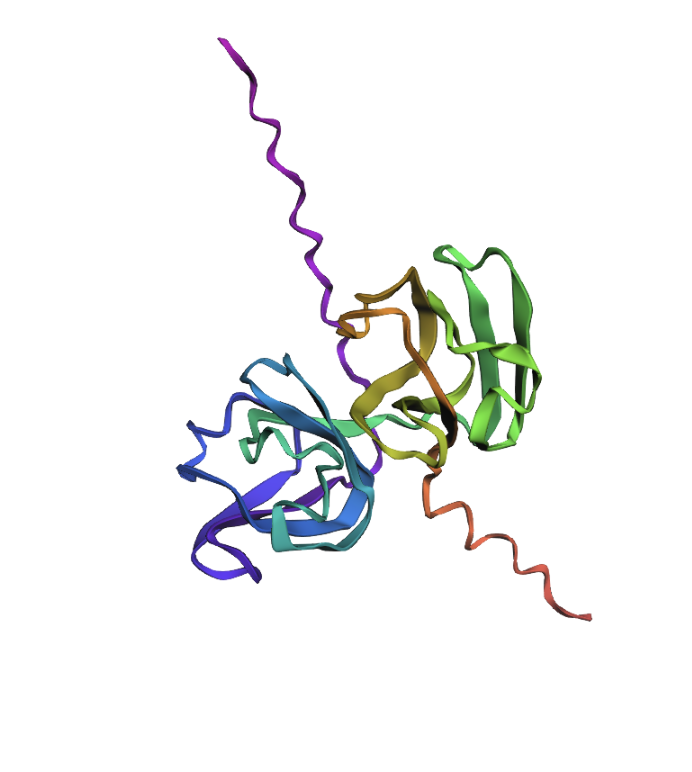
So I would conclude that the model was quite resilient to mutations (for these positions at least).
C3. Protein Generation
Inverse-Folding a protein:
Here are the input options I introduced in the Helper section of the code, which are identifiers and characteristics of my protein, human beta-crystallin B3:
This is the output:

1. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
For the original protein sequence the score is 1.5725, and for the new generated sequence the score is 0.8219, which is lower. Lower means the model is more confident in this new generated sequence. Also, the seq_recovery is at 0.4000 which means 40% of the amino acids match the original sequence.
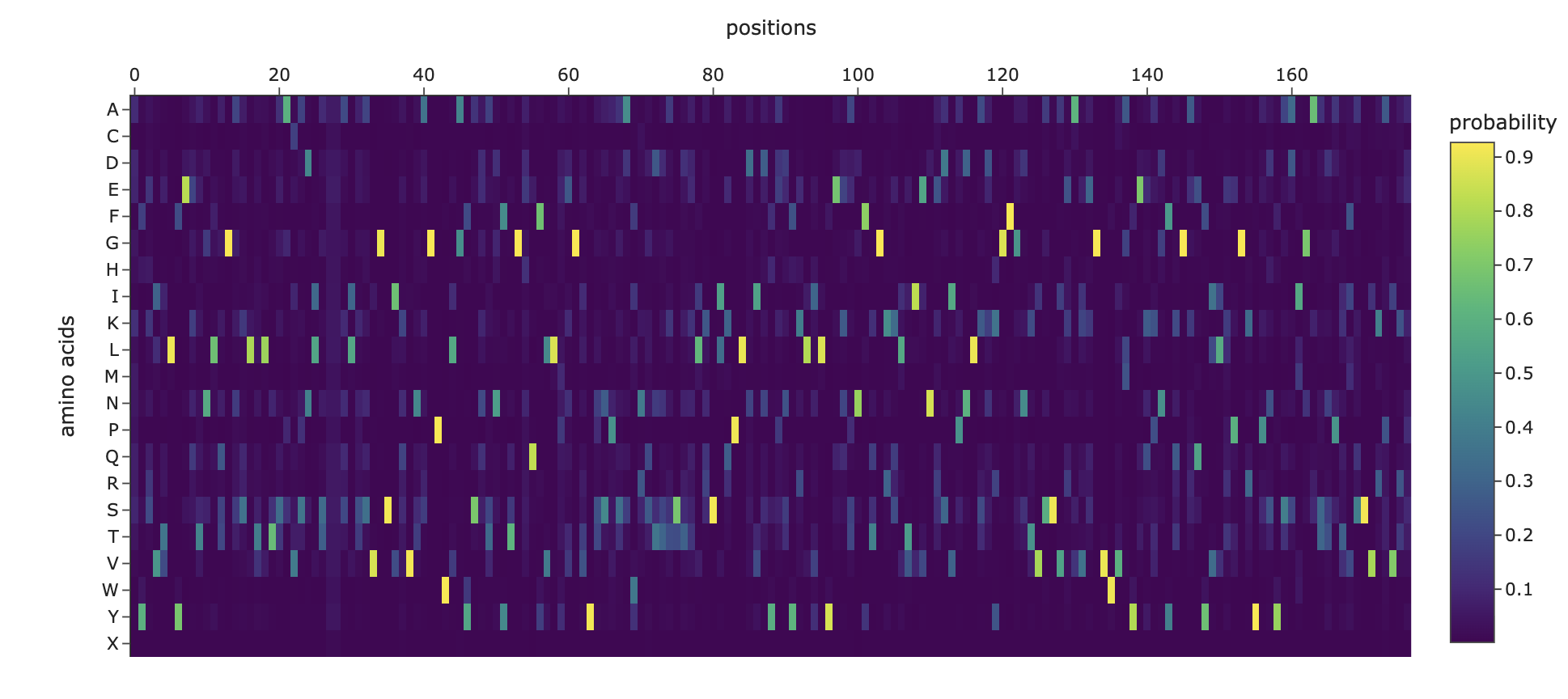
And this is the heat map for the amino acid probabilities:
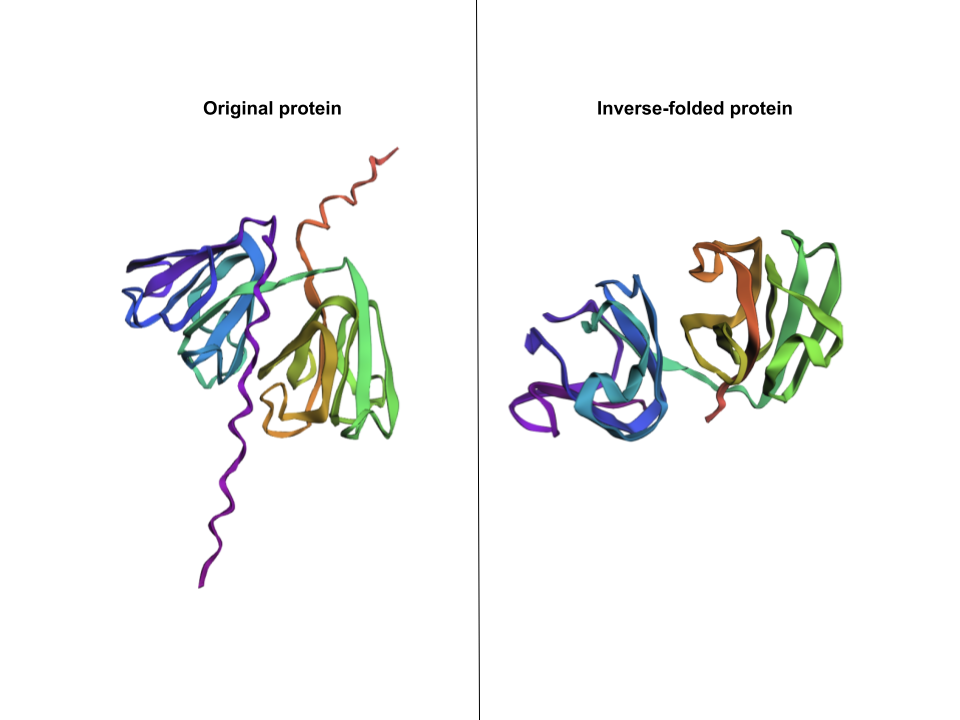
2. Input this sequence into ESMFold and compare the predicted structure to your original.
The new generated sequence is the following: “DYEIILYEKENLQGNSLTLTSAVSDLSXXKLSSVGSIKVVKGPWLAYSNKNYTGEQFVLPEGVYNSISDIRQDTSSTEIKSIKPLDIDYDTFELVLYEEENFQGKKLTIVNESVPNLADKGFGNTVSSAEAKKGVWVLYEKPNYQGRQFVLEPGKYPNYKDMGMSTPTVSSVKPVKK”
I pasted that one onto ESMFold and here’s the comparison: This structure does capture beta-crystallin B3’s two beta-sheet domains, but is missing the two coils that go through the protein.
References
CRYBB3 crystallin beta B3 [Homo sapiens (human)] - Gene - NCBI. (2026). Nih.gov. https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=1417
Piumetti, M. (2022). Structure of Proteins. In: Molecular Dynamics and Complexity in Catalysis and Biocatalysis. Springer, Cham. https://doi.org/10.1007/978-3-030-88500-7_1
Part D. Group Brainstorm on Bacteriophage Engineering
Proposal: Engineering the MS2 Lysis Protein L to Enhance Stability
Background
The MS2 bacteriophage lysis (L) is a 75 amino acid long-protein, and it is responsible for triggering host cell lysis, this is why it is also called a toxin from the group of bacteriophages (Mezhyrova, 2023). It is a powerful protein that has been used widely in studies where researchers seek to control cell death, but it is difficult to do so due to its instability (Mylon, 2010).
Objectives
To use computational protein design tools to engineer possible stable variants of the MS2 L-protein.
To identify variants where structure is preserved but higher stability is shown
Analyze if the variants can fold like the original protein and if they interact correctly with DnaJ, its chaperone
Methods
- Obtain an initial protein backbone for MS2 L-protein using ESMFold
- Obtain alternative sequences to the protein using ProteinMPNN
- Mutate the alternative sequences to the protein using ESM-2
- Model the variants using ESMFold and analyze if the folding is maintained in comparison to the initial protein (MS2 L-protein). Create a ranking based on confidence metrics
- Assess the top 3 variant’s interactions with DnaJ using AlphaFold-Multimer to predict 3D structures of protein complexes (co-folding multiple chains)
Expected Outcomes
Hopefully, these methods are able to identify some stabilized MS2 L-protein variants with correct folding and interaction with its chaperone, DnaJ. If successful, these designs could serve as templates for further experimental testing in E. coli and provide a methodology adaptable to other phage‑derived membrane proteins that also show decreased stability.
Potential Challenges
The main concern would be generating variants with correct folding, but incorrect interaction with DnaJ, as computational models sometimes can not predict the dynamics of those interactions (Chamakura et al., 2017 & Mondal et al., 2024), thus disrupting the lysis mechanism.
References
Mezhyrova, J., Martin, J., Börnsen, C., Dötsch, V., Frangakis, A. S., Morgner, N., & Bernhard, F. (2023). In vitro characterization of the phage lysis protein MS2-L. Microbiome Research Reports, 2(4), 28.
Mylon, S. E., Rinciog, C. I., Schmidt, N., Gutierrez, L., Wong, G. C., & Nguyen, T. H. (2010). Influence of salts and natural organic matter on the stability of bacteriophage MS2. Langmuir, 26(2), 1035-1042.
Chamakura, K. R., Tran, J. S., & Young, R. (2017). MS2 lysis of Escherichia coli depends on host chaperone DnaJ. Journal of Bacteriology, 199(12), 10-1128.
Mondal, A., Singh, B., Felkner, R. H., De Falco, A., Swapna, G. V. T., Montelione, G. T., … & Perez, A. (2024). A Computational Pipeline for Accurate Prioritization of Protein‐Protein Binding Candidates in High‐Throughput Protein Libraries. Angewandte Chemie International Edition, 63(24), e202405767.